

Abstract
We previously identified two novel endogenous murine leukemia virus proviruses, PreXMRV-1 and PreXMRV-2, and showed that they most likely recombined during xenograft passaging of a human prostate tumor in mice to generate xenotropic murine leukemia virus-related virus (XMRV). To determine the recombination potential of PreXMRV-1 and PreXMRV-2, we examined the generation of replication-competent retroviruses (RCRs) over time in a cell culture system. We observed that either virus alone was noninfectious and the RNA transcripts of the viruses were undetectable in the blood and spleen of nude mice that carry them. To determine their potential to generate RCRs through recombination, we transfected PreXMRV-1 and PreXMRV-2 into 293T cells and used the virus produced to infect fresh cells; the presence of reverse transcriptase activity at 10 days postinfection indicated the presence of RCRs. Population sequencing of proviral DNA indicated that all RCRs contained the gag and 5′ half of pol from PreXMRV-2 and the long terminal repeat, 3′ half of pol (including integrase), and env from PreXMRV-1. All crossovers were within sequences of at least 9 identical nucleotides, and crossovers within each of two selected recombination zones of 415 nucleotides (nt) in the 5′ untranslated region and 982 nt in pol were required to generate RCRs. A recombinant with the same genotype as XMRV was not detected, and our analysis indicates that the probability of generating an identical RCR is vanishingly small. In addition, the studies indicate that the process of RCR formation is primarily driven by selection for viable cis and trans elements from the parental proviruses.
INTRODUCTION
Endogenous retroviruses (ERVs) are RNA viruses that have integrated their DNA copies into the genome of the host germ line. The integrated copy is passed down through successive generations and established as an endogenous element (1). All vertebrates possess ERVs, and ERVs constitute ∼10% of the mouse genome (2). Murine ERVs are divided into three major categories: class I ERVs (0.68% of the murine genome) are closely related to gammaretroviruses, specifically, type C murine leukemia viruses (MLVs); class II ERVs (3.14% of the murine genome) include those that share close similarity to betaretroviruses, such as mouse mammary tumor virus and intracisternal A-type particles (IAPs); and class III ERVs (5.4% of the murine genome) show close similarity to spumaviruses (reviewed in reference 3).
Murine ERVs are subject to host gene silencing, and thus, most remain transcriptionally inactive in the genome. DNA methylation of ERV promoters, the site of ERV DNA integration, overall methylation of the host genome (CpG islands), as well as other specific factors can silence ERV transcription (reviewed in reference 4). However, in contrast to humans, where almost all endogenous elements are extinct, in that no provirus is competent for replication, many murine ERVs are still active and capable of replication. Murine endogenous retroviruses that are unable to reinfect mouse host cells on the basis of their cell receptor tropism are termed xenotropic (5). However, in the context of xenografts, where foreign tissue (i.e., human tumor tissue) has been grafted into an immunocompromised mouse, expressed xenotropic murine endogenous retroviruses (X-MLVs) have the potential to infect and spread in the foreign tissue. This phenomenon was observed as early as the 1970s, when subtype C particles were found to be associated with tumor tissues passaged in mice (6–10). Surveys of currently used human cell lines have found that some are positive for MLVs, as a result of either xenografting, cross-contamination, recombination of endogenous retroviruses during passage of cultured cells, or recombination of plasmids in helper cell lines (11–14).
Identification of the xenotropic murine leukemia virus-related virus (XMRV) in 2006 (15) and its potential association with prostate cancer and chronic fatigue syndrome (CFS) (15–17) led to much controversy over whether this virus was a new human pathogen which had crossed species from mice to humans. These findings have been refuted by numerous independent laboratories, debunking any disease association with XMRV (18–21). We previously reported that XMRV was a laboratory-derived virus which resulted from recombination between two endogenous proviruses, PreXMRV-1 and PreXMRV-2 (18, 22), present in Harlan Sprague-Dawley (Hsd) nude mice, in which a human prostate tumor (CWR22) was repeatedly passaged as a xenograft. During this process, progeny of these two proviruses recombined to form a replication-competent xenotropic MLV that was able to spread throughout the human tumor tissue present in the mouse. Consequently, the 22Rv1 cell line derived from the CWR22 xenograft was also positive for XMRV (23). Since the genome sequence of XMRV from the 22Rv1 cell line was nearly identical to the genome sequences of all published patient XMRVs (24, 25), it was concluded that contamination by mouse DNA, XMRV, or XMRV RNA/DNA, often due to contamination of common laboratory reagents with mouse DNA, could explain why positive associations between XMRV and CFS or prostate cancer had been previously reported (26–33). Indeed, the association between XMRV and human prostate cancer was recently shown by the authors of the original study to be the result of contamination (34).
Although XMRV is no longer believed to be a human pathogen, the origin of XMRV provides an excellent opportunity to elucidate the mechanisms by which novel replication-competent retroviruses (RCRs) arise via retroviral recombination events. Novel viruses generated via recombination can become human pathogens; human immunodeficiency virus type 1 (HIV-1), the causative agent of AIDS, is thought to be the result of zoonotic transmission of a simian immunodeficiency virus (SIV) from chimpanzees (SIVcpz), which itself arose through recombination between two different SIVs (35). Retroviral recombination can greatly impact the evolution of viral drug resistance, immune evasion, tissue tropism, and expansion of the host range. In addition, recombination between endogenous proviruses can result in the formation of novel RCRs that can infect xenograft tissues in immunocompromised mice. Xenografting of human tumor tissues in nude mice remains an important model system in cancer biology and cancer drug discovery, and the generation and presence of RCRs in these in vivo models can complicate these studies (36). For example, it was recently shown that PreXMRV-1 contributed to the generation of another RCR, B4rv, during xenograft passaging of another human prostate tumor; furthermore, the PreXMRV-1 envelope was shown to trigger signaling in the host mouse cells that resulted in depletion of smooth muscle cells from blood vessels that infiltrated the tumor, leading to the formation of larger hemorrhagic tumors (37).
Having identified the two parental proviruses that gave rise to XMRV, we further explored the recombination potential of PreXMRV-1 and PreXMRV-2 and the probability for XMRV, or any RCR, to be generated. In the results presented in this report, we show that while RCRs could be detected, we could not detect any recombinant with the same sequence as XMRV, confirming that the probability of generating XMRV independently is extremely low.
MATERIALS AND METHODS
Plasmids and cell lines.
Plasmid VP62 (38), which expresses a full-length clone of XMRV, was a kind gift from Robert Silverman at the Cleveland Clinic. Construction of plasmid clone PreXMRV-1 was previously described (18). For the construction of plasmid clone PreXMRV-2, two overlapping halves of the provirus were amplified using flanking primers (C12-1F, C12-4R [22]) and provirus primers (M19F [5′-TGGCCTTACTGAAAGCTCTCTTCC-3′], 4438R [5′-TACTGAGTCTCTTGGGCAGGA-3′]). The two halves were digested by the SalI restriction enzyme, and the full-length provirus was cloned into pCR-Blunt (Invitrogen).
Human 293T cells (American Type Culture Collection) were maintained at 5% CO2 and 37°C in Dulbecco's modified Eagle's medium (CellGro) supplemented with 10% fetal calf serum (HyClone), penicillin (50 U/ml; Gibco), and streptomycin (50 μg/ml; Gibco). DIG (D17 indicators of gammaretroviruses) is a previously described D17-based canine osteosarcoma cell line that contains an integrated MLV-based reporter virus which contains overlapping FP and GF fragments of the green fluorescent protein (GFP) gene (GF refers to the first two-thirds of gfp, and FP refers to the last two-thirds of gfp) in the 5′and 3′ long terminal repeats (LTRs), respectively; gfp is reconstituted upon reverse transcription, and mobilization of the GFP-expressing MLV vector by a replication-competent virus increases the proportion of GFP-positive cells in the culture (39).
Hsd mouse RNAs and assay for PreXMRV-1 and PreXMRV-2 in vivo transcription.
