

Abstract
Human adenovirus (Ad) serotypes Ad3, Ad7, Ad11, and Ad14, as well as a recently emerged strain of Ad14 (Ad14p1), use the epithelial junction protein desmoglein 2 (DSG2) as a receptor for infection. Unlike Ad interaction with CAR and CD46, structural details for Ad binding to DSG2 are still elusive. Using an approach based on Escherichia coli expression libraries of random Ad3 and Ad14p1 fiber knob mutants, we identified amino acid residues that, when mutated individually, ablated or reduced Ad knob binding to DSG2. These residues formed three clusters inside one groove at the extreme distal end of the fiber knob. The Ad3 fiber knob mutant library was also used to identify variants with increased affinity to DSG2. We found a number of mutations within or near the EF loop of the Ad3 knob that resulted in affinities to DSG2 that were several orders of magnitude higher than those to the wild-type Ad3 knob. Crystal structure analysis of one of the mutants showed that the introduced mutations make the EF loop more flexible, which might facilitate the interaction with DSG2. Our findings have practical relevance for cancer therapy. We have recently reported that an Ad3 fiber knob-containing recombinant protein (JO-1) is able to trigger opening of junctions between epithelial cancer cells which, in turn, greatly improved the intratumoral penetration and efficacy of therapeutic agents (I. Beyer, et al., Clin. Cancer Res. 18:3340–3351, 2012; I. Beyer, et al., Cancer Res. 71:7080–7090, 2011). Here, we show that affinity-enhanced versions of JO-1 are therapeutically more potent than the parental protein in a series of cancer models.
INTRODUCTION
We recently identified DSG2 as the main receptor for a group of species B adenoviruses, including adenovirus serotype 3 (Ad3), a serotype which is widely distributed in the human population (1). We found that the DSG2-interacting domain(s) within Ad3 is formed by several fiber knobs (2). This specific mode of Ad3 fiber knob-DSG2 interaction provides a high avidity and is functionally relevant for opening of epithelial junctions (1, 2). The latter involves clustering of DSG2 and activation of pathways that are reminiscent of an epithelial-to-mesenchymal transition, including the phosphorylation of mitogen-activated protein kinase (MAPK) and the downregulation of junction protein expression (1, 3, 4). The ability to open epithelial junctions appears to be important for Ad3 penetration into and spread within airway epithelial cells (1, 2, 4). In a recent study, we attempted to find the minimal moiety within the Ad3 capsid that confers efficient binding to DSG2 (2). We generated a small recombinant protein which contains the Ad3 fiber knob and a domain that allows for the self-dimerization of trimeric Ad3 fiber knobs (JO-1) (2). JO-1 can be readily produced in Escherichia coli and purified by affinity chromatography. In polarized epithelial cell cultures, JO-1 triggered the opening of intercellular junctions, while intravenous injection of JO-1 into mice with epithelial tumors allowed for better penetration of anti-cancer drugs (3, 5).
The first goal of the present study was to further delineate structural features of the Ad3 fiber knob-DSG2 interaction. This included identifying the amino acid residues within the Ad3 fiber knob that are involved in binding to DSG2 and creating JO-1 mutants with reduced and ablated binding to DSG2. The second goal of this study, which has translational relevance, was to further improve JO-1 by enhancing its affinity to DSG2, thereby increasing its therapeutic effect. This was done by identifying mutants with increased binding to DSG2.
Both goals were achieved using an E. coli expression library of Ad3 fiber knob mutants. We have identified residues in three different clusters within the Ad3 fiber knob that are critically involved in binding to DSG2. All residues are localized within one groove at the distal end of the fiber knob facing the receptor. We then assessed the effect of these mutations on the fiber knob's ability to open epithelial junctions by measuring the transepithelial electrical resistance in polarized epithelial cells in vitro and the ability to enhance the efficacy of a chemotherapy drug in mice with epithelial xenograft tumors. As expected, when mutations with reduced affinity to DSG2 were introduced into JO-1, the resulting proteins were less capable of opening epithelial junctions. On the other hand, a number of mutations that increased the affinity of JO-1 to DSG2 displayed a stronger activity in opening of epithelial junctions. Overall, these studies indicate a correlation between the affinity of Ad3 fiber knobs to DSG2 and subsequent effects on epithelial junctions.
The third goal of this study was to delineate the DSG2-interacting fiber knob residues of another DSG2-targeting Ad serotype, the newly emerged strain Ad14p1 (6), which is considered to be more pathogenic/virulent than the parental strain (Ad14-deWit) (7–9). The beta sheet distribution of Ad14p1 differs from that of Ad3, which could result in differences in the mode of DSG2 binding. Therefore, we generated an E. coli expression library of Ad14p1 fiber knob mutants to identify the DSG2-interacting residues of Ad14p1.
MATERIALS AND METHODS
Proteins.
Recombinant human DSG2 (hDSG2) protein was from Leinco Technologies, Inc. (St. Louis, MO). The Ad3 fiber knob was derived from Ad3 virus, GB strain, obtained from the ATCC. The Ad14p1 fiber knob is derived from Ad14p1 virus, strain Portland 2971/2007, provided by the Centers for Disease Control and Prevention (Atlanta, GA) (6). The fiber knobs were produced in E. coli with N-terminal 6-His tags using the pQE30 expression vector (Qiagen, Valencia, CA) and purified by nickel-nitrilotriacetic acid (Ni-NTA) agarose chromatography as described elsewhere (10).
Cell lines.
293, HeLa, and A549 cells were maintained in Dulbecco's modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum (FBS), a mix of 100 U/ml penicillin and 100 μg/ml streptomycin (P/S), 2 mM glutamine (Glu), and 1× MEM nonessential amino acid solution (Invitrogen, Carlsbad, CA). Colon cancer T84 cells (ATCC CCL-248) were cultured in a 1:1 mixture of Ham's F12 medium and DMEM, 10% FBS, Glu, and P/S. Ovc316 cells are Her2/neu-positive epithelial tumor cells derived from an ovarian cancer biopsy specimen (11). Ovc316 cells were cultured in mammary epithelial cell growth medium (MEGM) containing 3 μg/liter human epidermal growth factor (hEGF), 5 μg/liter insulin, 5 mg/liter hydrocortisone, 26 mg/liter bovine pituitary extract, 25 mg/liter amphotericin B (Lonza, Mapleton, IL), 1% FBS, 100 IU penicillin, 100 μg/liter streptomycin, and 10 mg/liter ciprofloxacin. MDA-MB-231 cells, a triple-negative breast cancer cell line (ATCC-HTB-26), were cultured in Leibovitz's L-15 medium supplemented with 10% FBS, 100 IU penicillin, and 100 μg/liter streptomycin. TC1-DSG2 cells were derived from TC1 cells, a C57BL/6 lung cancer cell line that expresses HPV16 E6 and E7 (12). TC1 cells were transduced with a vesicular stomatitis virus protein G (VSV-G)-pseudotyped lentivirus vector expressing human DSG2 (1). A clone that expressed human DSG2 at a level seen in human tumors was selected for in vivo studies.
Adenoviruses.
Propagation, [methyl-3H]thymidine labeling, purification, and titer determination for wild-type Ad3 (wt Ad3) was performed as described elsewhere (13). Ad3-GFP is a wild-type Ad3-based vector containing a cytomegalovirus-green fluorescent protein (CMV-GFP) expression cassette inserted into the E3 region (1). Viral particle (VP) concentrations were determined spectrophotometrically by measuring the optical density at 260 nm (OD260). Titers of PFU were performed using 293 cells as described elsewhere (14). The VP/PFU ratio was 20:1 for all virus preparations.
Ad3 knob library.
The coding sequence of the Ad3 knob (amino acids [aa] 108 to 319) containing the last two shaft repeats was obtained by PCR from Ad3 DNA using primers P1 (5′ATCACGGATCCGGTGGCGGTTCTGGCGGTGGCTCCGGTGGCGGTTCTAACAAACTTTGCAGTAAACTC 3′) and P2 (5′CTCAGCTAATTAAGCTTAGTCATCTTCTCTAATATAGGA3′) and cloned into pQE30 (Qiagen, Valencia, CA) for expression in E. coli. The resulting plasmid was called pQE-Ad3knob. Random mutagenic PCR was performed based on a protocol published elsewhere (15, 16). Briefly, 20 fmol pQE-Ad3knob DNA template; 30 pmol (each) PCR primers (Pmut1, 5′-CCAATTCTATTGCACTTAAGAATAACACTTTATGGACAGGT-3′; Pmut2, 5′-GTCCAAGCTCAGCTAATTAAGCTTAGTCATCTTC-3′); 2.5 μl, 3.5, 5, or 10 μl of 10× mutagenic buffer (70 mM MgCl2, 500 mM KCl, 100 mM Tris [pH 8.3 at 25°C], 0.1% [wt/vol] gelatin); 10 μl 5 mM MnCl2; 10 μl deoxynucleoside triphosphate (dNTP) mix (2 mM dGTP, 2 mM dATP, 10 mM dCTP, 10 mM dTTP); and 5 U of Taq polymerase (Promega, Madison, WI) were mixed in a final volume of 100 μl. PCR conditions were 94°C for 1 min, 45°C for 1 min, and 72°C for 1 min (30 cycles). The mutant PCR products (615 bp in length containing mutations only in the reading frame of the fiber knob head) were purified, digested with appropriate enzymes, and cloned into the plasmid pQE-Ad3knob. For quality control of the random mutagenic library, the ligation product was transformed into E. coli M15 (Qiagen, Valencia, CA) and plated on kanamycin and ampicillin plates, and 50 colonies were randomly picked for sequencing.
Ad14 library.
