

Abstract
The genomes of RNA viruses often contain RNA structures that are crucial for translation and RNA replication and may play additional, uncharacterized roles during the viral replication cycle. For the picornavirus family member poliovirus, a number of functional RNA structures have been identified, but much of its genome, especially the open reading frame, has remained uncharacterized. We have now generated a global RNA structure map of the poliovirus genome using a chemical probing approach that interrogates RNA structure with single-nucleotide resolution. In combination with orthogonal evolutionary analyses, we uncover several conserved RNA structures in the open reading frame of the viral genome. To validate the ability of our global analyses to identify functionally important RNA structures, we further characterized one of the newly identified structures, located in the region encoding the RNA-dependent RNA polymerase, 3Dpol, by site-directed mutagenesis. Our results reveal that the structure is required for viral replication and infectivity, since synonymous mutants are defective in these processes. Furthermore, these defects can be partially suppressed by mutations in the viral protein 3Cpro, which suggests the existence of a novel functional interaction between an RNA structure in the 3Dpol-coding region and the viral protein(s) 3Cpro and/or its precursor 3CDpro.
INTRODUCTION
The genomes of RNA viruses, such as poliovirus (PV), often contain complex RNA secondary and tertiary structures. These structures are crucial for translation and replication of the viral genome and may play additional roles in other processes, such as genome packaging and modulation of the host antiviral response.
Poliovirus, the prototypical picornavirus and causative agent of poliomyelitis, is a nonenveloped virus with a single-stranded RNA genome of positive polarity. The virion consists of an icosahedral protein shell, composed of four capsid proteins (VP1, VP2, VP3, and VP4), which encapsidates the RNA genome (1). Poliovirus has a rapid replication cycle, with approximately 8 h elapsing between infection and release of progeny virions upon host cell lysis. During an infection, high yields of both viral proteins and genomes are produced. These yields ensure synthesis of up to 10,000 virions per cell (2), which can be widely disseminated to neighboring cells and/or new hosts. The compact nature of the viral genome (less than 7,500 nucleotides [nt] long) facilitates this rapid exponential growth. The viral genome acts as an mRNA and can be divided into a highly conserved 742-nt 5′ untranslated region (5′ UTR), a single long open reading frame encoding the viral polyprotein, a 68-nt 3′ untranslated region (3′ UTR), and a polyadenosine tract of a variable length (see Fig. 2A). A small viral protein of 22 amino acids, VPg (3B), is covalently attached to the 5′ end of the RNA. The 5′ UTR contains a structure critical for viral translation (the internal ribosome entry site or IRES) (3, 4), as well the 5′-cloverleaf (5′-CL) structure, which is involved in both positive- and negative-strand RNA synthesis (5–7). The 3′ UTR also contains a pseudoknot RNA structure involved in replication (8). Within the open reading frame, two RNA structures have been previously identified: the cis-acting replication element in the 2C coding region (2C-CRE) (9) and a structure in the 3Cpro coding region that inhibits RNase L (RNase L competitive inhibitor RNA [ciRNA]) (10, 11). However, until recently (12), it was not known whether any additional RNA structures were present in the viral genome or what the function(s) of any such structures might be.
Fig 2.

Organization, SHAPE reactivity, and pairing probability of the poliovirus genome and bootstrap analysis of protein linker regions. (A) Poliovirus genome organization. Untranslated regions are shown as lines, and the open reading frame as a box, with length proportional to nucleotide length. The open reading frame is divided (solid lines) by convention into three domains: P1 (structural genes; light green) and P2 and P3 (nonstructural genes; both light blue). Domains are further divided (dashed lines) into the 11 major viral proteins. (B) Median SHAPE reactivity of native virion RNA. Median SHAPE reactivity is calculated over a sliding 75-nt window (black line). The global median is 0.38 (red line). Regions representing putative local structures identified by SHAPE are marked by dashed red lines (see also Table 1). Regions representing previously described RNA structures, and the 3D-7000 element region targeted by mutagenesis in the current work, are marked by solid black brackets and labeled. (C) Median pairing probability. Median pairing probability is calculated over a sliding 75-nt window (black line). The global median is 1.9 × 10−4 (red line). Probabilities of <1.0 × 10−6 are plotted at 1.0 × 10−6. (D) Median SHAPE reactivity of total viral RNA at 7 h postinfection. Median SHAPE reactivity is calculated over a sliding 75-nt window (black line). The global median is 0.39 (red line).