Total DNA from mouse tail snips from 10 Hsd nude mice were prescreened for the presence of PreXMRV-1 and PreXMRV-2 using PCR primer pairs XF6 and C3-4R for PreXMRV-1 and PCR primer pairs C12_1f and 129_1r for PreXMRV-2, as previously described (22). Three mice that were positive for PreXMRV-2 only and 3 mice that were positive for both PreXMRV-1 and PreXMRV-2 were then sacrificed, and total RNA was extracted from the blood and spleens of these 6 animals, as well as two negative-control mice (BALB/cAnNHsd) obtained from Harlan Laboratories. All animal procedures used in this paper were approved by the Harlan Laboratories Institutional Animal Care and Use Committee.
Isolated RNA was treated for 30 min with DNase I using a Turbo DNA-free kit (Ambion) to remove any remaining genomic DNA (gDNA) contamination. RNAs were further confirmed to be free of gDNA contamination by quantitative PCR with a mouse β-actin probe/primer set (40) and Roche LightCycler 480 DNA Probes Master reaction mix with a LightCycler 480 Roche instrument (Roche Diagnostics), according to the manufacturer's instructions.
To determine transcription levels, RNAs were converted to cDNA using random hexamers (SuperScript III First Strand; Invitrogen); reactions with and without reverse transcriptase (RT) further confirmed a lack of DNA contamination. Quantitative PCR was performed using an mRNA β-actin primer/probe set (40) and mRNA DPPK (Mus musculus protein kinase, DNA activated, catalytic polypeptide [Prkdc]) primer/probe set: forward primer 5′-TCAAATGGTCCATTAAGCAAACAA-3′, reverse primer 5′-GCTGCACCTAGCCTCTTGAAA-3′, and probe 5′-FAM-AGCAAGTCACTTTTCAAGCGGCT-3′-TAMARA, where FAM is 6-carboxyfluorescein and TAMRA is 6-carboxytetramethylrhodamine. For PreXMRV-1 detection, the following primer/probe set was used: forward primer 5′-CCTACTACATGTTAAACCGG-3′, reverse primer 5′-ACCCCTGCCCCAATT-3′, and probe 5′-FAM-TATGTGACTGAGACCTGCACC-3′-TAMARA. For PreXMRV-2 detection, the following primer/probe set was used: forward primer 5′-AAAAGAGACCTGGACACTCT-3′, reverse primer 5′-CCACCTCGTGGAGGATCTTATCTTT-3′, and probe 5′-FAM-TGCCCAGTCAAGACACCTTCGAACTT-3′-TAMARA.
Assay for PreXMRV-1 and PreXMRV-2 ex vivo transcription.
PreXMRV-1, PreXMRV-2, or XMRV was transfected (3 independent transfections) by calcium phosphate precipitation into 293T cells plated at 5 × 106 cells per 100-mm-diameter dish. Forty-eight hours later, total RNA was extracted using an RNeasy minikit (Qiagen) and treated with DNase I using a Turbo DNA-free kit (Ambion). RNA was converted to cDNA using random hexamers and confirmed to be negative for gDNA contamination by carrying out the reaction in the absence of reverse transcriptase. Total RNA input was normalized using the following mRNA glyceraldehyde 3-phosphate dehydrogenase (GAPDH) primer/probe set: forward primer 5′-TCCACCCATGGCAAATTC-3′, reverse primer 5′-AGACGCCAGTGGACTC-3′, and probe 5′-FAM-TTTCCATTGATGACAAGCTTCC-3′-TAMARA. GAPDH cDNA plasmid DNA was used to generate a standard curve to estimate RNA copy numbers. To determine PreXMRV-1 and PreXMRV-2 RNA copy numbers, the following universal envelope primer/probe set was used with VP62 plasmid DNA as the standard curve: forward primer 5′-TCAGGACAAGGGTGGTTTGAG-3′, reverse primer 5′-GGCCCATAATGGTGGATATCA-3′, and probe 5′-FAM-TTAACAGGTCCCCATGGTTCACGACC-3′-TAMARA.
Western blot analysis.
293T cells transfected with PreXMRV-1, PreXMRV-2, or XMRV were lysed for Western blot analysis at 48 h posttransfection, as previously described (41). Supernatants were also collected, filtered through a Millex GS 0.45-μm-pore-size filter (Nalgene), concentrated 100-fold by centrifugation at 25,000 rpm for 90 min (Surespin; Sorvall), and stored at −80°C. Cell lysates and viral pellets were assayed using an anti-MLV capsid monoclonal antibody at a 1:200 dilution (42). Total protein in the cell lysate was assessed using mouse antitubulin antibody (Sigma) at a 1:10,000 dilution. The primary antibodies were detected with horseradish peroxidase-labeled goat antimouse secondary antibody at a 1;10,000 dilution (Sigma), and the proteins were visualized using a Western Lightning Plus chemiluminescence reagent kit (PerkinElmer).
Assay for infectious PreXMRV-1 or PreXMRV-2.
Filtered viral supernatants obtained at 48 h posttransfection from 293T cells transfected with PreXMRV-1, PreXMRV-2, or XMRV were used to infect DIG cells. Cells were monitored for 1 week to 1 month by fluorescence-activated cell sorter (FACS) analysis for the presence of GFP-positive cells.
PreXMRV-1 and PreXMRV-2 cotransfection.
293T cells plated at 2 × 105 cells/well of a 6-well plate were cotransfected with PreXMRV-1 and PreXMRV-2 plasmid DNA via calcium phosphate precipitation. Six independent transfections (transfections A to F) were performed and allowed to expand for 1 week before collecting virus. Cell supernatants were then harvested, filtered, and treated at 37°C with DNase I (5 U) plus DpnI (10 U) for 2.5 h. DNA was extracted (QIAamp DNA blood minikit; Qiagen) from an aliquot of the final supernatant and tested for plasmid or gDNA contamination using PCR primers for GAPDH (intron primers GAPDHfor [5′-CCCCACACACATGCACTTACC-3′] and GAPDHrev [5′-CCTACTCCCAGGGCTTTGATT-3′]) and universal PCR primers for PreXMRV-1 and PreXMRV-2 (primers univPol For [5′-CCGGATCCAGCACCCAGACTTG-3′] and univEnvRev [5′-CACGGGTTAATCTTATCTTTAAGGG-3′]). Target DIG or 293T cells were plated at 2 × 105 cells/well of a 60-mm-diameter dish and infected with 2 ml of undiluted supernatant in the presence of 60 μl Polybrene (1 mg/ml) from each transfection for 3 h. Cells were then washed with medium and fresh medium was added. Every 3 days, the infected DIG cells were analyzed by FACS for the presence of GFP-positive cells. Concurrently, every 3 days, a portion of the infected 293T cells was lysed for DNA extraction. Primers used for bulk sequencing and single clones were as follows: univRfor (5′-CCCGTGTTCCCAATAAAGCC-3′), midGagrev (5′-CCAGCGATACCGCTTTCCTCCAGTAGCC-3′), midGagfor (5′-GGCTACTGGAGGAAAGCGGTATCGCTGG-3′), univPolrev (5′-CAAGTCTGGGTGCTGGATCCGG-3′), univPolfor (5′-CCGGATCCAGCACCCAGACTTG-3′), univEnvrev (5′-CACGGGTTAATCTTATCTTTAAGGG-3′), univEnvfor (5′-CCCTTAAAGATAAGATTAACCCGTG-3′), and univRrev (5′-GGCTTTATTGGGAACACGGG-3′).
Nucleotide sequence accession numbers.
GenBank accession numbers for the recombinant clones presented in Fig. 5A to D are as follows: panel A, KF584043 to KF584051; panel B, KF584023 to KF584028; panel C, KF584029 to KF584042; and panel D, KF584014 to KF584022.
Fig 5.