The coding sequence of the Ad14p1 knob (aa 108 to 323) containing the last two shaft repeats was obtained by PCR from Ad14p1 DNA using primers P1 (5′CATCACGGATCCGGTGGCGGTTCTGGCGGTGGCTCCGGTGGCGGTTCTAATAAACTTTGTACCAAATTGGGAGAAGG 3′) and P2 (5′ GCTAATTAAGCTTAGTCGTCTTCTCTGATGTAGTAAAAGG 3′) and cloned into pQE30 (Qiagen, Valencia, CA) for expression in E. coli. The resulting plasmid was called pQE-Ad14p1knob. Random mutagenic PCR was performed by using PCR primers (Pmut1, 5′-AACACCCTGTGGACAGGAGTTAACCC−3′; Pmut2, 5′-CTCAGCTAATTAAGCTTAGTCGTC-3′). The mutant PCR products (594 bp in length, containing mutations only in the reading frame of the fiber knob head) were purified, digested with appropriate enzymes, and cloned into the plasmid pQE-Ad14p1knob. For quality control of the random mutagenic library, the ligation product was transformed into E. coli M15 (Qiagen, Valencia, CA) and plated on kanamycin and ampicillin plates, and 50 colonies were randomly picked for sequencing.
Colony assays.
The Ad3 or Ad14p1 knob mutant plasmid library was transformed into XL-1 Blue or M15 E. coli host strains and plated on LB plates with appropriate antibiotics, i.e., ampicillin or ampicillin and kanamycin, respectively. After overnight growth, a 0.45-μm Durapore filter membrane (Millipore, Billerica, MA) was placed on top of the colonies. The membrane was peeled off and placed carefully, with the colonies facing upwards, on two sheets of Whatman 3MM paper soaked in LB medium supplemented with antibiotics and 1 mM isopropyl-β-d-thiogalactopyranoside (IPTG). Protein expression of the colonies was induced for 6 h at 30°C, after which the filter with the colonies was placed on top of a nitrocellulose filter and Whatman 3MM paper soaked in native lysis buffer (20 mM Tris-Cl [pH 8], 300 mM NaCl, 50 mM MgCl2, 0.1 mg/ml lysozyme, 0.75 mg/ml DNase I, and half of a Complete EDTA-free protease inhibitor cocktail tablet/10 ml [Roche, Palo Alto, CA]). The filter sandwich was incubated at room temperature for 10 min and then freeze-thawed 4 times for 10 min at −80°C and 10 min at 30°C. The nitrocellulose membrane was removed from the sandwich and blocked with 3% bovine serum albumin (BSA) in Tris-buffered saline-Tween 20 (TBST) at 4°C overnight. The blot was then incubated with 0.1 ng/ml of recombinant DSG2 protein (Leinco, St. Louis, MO) in TBST-BSA, followed by the addition of mouse anti-DSG2 monoclonal antibodies (MAbs) (clone 6D8; SeroTec Ltd., Oxford, United Kingdom) and anti-mouse IgG horseradish peroxidase (HRP) conjugate. Colonies without DSG2 binding were picked and cultured in 3 ml LB medium overnight. Protein expression was induced with 1 mM IPTG for 5 h, and the bacteria were then pelleted, resuspended in SDS loading buffer, and freeze-thawed 3 times. After electrophoresis, proteins were transferred to nitrocellulose and incubated with anti-His antibodies (MCA1396; Sertec) to assess Ad knob trimerization. To screen for mutants with stronger binding to DSG2, the Ad3 knob mutant library was transformed into the M15 E. coli host strain. Protein expression of the colonies was induced for only 20 min at room temperature. The colonies that showed the most intense DSG2 binding signal were picked.
Western blotting.
Mini-Protean precast gels (Bio-Rad, Hercules, CA) with 4 to 15% gradient polyacrylamide were used. A total of 1 μg protein mixed with 2× loading buffer (10 mM Tris-HCl, pH 6.8, 200 mM dithiothreitol [DTT], 4% SDS, 20% glycerol, 0.2% bromophenol blue) was loaded per lane. Samples were either boiled (B) for 5 min or loaded unboiled (UB). The following running buffer was used: 25 mM Tris, pH 8.3, 0.192 M glycine, 0.1% SDS. After electrophoresis, proteins were transferred to nitrocellulose and incubated with recombinant human DSG2 protein and anti-DSG2 antibodies as described previously (1). The Western blots were scanned and quantified using ImageJ 1.32 software (National Institutes of Health, Bethesda, MD). JO-1 band intensity was set as 100%. For analysis of MAPK activity, polarized T84 cultures were lysed in 20 mM HEPES (pH 7.5), 2 mM EGTA, 10% glycerol, 1% Triton X-100, 1 mM phenylmethylsulfonyl fluoride (PMSF), 200 μM Na3VO4, and protease inhibitors on ice. After sonication, samples were pelleted and protein containing supernatant stored at −80°C. Fifteen μg of total protein was used for Western blotting with MAb against phospho-p44/42 MAPK (Erk1/2; Thr202/Tyr204) (Cell Signaling Danvers, MA) or MAb against mouse anti-Erk1/2 (Cell Signaling).
Competition assays.
HeLa cells were detached from culture dishes by incubation with Versene and washed with phosphate-buffered saline (PBS). A total of 105 cells per tube were resuspended in 50 μl of ice-cold adhesion buffer (DMEM supplemented with 2 mM MgCl2, 1% FBS, and 20 mM HEPES) containing different concentrations of Ad3 fiber knob protein and incubated on ice for 1 h. 3H-labeled wild-type Ad3 virus then was added in adhesion buffer at a multiplicity of infection (MOI) of 8,000 VP per cell to a final volume of 100 μl. After 1 h of incubation on ice, cells were pelleted and washed twice with 0.5 ml of ice-cold PBS. After the last wash, the supernatant was removed and the cell-associated radioactivity was determined by a scintillation counter. The number of VP bound per cell was calculated by using the virion-specific radioactivity and the number of cells.
Surface plasmon resonance.
Acquisitions were done on a Biacore 3000 instrument. HBS-N (GE Healthcare, Pittsburgh, PA) supplemented with 2 mM CaCl2 was used as the running buffer in all experiments at a flow rate of 5 μl/min. Immobilization on a CM4 sensorchip (Biacore) was performed using DSG2 at 10 μg/ml diluted in 10 mM acetate buffer, pH 4.5, injected for 10 min on an ethyl(dimethylaminopropyl) carbodiimide-N-hydroxysuccinimide (EDC-NHS)-activated flow cell. A control flow cell was activated by EDC-NHS and inactivated by ethanolamine. Different concentrations of Ad3 fiber knob proteins were injected for a 3-min association time followed by a 2.5-min dissociation time, and the signal was automatically subtracted from the background of the ethanolamine-deactivated EDC-NHS flow cell. Kinetic and affinity constants were calculated using BIAeval software.
Crystallography.
Crystallization conditions for wt Ad3 and K217E/F224S knob mutants were from the service of the High-Throughput Screening Laboratory at Hauptman Woodward Medical Research Institute. For diffraction studies, wt Ad3 and the K217E/F224S knob mutant were crystallized using the hanging-drop method. Crystals were grown using a reservoir solution of 1.65 M MgSO4(7H2O) in 0.1 M TAPS buffer, pH 9.0, and a protein solution of 15 mg/ml. Crystals were frozen using a cryoprotectant composed of 85% reservoir and 15% glycerol (vol/vol). Data collection was performed at 100 K on ID14-4 of the ESRF using the EDNA pipeline (17). Data were indexed and scaled using XDS/XSCALE (18, 19), and the structure was solved by molecular replacement (Protein Data Bank [PDB] code 1H7Z) with the program PHASER (20). The model was built and refined using COOT (21) and PHENIX (22), respectively (Table 1). The structure of the adenovirus 3 knob domain K217E and F224S mutant has been assigned the RCSB ID code rcsb080687 and PDB code 4LIY.
Table 1.
Data collection and refinement statistics
| Parameter | Value for Ad3 knoba (K217E/F224S mutant) |
|---|---|
| Data collection statistics | |
| Wavelength (Å) | 0.972 |
| Resolution range (Å) | 48.0–2.1 (2.175–2.1) |
| Space group | P3221 |
| Unit cell dimensions | |
| a, b, c (Å) | 96.663, 96.663, 156.399 |
| α = β (°) | 90 |
| γ (°) | 120 |
| Total no. of reflections | 222,816 (21,396) |
| No. of unique reflections | 49,784 (4,831) |
| Multiplicityf | 4.5 (4.4) |
| Completeness (%) | 99.61 (98.96) |
| Avg I/σ(I) | 11.46 (1.89) |
| Wilson B-factor (Å2) | 40.48 |
| Rmerge (%) | 0.07161 (0.6146) |
| Rmeas (%) | 0.08092 (0.72) |
| Refinement statistics | |
| CC1/2c | 0.998 (0.801) |
| CC*d | 1 (0.943) |
| Rwork (%) | 0.1759 (0.2670) |
| Rfree (%) | 0.2012 (0.3133) |
| No. of atoms: | |
| Macromolecules | 4,310 |
| Ligands | 5 |
| Water molecules | 272 |
| Protein residues | 553 |
| Total | 4,587 |
| RMSDb | |
| Bond length (Å) | 0.011 |
| Bond angle (°) | 1.27 |
| Ramachandran plot (%) | |
| Favored regions | 96 |
| Outliers | 0 |
| Clashscoree | 4.58 |
| B-factor | |
| Avg | 50.0 |
| Macromolecules | 49.8 |
| Ligands | 126.6 |
| Solvent | 51.0 |
Statistics for the highest-resolution shell are shown in parentheses.
RMSD, root mean square deviations.
CC(1/2) is the percentage of correlation between intensities from random half data sets.
CC* is the estimate of the correlation coefficient of the data to the true intensities.
The clashscore is the number of overlaps greater than 0.4 Å per 1,000 atoms.
Multiplicity is the average number of observations for each reflection.