To identify potential structures, we used a method, selective 2′-hydroxyl acylation analyzed by primer extension (SHAPE) (13), which enables high-throughput chemical probing and analysis of RNA structure. Using this method, the genomes of a few other RNA viruses have been analyzed (14–16). We have used SHAPE to interrogate authentic full-length poliovirus genomic RNA extracted, in a gentle and nondenaturing manner, from purified virions. We identified many regions of the poliovirus genome where SHAPE data indicate that nucleotide flexibility is constrained, suggesting that the RNA is folded into secondary and/or tertiary structures. We then complemented our chemical probing with a parallel, independent evolutionary analysis of pairing probability, which demonstrated that several of the newly identified structures are also highly conserved. Two of the structures that we identified were independently identified by another group (12), validating this method of global analysis as a means of identifying functionally important RNA elements. To determine the function of one such RNA structure in the 3Dpol coding region, we introduced synonymous mutations, which disrupted the structure and revealed that it is critically involved in viral replication and infectivity. We further showed that the defects caused by mutations in this RNA structure can be partially suppressed by mutations in the viral protein 3Cpro, which suggests that this RNA structure interacts directly with 3Cpro and/or its precursor 3CDpro. The location of these suppressor mutations potentially identifies a novel RNA-protein interface.
MATERIALS AND METHODS
Cells.
HeLa S3 cells were grown either in tissue culture flasks in Dulbecco modified Eagle medium (DMEM) high-glucose 50%–F-12 50% mix supplemented with 1× penicillin-streptomycin-glutamine (PSG) and 10% newborn calf serum (NCS) at 37°C with 5% CO2 or in suspension in suspension minimum essential medium Eagle spinner modification (SMEM) (Joklik-modified) supplemented with 1× PSG and 10% fetal bovine serum (FBS) at 37°C.
Virus production.
Infectious wild-type (WT) (or mutant) poliovirus 1 (Mahoney) RNA was produced by in vitro transcription of linearized prib(+)XpA plasmid (17) (or plasmids derived therefrom; see below). HeLa S3 cells were trypsinized, washed three times with Dulbecco's phosphate-buffered saline (D-PBS), and adjusted to 5 × 106 cells/ml. An 800-μl amount of cells was transfected with 20 μg RNA in 0.4-cm cuvettes using an Electro cell manipulator 600 (BTX Inc.) with voltage of 300 V, capacitance of 1,000 μF, and resistance of 24 Ω. Cells were recovered in 7.5 ml tissue culture medium supplemented with 1× PSG and 2.5% NCS, transferred to tissue culture flasks, and incubated overnight at 37°C with 5% CO2. The P0 virus stock was then obtained after three freeze/thaw cycles, followed by centrifugation at 3,500 rpm for 5 min in a Legend T centrifuge (Sorvall), and stored at −20°C. Titers of virus stocks were determined by standard plaque assay and/or 50% tissue culture infective dose (TCID50). Subsequent (P1 and later) virus stocks were obtained by washing HeLa S3 cell monolayers with PBS and infecting with the previous stock diluted to the desired multiplicity of infection (MOI) in tissue culture medium supplemented with 1× PSG for 30 min at 37°C with occasional rocking. The inoculum was then removed, and the cells were recovered in tissue culture medium supplemented with 1× PSG and 2.5% NCS and incubated at 37°C and 5% CO2 until cytopathic effect (CPE) was observed, when the virus stock was harvested as described for P0. To obtain large amounts of virus for virion purification, HeLa S3 cells were infected in suspension. P3 or later virus stock was adjusted to ∼5 × 108 PFU/ml in suspension medium supplemented with 1× PSG. Three liters of suspension culture (∼5 × 105 cells/ml) was subjected to centrifugation at 1,000 rpm for 5 min in a Legend T centrifuge (Sorvall). Cells were resuspended in 30 ml inoculum (MOI of ∼10) and incubated for 30 min at 37°C. The infection mixture was then diluted to 300 ml in suspension medium supplemented with 1× PSG and incubated in suspension at 37°C until CPE was observed (∼12 h), when the virus stock was harvested as described above. For total RNA extraction, 1 liter of suspension culture was infected in a similar manner for 7 h. Cells were then were subjected to centrifugation at 1,000 rpm for 5 min in a Legend T centrifuge (Sorvall), washed twice in PBS, and resuspended in 10 ml PBS with 1% Triton X-100. Cell lysis was confirmed by staining a small aliquot with trypan blue. The lysate was then subjected to centrifugation at 14,000 rpm for 1 min in a 5415 C centrifuge (Eppendorf) to pellet nuclei, and the supernatant was stored at −80°C.