Sequencing of recombinant clones. Proviral clones from infections A to F were sequenced in four regions: R/U5 to protease (A), protease to the 5′ half of pol (B), the 3′ half of pol to the end of IN (C), and the end of IN to U3/R (D). For ease of comparison, each sequence is represented as Hypermut plots; colored vertical hash marks represent nucleotide differences from PreXMRV-1, as defined by Hypermut (red, GG → GA; cyan, GA → AA; green, GC → AC; magenta, GT → AT; black, no G → A transition; yellow, gaps). The PreXMRV-1 sequence is depicted as a solid black line. For each infection, the different proviral clones (lowercase letters) and the number of each type of clone are shown. The nucleotide sequence is numbered according to the PreXMRV-1 sequence (GenBank accession number NC_007815) when aligned against PreXMRV-2 (GenBank accession number FR871850). The 415-nt and 982-nt selected recombination zone used by the RCRs in the UTR (A) and pol domain (B), respectively, are shown as a black box. The relative positions of R/U5, gag, protease (pro), reverse transcriptase (RT), connection subdomain (CN), RNase H (RH), integrase (IN), envelope (env), envelope surface unit (SU) glycoprotein, envelope transmembrane subunit (TM), and U3/R are labeled. Sequences of clones presented in this figure were deposited in GenBank.
RESULTS
PreXMRV-1 and PreXMRV-2 RNA transcripts are undetectable in the blood and spleen of Hsd nude mice.
Previously, it was determined by our groups that the Hsd nude mouse population (which is maintained as an outbred stock) harbors integrated copies of PreXMRV-1 and/or PreXMRV-2, with 53% of the mice being positive for both PreXMRV-1 and PreXMRV-2 (22). To determine the levels of expression of these proviruses in mouse tissues, total RNA was extracted from the blood and spleen of three Hsd nude mice positive for both proviruses, three Hsd nude mice positive for PreXMRV-2 only, and two BALB/cAnNHsd mice negative for both proviruses. High-level transcripts, such as the β-actin transcript (average ± standard deviation, 2,461 ± 1,084 copies/ng cDNA), and rare transcripts, such as the DPPK transcript (average ± standard deviation, 1.9 ± 1.3 copies/ng cDNA), were detected at their expected high and low levels, respectively, from either the blood or spleen of all mice, suggesting that the purified RNA was of good quality and abundant and rare RNA species were well represented (Fig. 1). However, primer/probe sets specific for PreXMRV-1 or PreXMRV-2 were unable to detect any PreXMRV-1 or PreXMRV-2 RNA transcripts (<0.2 copies/ng cDNA). Assuming, on average, 10 pg of total RNA per cell (43), PreXMRV-1 and PreXMRV-2 were present at less than <0.002 copy/cell in either the spleen or blood.
Fig 1.
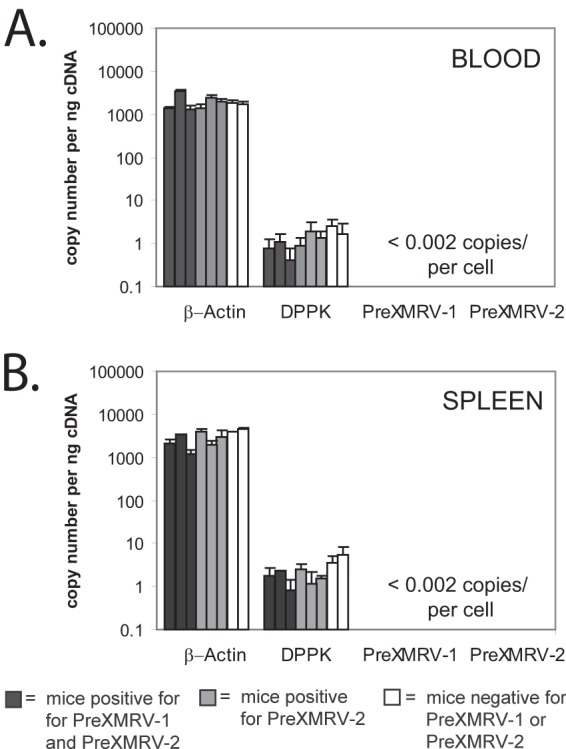
PreXRMV-1 and PreXMRV-2 RNA quantitation in Hsd nude mouse cells. Quantitative real-time reverse transcription-PCR analysis of RNA transcripts from the blood (A) and spleen (B) of Hsd nude mice carrying both PreXMRV-1 and PreXMRV-2 (dark gray bars), Hsd nude mice carrying only PreXMRV-2 (light gray bars), and BALB/cAnNHsd mice negative for both PreXMRV-1 and PreXMRV-2 (white bars). Nucleic acid quality and quantity were verified by testing for highly expressed β-actin RNA and poorly expressed DPPK RNA. Using an estimate of 10 picograms of RNA per cell, blood and spleen cells of Hsd mice contained <0.002 copies of PreXMRV-1 and PreXMRV-2 RNA.
PreXMRV-1 and PreXMRV-2 are noninfectious.
Sequence analysis of PreXMRV-1 indicated that a 16-nucleotide (nt) deletion leading to a frameshift in the capsid gene and a single-nucleotide insertion leading to a frameshift in pol rendered this virus replication incompetent (18). However, the gag-pro-pol and env reading frames are open in PreXMRV-2, suggesting that it has the potential to be replication competent (18). To determine their replication potential, proviral molecular clones of PreXMRV-1, PreXMRV-2, and XMRV, as a control, were independently transfected into 293T cells. Using Western blot analysis, we were unable to detect capsid proteins in either the cell lysates or viral pellets from PreXMRV-2; however, we were able to detect the presence of XMRV (Fig. 2A). Since PreXMRV-1 has a frameshift mutation in the capsid gene, only the first 101 amino acids of the capsid (total length, 263 amino acids) would be translated, and the capsid may not react with the anti-MLV capsid monoclonal antibody. However, despite the lack of protein detection on the Western blot, both PreXMRV-1 and PreXMRV-2 RNA were found in 293T cells at similar levels following transfection (Fig. 2B). A real-time reverse transcription-PCR primer/probe set designed to detect env sequences was used to detect both unspliced and spliced transcripts. PreXMRV-1 and PreXMRV-2 transcripts were detected at levels of 38% and 30%, respectively, compared to the level of XMRV. Of note, the XMRV positive control was transcribed from the cytomegalovirus (CMV) promoter, whereas PreXMRV-1 and PreXMRV-2 were transcribed from their LTR promoters. Regardless, the results suggest that there was a posttranscriptional block to stable expression of viral capsid protein from the PreXMRV-2 provirus, the reasons for which may include aberrant splicing, inhibition of translation, and/or instability of the protein.
Fig 2.