Three-dimensional (3D) structure.
Pymol software was used to analyze the structure. Mutations in the Ad3 knob domain (PDB code 1H7Z) were stained using different colors on the purple isosurface. Monomers of Ad3 knob mutant K217E/F224S were drawn in colored cartoons with mutations in sticks and overlaid on the gray cartoon view of the wild-type Ad3 fiber knob.
Negative-stain electron microscopy.
Recombinant JO-2 protein was visualized by negative-stain EM to assess its assembly status. The standard mica-carbon preparation was used with protein at 0.1 mg/ml. Sample was stained using 1% (wt/vol) sodium silicotungstate (pH 7.0) and visualized on a JEOL-1200 electron microscope at 100 kV.
Permeability assay.
A total of 5 × 105 T84 cells were seeded in 12-mm transwell inserts (polyethylene terephthalate [PET] membrane with 0.4-μm pore size; Corning, NY) and cultured for >14 days until transepithelial electrical resistance (TEER) was stable. Culture medium was changed every 2 to 3 days. The cells were exposed to DSG2 ligands (20 μg/ml) in adhesion medium (DMEM, 1% FBS, 2 mM MgCl2, 20 mM HEPES) for 15 min at room temperature, and TEER was measured and calculated as described elsewhere (23).
Animal studies.
All experiments involving animals were conducted in accordance with the institutional guidelines set forth by the University of Washington. Mice were housed in specific-pathogen-free facilities. Immunodeficient (CB17) mice (strain NOD.CB17-Prkdcscid/J) were obtained from the Jackson Laboratory. Human DSG2 transgenic mice contain 90 kb of the hDSG2 locus and express hDSG2 at a level and in a pattern similar to that of humans (4).
A549, MDA-MB-231, and Ovc316 xenograft tumors were established by injection of the corresponding tumor cells into the mammary fat pad (1:1 with Matrigel) of CB17 mice. TC1-DSG2 tumors were established by subcutaneous injection of TC1-DSG2 cells into DSG2 transgenic mice. JO-0, JO-1, JO-2, or JO-4 was intravenously injected 1 h before the application of chemotherapeutic drugs: irinotecan (Camptosar; Pfizer Inc., Groton, CT), pegylated liposomal doxorubicin (Lipodox; Sun Pharmaceuticals IN, India), cetuximab (Erbitux; ImClone, Somerville, NJ), and nanoparticle albumin-conjugated paclitaxel (nab-paclitaxel; Abraxane; Abraxis Biosciences, Summit, NJ). Tumor volumes were measured three times a week. Each treatment group consisted of a minimum of 5 mice. Animals were sacrificed and the experiment terminated when tumors in one of the groups reached a volume of 800 mm3 or tumors displayed ulceration.
Anti-JO-4 antibodies.
Anti-JO-4 antibody concentrations in human serum samples were measured by enzyme-linked immunosorbent assay (ELISA). Plates were coated with rabbit polyclonal anti-Ad3 fiber antibodies (1), followed by recombinant JO-4, human serum samples (1:2 to 1:1,000 dilution), and anti-human IgG-HRP. Serum samples from ovarian cancer patients were provided by the Pacific Ovarian Cancer Research Consortium.
3D structure.
Pymol software was used to visualize the 3D structure of the Ad3 fiber knob (MMDB code 16945, PDB code 1H7Z) (24).
Statistical analysis.
All results are expressed as means ± standard deviations (SD). Two-way analysis of variance (ANOVA) for multiple testing was applied. Animal numbers and P values are indicated in the figure legends.
PDB accession number.
The Ad3 fiber knob domain of the K217E and F224S mutant was deposited in PDB under code 4LIY.
RESULTS
Residues critical for DSG2 binding.
We first focused our work on Ad3. High-affinity binding to DSG2 and subsequent epithelial junction opening requires several trimeric fiber knobs in a spatial constellation present in the virion, PtDd, or dimerized (trimeric) Ad3 fiber knob (e.g., JO-1) (2). The recombinant (trimeric) fiber knob with two shaft motifs but without the dimerization domain (Ad3 knob monomer) binds to DSG2 with an affinity that is orders of magnitude less than that of JO-1, is not able to block Ad3 infection, and does not trigger junction opening (1, 2, 13). However, the affinity of Ad3 knob monomer is high enough to detect binding in Western blot analyses in which soluble DSG2 is used as a probe. Therefore, we used an E. coli expression library of His-tagged Ad3 knob monomer mutants to identify the amino acid residues within the Ad3 fiber knob that are critical for DSG2 binding. To generate this library, we employed mutagenic PCR (15, 16) in a protocol that generated, on average, one to two amino acid substitutions per knob. The Ad3 fiber knob library in E. coli XL-1 Blue was plated on agar plates, knob expression was induced by IPTG, and colonies were screened for DSG2 binding using recombinant DSG2 and anti-DSG2 antibodies. A first screening round of ∼10,000 colonies for variants that did not bind to DSG2 revealed 240 candidate colonies. When analyzed by Western blotting for the 6×His tag, 40 of the 240 colonies showed expression of trimeric fiber knob, indicating the absence of major conformational changes. The remaining variants had truncated fiber knobs or did not form trimers. The corresponding 40 plasmids were sequenced. The vast majority of colonies had single-amino-acid substitutions within the fiber knob. If multiple amino acid substitutions per knob were encountered, new Ad3 knob genomes containing the corresponding mutations individually were synthesized. Further rounds of colony screening did not uncover other regions, indicating that all of the DSG2-interacting residues had been found. A total of 8 independent mutants then were used for subsequent studies (Fig. 1A). For all subsequent studies, we generated self-dimerizing forms of the Ad3 fiber mutants (Fig. 1B) and purified them by affinity chromatography using Ni-NTA columns. The purified dimerized knob mutants were analyzed for DSG2 binding by Western blotting (Fig. 1C to F). All mutants were severely reduced in binding compared to JO-1 (i.e., the dimerized form containing the wt Ad3 fiber knob). Mutants D261N and F265L were almost completely ablated for DSG2 binding, while the other mutants had different levels of residual DSG2 binding (3.6 to 20% of the wt Ad3 knob level) (Table 2). The residues identified as being critical for Ad3 knob binding to DSG2 were in three different areas of the Ad3 fiber knob within the CD-loop/D-beta sheet (N186D, V189G, and S190P), the FG-loop/G-beta sheets (D261N and F265L), and the H-beta sheet/HI-loop (L292A, L296R, and E299V) (Fig. 1A). In a 3D model of the Ad3 fiber knob (PDB accession number 1H7Z_A), all identified residues were located at the apical side of the fiber knob and followed one specific groove in the knob (Fig. 2). It is noteworthy that, except for F265L, the substitutions resulted in a change of charge in the corresponding residue. Differences in migrations patterns in polyacrylamide gels (for example, that for D261N) indicate that the substitutions have caused conformational changes. We are currently attempting to crystallize these mutants to analyze their 3D structure.
Fig 1.

Residues found to be critically involved in binding to DSG2. (A) Shown are the amino acid sequences of the Ad3 and Ad14p1 fiber knob. Beta sheets present in the Ad3 knob (PDB code 1H7Z_A) and Ad14 knob (PDB code 3F0Y_A) are indicated by blue lines. Black arrows indicate residues within the Ad3 fiber knob which, when mutated individually, ablate or reduce binding to DSG2. Compared to the parental strain of Ad14 (deWit), Ad14p1 had a deletion of two amino acid residues within the FG loop of the fiber protein knob (45), indicated by a red triangle. (B) Schematic structure of dimeric Ad3 fiber knob mutants. The fiber knob domain and one shaft motif was fused through a flexible linker to a homodimerizing K-coil domain (2). The proteins are self-dimerizing and can be purified by His-Ni-NTA affinity chromatography. (C to F) Analysis of binding of dimeric Ad3 fiber knob mutants to soluble DSG2. (C and D) Coomassie staining. Ten μg of purified Ad3 fiber knob (unboiled) was loaded per lane. Trimeric forms of the fiber knobs are indicated by an arrow. The gel contained SDS and the loading buffer containing DTT, which caused the disassembly of dimers of trimeric fiber knobs, as previously reported (2). (E and F) Western blot using soluble recombinant DSG2 as a probe, followed by anti-DSG2-MAb and anti-mouse IgG-HRP. For comparison, JO-1 (0.5 μg/lane) is shown. The Western blots were scanned and signals were quantified.
Table 2.
Analysis of Ad3 fiber mutants
| Mutant | % Residual DSG2 binding by Western blottinga | % Inhibition of: |
|
|---|---|---|---|
| Infection in the presence of dimeric knobb | Attachment in the presence of dimeric knobc | ||
| N186D | 5.3 | 32.7 | 56.5 |
| V189G | 14 | 54 | 81 |
| S190P | 7.1 | 30.9 | 73.4 |
| D261N | 0 | 5.3 | 45 |
| F265L | 0 | 17.6 | 55.6 |
| L296R | 3.6 | 5.1 | 73.7 |
| E299V | 20 | 50.7 | 97.3 |
| N186D, D261N | 7.0 | 5.2 | 23.5 |
| D261, L296R | 7.0 | 1.5 | 20.1 |
| N186D, D261N, L296R | 0 | 0 | 18.5 |
Quantitative analysis of Western blot bands corresponding to Ad3 knob trimers. The intensity of the wt Ad3 fiber knob was taken as 100%.
The data reflect the ability of dimeric Ad3 fiber knob mutants to inhibit Ad3-GFP infection (Fig. 3B). The higher the percentage, the stronger the inhibition. Inhibition by JO-1 (dimeric wt Ad3 knob) is taken as 100%.
Corresponding data for Ad3 virus attachment. n = 3. Averages are shown. The standard deviations were less than 10%.
Fig 2.