Virion purification and RNA extraction.
For a single virion prep, ∼200 ml of virus stock prepared as described above was subjected to ultracentrifugation in a Beckman SW28 rotor at 14,000 rpm for 10 min at 20°C. The supernatant was treated with 10 μg/ml RNase A for 1 h at 20°C and then adjusted to 0.5% SDS and 2 mM EDTA. It was then divided into six equal parts, and each part was overlaid onto a 6 ml sucrose cushion (30% sucrose in Hanks buffered salt solution [HBSS]). Virions were then purified through the sucrose cushion by ultracentrifugation in a Beckman SW28 rotor at 28,000 rpm for 4 h at 20°C with slow acceleration and deceleration. After removal of the supernatant and sucrose cushion, virions were resuspended in 1.2 ml virion lysis buffer (VLB) (50 mM HEPES [pH 8.0], 200 mM NaCl, and 3 mM MgCl2) (14) and stored at −20°C. RNA was extracted in a gentle manner, largely as described for SHAPE of HIV genomic RNA (14), in the presence of mono- and divalent ions and the absence of heat, chemical denaturants, or chelating agents. Briefly, the purified virions in VLB were lysed with 1% SDS and 100 μg/ml proteinase K at 37°C for 1 h. The digest was then extracted with Tris-saturated phenol and the aqueous phase adjusted to 300 mM NaCl and precipitated with 75% (vol/vol) ethanol. The RNA pellet was dissolved in RNA modification buffer (RMB) (50 mM HEPES [pH 8.0], 200 mM potassium acetate [KOAc] [pH 8.0], and 3 mM MgCl2) (14) and stored at −80°C.
Total RNA extraction.
Infection lysate supernatant prepared as described above was adjusted to 1% SDS and 100 μg/ml proteinase K and incubated at 37°C for 1 h. The digest was then extracted with Tris-saturated phenol, and the aqueous phase was adjusted to 300 mM NaCl and precipitated with 75% (vol/vol) ethanol. The RNA pellet was dissolved in RMB (14) and stored at −80°C.
RNA modification.
The virion RNA concentration was adjusted to 267 ng/μl in RMB or the total RNA concentration was adjusted to 1.6 μg/μl in RMB, and the RNA was divided into two equal parts and incubated at 37°C for 20 min. One volume of dimethyl sulfoxide (DMSO) with or without 32.5 mM N-methylisotoic anhydride (NMIA) was then added to 9 volumes of RNA and incubated at 37°C for 45 min. The reaction mixtures were then adjusted to 0.2 μg/μl glycogen, 4 mM EDTA (pH 8.0), and 300 mM NaCl and precipitated with 75% (vol/vol) ethanol. The RNA pellets were dissolved in 9 volumes 0.5× TE (5 mM Tris [pH 8.0] and 0.5 mM EDTA), divided into 9-μl (1-pmol) aliquots, and stored at −80°C.
Primer extension and capillary electrophoresis.