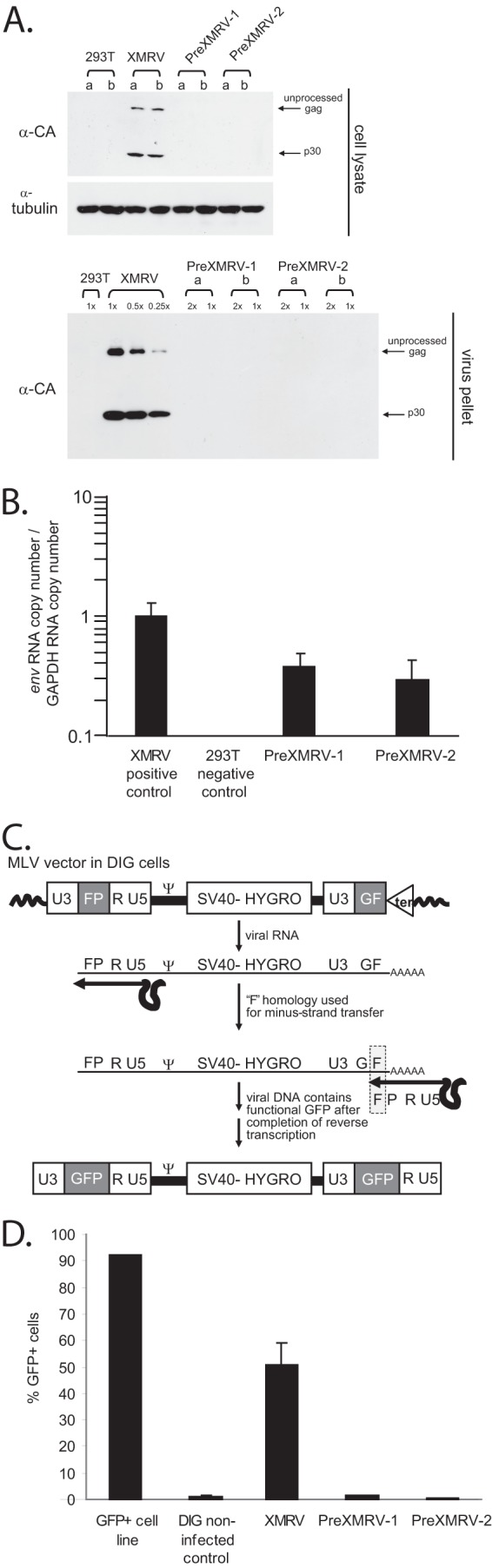
Absence of infectious PreXMRV-1 and PreXMRV-2 virus. (A) Western blotting of cell lysates and virus from 293T cells transfected with XMRV (VP62), PreXMRV-1, or PreXMRV-2. MLV unprocessed Gag and p30 capsid were detected in XMRV-transfected cells using the monoclonal MLV anti-capsid antibody (α-CA) but not in PreXMRV-1- or PreXMRV-2-transfected 293T cells (top). a and b, two independent transfections. α-Tubulin was used as a loading control. Viral supernatants were concentrated 100-fold and analyzed at the indicated dilutions. (B) Relative env RNA copy numbers normalized to GAPDH copy numbers for XMRV, PreXMRV-1, and PreXMRV-2. Quantitative PCR using a universal env primer/probe set able to detect VP62, PreXMRV-1, and PreXMRV-2 was performed after conversion of total cytoplasmic RNA to cDNA using random hexamer primers. (C) Monitoring the presence of RCRs using DIG cells. During reverse transcription, the F portion of gfp is used for minus-strand transfer, resulting in reconstitution of a functional gfp to generate GFP-positive cells; furthermore, the retroviral vector containing the reconstituted gfp can spread through the culture in the presence of an RCR. Ψ, MLV packaging signal. (D) PreXMRV-1 and PreXMRV-2 are noninfectious. Supernatant harvested from 293T cells transfected with XMRV, PreXMRV-1, or PreXMRV-2 was used to infect target DIG cells. A GFP-positive D17 cell line, A3GFP11, was used as a positive control for FACS analysis (52).
To use a more sensitive assay to test for viral production, supernatants from the PreXMRV-1-, PreXMRV-2-, and XMRV-transfected 293T cells were used to infect DIG reporter cells. Briefly, DIG cells contain a Ψ-positive MLV (Δgag, Δpol, and Δenv) with partially redundant portions of the gfp gene in both LTRs (shown in Fig. 2C as GF and FP). In the presence of a replicating virus, these RNA transcripts are packaged and reconstitute GFP upon reverse transcription. The replicating virus also serves as a helper virus to mobilize the new GFP-positive provirus and subsequently detect GFP expression throughout the DIG cell culture. Thus, DIG cells treated with supernatants from cells transfected with PreXMRV-1, PreXMRV-2, or XMRV were monitored for increases in GFP expression and fluorescence by FACS analysis from 1 week to 1 month. A high proportion of DIG cells infected with XMRV (50%) was GFP positive, confirming that XMRV is replication competent; however, GFP-positive cells could not be detected after infection with PreXMRV-1 or PreXMRV-2 (Fig. 2C), indicating that PreXMRV-1 and PreXMRV-2 alone were not able to generate infectious virions. Similar results were previously obtained by transfection of another XMRV-susceptible cell line with cloned full-length PreXMRV-2 provirus (22) (data not shown).
Recombination potential of PreXMRV-1 and PreXMRV-2.
To assess the recombination potential of PreXMRV-1 and PreXMRV-2, 293T cells were cotransfected with their proviral clones. One week later, supernatants were harvested and used to infect fresh 293T or DIG target cells (Fig. 3). Supernatant was harvested at 1 week posttransfection to give the cultures ample time to produce virus. FACS analysis of the DIG cell cultures was performed to monitor the appearance of GFP-positive cells, indicating the production of an RCR. At 3 days postinfection, there was a negligible GFP signal, but at 6 days postinfection, 4 to 16% of the cells were GFP positive in the 6 independent infections (infections A to F), and at 10 days postinfection, 64 to 73% of cells were GFP positive. Thus, recombination between PreXMRV-1 and PreXMRV-2 in each culture gave rise to one or more RCRs that mobilized the MLV vector in the DIG cells to reconstitute the gfp gene.
Fig 3.

Cotransfection of PreXMRV-1 and PreXMRV-2 produces replication-competent virus. (A) Protocol. Supernatants were harvested at 7 days postcotransfection of 293T cells with PreXMRV-1 and PreXMRV-2 (6 independent cotransfections) and then split to infect either DIG or 293T target cells. The DIG cultures were monitored every 3 days for GFP-positive cells; concurrently, 293T cells were split every 3 days and frozen, and DNA was extracted. (B) FACS analysis of DIG cells at 3, 6, and 10 days postinfection for infections labeled A to F.
Population sequencing analysis of proviruses.
Since each of the six replicates likely contained numerous viral variants, we amplified different regions of the proviruses by PCR and directly sequenced the PCR products to obtain an average sequence of the proviral population. The population sequencing was initially performed for a region spanning the polymerase, connection subdomain, and RNase H domain of pol, as well as env (Fig. 4). Figure 4 plots the percentage of PreXMRV-1 and PreXMRV-2 signals from the sequencing chromatograms at each position where PreXMRV-1 and PreXMRV-2 differ in nucleotide sequence. The population sequencing plots for the pol regions in Fig. 4A show that infections A, B, and D contained pools of viral variants, while infections E and F likely contained only 2 viral variants that differed at a single nucleotide position at the crossover site. For infection C, only one viral variant was detectable by bulk sequencing, with the crossover occurring in a region of nucleotide similarity between PreXMRV-1 and PreXMRV-2. Interestingly, for env in Fig. 4B, infections A, C, D, and F showed that all the viral variants in the population shared 100% sequence identity with PreXMRV-1, while infections B and E showed that the viral variants shared some short stretches of identity with PreXMRV-2. These results show that, while env from PreXMRV-1 is predominantly present in the RCRs, replacement of some of these regions of PreXMRV-1 env with the homologous regions of PreXMRV-2 env is compatible with preserving PreXMRV-1 env function.
Fig 4.

Recombinant populations scanned by population sequencing. The percentage of the PreXMRV-1 sequence (blue diamonds) and the percentage of the PreXMRV-2 sequence (purple squares) in the amplified provirus pools from the experiment whose results are shown in Fig. 3 were plotted. (A) pol (3′ half of pol and connection and 5′half of RNase H) for infections (inf) A to F. cn, connection subdomain; rh, RNase H. (B) env regions of the recombinant populations for infections A to F. Every blue diamond or purple square represents a position where PreXMRV-1 and PreXMRV-2 differ in nucleotide sequence. The percentage of each base corresponding to PreXMRV-1 or PreXMRV-2 is plotted on the basis of peak heights from the raw chromatograms.
Sequence analysis of cloned PCR products confirms population sequencing data.