3D model of the Ad3 fiber knob. The structure is based on that of PDB accession number 1H7Z_A. (Upper) Four critical areas involved in DSG2 binding. The critical residues are shown on the pink isosurface of the trimeric fiber knob. The view is from the top (apical side) facing the receptor. (Lower) All critical residues combined. On the right is an enlargement of the groove after a slight side rotation.
To create an Ad3 fiber knob and eventually an Ad3 virus with maximum ablation of DSG2 binding, we introduced multiple mutations in the three identified areas, specifically a combination of N186D and D261N, a combination of D261N and L296R, and a combination of N186D, D261N, and L296R. As expected, the combination of mutations in all three critical regions conferred the highest level of ablation (Table 2 and Fig. 1F).
Because of its relevance as a recently emerged pathogen, we also generated a library of (monomeric) Ad14p1 fiber knob mutants. A first screening revealed ∼300 candidate colonies for variants that did not bind to DSG2. When analyzed by Western blotting for the 6×His tag, 45 of the 300 colonies showed expression of trimeric fiber knob. Sequencing of these variants revealed 15 independent mutants with reduced binding to DSG2 (Fig. 1A). Interestingly, in spite of a different beta sheet distribution, the amino acid residues that were critical for Ad14p1 knob binding were in the same three regions that were identified for the Ad3 fiber knob. Because of these similarities, we performed further studies only with selected Ad3 fiber knob mutants.
Functional validation.
Competition studies were performed on HeLa cells, which express DSG2 (1). We first studied the attachment of 3H-labeled Ad3 virus after preincubation of cells with dimeric Ad3 fiber knobs (Fig. 3A). Reduction in Ad3 virus binding was compared to that at preincubation with JO-1, i.e., the dimeric protein that contained the wild-type Ad3 fiber knob. Inhibition of binding by JO-1 was taken as 100%. The mutants L296R, D261N, and F265L blocked Ad3 virus binding the least (5.1, 5.3, and 17.6%), followed by mutants S190P, N186D, and E299V (30.9, 32.7, and 50.7% reduced binding, respectively) (Table 2). A similar assay setup was used to measure the ability of dimeric Ad3 knob mutants to block transduction of HeLa cells by an Ad3-GFP vector. Transduction was measured based on GFP expression (Fig. 3B). Similar to what we observed in the attachment study, Ad3-GFP infection was least reduced by preincubation with mutants D261N and F265L, followed by mutants N186D, S190P, L296R, and V189G. Taking the DSG2 binding (Western blotting), attachment, and infection competition data together, we concluded that the area containing residues 261 to 265 is the most critical area in DSG2 binding. The region around residues 186 to 190 also contributes to binding, while the region containing residue 299 appears to be only marginally involved in binding. Dimeric Ad3 knob mutants with combined mutations were also analyzed for their ability to compete with Ad3 virus for attachment (Fig. 3C) and infection (Fig. 3D). The mutant with mutations in all three areas (N185, D261, and L296) did not block Ad3 binding or infection even at concentrations of 200 μg/ml, indicating that it is nearly ablated for DSG2 binding. Notably, when using soluble CD46 (sCD46) as a probe in the Western blot analysis of wild-type Ad3 fibers, no specific binding was observed (Fig. 4). This indicates that Ad3 only inefficiently binds to CD46.
Fig 3.

Competition of Ad3 virus by dimerized Ad3 knob mutants. (A) Relative attachment of 3H-labeled Ad3 virus in the presence of dimeric fiber knob mutants. A total of 1.8 × 105 HeLa cells were incubated with Ad3 knob mutants at concentrations of 2.5 and 100 μg/ml on ice for 1 h. Four hundred PFU/cell of 3H-Ad3 virus was added on ice for another hour. Unbound virus particles were washed away. Attachment of virus particles incubated with PBS was taken as 100%. n = 3. (B) Competition of Ad3-GFP virus infection on HeLa cells. A total of 1.5 × 105 HeLa cells were seeded into 24-well plates. Cells were incubated with the Ad3 knob mutants at increasing concentrations for 1 h at room temperature. One hundred PFU/cell of Ad3-GFP virus was added, and GFP expression was analyzed 18 h later by flow cytometry. (Left) Percentage of GFP-positive cells. (Right) Mean fluorescence intensity (MFI). n = 3. The standard deviations were less than 10%. (C) Relative attachment of 3H-labeled Ad3 virus in the presence of dimeric fiber knob mutants with multiple mutations. The study was performed as described for panel B. The standard deviations were less than 10%. (D) Competition of Ad3-GFP virus infection on HeLa cells. Geo MFI, geometric mean fluorescence intensity. The study was performed as described for panel C. The standard deviations were less than 10%.
Fig 4.
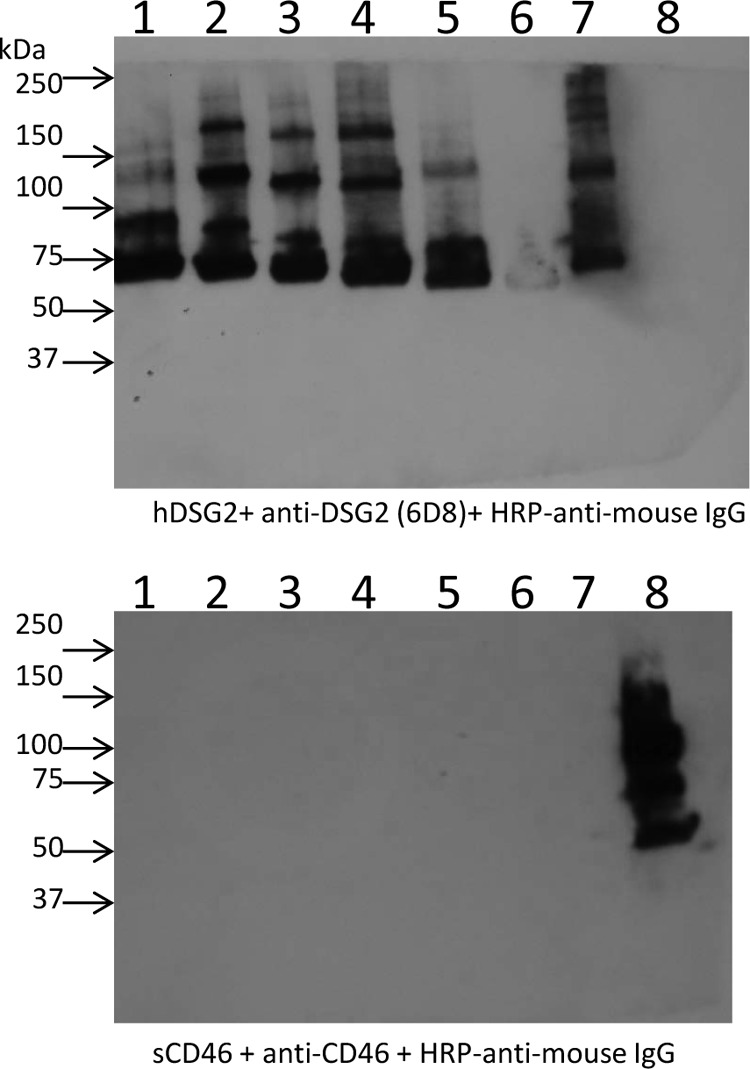
Analysis of Ad3 fiber knob binding to soluble CD46. Ad3 fiber knobs containing different numbers of shaft motifs and either the wild-type Ad3 fiber knob (lane 1, Ad3-S6/Kn; lane 2, Ad3-S5/Kn; lane 3, Ad3-S4/Kn; lane 4, Ad3-S3/Kn; lane 5, Ad3-S2/Kn; lane 6, Ad3-S/Kn), JO-1 (lane 7) or the CD46-binding Ad35 fiber knob (lane 8) were blotted and hybridized with soluble DSG2 (upper) or soluble CD46 (lower). Binding was detected by anti-DSG2 MAb or anti-CD46 MAb.
Correlation of reduced DSG2 binding and weaker ability to open epithelial junctions.
A straightforward assay for the junction-opening function of dimeric Ad3 knob mutants is based on measuring the transepithelial electrical resistance (TEER) in transwell cell cultures. Epithelial cancer cells are cultured until the TEER is constant, when major intercellular junctions are formed. Addition of JO-1 for 1 h to the apical side of the transwell cultures resulted in a rapid decrease in the TEER, indicating opening of junctions (Fig. 5A). Incubation with mutant D261N had no effect on the TEER. N186D and E299V had intermediate effects that correlated with the residual binding of the corresponding fiber knobs to DSG2. In previous studies, we have also established that JO-1 triggered changes in epithelial junctions of xenograft tumors and increased the anti-tumor efficacy of chemotherapeutics with high molecular masses, for example, irinotecan. (Irinotecan has a molecular mass of 586.7 Da and is used to treat colon and lung cancer). We used this effect to assess the function of dimeric Ad3 knob mutants in vivo (Fig. 5B). Similar to what we observed in vitro, JO-1 enhanced irinotecan therapy while mutants with reduced DSG2 binding had no significant effect on irinotecan efficacy.
Fig 5.

Correlation of reduced DSG2 binding with the ability to open epithelial junctions. (A) Transepithelial electrical resistance (TEER) measured on polarized colon cancer T84 cells. Cells were cultured in transwell chambers until the TEER was constant, i.e., tight junctions had formed. A total of 5 μg of dimeric Ad3 fiber knobs in PBS was then added for 1 h to the apical chamber. TEER was measured at the indicated time points. n = 6. For time points 1.5 and 4 h, the difference between JO-1 and the D261N and N186D mutants was significant (P < 0.01). The arrows indicate the addition and removal of Ad3 fiber knobs. (B) Enhancement of irinotecan therapy. A total of 4 × 106 A549 cells were injected subcutaneously into CB17-SCID/beige mice. Once the tumor reached a volume of ∼100 mm3 (day 15 after implantation), the mice were injected intravenously with 2 mg/kg JO-1, E299V, N186D, or PBS, followed by an intravenous injection of irinotecan (37.5 mg/kg) 1 h later. The treatment was repeated on day 25. n = 5. The differences between the irinotecan versus E299V plus irinotecan groups or irinotecan versus N186 plus irinotecan groups were not significant. The difference between irinotecan and JO-1 plus irinotecan was significant (P < 0.01) from day 20 on.