Twenty-eight primers spanning the poliovirus genome were designed manually or with the aid of the software program Primer3Plus (http://www.bioinformatics.nl/cgi-bin/primer3plus/primer3plus.cgi) (sequences are available upon request). Two 5′-fluorescently labeled versions (6-carboxyfluorescein [6-FAM] and VIC) of each primer were ordered from Life Technologies. The two-dye approach described here, rather than the four-dye approach published elsewhere (14, 18), was based on the kind advice of David Mauger. Along with the experimental (+) and (−) RNA, dideoxy sequencing reactions were performed to enable accurate alignment of the chromatograms in the software program ShapeFinder (see below). These sequencing reactions used in vitro-transcribed poliovirus RNA at 267 ng/μl as the template. For each experiment, eight primer extension reactions were performed: 1 (+), 2 (−), and 3 to 8 sequencing. To denature the RNA, 9 μl (1 pmol) was incubated at 95°C for 3 min and placed on ice. Two microliters of 1 μM VIC-labeled primer was added to reactions 1 to 4, and the same amount of 6-FAM-labeled primer was added to reactions 5 to 8. To anneal the primers to the RNA, the reaction mixtures were incubated 65°C for 5 min and 35°C for 10 min and placed on ice. Nine microliters of the appropriate reverse transcriptase (RT) mix was added. The basic RT mix (SuperScript III, 5× buffer, dithiothreitol [DTT], and deoxynucleoside triphosphates [dNTPs]; Life Technologies) was added to reactions 1 and 2. The basic mix was supplemented with ddATP (TriLink BioTechnologies) for reactions 3 and 5 to 8 and ddCTP (TriLink BioTechnologies) for reaction 4. The reaction mixtures were incubated at 52°C for 15 min and placed on ice. To degrade the RNA template, 2.5 μl of 1 M NaOH was added to each reaction mixture, and the mixtures were incubated at 98°C for 15 min and placed on ice. To neutralize the reaction, 2.5 μl of 1 M HCl was added. The VIC and 6-FAM reaction mixtures were pooled in a pairwise manner (1 and 5, 2 and 6, 3 and 7, and 4 and 8) and diluted 2-fold with water. These four pooled reactions were adjusted to 0.2 μg/μl glycogen and 300 mM NaOAc (pH 5.2) and precipitated with 75% (vol/vol) ethanol. cDNA pellets were washed twice with 75% ethanol and dissolved in 10 μl HiDi formamide (Life Technologies). Capillary electrophoresis was performed by the UC Berkeley DNA Sequencing Facility (http://mcb.berkeley.edu/barker/dnaseq/) according to their Fragment Analysis protocol. Each of the four pooled reactions was analyzed in a separate capillary.
SHAPE data processing.
Raw electropherograms, which contain data for fluorescence intensity versus elution time, were analyzed in ShapeFinder (18). The early steps of data processing in ShapeFinder for the two-dye approach used here differ somewhat from the “standard” four-dye approach and were again based on the kind advice of David Mauger. Data from the 6-FAM and VIC channels for each of the four capillaries were first combined into a single file. A fitted baseline adjustment with a window width of 200 was applied to all eight channels. Cubic mobility shifts were performed manually on the four 6-FAM channels to align these identical ddATP sequencing reactions. The shift values from these alignments were then applied to the appropriate VIC channels, aligning the two experimental (+) and (−) channels and the ddATP and ddCTP sequencing channels to one another. The data from the four 6-FAM channels were then removed before further analysis. Signal decay correction was applied to each of the four remaining VIC channels. The region of interest was determined based on the (+) channel, and the same region was applied to all four channels (typically from between 1,000 and 2,000 to between 5,500 and 7,000). The rescale factor and equation parameters, A, q, and C, were kept at their default values of 10,000, 1,000,000, 0.999, and 10,000, respectively. A scale factor was then applied to the (−) channel such that most peaks were of a height equal to or lesser than that of the corresponding (+) peak. This factor was typically between 0.3 and 0.7. Alignment and integration were then performed over a range equal to or narrower than that used for signal decay correction. After manual inspection and correction of peaks, the ShapeFinder software performed a whole-channel Gaussian integration to quantify all individual peak areas, and the (−) peak areas were subtracted from the (+) areas. The output raw reactivity data for each primer were then normalized using a model-free box plot analysis (19, 20). Normalized reactivity values between −0.5 and 0 were set to 0. Normalized reactivity values of less than −0.5 or greater than 3.0 were discarded as extreme outliers. Replicate experiments with the same primer were highly reproducible. In this manner, we were able to obtain SHAPE reactivity values for 98% of the nucleotides in the poliovirus genome (7,298 out of 7,440). Sixty-four nucleotides at the extreme 3′ end were impossible to examine due to the need for a primer binding site, and a further 78 were discarded as outliers and not analyzed further.