Since minor viral variants would not be detectable by population sequencing, single PCR-amplified molecules were cloned and sequenced for all infections across the entire genome. The genome was divided into 4 regions starting with R/U5 to the beginning of pro, the beginning of pro to the conserved YVDD box in pol, the YVDD box in pol to the end of the integrase (IN)/start of env, and the start of env to U3/R. Figure 5 shows individual clones sequenced for each region and for each infection (infections A to F), plotted using the Hypermut HIV sequence database to facilitate visualization of PreXMRV-1 and PreXMRV-2 sequence differences (http://www.hiv.lanl.gov/content/sequence/HYPERMUT/hypermut.html). For all clones, a minimum of 9 nt of sequence identity was present at the site of a crossover between PreXMRV-1 and PreXMRV-2. All of the analyzed clones exhibited recombination events within the region from R/U5 to the beginning of gag (Fig. 5A and 6A). All clones from infections D, E, and F contained sequences matching the XMRV sequence 100%, indicating that these clones used the same 27-nt window in the untranslated region (UTR) upstream of the Gag start codon to cross over between PreXMRV-1 and PreXMRV-2 (Fig. 6A). The precise location within the 27-nt window in which the crossover occurred cannot be determined, since the two parent viruses have the same nucleotide sequence in this region. A total of 6 different recombination junctions was observed among the 6 infections for this region (Fig. 5A and 6A). As shown in Fig. 5B, all clones from all infections maintained 100% PreXMRV-2 sequence identity in the region from the beginning of protease to the YVDD box in pol. For the region starting with the YVDD box in pol to the end of IN (Fig. 5C and 6B), 14 different recombination junctions were observed among the 6 infections, with only one clone in infection B using the same 46-nt region of identity between PreXMRV-1 and PreXMRV-2 that XMRV used to undergo a crossover event. Lastly, the region from the end of IN to U3/R (Fig. 5D and 6C) showed no clones with 100% identity to XMRV. Most clones maintained the PreXMRV-1 sequence for all of env and U3/R, with clones from infections B and E containing stretches of PreXMRV-2 sequence. A total of 6 crossover events was observed among the clones from the 6 infections (Fig. 6C). In infection C, one minor recombinant (1 of 6) was detected by sequencing of cloned PCR products (Fig. 5C) but was not detected by bulk sequencing (Fig. 4A). With the exception of the results for this clone, the results of the single-clone sequencing analysis matched the population sequencing data, indicating that the population sequencing results were generally representative of those for the viral variants present in each population. Additionally, analysis of population sequencing data from days 3, 6, and 10 for reverse transcriptase (RT) or env in the recombinants did not vary (data not shown), suggesting that selection for the predominant RCRs in each infection had already occurred by day 3. Finally, these data show that the recombinants preferred the gag and 5′ half of pol from PreXMRV-2 and the LTR, 3′ half of pol (including IN), and env from PreXMRV-1.
Fig 6.



Detailed mapping of the recombination junctions for each recombinant clone. (A) 5′ UTR nucleotide sequence for proviral clones from infections A to F, with the 415-nt selected recombination zone used by the clones marked with arrows. Characteristic markings for Ψ are noted (PAL1, PAL2, SL1, SL2), as is the characteristic XMRV 24-nt deletion in the 5′ UTR. Glyco-gag, the gag start, and the splice donor (SD) are marked. (B) RT sequence for proviral clones from infections A to F, with the 982-nt selected recombination zone used by the clones marked with arrows. The nucleotide and amino acid (aa) sequences of the 3′ pol (POL), connection subdomain (CN), and RNase H (RH) are shown for PreXMRV-1 and PreXMRV-2, with amino acid changes between PreXMRV-1 and PreXMRV-2 shaded in gray boxes. The polymerase active site, YVDD, is boxed in black. (C) Envelope nucleotide and amino acid sequences for proviral clones b2, b3, and e1 from infections B and E are shown; the sequences of the remainder of the clones are identical to the sequence of PreXMRV-1. The portions of the surface unit and transmembrane domains are noted. Purple, blue, and yellow boxes, the 3 clones (b2, b3, and e1, respectively; see also Fig. 5D) that underwent two crossovers in env between PreXMRV-1 and PreXMRV-2 (noted as bent arrows). For panels A to C, the regions in which template switching occurred during the formation of XMRV are highlighted with gray shaded boxes (crossover numbers are labeled as in Fig. 7 and reference 18). Black boxes (or colored boxes in panel C) highlight the crossover points used by each clone (lowercase letter) from each infection (infections A to F) (also cross-reference Fig. 5). The nucleotide sequence is numbered according to the sequence of PreXMRV-1 (GenBank accession number NC_007815). Identical nucleotides or amino acids between PreXMRV-1 and PreXMRV-2 are noted by a dot.
Estimation of the probability of generating an RCR with the same sequence as XMRV.
On the basis of data from the analyzed recombinant clones, the probability that a recombinant with the same genotype as XMRV would be generated can be calculated by asking how many of the recombinants used the same regions of identity as XMRV to undergo a crossover event (Fig. 7). None of the recombinant clones used crossover 1, 2, 3, or 4 that XMRV used in env (<1/34 total clones analyzed), and only 1/26 total clones used the same region of identity in crossover 5 in pol as XMRV. A total of 12/26 clones used the same identity region in the UTR before the start of gag that XMRV used. Multiplying these numbers together (12/26 × 1/26 × <1/34 × <1/34 × <1/34 × <1/34), on the basis of the results obtained for the clones analyzed, the probability that XMRV would be generated is <1.3 × 10−8. However, it must be noted that the actual probability is likely to be much lower, in view of the following considerations: (i) the probability that an Hsd nude mouse possesses both PreXMRV-1 and PreXMRV-2 is approximately 50%; (ii) the probability that PreXMRV-1 and PreXMRV-2 would be transcribed in the same mouse cell at the same time is unknown but is likely to be low, on the basis of the undetectable levels of RNA transcripts in blood and spleen; (iii) the probability of copackaging of the PreXMRV-1 and PreXMRV-2 RNAs to form a heterozygous virion is low, since MLVs have a lower frequency of heterozygous virion formation than expected by random copackaging (44, 45); (iv) the probability that proteins expressed from PreXMRV-1 and PreXMRV-2 do not complement to generate an infectious virus, and a third helper virus is needed to copackage the two parental genomes is low; and (v) the probability for a heterozygous virus to infect xenograft tissue, recombine to form an RCR, and spread in the xenograft is unknown but is likely to be low, since XMRV was not detected for at least three passages in nude mice (18), constituting several months of xenograft passage in mice.
Fig 7.
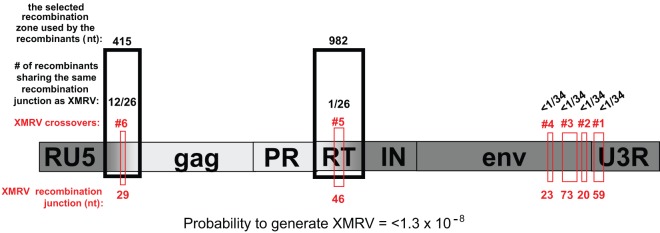
Selected recombination zones and the probability of generating a recombinant with exactly the same sequence as XMRV. A 415-nt region in the 5′ UTR and a 982-nt region in RT are defined as the selected recombination zones. A crossover within each of these selected recombination zones was observed in all RCRs and is postulated to be essential for formation of an RCR. Using the data from the recombinants from infections A to F, the number of recombinants that used the same region for any of the 6 crossover switches between PreXMRV-1 and PreXMRV-2 that generated XMRV is listed (red boxes). For example, in the 5′ UTR, 15 of the 26 clones analyzed used the same region as XMRV for a crossover. Multiplying together the number of clones that used the same homology region as XMRV for crossover events (12/26 × 1/26 × <1/34 × <1/34 × <1/34 × <1/34), the probability of generating XMRV is <1.3 × 10−8. Dark gray shading, PreXMRV-1 sequences; light gray shading, PreXMRV-2 sequences.