Affinity-enhanced dimeric Ad3 fiber knobs.
As outlined above, JO-1 is relevant for cancer therapy. Therefore it is important to better understand structural details of its interaction with DSG2 and create JO-1 mutants with increased affinity to DSG2. Affinity enhancement of biologics is used to (i) decrease their effective dose, (ii) increase their half-lives, (iii) potentially increase their therapeutic effects, and (iv) circumvent the adverse effects of antibodies generated by patients against the biologic (e.g., neutralization or changes to the pharmokinetics). To make JO-1 analogues with increased affinity, we screened the E. coli expression library with random mutations within JO-1 for variants with increased binding to DSG2. Out of 10,000 colonies plated, 20 colonies with the most intense DSG2 signals were picked, and plasmid DNA was sequenced. Seven different mutants with one or two amino acid substitutions were identified: Y250F, K217E+F224S, N293S, V239D, F224L, E248G+K258E, and L277R+N293D. The localizations of the residues in the primary and 3D structures of the Ad3 fiber knob are shown in Fig. 6. Notably, most of the mutations were localized within the EF loop, indicating that this loop is involved in stabilizing the interaction between Ad3 and DSG2. V239 and Y250 are not exposed at the knob surface, suggesting a structural change in the knob rather than an involvement in direct binding to DSG2 (Fig. 6B, right). Recombinant mutant dimeric Ad3 knob proteins then were purified. To measure the affinity of the mutants to DSG2, we performed surface plasmon resonance studies. The outcome of studies with knobs containing the dimerization domain was complex, most likely due to the fact that these mutants formed multimeric complexes. Therefore, we performed studies with knob proteins lacking the dimerization domain (noDD). The association rate constant (Ka or Kon) and the dissociation rate constant (Kd or koff), as well as the KD (equilibrium dissociation constant) of wt Ad3 knob and all knob mutants, are shown in Fig. 7. In agreement with previous studies (2), we found that the wt Ad3 knob without the dimerization domain bound to DSG2 with only relatively low affinity (KD = 10 μM). With the exception of mutant L227R/noDD plus N293D/noDD, all mutants identified in the colony blot screen had higher affinities to DSG2. Notably, the affinities of mutant Y250F/noDD or V239D/noDD were 885- or 405-fold higher than those of wt Ad3knob/noDD. The high affinity of the different mutants was mainly due to a faster association with DSG2 rather than a change in the dissociation rate. The only exception to this trend was mutant N293S/noDD, for which the association rate was the lowest of the mutants. However, this was partially compensated for by a slower dissociation rate. Together, these results indicate that wt Ad3 knob (noDD) binding to DSG2 is mostly limited by a slow association rate that can be improved by a panel of mutations. These mutations do not appear to modify the stability of this interaction but the balance of association versus dissociation, resulting in higher affinities of ligands to the receptor.
Fig 6.

Amino acid substitutions that increase the binding to DSG2. (A) Shown is the amino acid sequence of the Ad3 fiber knob. Beta sheets are indicated by blue lines. Arrows indicate residues within the Ad3 fiber knob which, when mutated, yielded stronger signals in colony blot assays, indicating stronger binding to DSG2. (B) The isosurface of the three knob monomers is colored in gray, pink, and light brown. Mutants involved in DSG2 recognition are shown in the same color as Fig. 1B (yellow, green, blue, and orange), and mutations enhancing the binding are shown in red. (Left) Top view; (right) side view. V239 and Y250 are not exposed at the top, suggesting a structural change in the knob rather than an involvement in direct binding to DSG2. (C) Localization of all mutations that enhance the binding to DSG2. Residues are show in magenta in two knob monomers. The isosurface of one monomer is shown in gray transparency.
Fig 7.
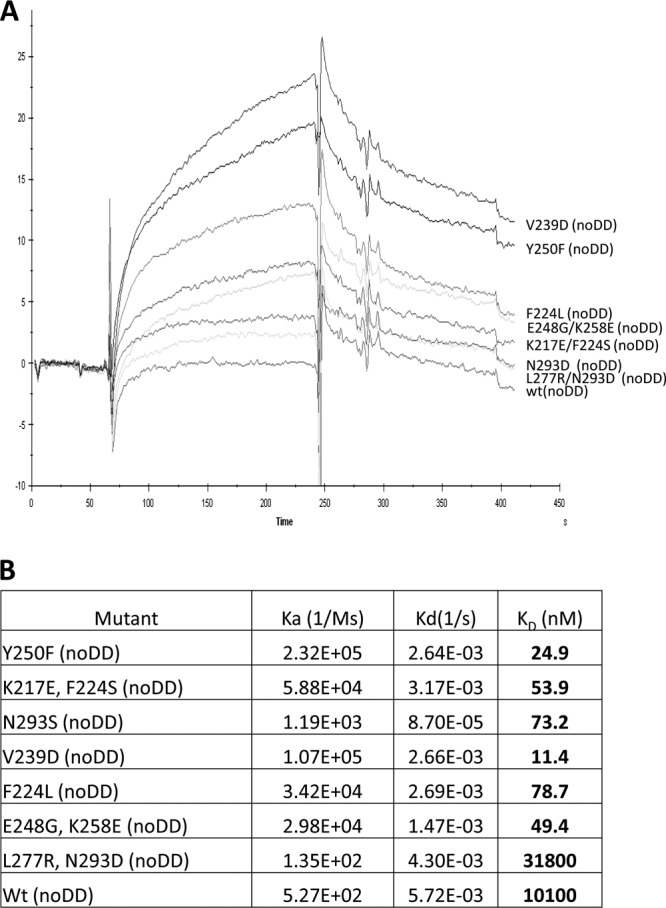
SPR analysis of nondimerized Ad3 fiber knob interactions with DSG2. (A) DSG2 was immobilized on sensorchips, and background was automatically subtracted from the control flow cell. The Ad3 fiber knobs (without a dimerization domain, termed noDD) were injected for 3 min at 2.5 μg/ml, followed by a 2.5-min dissociation period. (B) Summary of SPR data. A concentration range from 2.5 to 10 μg/ml of the knobs was injected, and kinetics and affinity parameters were evaluated using BIAeval software. The extracted data are shown in the table. Wt, Ad3 fiber knob without mutations.
To better understand structural elements that enhance binding to DSG2, we performed a more detailed analysis with mutant K217E/F224S. Transmission electron microscopy with uranyl acetate-stained K217E/F224S fiber knobs containing the dimerization domain showed particles with 6 knobs representing dimerized trimeric fibers (Fig. 8A, thick arrows, and B). Interestingly, under these conditions, fiber knobs also formed regularly shaped arrogates (with ∼30-nm diameter) resembling collapsed PtDd (Fig. 8A, thin arrows, and C). We then performed X-ray crystallography studies to resolve the structure of the K217E/F224S mutant at the atomic level (Fig. 8D to H). As expected, the K217E/F224S mutant formed a monotrimer of fiber knobs (Fig. 8E). The 3D structure of the mutant was overlaid with that of the wild-type Ad3 fiber knob (Fig. 8F to H). This revealed that the EF loop in the K217E/F224S mutant was completely disordered. This loop is at the base of the knob domain at the junction with the fiber shaft. Therefore, the K217E/F224S mutations may allow for easier binding by increasing the flexibility of this loop region.
Fig 8.

Electron microscopy and 3D structure of Ad3 fiber knob mutant JO-2. (A to C) Negative staining of JO-2 with SST. Dimeric forms can be seen, but higher organizations are also visible. A heterogeneous complex of around 50 nm is depicted by thin arrows, and a smaller, regular dodecahedral-like particle is depicted by thick arrows. Closeup views are presented in panels B and C. (D to G) Crystallographic structure of the nondimerized form of the K217E/F224S mutant. (D) Protein crystals. (E) The wild-type Ad3 knob is colored in gray, and the EF loop 217-224 is in cyan. This is the loop which becomes disordered in the mutant. There is no density for these residues in the mutant structure. (F) The mutant is displayed as a cartoon, and each monomer is colored red, blue, or green. (G) Overlay of these two structures shows that the EG loop is completely disordered in the K217E/F224S mutant. The bottom panels show closeup views of one monomer. K217 and F224 appear as blue sticks.
Correlation of increased affinity with stronger ability to open epithelial junctions.
For the following studies, we used Ad3 fiber knob forms containing the dimerization domain. To analyze the selected high-affinity mutants, we performed competition infection studies with Ad3-GFP on HeLa cells and the dimeric forms of the affinity-enhanced Ad3 fiber mutants (Fig. 9A). Based on GFP expression, all dimeric mutants except mutant L277R+N293D inhibited Ad3-GFP infection significantly more than JO-1. Notably, the nondimerized forms of Ad3 fiber knobs with increased affinity to DSG2 were unable to act as competitors in transduction studies (Fig. 9B). Higher affinity to DSG2 resulted in an increased capability to open epithelial junctions in transwell cultures (Fig. 9C). Compared to JO-1, the TEER in cultures incubated with the mutants V250F, V239D, and K217E+F224S was significantly higher.
Fig 9.