Pairing probability analysis.
XDecoder, a phylogrammar modeling the evolution of RNA structures that may partially overlap protein coding sequence, was implemented in the XRate software program as previously described (21). A multiple alignment of poliovirus genomes was annotated with the reading frame from poliovirus 1 (Mahoney). Summing over all possible structures using the Inside/Outside recursions (implemented within XRate), the cumulative posterior probability of all pairings for each position was tabulated (using the Wiggle output format option to XRate). Structures with greater than 300 bases separating paired positions were excluded. The XDecoder grammar, the sequence alignment used, and the commands used to generate the Wiggle data are available as information supplementary to the work of Westesson and Holmes (21).
Bootstrap analysis.
A bootstrap analysis comparing the SHAPE reactivities of 60-nt regions to random regions of the same length from the poliovirus genome was performed as described for HIV-1 (14).
Mutagenesis and cloning.
Mutant viruses and replicons were generated by site-directed mutagenesis and cloning of the prib(+)XpA and prib(+)RLucpA plasmids (17), followed by in vitro transcription of linearized plasmid and transfection as described above for viral RNA and as described below for replicon RNA. Mutagenesis was achieved by overlap PCR using mutagenesis primers (sequences are available upon request) and the PCR primers 5434For (5′-ACAAGGACCAGGGTTCGATTACGCAGTGGCTATGGCT-3′) and 7662Rev (5′-GGGGTTCCGCGCACATTTC-3′). Briefly, the appropriate mutagenesis and PCR primers were combined with plasmid template for 30 cycles of PCR (30-s extension time) with Herculase II Fusion DNA polymerase (Agilent). The first-round PCR products were purified (NucleoSpin Extract II; Macherey-Nagel), and 0.3 pmol of each was combined as the template for the second round of PCR. Ten cycles of PCR (30-s extension time) were performed without PCR primers, and then the PCR primers were added and a further 20 cycles (40-s extension time) were performed. The second-round PCR products were purified and digested with BglII and MluI (NEB). The plasmids vectors were similarly digested and also dephosphorylated (Antarctic Phosphatase; NEB). Insertion was achieved using the Quick Ligation kit (NEB). Plasmids were transformed into Sure electroporation-competent cells (Agilent), and plasmid preps were sequenced (Elim Biopharm) to confirm the presence of the correct insert.
Plaque assays.
Plaque assays were performed using clonal stocks of mutant and wild-type virus. Ten-centimeter plates of confluent (∼1.5 × 107 cells) HeLa S3 cells were washed with phosphate-buffered saline (PBS) and infected with ∼40 PFU or ∼ 40,000 PFU in 1.5 ml tissue culture medium supplemented with 1× PSG. Plates were incubated for 30 min at 37°C and 5% CO2 with occasional rocking. Cells were overlaid with growth medium containing 1% (wt/vol) agarose and incubated at 37°C and 5% CO2. At 48 h postinfection, plaque assays were fixed through the overlays with 2% (vol/vol) formaldehyde for 15 min at 20°C. The overlays were removed, and the cells were stained with 0.1% (wt/vol) crystal violet. For plaque area quantification, plates were individually scanned and plaques were measured as described elsewhere (22).
Luciferase replicons.