DISCUSSION
Retroviral recombination is a powerful mechanism that can shuffle genomes to generate novel viruses. The identification of PreXMRV-1 and PreXMRV-2 as the parental endogenous proviruses that recombined to generate XMRV provides a rare opportunity to elucidate the process and mechanisms by which RCRs are generated from endogenous proviruses. We previously reported that XMRV was not present in the 3rd-passage CWR22 xenografts but was detectable in 7th-passage xenografts (18); since each passage represented several months in a mouse, the xenograft was in nude mice for more than a year before XMRV could be detected. Thus, the formation of an RCR through recombination of PreXMRV-1 and PreXMRV-2 and its successful infection of the human tumor tissue were rare events. The results of studies presented here suggest at least two important factors that acted as impediments to the formation of XMRV and its spread in the human tumor tissues. First, undetectable levels of PreXMRV-1 or PreXMRV-2 RNA in the Hsd nude mouse blood or spleen suggest that these proviruses are rarely transcribed. Second, neither PreXMRV-1 nor PreXMRV-2 alone can form infectious virions, suggesting that either their expressed proteins complement each other to form infectious virions or they undergo recombination to generate an RCR. As previously proposed (22), assuming that they do not complement each other to from infectious virions, a third endogenous provirus may have been needed as a helper virus to copackage PreXMRV-1 and PreXMRV-2 RNAs so that an RCR could be generated.
The low undetectable transcript level of PreXMRV-1 and PreXMRV-2 in blood and spleen is not unusual, since most murine endogenous proviruses are transcriptionally silenced during early embryonic stages and usually remain inactive in the host genome throughout the life of the mouse. However, the possibility that PreXMRV-1 and PreXMRV-2 are transcriptionally active in some mouse tissue or at some stage in the life of the mouse cannot be excluded. It is also possible that treatment of Hsd nude mice with androgens during the xenograft passaging led to the activation of these proviruses, thereby contributing to the formation of XMRV.
We expected PreXMRV-1 to be replication defective because it has a deletion/frameshift mutation in gag and an insertion/frameshift mutation in pol. However, we were surprised that PreXMRV-2 is also replication defective, even though its gag, pol, and env reading frames are open and have no obvious missense mutations. Furthermore, PreXMRV-2 did not express any detectable capsid protein in transfected 293T cells, even though PreXMRV-2 RNA could be detected at about 30% of the level observed for VP62, an XMRV molecular clone that transcribes viral RNA from a CMV promoter (38). PreXMRV-2 RNA was quantified using a primer/probe set targeting env and can detect both spliced and unspliced RNA; thus, it is possible that Gag was not expressed because of aberrant splicing that results in the loss of full-length RNA, which is used to express Gag-Pol, but not spliced RNA, which is used to express Env. Other possible reasons for the lack of Gag expression include suppression of Gag translation or microRNA-induced degradation of full-length RNA.
Our results showed that even though PreXMRV-1 and PreXMRV-2 were replication defective, they could readily recombine to generate RCRs. Although recombination of the endogenous proviruses in mice most likely occurs only during reverse transcription, the RCRs analyzed in this experiment could have resulted from recombination during transfection or reverse transcription. It is likely that recombination events can occur anywhere in the viral genome during either transfection or reverse transcription and RCRs with the same structures can form regardless of the mechanism of recombination. Another possibility is that in mice a third provirus acts as a helper virus to copackage PreXMRV-1 and PreXMRV-2 RNAs, which is a prerequisite to their recombination during reverse transcription. However, since there are no potential helper viruses in DIG cells, recombination during transfection was required to generate the RCRs. Regardless of the mechanism, the recombinants generated in vivo and in this cell culture system were selected for their ability to replicate efficiently and become RCRs; hence, the RCRs selected in this study reflect the RCRs that are likely to be subjected to selection in mice and provide insights into the capacity of PreXMRV-1 and PreXMRV-2 to generate RCRs.
The structures of the RCRs indicate that all of the recombinants had a crossover within a 415-nt selected recombination zone within the 5′ UTR before the start of gag and another crossover within a 982-nt selected recombination zone in pol. The term “selected recombination zone” does not imply that this region was a hot spot for RT template switching. Recombination events in these two zones were strongly selected and were presumably essential for the formation of the RCRs. Thus, there was a strong selection for the recombinants to maintain the LTR, 3′ pol (C-terminal portions of RT and IN), and env of PreXMRV-1 origin and the gag, pro, and 5′ pol (N-terminal half of RT) of PreXMRV-2 origin. The reason for the selection of the PreXMRV-1 LTR is not clear, since the PreXMRV-2 LTR appears to be functional in directing transcription in transfected 293T cells. While the selection for PreXMRV-2 gag-pol was expected because of mutations in gag and the beginning of pol in the PreXMRV-1 genome, it appears that the PreXMRV-2 IN may be defective or poorly functional in the recombinants since PreXMRV-1 IN was strongly selected. The attachment sites for integration in PreXMRV-1, which are present in all RCRs, and PreXMRV-2 are identical, making it unlikely that these elements contributed to the selection of PreXMRV-1. PreXMRV-1 and PreXMRV-2 IN primary sequences are quite divergent and share only 84% nucleotide sequence identity (87% amino acid sequence identity). It is possible that the PreXMRV-2 IN is either defective or has low activity, leading to the selection of PreXMRV-1 IN; however, additional studies are needed to test this hypothesis.
Closer examination of the 5′ UTR shows that all RCRs had a recombination junction within the 415-nt selected recombination zone in the 5′ UTR (Fig. 6A), which contains important cis elements, such as the primer binding site (PBS), packaging signal, splice donor site, and N-terminal region of glyco-Gag. Nearly all of the 5′ UTRs from PreXMRV-1 and PreXMRV-2 were present in at least one RCR, suggesting that these cis elements are functional in both parental proviruses. For example, while most RCRs contained the PBS from PreXMRV-1, which is complementary to proline tRNA, one RCR contained the PBS from PreXMRV-2, which is complementary to glutamine tRNA. Both tRNAs are known to support MLV replication (46–48). It was recently shown the glyco-Gag potentially expressed from XMRV is a truncated nonfunctional protein and that replication of XMRV was enhanced by expression of MLV glyco-Gag (49). Most RCRs as well as XMRV retained the PreXMRV-2 glyco-Gag open reading frame (ORF; 53 amino acids), but one recombinant did contain the PreXMRV-1 glyco-Gag ORF (26 amino acids).
It is interesting to note that none of the RCRs recombined within the 943-nt region between the Gag start codon and the frameshift/deletion mutation in the capsid gene of PreXMRV-1, suggesting that recombinant Gag proteins may be defective or have reduced function. It is also noteworthy that the majority of recombinants had crossovers within palindrome 2 (PAL2) (Fig. 6A) of the Ψ sequence (50, 51), suggesting that the RNA structure within this region may have influenced the crossover frequency.
Although there are many nucleotide differences between PreXMRV-1 and PreXMRV-2 within the 982-nt selected recombination zone (Fig. 6B), there are only 4 amino acid differences within RT, 3 amino acid differences within the connection subdomain, and 5 amino acid differences within the RNase H domain. Since the amino acids from both parental proviruses were present in one or more RCRs, we conclude that none of these amino acid differences is detrimental to RT function. Since regions of PreXMRV-2 RNase H downstream of the 982-nt selected recombination zone, as well as the PreXMRV-2 IN, were not present in any of the RCRs, we assume that one or more of the amino acid differences present in PreXMRV-2 are detrimental for function. On the other hand, PreXMRV-2 sequences within and upstream of the 982-nt selected recombination zone are functional and present in some or all RCRs.
The observation that all of the RCRs maintained PreXMRV-1 env indicates that the PreXMRV-1 env is fully functional and that the PreXMRV-2 env is most likely nonfunctional, although some portions of PreXMRV-1 env can be replaced with PreXMRV-2 env without a loss of function (Fig. 6C). Interestingly, two of the three RCRs resulted in one env amino acid change as a result of recombination between PreXMRV-1 and PreXMRV-2. Furthermore, no recombinant used the same regions of identity between PreXMRV-1 and PreXMRV-2 in env that were used to generate XMRV, again confirming that the likelihood of generating a recombinant with the exact XMRV sequence is very low. Based on the analysis of a limited number of recombinants generated during this study, we estimated that less than 1 in 1.3 × 108 RCRs resulting from recombination between the two proviruses would have a structure that is identical to that of XMRV (Fig. 7).