Analysis of dimeric Ad3 fiber knob mutants with increased affinity to DSG2. (A) Competition of Ad3-GFP virus infection on HeLa cells with dimeric affinity-enhanced mutant Y250F and JO-1 (dimeric wt Ad3 fiber knob). The experimental settings are the same as those described for Fig. 3C. (Left) Percentage of GFP-positive cells. (Right) Mean fluorescence intensity. n = 3. The standard deviations were less than 10%. (B) Competition of Ad3-GFP virus infection on HeLa cells by Ad3 knob mutants with enhanced DSG2 binding but without a dimerization domain. A total of 1.5 × 105 HeLa cells were seeded into a 24-well plate. Cells were incubated with the Ad3 knob mutants at increasing concentrations for 1 h at room temperature. One hundred PFU/cell of Ad3GFP virus was then added, and GFP expression was analyzed 18 h later. (C) TEER on colon cancer T84 cells. The experimental settings were the same as those described for Fig. 5A. The TEER at 4 h is shown. n = 3.
Two of the affinity-enhanced versions of JO-1, V293D and Y250F, and V250F were analyzed in an in vivo assay. We called these mutants JO-2 and JO-4, respectively. The first study was performed in a xenograft model derived from A549 cells, similar to the study described for Fig. 5B. Figure 10A shows that the affinity-enhanced mutants JO-2 and JO-4 increased irinotecan therapy significantly more than JO-1 (P < 0.05 starting from day 27). Furthermore, JO-4 (KD, 11.4 nM) was significantly more efficient than JO-2 (KD, 24.9 nM), indicating a correlation between affinity to DSG2 and therapeutic effect. Additional studies were performed in xenograft tumors derived from Ovc316 cells (11, 25). Ovc316 cells are Her2/neu-positive epithelial tumor cells derived from an ovarian cancer biopsy specimen. These cells can undergo epithelial-to-mesenchymal transition (EMT) and the reverse process, mesenchymal-to-epithelial-transition (MET), under specific conditions in vitro and in vivo. A subfraction of Ovc316 cells that is positive for Nanog, CD133, and E-cadherin is enriched for cancer stem cells, i.e., self-renewing cells with pluripotent potential and tumor-forming ability (25). Therefore, Ovc316 cells closely model the heterogeneity and plasticity seen in tumors in situ. Intravenous injection of JO-1 at a dose of 2 mg/kg of body weight 1 h before injection of pegylated liposomal doxorubicin (PLD), a drug that is widely used for chemotherapy of ovarian cancer, significantly increased the treatment efficiency (Fig. 10B). Importantly, at a dose of 0.5 mg/kg, JO-4 had an even greater stimulating effect on PLD therapy. Finally, we tested JO-4 in a model for triple-negative breast cancer (TNBC). TNBC is characterized by a lack or minimal expression of estrogen receptor (ER) and progesterone receptor (PR) and the absence of Her2/neu overexpression. TNBC accounts for 15% of all breast cancers. Overall survival is poor compared to that of patients who have other phenotypes. A characteristic feature of TNBC is high levels of DSG2 and epithelial junctions. Promising clinical results in the treatment of TNBC have been achieved with nanoparticle albumin-conjugated paclitaxel (nab-paclitaxel) alone or in combination with the EGF receptor (EGFR)-targeting mAb cetuximab (26). Our study showed that JO-4 significantly increased nab-paclitaxel/cetuximab efficacy in a mouse model with orthotopic TNBC tumors (Fig. 10C). Because of its therapeutic relevance, we further studied JO-4 in an adequate mouse tumor model. Because Ad3 virus and Ad3 fiber knob derivatives do not bind to mouse cells and tissues, we used human transgenic mice that expressed human DSG2 in a pattern and at a level similar to that of humans (4). These mice were subcutaneously implanted with syngeneic TC1-hDSG tumors. When tumors reached a volume of ∼600 mm3, JO-1 or JO-4 was intravenously injected for safety and efficacy studies. Both JO-1 and JO-4 serum concentrations declined more than one order of magnitude within an hour after injection, and the decline was significantly greater for JO-4 (P < 0.01 for 1 h postinjection) (Fig. 11A). After 1 h postinjection, JO-1 and JO-4 concentrations reached a plateau with ∼100 ng/ml. We also analyzed hematological parameters after intravenous JO-1 and JO-4 injection. Blood chemistry did not show abnormal changes. Blood cell counts were normal except for lymphocyte and platelet numbers, which decreased early after injection (Fig. 11B). Lymphocyte and platelet counts reached a nadir at 24 h postinfection with significantly lower numbers for JO-4 (P < 0.01). Interestingly, lymphocyte and platelet counts returned to normal levels faster in JO-4-injected mice than in JO-1-treated animals.
Fig 10.

Combination of affinity-enhanced JO-1 versions with chemotherapy. (A) Enhancement of irinotecan (I) therapy. The experimental settings were the same as those described for Fig. 5B. The differences in the groups JO-1 plus I versus JO-2 plus I and JO-2 plus I versus JO-4 plus I were significant from day 20 on. n = 5. (B) JO-4 enhances PLD therapy in an ovarian cancer model at a lower dose than JO-1. Mammary fat pad tumors were established from primary ovarian cancer Ovc316 cells. Treatment was started when tumors reached a volume of 100 mm3. Mice were injected intravenously with 2 mg/kg JO-1 or with 0.5 mg/kg JO-4, followed by an intravenous injection of PLD (1 mg/kg) 1 h later. Treatment was repeated weekly. (C) JO-4 enhances therapy in poor-prognosis triple-negative breast cancer (TNBC). A total of 4 × 106 TNBC MDA-MB-231 cells were injected into the mammary fat pad of CB17 SCID/beige mice. JO-4 (2 mg/kg) was intravenously injected 1 h before the application of cetuximab (C) (10 mg/kg, intraperitoneal) and nab-paclitaxel (nab-P) (5 mg/kg, intravenous). Treatment was given weekly. n = 10. P < 0.01 at day 25 for nab-P plus C versus JO-4 plus nab-P plus C.
Fig 11.

Pharmacokinetics, toxicity, and immunogenicity of JO-4. (A) Serum clearance of JO-1 and JO-4. hDSG2 transgenic mice with subcutaneous TC1-hDSG2 tumors (∼600 mm3) were intravenously injected with JO-1 or JO-4 (2 mg/kg), and serum samples were analyzed by ELISA. n = 3. Note that the y axis has a log scale. (B) Lymphocyte and platelet counts in hDSG2/TC1-hDSG2 transgenic mice after JO-1 or JO-4 injection. n = 3. (C) Therapy studies in immunocompetent hDSG2 transgenic mice with TC1-hDSG2 tumors. When tumors reached a volume of ∼80 mm3, JO-1 or JO-4 (2 mg/kg) or PBS was injected intravenously, followed 1 h later by PLD (Doxil; 1.5 mg/kg, intravenous). Treatment was repeated as indicated by arrows. Tumors were then allowed to regrow for about 2 weeks. From day 15 on, serum anti-JO-1/JO-4 antibodies were detectable by ELISA. Two more treatment cycles were performed at day 28 and day 35. JO-1 and JO-4 continued to be effective after multiple treatment cycles, even in the presence of detectable antibodies. The difference between JO-1/PLD and JO-4/PLD is significant from day 31 on. n = 10.
JO-1 and JO-4 are virus-derived proteins and are immunogenic. In immunocompetent mice, serum IgG antibodies against these proteins can be detected by ELISA 2 weeks after injection (5). One theoretical premise for affinity enhancement of therapeutic proteins is that it circumvents neutralizing serum antibodies. To test this, we performed repeated injections of JO-1 and JO-4 in an immunocompetent hDSG2 mouse tumor model with TC1-hDSG2 tumors (Fig. 11C). After two treatment cycles of JO-1 and PLD, treatment was stopped and tumors were allowed to regrow. The 3rd and 4th treatment cycles were started on days 28 and 35, respectively. At the time of the 3rd cycle, serum anti-JO-1 antibodies were detectable by ELISA. Importantly, in both the 3rd and 4th treatment cycles, JO-1 and JO-4 had an enhancing effect on PLD therapy, whereby the enhancing effect was significantly stronger for JO-4.
Overall, our functional studies with affinity-enhanced dimeric Ad3 fiber mutants demonstrate a correlation between DSG2 affinity and epithelial junction opening/therapeutic effects.
DISCUSSION
Residues involved in Ad3 knob binding to DSG2.
Unlike Ad interaction with CAR and CD46 (27, 28), structural details of Ad interaction with DSG2 are still elusive. Although the crystal structure of the Ad3 fiber knob has been resolved, for DSG2, the 3D structure of only the most distal of the four extracellular domains (ECD) is available (MMDB code 59843). However, our previous competition studies with monoclonal antibodies against different DSG2 domains indicated that ECDs 3 and 4 are involved in binding to Ad3 (1). In this study, we used mutagenesis-based analyses to identify the amino acid residues within the Ad3 fiber knob that are critical for binding to DSG2. Mutagenic analysis of DSG2 was not possible, because, when expressed in E. coli, the protein did not bind to Ad3, indicating that posttranslational processing is required to create active Ad3 binding sites within DSG2 (data not shown). The identified residues, critical for Ad3 knob binding to DSG2, were in three different areas of the Ad3 fiber knob and formed a potential binding pocket localized in a groove at the distal end of the fiber knob facing the receptor. Notably, binding of other Ad serotypes to CAR or CD46 primarily involves regions at the lateral or basal side of the corresponding fiber knobs (10, 29). Our data indicate that Ad3 uses a different binding strategy. We are currently performing crystallography studies with dimeric Ad3 fiber knobs and DSG2. Considering that multimeric Ad3 fiber knobs cluster several DSG2 molecules (2), it is expected that the 3D structure of this complex will be complicated. It remains to be studied whether the residues critical for Ad3 fiber knob binding to DSG2 will also be involved in binding of other species B Ads to DSG2. Notably, while D261, F265, and E299 are conserved in all four DSG2-interacting Ads (Ad3, Ad7, Ad11, and Ad14), other critical residues (N186, V189, and L296) differ between these serotypes (Fig. 12).