Wild-type (or mutant) PLuc RNA was produced by in vitro transcription of linearized prib(+)RLucpA plasmid (17) (or plasmids derived therefrom; see above). HeLa S3 cells were trypsinized, washed three times with D-PBS, and adjusted to 1 × 107 cells/ml. Eight hundred microliters of cells were transfected with 20 μg RNA in 0.4-cm cuvettes using an Electro cell manipulator 600 (BTX Inc.) with voltage of 300 V, capacitance of 1,000 μF, and resistance of 24 Ω. Cells were recovered in 13 to 15 ml tissue culture medium supplemented with 1× PSG and 10% NCS and split into two equal parts. Guanidine hydrochloride (GdnHCl) was added to one part to a final concentration of 2 mM, and cells were incubated at 37°C. Samples were taken every 30 to 60 min. At each time point, 0.5 to 1.0 ml of cells was removed and subjected to centrifugation at 6,000 rpm for 1 min in a 5415 C centrifuge (Eppendorf). The cells were resuspended in 60 to 120 μl 1× cell culture lysis reagent (Promega) and incubated for 1 min at 20°C. The lysate was then subjected to centrifugation at 14,000 rpm for 1 min, and 50 to 100 μl of supernatant was removed and stored at −20°C. Luciferase activity was assayed by mixing 10 μl of lysate with 100 μl luciferase assay reagent (Promega). Luminescence was read on either an Optocomp I luminometer (MGM Instruments) or an Ultra Evolution plate reader (Tecan).
One-step growth analysis.
Wild-type, 3D4A, and 3D4A-3CY113F viruses (P1 stocks) were diluted to 1.25 × 108 PFU/ml in tissue culture medium supplemented with 1× PSG, treated with 10 μg/ml RNase A for 1 h at 20°C to remove unpackaged viral RNA, and frozen at −20°C overnight. Confluent (∼5 × 105 cells/well) HeLa S3 cells in 24-well plates were washed with cold PBS and infected in sextuplicate at an MOI of ∼25 for 30 min at 4°C with occasional rocking. The inoculum was then removed, and the cells were washed twice in cold PBS and recovered in tissue culture medium supplemented with 1× PSG and incubated at 37°C and 5% CO2. Samples were taken every hour. At each time point, the plate was frozen at −80°C. RNA was extracted from three of the six replicate wells using the PureLink RNA microkit (Life Technologies) and diluted for strand-specific qPCR based on expected RNA levels. After three freeze/thaw cycles, the remaining three replicates were subjected to centrifugation at 14,000 rpm for 1 min in a 5415 C centrifuge (Eppendorf), and supernatants stored at −20°C. Titers of each replicate were determined by TCID50.
Strand-specific RT-qPCR.
Strand-specific quantitative PCR (qPCR) was based on a published protocol (23). Briefly, cDNA was synthesized from diluted total RNA using SuperScript III reverse transcriptase (Life Technologies), and 125 nM strand-specific RT primer (+strand_RT, 5′-GGCCGTCATGGTGGCGAATAATGTGATGGATCCGGGGGTAGCG-3′; -strand_RT, 5′-GGCCGTCATGGTGGCGAATAACATGGCAGCCCCGGAACAGG-3′) in a 5-μl reaction mixture. Separate RT reactions for positive and negative strands were performed for each sample. RT products were treated with 0.5 U of exonuclease I (Fermentas) to remove excess RT primer prior to qPCR. cDNAs were diluted 10-fold and analyzed by qPCR using 2× SYBR Fast master mix (Kapa Biosystems), 200 nM strand-specific qPCR primer (+strand_For, 5′-CATGGCAGCCCCGGAACAGG-3′; -strand_Rev, 5′-TGTGATGGATCCGGGGGTAGCG-3′), and 200 nM Tag primer (5′-GGCCGTCATGGTGGCGAATAA-3′) in a 10-μl reaction mixture. A 10× dilution series of in vitro-transcribed positive- and negative-strand RNA standards was run alongside experimental samples and used to construct a standard curve.
Virion immunoprecipitation.
Supernatant from the 8-h-postinfection time point of the one-step growth curves were applied to 10 μl protein A-coupled Dynabeads (Life Technologies) bound to 2 μg anti-poliovirus type 1 monoclonal antibody (Pierce Biotechnology). Immunoprecipitation was performed according to the manufacturer's protocol. RNA was extracted from eluates using the PureLink RNA microkit (Life Technologies) and subjected to positive-strand RT-qPCR.
Western blots.