Retroviral recombination allows the reassortment of mutations and genetic polymorphisms, leading to the generation of RCRs from otherwise defective parental viruses. The number of recombinants generated from PreXMRV-1 and PreXMRV-2 in this study could have been quite large, but only those that were able to replicate were identified in this system. Analysis of the recombinants between these parental proviruses has provided insights into the structure and function of the xenotropic Gag-Pol and Env proteins encoded by PreXMRV-1 and PreXMRV-2. In addition, these studies indicate that retroviral recombination is crucial for generating recombinants by combining functional cis and trans elements from the parental proviruses and that selection for these functional elements results in the formation of novel RCRs.
ACKNOWLEDGMENTS
The content of this publication does not necessarily reflect the views or policies of the U.S. Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
This work was supported in part by a Bench-to-Bedside Award to V.K.P. and grant R37 CA 089441 to J.M.C. from the National Cancer Institute. J.M.C. was a research professor of the American Cancer Society with support from the F. M. Kirby Foundation.
Footnotes
Published ahead of print 21 August 2013
REFERENCES
- 1.Cohen JC, Varmus HE. 1979. Endogenous mammary tumour virus DNA varies among wild mice and segregates during inbreeding. Nature 278:418–423 [DOI] [PubMed] [Google Scholar]
- 2.Waterston RH, Lindblad-Toh K, Birney E, Rogers J, Abril JF, Agarwal P, Agarwala R, Ainscough R, Alexandersson M, An P, Antonarakis SE, Attwood J, Baertsch R, Bailey J, Barlow K, Beck S, Berry E, Birren B, Bloom T, Bork P, Botcherby M, Bray N, Brent MR, Brown DG, Brown SD, Bult C, Burton J, Butler J, Campbell RD, Carninci P, Cawley S, Chiaromonte F, Chinwalla AT, Church DM, Clamp M, Clee C, Collins FS, Cook LL, Copley RR, Coulson A, Couronne O, Cuff J, Curwen V, Cutts T, Daly M, David R, Davies J, Delehaunty KD, Deri J, Dermitzakis ET, et al. 2002. Initial sequencing and comparative analysis of the mouse genome. Nature 420:520–562 [DOI] [PubMed] [Google Scholar]
- 3.Stocking C, Kozak CA. 2008. Murine endogenous retroviruses. Cell. Mol. Life Sci. 65:3383–3398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Maksakova IA, Romanish MT, Gagnier L, Dunn CA, van de Lagemaat LN, Mager DL. 2006. Retroviral elements and their hosts: insertional mutagenesis in the mouse germ line. PLoS Genet. 2:e2. 10.1371/journal.pgen.0020002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Levy JA. 1973. Xenotropic viruses: murine leukemia viruses associated with NIH Swiss, NZB, and other mouse strains. Science 182:1151–1153 [DOI] [PubMed] [Google Scholar]
- 6.Todaro GJ, Arnstein P, Parks WP, Lennette EH, Huebner RJ. 1973. A type-C virus in human rhabdomyosarcoma cells after inoculation into NIH Swiss mice treated with antithymocyte serum. Proc. Natl. Acad. Sci. U. S. A. 70:859–862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Achong BG, Trumper PA, Giovanella BC. 1976. C-type virus particles in human tumours transplanted into nude mice. Br. J. Cancer 34:203–206183801 [Google Scholar]
- 8.Suzuki T, Yanagihara K, Yoshida K, Seido T, Kuga N. 1977. Infectious murine type-C viruses released from human cancer cells transplanted into nude mice. Gann 68:99–106 [PubMed] [Google Scholar]
- 9.Gautsch JW, Knowles AF, Jensen FC, Kaplan NO. 1980. Highly efficient induction of type C retroviruses by a human tumor in athymic mice. Proc. Natl. Acad. Sci. U. S. A. 77:2247–2250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ito YZ, Nakazato Y. 1984. A new serially transplantable human prostatic cancer (HONDA) in nude mice. J. Urol. 132:384–387 [DOI] [PubMed] [Google Scholar]
- 11.Williams DK, Galvin TA, Ma H, Khan AS. 2011. Investigation of xenotropic murine leukemia virus-related virus (XMRV) in human and other cell lines. Biologicals 39:378–383 [DOI] [PubMed] [Google Scholar]
- 12.Zhang YA, Maitra A, Hsieh JT, Rudin CM, Peacock CD, Karikari C, Brekken RA, Stastny V, Gao B, Girard L, Wistuba I, Frenkel E, Minna JD, Gazdar AF. 2011. Frequent detection of infectious xenotropic murine leukemia virus (XMLV) in human cultures established from mouse xenografts. Cancer Biol. Ther. 12:617–628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sfanos KS, Aloia AL, Hicks JL, Esopi DM, Steranka JP, Shao W, Sanchez-Martinez S, Yegnasubramanian S, Burns KH, Rein A, De Marzo AM. 2011. Identification of replication competent murine gammaretroviruses in commonly used prostate cancer cell lines. PLoS One 6:e20874. 10.1371/journal.pone.0020874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Takeuchi Y, McClure MO, Pizzato M. 2008. Identification of gammaretroviruses constitutively released from cell lines used for human immunodeficiency virus research. J. Virol. 82:12585–12588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Urisman A, Molinaro RJ, Fischer N, Plummer SJ, Casey G, Klein EA, Malathi K, Magi-Galluzzi C, Tubbs RR, Ganem D, Silverman RH, DeRisi JL. 2006. Identification of a novel Gammaretrovirus in prostate tumors of patients homozygous for R462Q RNASEL variant. PLoS Pathog. 2:e25. 10.1371/journal.ppat.0020025 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 16.Schlaberg R, Choe DJ, Brown KR, Thaker HM, Singh IR. 2009. XMRV is present in malignant prostatic epithelium and is associated with prostate cancer, especially high-grade tumors. Proc. Natl. Acad. Sci. U. S. A. 106:16351–16356 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 17.Lombardi VC, Ruscetti FW, Das Gupta J, Pfost MA, Hagen KS, Peterson DL, Ruscetti SK, Bagni RK, Petrow-Sadowski C, Gold B, Dean M, Silverman RH, Mikovits JA. 2009. Detection of an infectious retrovirus, XMRV, in blood cells of patients with chronic fatigue syndrome. Science 326:585–589 [DOI] [PubMed] [Google Scholar]
- 18.Paprotka T, Delviks-Frankenberry KA, Cingoz O, Martinez A, Kung HJ, Tepper CG, Hu WS, Fivash MJ, Jr, Coffin JM, Pathak VK. 2011. Recombinant origin of the retrovirus XMRV. Science 333:97–101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Knox K, Carrigan D, Simmons G, Teque F, Zhou Y, Hackett J, Jr, Qiu X, Luk KC, Schochetman G, Knox A, Kogelnik AM, Levy JA. 2011. No evidence of murine-like gammaretroviruses in CFS patients previously identified as XMRV-infected. Science 333:94–97 [DOI] [PubMed] [Google Scholar]
- 20.Delviks-Frankenberry K, Cingoz O, Coffin JM, Pathak VK. 2012. Recombinant origin, contamination, and de-discovery of XMRV. Curr. Opin. Virol. 2:499–507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Alter HJ, Mikovits JA, Switzer WM, Ruscetti FW, Lo SC, Klimas N, Komaroff AL, Montoya JG, Bateman L, Levine S, Peterson D, Levin B, Hanson MR, Genfi A, Bhat M, Zheng H, Wang R, Li B, Hung GC, Lee LL, Sameroff S, Heneine W, Coffin J, Hornig M, Lipkin WI. 2012. A multicenter blinded analysis indicates no association between chronic fatigue syndrome/myalgic encephalomyelitis and either xenotropic murine leukemia virus-related virus or polytropic murine leukemia virus. mBio 3(5):e00266–12. 10.1128/mBio.00266-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cingoz O, Paprotka T, Delviks-Frankenberry KA, Wildt S, Hu WS, Pathak VK, Coffin JM. 2012. Characterization, mapping, and distribution of the two XMRV parental proviruses. J. Virol. 86:328–338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Knouf EC, Metzger MJ, Mitchell PS, Arroyo JD, Chevillet JR, Tewari M, Miller AD. 2009. Multiple integrated copies and high-level production of the human retrovirus XMRV (xenotropic murine leukemia virus-related virus) from 22Rv1 prostate carcinoma cells. J. Virol. 