Fig 12.

Alignment of fiber knob sequences. The residues that ablate/reduce Ad3 knob binding to DSG2 are indicated.
Ad14 is an important research object because of the recent appearance of a new strain (Ad14p1). Having never been previously documented in the United States, Ad14p1 was reported in March and April 2006 during routine surveillance at several U.S. military recruit training centers (30). During March to June of the following year, a total of 140 additional cases of confirmed HAdV-B14p1 respiratory illness were reported in patients in Oregon, Washington, and Texas (31). Thirty-eight percent of these patients were hospitalized, including 17% who were admitted to intensive care units; 5% of patients died. Outbreaks of HAdV-B14p1 were subsequently detected in the other 5 bases and in civilian populations in Washington, Oregon (32), Alaska (33), Wisconsin, and Pennsylvania (7, 9), as well as in Canada (8), China (34), and South Korea (35). At this point, the molecular basis for the high pathogenicity and/or virulence of Ad14p1 is unclear. We attempted to delineate the structural components for Ad14p1 binding to DSG2. The beta sheet distribution of Ad14p1 differs from that of Ad3 (Fig. 1A).Therefore, similar to CD46-interacting serotypes (36, 37), it is possible that DSG2-interacting Ads vary in their binding strategy to DSG2, which could result in different DSG2 binding areas. However, the screening of an Ad14p1 fiber knob mutant library did not support this hypothesis. The areas involved in DSG2 binding were essentially the same for Ad3 and Ad14p1 fiber knobs. Nevertheless, our finding are relevant for the treatment of Ad14p1 viremia, specifically for the production of Ad14p1 inhibitors or high-affinity decoys that can trigger the opsonization of virus present in the blood circulation or airway.
It has been reported that, in addition to DSG2, Ad3 can use CD46 as a receptor to infect cells if DSG2 is absent (38). Previously, we found that in polarized normal epithelial cells, DSG2 is trapped in tight junctions and is not accessible from the apical side, while CD46 is present on both membrane sides (1). Therefore, we speculate that CD46 can serve as a relatively inefficient entry receptor for Ad3, while de novo-produced Ad3 and Ad3 penton-dodecahedra interact with DSG2, open epithelial junctions, and allow for efficient lateral spread of Ad3 or penetration into deeper tissue layers and blood circulation. The ability to individually ablate the Ad3 knob residues that are critical for DSG2 and CD46 binding should make it possible to prove this hypothesis.
Affinity-enhanced fiber knobs.
Most of the mutations that increased the affinity to DSG2 were localized within the EF loop, indicating that this loop is involved in stabilizing the interaction between Ad3 and DSG2. Interestingly, unlike Ad7, Ad11, and Ad14, the Ad3 fiber knob has two additional residues (VL) followed by a proline in this area. Therefore, this loop could be extended further and the proline could orient it in a way that might allow for better interaction with the receptor. The analysis of the 3D structure of one of these mutants at the atomic level supports this conclusion. These studies indicate that the introduced mutations make the loop more flexible, which might facilitate the interaction with DSG2.
The identification of Ad3 knobs with higher affinity than the wt Ad3 knob has implications for Ad3-mediated gene therapy. Recently, gene transfer vectors based on Ad3 have shown promise for cancer therapy in clinical trials (39). Theoretically, affinity-enhanced Ad3 vectors could be used at lower doses and outcompete neutralizing antibodies. Recently, attempts were undertaken to incorporate high-affinity ligands into measles virus (40) and Ad5-based vectors (31, 41, 42) in order to increase efficacy and specificity of target cell infection in vivo. Based on our findings in this study, a similar strategy can now be pursued for Ad3 vectors.
In addition to improving Ad3 vectors, affinity-enhanced versions of JO-1 have translational relevance. Most solid tumors are of epithelial origin, and although malignant cells are dedifferentiated, they maintain intercellular junctions, a key feature of epithelial cells, both in the primary tumor as well as in metastatic lesions (5, 43). These intercellular junctions represent a protection mechanism against attacks by the host immune system and pose physical barriers that prevent intratumoral penetration and dissemination of cancer therapeutics, including monoclonal antibodies and chemotherapy drugs (5, 43). When injected intravenously into mice with xenograft or syngeneic DSG2 transgenic tumors, JO-1 markedly enhanced therapeutic effects with a variety of chemotherapy drugs as well as monoclonal antibodies (3, 5). In this study, we have shown that new affinity-enhanced versions of JO-1 (e.g., JO-4) increased the efficacy of cancer therapeutics significantly more than JO-1 (irinotecan, nab-paclitaxel, pegylated liposomal doxorubicin, and cetuximab) in four tumor models (A549, Ovc316, MDA-MB231, and TC1-DSG2). Studies of DSG2 transgenic mice with syngeneic tumors showed that serum JO-4 levels rapidly decreased, most likely due to binding to DSG2 on tissues. Previous studies showed that, in addition to tumors, lymphocytes and platelets of transgenic hDSG2 mice express hDSG2 (similar to human and monkeys) (1, 4). Along these lines, we found that JO-4 injection resulted in a transient reduction of lymphocyte and platelet counts.
Despite the fact that approximately one-third of humans have neutralizing antibodies against Ad3 (1), in a recent study with serum from ovarian cancer patients we found detectable (binding) antibodies against JO-4 in only 10% of patients (n = 38) (Fig. 13). However, it is certain that adaptive immune responses against intravenously administered JO-4 will develop in humans, particularly after repeated injection. In this context, it is noteworthy that anti-JO-4 antibodies generated after injection into immunocompetent mice appeared not to critically inhibit the function of JO-4. The data shown in Fig. 11C demonstrate that JO-1 and JO-4 continue to be effective after multiple treatment cycles, even in the presence of detectable antibodies. Because the therapeutic effect after repeated injection was significantly greater for JO-4, we speculate that JO-4 is more potent not only in junction opening but also in disrupting complexes between the junction opener and serum antibodies.
Fig 13.
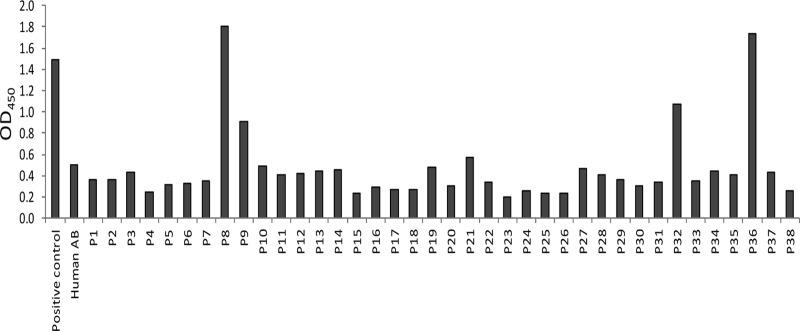
Sera from humans and hypervaccinated mice do not inhibit activity of JO-4. Analysis of human serum for binding with JO-4 by ELISA is shown. Rabbit polyclonal antibodies against the Ad3 fiber knob were used for capture, followed by recombinant JO-1 protein, human serum (dilutions 1:20 to 1:1,000), and anti-human IgG-HRP. Commercial human Ab serum depleted for IgG was used as a negative control (blue horizontal line). Serum from a scientist who routinely works with Ad3 virus was used as a positive control. P1 to P38 are serum samples from ovarian cancer patients obtained from the Pacific Ovarian Cancer Research Consortium.
In summary, our studies uncover important structural details of Ad3 and Ad14p1 fiber knob binding to DSG2. Furthermore, it shows a correlation between the affinity of Ad3 fiber knobs to DSG2 and subsequent effects on epithelial junctions. Finally, the generation of affinity-enhanced recombinant dimeric Ad3 fiber knobs has implications for cancer therapy.
ACKNOWLEDGMENTS
The study was supported by NIH grants R01 CA080192 and R01 HLA078836.
We thank the Pacific Ovarian Cancer Research Consortium for providing patient serum samples.