Fifty percent confluent (∼1 × 106 cells/well) HeLa S3 cells in 6-well plates were washed with PBS and infected with 3D4, 3D4A, 3D4A, SL IV 298 (24), or wild-type virus (P1 stock) at an MOI of ∼20 or mock infected for 30 min at 37°C and 5% CO2 with occasional rocking. The inoculum was then removed, and the cells were washed three times with PBS and recovered in 1 ml/well tissue culture medium supplemented with 1× PSG and 10% FBS and incubated at 37°C and 5% CO2. At 5 h postinfection, cells were collected and subjected to centrifugation at 3,000 rpm for 2 min in a 5415 C centrifuge (Eppendorf). The cells were resuspended in 50 μl lysis buffer (50 mM Tris-HCl [pH 7.5], 150 mM NaCl, 0.5% NP-40, 10 mM DTT, and 1× Complete mini protease inhibitor cocktail [Roche]) and incubated for 30 min on ice with occasional mixing. The lysate was then subjected to centrifugation at 14,000 rpm for 1 min in a 5415 C centrifuge (Eppendorf), and the supernatant was removed and stored at −20°C. Five microliters of lysate was run for each sample. Samples were run and transferred on standard gels, membranes, and equipment (Bio-Rad). Membranes were probed with primary mouse anti-3D (25, 26) and secondary ECL anti-mouse IgG (GE Healthcare) or primary rabbit anti-2C (27) and secondary ECL anti-rabbit IgG (GE Healthcare). Horseradish peroxidase (HRP) activity was assayed using Pierce ECL Plus Western blotting substrate (Thermo Scientific). Membrane was then stripped with Restore PLUS Western blot stripping buffer (Thermo Scientific) and reprobed with primary rabbit anti-glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (Imgenex) and secondary ECL anti-rabbit IgG (GE Healthcare).
Plaque purification and sequencing.
Plaque assays were performed using a P0 stock of 3D3 virus as described above. At 72 h post-infection with the 3D3 mutant, plaques were identified through the agarose overlay, marked, and “picked” using a 10-μl pipette tip. Plates were then incubated a further 24 h, and plaques that had not been visible earlier were picked. Picked plaques were amplified by infecting individual wells of 24-well tissue culture plates containing confluent (∼5 × 105 cells/well) HeLa S3 cells in 0.25 ml tissue culture medium supplemented with 1× PSG. Plates were incubated at 37°C and 5% CO2 until CPE was observed, ∼48 h postinfection. After three freeze/thaw cycles, samples were subjected to centrifugation at 14,000 rpm for 1 min in a 5415 C centrifuge (Eppendorf), and supernatants were stored at −20°C. RNA was extracted from 100 μl plaque-purified supernatant and eluted in 10 μl water (ZR viral RNA kit; Zymo Research). cDNA was synthesized from 9 μl RNA template primed with oligo(dT) (ThermoScript RT-PCR system for first-strand cDNA synthesis; Life Technologies). PCR was performed using the primers 6736For (5′-GATGGTGCTTGAGAAAATCGGATTCGGAGACAGAGTT-3′) and 7400Rev (5′-CCAATCCAATTCGACTGAGGTAGGG-3′). PCR products were purified (NucleoSpin Extract II; Macherey-Nagel) and sequenced using the PCR primers (Elim Biopharm).
Suppressor isolation and characterization.
The amplified 3D3 “72 h” plaques that lacked the A7006C mutation were subject to standard plaque assay to confirm a reproducible large-plaque phenotype. Three out of twenty-one clones exhibited the phenotype. The cDNA from these clones was subjected to PCR, and the entire viral genome was sequenced as described previously (28). A 3D4A P0 stock was subject to limiting dilution, and 16 clones were sequenced as described. Seven of the sixteen possessed the 3D4A mutations and no others. These clones were divided and passaged twice at 37 and 39°C. Viral RNA was extracted and subjected to RT-PCR and sequencing as described above. Once the suppressor mutations were identified, new clonal stocks were generated from the original mutants and sequenced to confirm the presence of the original RNA mutation and the 3C suppressor mutation and no other mutations. These stocks were used for plaque assays and/or to generate P1 stocks for growth curves as described above.
RESULTS
Mapping RNA structures in the poliovirus genome.