83:7353–7356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hue S, Gray ER, Gall A, Katzourakis A, Tan CP, Houldcroft CJ, McLaren S, Pillay D, Futreal A, Garson JA, Pybus OG, Kellam P, Towers GJ. 2010. Disease-associated XMRV sequences are consistent with laboratory contamination. Retrovirology 7:111. 10.1186/1742-4690-7-111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Katzourakis A, Hue S, Kellam P, Towers GJ. 2011. Phylogenetic analysis of murine leukemia virus sequences from longitudinally sampled chronic fatigue syndrome patients suggests PCR contamination rather than viral evolution. J. Virol. 85:10909–10913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Smith RA. 2010. Contamination of clinical specimens with MLV-encoding nucleic acids: implications for XMRV and other candidate human retroviruses. Retrovirology 7:112. 10.1186/1742-4690-7-112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Oakes B, Tai AK, Cingoz O, Henefield MH, Levine S, Coffin JM, Huber BT. 2010. Contamination of human DNA samples with mouse DNA can lead to false detection of XMRV-like sequences. Retrovirology 7:109. 10.1186/1742-4690-7-109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Robinson MJ, Erlwein OW, Kaye S, Weber J, Cingoz O, Patel A, Walker MM, Kim WJ, Uiprasertkul M, Coffin JM, McClure MO. 2010. Mouse DNA contamination in human tissue tested for XMRV. Retrovirology 7:108. 10.1186/1742-4690-7-108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sato E, Furuta RA, Miyazawa T. 2010. An endogenous murine leukemia viral genome contaminant in a commercial RT-PCR kit is amplified using standard primers for XMRV. Retrovirology 7:110. 10.1186/1742-4690-7-110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tuke PW, Tettmar KI, Tamuri A, Stoye JP, Tedder RS. 2011. PCR master mixes harbour murine DNA sequences. Caveat emptor! PLoS One 6:e19953. 10.1371/journal.pone.0019953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wolff D, Gerritzen A. 2011. Presence of murine leukemia virus (MLV)-related virus gene sequences in a commercial RT-PCR reagent. Clin. Lab. 57:631–634 [PubMed] [Google Scholar]
- 32.Zheng H, Jia H, Shankar A, Heneine W, Switzer WM. 2011. Detection of murine leukemia virus or mouse DNA in commercial RT-PCR reagents and human DNAs. PLoS One 6:e29050. 10.1371/journal.pone.0029050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Erlwein O, Kaye S, McClure MO, Weber J, Wills G, Collier D, Wessely S, Cleare A. 2010. Failure to detect the novel retrovirus XMRV in chronic fatigue syndrome. PLoS One 5:e8519. 10.1371/journal.pone.0008519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lee D, Das Gupta J, Gaughan C, Steffen I, Tang N, Luk KC, Qiu X, Urisman A, Fischer N, Molinaro R, Broz M, Schochetman G, Klein EA, Ganem D, Derisi JL, Simmons G, Hackett J, Jr, Silverman RH, Chiu CY. 2012. In-depth investigation of archival and prospectively collected samples reveals no evidence for XMRV infection in prostate cancer. PLoS One 7:e44954. 10.1371/journal.pone.0044954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sharp PM, Hahn BH. 2011. Origins of HIV and the AIDS pandemic. Cold Spring Harb. Perspect. Med. 1:a006841. 10.1101/cshperspect.a006841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Langdon SP. 2012. Animal modeling of cancer pathology and studying tumor response to therapy. Curr. Drug Targets 13:1535–1547 [DOI] [PubMed] [Google Scholar]
- 37.Murgai M, Thomas J, Cherepanova O, Delviks-Frankenberry K, Deeble P, Pathak VK, Rekosh D, Owens G. 2013. Xenotropic MLV envelope proteins induce tumor cells to secrete factors that promote the formation of immature blood vessels. Retrovirology 10:34. 10.1186/1742-4690-10-34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dong B, Kim S, Hong S, Das Gupta J, Malathi K, Klein EA, Ganem D, Derisi JL, Chow SA, Silverman RH. 2007. An infectious retrovirus susceptible to an IFN antiviral pathway from human prostate tumors. Proc. Natl. Acad. Sci. U. S. A. 104:1655–1660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chaipan C, Dilley KA, Paprotka T, Delviks-Frankenberry KA, Venkatachari NJ, Hu WS, Pathak VK. 2011. Severe restriction of xenotropic murine leukemia virus-related virus replication and spread in cultured human peripheral blood mononuclear cells. J. Virol. 85:4888–4897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Overbergh L, Valckx D, Waer M, Mathieu C. 1999. Quantification of murine cytokine mRNAs using real time quantitative reverse transcriptase PCR. Cytokine 11:305–312 [DOI] [PubMed] [Google Scholar]
- 41.Paprotka T, Venkatachari NJ, Chaipan C, Burdick R, Delviks-Frankenberry KA, Hu WS, Pathak VK. 2010. Inhibition of xenotropic murine leukemia virus-related virus by APOBEC3 proteins and antiviral drugs. J. Virol. 84:5719–5729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chesebro B, Britt W, Evans L, Wehrly K, Nishio J, Cloyd M. 1983. Characterization of monoclonal antibodies reactive with murine leukemia viruses: use in analysis of strains of friend MCF and Friend ecotropic murine leukemia virus. Virology 127:134–148 [DOI] [PubMed] [Google Scholar]
- 43.Alberts B, Dennis B, Lewis J, Raff M, Roberts K, Watson JD. 1994. Molecular biology of the cell, 3rd ed. Garland Publishing Inc., New York, NY [Google Scholar]
- 44.Hu WS, Temin HM. 1990. Genetic consequences of packaging two RNA genomes in one retroviral particle: pseudodiploidy and high rate of genetic recombination. Proc. Natl. Acad. Sci. U. S. A. 87:1556–1560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Flynn JA, An W, King SR, Telesnitsky A. 2004. Nonrandom dimerization of murine leukemia virus genomic RNAs. J. Virol. 78:12129–12139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nikbakht KN, Ou CY, Boone LR, Glover PL, Yang WK. 1985. Nucleotide sequence analysis of endogenous murine leukemia virus-related proviral clones reveals primer-binding sites for glutamine tRNA. J. Virol. 54:889–893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Colicelli J, Goff SP. 1986. Isolation of a recombinant murine leukemia virus utilizing a new primer tRNA. J. Virol. 57:37–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lund AH, Duch M, Lovmand J, Jorgensen P, Pedersen FS. 1993. Mutated primer binding sites interacting with different tRNAs allow efficient murine leukemia virus replication. J. Virol. 67:7125–7130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nitta T, Lee S, Ha D, Arias M, Kozak CA, Fan H. 2012. Moloney murine leukemia virus glyco-gag facilitates xenotropic murine leukemia virus-related virus replication through human APOBEC3-independent mechanisms. Retrovirology 9:58. 10.1186/1742-4690-9-58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hibbert CS, Mirro J, Rein A. 2004. mRNA molecules containing murine leukemia virus packaging signals are encapsidated as dimers. J. Virol. 78:10927–10938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rein A. 2011. Murine leukemia viruses: objects and organisms. Adv. Virol. 2011:403419. 10.1155/2011/403419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhang WH, Svarovskaia ES, Barr R, Pathak VK. 2002. Y586F mutation in murine leukemia virus reverse transcriptase decreases fidelity of DNA synthesis in regions associated with adenine-thymine tracts. Proc. Natl. Acad. Sci. U. S. A. 99:10090–10095 [DOI] [PMC free article] [PubMed] [Google Scholar]