Footnotes
Published ahead of print 14 August 2013
REFERENCES
- 1.Wang H, Li ZY, Liu Y, Persson J, Beyer I, Moller T, Koyuncu D, Drescher MR, Strauss R, Zhang XB, Wahl JK, III, Urban N, Drescher C, Hemminki A, Fender P, Lieber A. 2011. Desmoglein 2 is a receptor for adenovirus serotypes 3, 7, 11 and 14. Nat. Med. 17:96–104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wang H, Li Z, Yumul R, Lara S, Hemminki A, Fender P, Lieber A. 2011. Multimerization of adenovirus serotype 3 fiber knob domains is required for efficient binding of virus to desmoglein 2 and subsequent opening of epithelial junctions. J. Virol. 85:6390–6402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Beyer I, van Rensburg R, Strauss R, Li Z, Wang H, Persson J, Yumul R, Feng Q, Song H, Bartek J, Fender P, Lieber A. 2011. Epithelial junction opener JO-1 improves monoclonal antibody therapy of cancer. Cancer Res. 71:7080–7090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang H, Beyer I, Persson J, Song H, Li Z, Richter M, Cao H, van Rensburg R, Yao X, Hudkins K, Yumul R, Zhang XB, Yu M, Fender P, Hemminki A, Lieber A. 2012. A new human DSG2-transgenic mouse model for studying the tropism and pathology of human adenoviruses. J. Virol. 86:6286–6302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Beyer I, Cao H, Persson J, Song H, Richter M, Feng Q, Yumul R, van Rensburg R, Li Z, Berenson R, Carter D, Roffler S, Drescher C, Lieber A. 2012. Coadministration of epithelial junction opener JO-1 improves the efficacy and safety of chemotherapeutic drugs. Clin. Cancer Res. 18:3340–3351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang H, Tuve S, Erdman DD, Lieber A. 2009. Receptor usage of a newly emergent adenovirus type 14. Virology 387:436–441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Carr MJ, Kajon AE, Lu X, Dunford L, O'Reilly P, Holder P, De Gascun CF, Coughlan S, Connell J, Erdman DD, Hall WW. 2011. Deaths associated with human adenovirus-14p1 infections, Europe, 2009–2010. Emerg. Infect. Dis. 17:1402–1408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Girouard G, Garceau R, Thibault L, Oussedik Y, Bastien N, Li Y. 2013. Adenovirus serotype 14 infection, New Brunswick, Canada, 2011. Emerg. Infect. Dis. 19:119–122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kajon AE, Lu X, Erdman DD, Louie J, Schnurr D, George KS, Koopmans MP, Allibhai T, Metzgar D. 2010. Molecular epidemiology and brief history of emerging adenovirus 14-associated respiratory disease in the United States. J. Infect. Dis. 202:93–103 [DOI] [PubMed] [Google Scholar]
- 10.Wang H, Liaw YC, Stone D, Kalyuzhniy O, Amiraslanov I, Tuve S, Verlinde CL, Shayakhmetov D, Stehle T, Roffler S, Lieber A. 2007. Identification of CD46 binding sites within the adenovirus serotype 35 fiber knob. J. Virol. 81:12785–12792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Strauss R, Sova P, Liu Y, Li Z-Y, Tuve S, Pritchard D, Brinkkoetter P, Moller T, Wildner O, Pesonen S, Hemminki A, Urban N, Drescher C, Lieber A. 2009. Epithelial phenotype of ovarian cancer mediates resistance to oncolytic adenoviruses. Cancer Res. 15:5115–5125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tuve S, Chen BM, Liu Y, Cheng TL, Toure P, Sow PS, Feng Q, Kiviat N, Strauss R, Ni S, Li ZY, Roffler SR, Lieber A. 2007. Combination of tumor site-located CTL-associated antigen-4 blockade and systemic regulatory T-cell depletion induces tumor-destructive immune responses. Cancer Res. 67:5929–5939 [DOI] [PubMed] [Google Scholar]
- 13.Tuve S, Wang H, Ware C, Liu Y, Gaggar A, Bernt K, Shayakhmetov D, Li Z, Strauss R, Stone D, Lieber A. 2006. A new group B adenovirus receptor is expressed at high levels on human stem and tumor cells. J. Virol. 80:12109–12120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shayakhmetov DM, Papayannopoulou T, Stamatoyannopoulos G, Lieber A. 2000. Efficient gene transfer into human CD34(+) cells by a retargeted adenovirus vector. J. Virol. 74:2567–2583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cadwell RC, Joyce GF. 1994. Mutagenic PCR. PCR Methods Appl. 3:S136–S140 [DOI] [PubMed] [Google Scholar]
- 16.Cadwell RC, Joyce GF. 1992. Randomization of genes by PCR mutagenesis. PCR Methods Appl. 2:28–33 [DOI] [PubMed] [Google Scholar]
- 17.Incardona MF, Bourenkov GP, Levik K, Pieritz RA, Popov AN, Svensson O. 2009. EDNA: a framework for plugin-based applications applied to X-ray experiment online data analysis. J. Synchrotron Radiat. 16:872–879 [DOI] [PubMed] [Google Scholar]
- 18.Kabsch W. 2010. Integration, scaling, space-group assignment and post-refinement. Acta Crystallogr. D Biol. Crystallogr. 66:133–144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kabsch W. 2010. Xds. Acta Crystallogr. D Biol. Crystallogr. 66:125–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. 2007. Phaser crystallographic software. J. Appl. Crystallogr. 40:658–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Emsley P, Lohkamp B, Scott WG, Cowtan K. 2010. Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66:486–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH. 2010. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66:213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Walters RW, Freimuth P, Moninger TO, Ganske I, Zabner J, Welsh MJ. 2002. Adenovirus fiber disrupts CAR-mediated intercellular adhesion allowing virus escape. Cell 110:789–799 [DOI] [PubMed] [Google Scholar]
- 24.Durmort C, Stehlin C, Schoehn G, Mitraki A, Drouet E, Cusack S, Burmeister WP. 2001. Structure of the fiber head of Ad3, a non-CAR-binding serotype of adenovirus. Virology 285:302–312 [DOI] [PubMed] [Google Scholar]
- 25.Strauss R, Li ZY, Liu Y, Beyer I, Persson J, Sova P, Moller T, Pesonen S, Hemminki A, Hamerlik P, Drescher C, Urban N, Bartek J, Lieber A. 2011. Analysis of epithelial and mesenchymal markers in ovarian cancer reveals phenotypic heterogeneity and plasticity. PLoS One 6:e16186. 10.1371/journal.pone.0016186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ueno NT, Zhang D. 2011. Targeting EGFR in triple negative breast cancer. J. Cancer 2:324–328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bewley MC, Springer K, Zhang YB, Freimuth P, Flanagan JM. 1999. Structural analysis of the mechanism of adenovirus binding to its human cellular receptor, CAR. Science 286:1579–1583 [DOI] [PubMed] [Google Scholar]
- 28.Persson BD, Reiter DM, Marttila M, Mei YF, Casasnovas JM, Arnberg N, Stehle T. 2007. Adenovirus type 11 binding alters the conformation of its receptor CD46. Nat. Struct. Mol. Biol. 14:164–166 [DOI] [PubMed] [Google Scholar]
- 29.Pache L, Venkataraman S, Nemerow GR, Reddy VS. 2008. Conservation of fiber structure and CD46 usage by subgroup B2 adenoviruses. Virology 375:573–579 [DOI] [PubMed] [Google Scholar]
- 30.Metzgar D, Osuna M, Kajon AE, Hawksworth AW, Irvine M, Russell KL. 2007. Abrupt emergence of diverse species B adenoviruses at US military recruit training centers. J. Infect. Dis. 196:1465–1473 [DOI] [PubMed] [Google Scholar]
- 31.Anonymous 2007. Acute respiratory disease associated with adenovirus serotype 14–four states, 2006–2007. MMWR Morb. Mortal. Wkly. Rep. 56:1181–1184 [PubMed] [Google Scholar]
- 32.Lewis PF, Schmidt MA, Lu X, Erdman DD, Campbell M, Thomas A, Cieslak PR, Grenz LD, Tsaknardis L, Gleaves C, Kendall B, Gilbert D. 2009. A community-based outbreak of severe respiratory illness caused by human adenovirus serotype 14. J. Infect. Dis. 199:1427–1434 [DOI] [PubMed] [Google Scholar]
- 33.Esposito DH, Gardner TJ, Schneider E, Stockman LJ, Tate JE, Panozzo CA, Robbins CL, Jenkerson SA, Thomas L, Watson CM, Curns AT, Erdman DD, Lu X, Cromeans T, Westcott M, Humphries C, Ballantyne J, Fischer GE, McLaughlin JB, Armstrong G, Anderson LJ. 2010. Outbreak of pneumonia associated with emergent human adenovirus serotype 14–southeast Alaska, 2008. J. Infect. Dis. 202:214–222 [DOI] [PubMed] [Google Scholar]
- 34.Tang L, An J, Xie Z, Dehghan S, Seto D, Xu W, Ji Y. 2013. Genome and bioinformatic analysis of a HAdV-B14p1 virus isolated from a baby with pneumonia in Beijing, China. PLoS One 8:e60345. 10.1371/journal.pone.0060345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Trei JS, Johns NM, Garner JL, Noel LB, Ortman BV, Ensz KL, Johns MC, Bunning ML, Gaydos JC. 2010. Spread of adenovirus to geographically dispersed military installations, May-October 2007. Emerg. Infect. Dis. 16:769–775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cupelli K, Muller S, Persson BD, Jost M, Arnberg N, Stehle T. 2010. Structure of adenovirus type 21 knob in complex with CD46 reveals key differences in receptor contacts among species B adenoviruses. J. Virol. 84:3189–3200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cupelli K, Stehle T. 2011. Viral attachment strategies: the many faces of adenoviruses. Curr. Opin. Virol. 1:84–91 [DOI] [PubMed] [Google Scholar]
- 38.Trinh HV, Lesage G, Chennamparampil V, Vollenweider B, Burckhardt CJ, Schauer S, Havenga M, Greber UF, Hemmi S. 2012. Avidity binding of human adenovirus serotypes 3 and 7 to the membrane cofactor CD46 triggers infection. J. Virol. 86:1623–1637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hemminki O, Diaconu I, Cerullo V, Pesonen SK, Kanerva A, Joensuu T, Kairemo K, Laasonen L, Partanen K, Kangasniemi L, Lieber A, Pesonen S, Hemminki A. 2012. Ad3-hTERT-E1A, a fully serotype 3 oncolytic adenovirus, in patients with chemotherapy refractory cancer. Mol. Ther. 20:1821–1830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hasegawa K, Hu C, Nakamura T, Marks JD, Russell SJ, Peng KW. 2007. Affinity thresholds for membrane fusion triggering by viral glycoproteins. J. Virol. 81:13149–13157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Campos SK, Parrott MB, Barry MA. 2004. Avidin-based targeting and purification of a protein IX-modified, metabolically biotinylated adenoviral vector. Mol. Ther. 9:942–954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zeng Y, Pinard M, Jaime J, Bourget L, Uyen Le P, O'Connor-McCourt MD, Gilbert R, Massie B. 2008. A ligand-pseudoreceptor system based on de novo designed peptides for the generation of adenoviral vectors with altered tropism. J. Gene Med. 10:355–367 [DOI] [PubMed] [Google Scholar]
- 43.Strauss R, Lieber A. 2009. Anatomical and physical barriers to tumor targeting with oncolytic adenoviruses in vivo. Curr. Opin. Mol. Ther. 11:513–522 [PubMed] [Google Scholar]