To map the global RNA structure of the poliovirus genome, we probed full-length poliovirus genomic RNA with the SHAPE reagent, N-methylisotoic anhydride (NMIA) (Fig. 1A). NMIA is a hydroxyl-selective electrophile that can acylate the ribose 2′-hydroxyl group to form a stable 2′-O-adduct, which can be detected as a stop to primer extension by reverse transcriptase (13). Like other SHAPE reagents, NMIA is relatively insensitive to base identity (29) but very sensitive to conformational dynamics. It reacts preferentially with flexible RNA nucleotides and poorly with those that are base paired or otherwise conformationally constrained (13). Thus, constrained (structured) nucleotides are generally protected from acylation and are said to have low SHAPE reactivity, while flexible (unstructured) nucleotides are unprotected and have high SHAPE reactivity.
Fig 1.

Experimental procedure and SHAPE reactivities of previously described RNA structures in the poliovirus genome. (A) Outline of SHAPE experimental approach. (B) 5′ UTR: 5′-cloverleaf (5′-CL) and internal ribosome entry site (IRES). Accepted RNA secondary structure model for nt 1 to 625 of the poliovirus 1 (Mahoney) genome colored by SHAPE reactivity (constrained nucleotides are black, moderately reactive nucleotides are green or yellow, and flexible nucleotides are red). A-U and C-G base pairing is indicated by lines, and G-U (wobble) base pairing is indicated by dots. (C) Agreement of SHAPE with accepted structure for IRES. Box plots indicate median SHAPE reactivity, 25% and 75% quartiles, and 1.5× interquartile range for nucleotides predicted to be paired or unpaired; outliers are shown as open circles. (D) 2C-CRE. Nucleotides 4444 to 4504 are colored by SHAPE reactivity. (E) RNase L ciRNA. Nucleotides 5742 to 5968 are colored by SHAPE reactivity; dashed lines indicate predicted tertiary interaction; arrowhead indicates nucleotide 5775, site of the 3D-7000 A-to-U suppressor mutation that encodes the 3Cpro Y113F change. (F) Agreement of SHAPE with accepted structure for RNase L ciRNA. Box plots are as for panel A.
We found that the method of RNA extraction was critical, since in vitro-transcribed RNA or denatured and refolded viral RNA gave SHAPE results different from those for the native genomic RNA when extracted in a gentle, nondenaturing manner from purified virions (data not shown). Thus, for our analysis, we employed native virion RNA, which was treated with NMIA and analyzed by primer extension and capillary electrophoresis (14, 18) (Fig. 1A). We also examined total viral RNA extracted from infected cells under gentle, nondenaturing conditions at 7 h postinfection. The global results were similar (compare Fig. 2B to D), but we observed a few differences, which likely represent a population average of genomes in distinct states—packaged, translating, replicating—so to simplify our analysis, we focused on the virion RNA, which is more likely to represent a uniform population.
To examine the accuracy of our analysis, we first examined four functional RNA structures that had been identified previously in the poliovirus genome. These included structures in the 5′ UTR (Fig. 1B) and in the open reading frame (Fig. 1D and E). Within the 5′ UTR (Fig. 1B), we examined both the poliovirus IRES, which is critical for viral translation (3, 4), and the 5′-CL, which facilitates both negative and positive-strand RNA synthesis (6, 7) and is also known to play a role in viral translation (30). Within the open reading frame, the 2C-CRE (Fig. 1D) templates the uridylylation of the peptide primer VPg (3B) (9, 31), and the RNase L ciRNA (Fig. 1E), which maps to the region that encodes 3Cpro, competitively inhibits host RNase L (10, 11).
Our analysis was able to accurately identify the RNA structures previously found in the poliovirus genome. Indeed, we observed a highly significant correlation of SHAPE reactivity with paired and unpaired nucleotides of the reported structures (Fig. 1C and F). However, there are several nucleotides that are inconsistent with the accepted structures. In general, these fall into two categories: protected nucleotides within predicted loops and unprotected nucleotides within predicted stems. Thus, it is likely that the accepted secondary structures fail to fully represent more intricate tertiary interactions. Of note, within the RNase L ciRNA structure, there are two predicted loops consisting entirely of protected nucleotides. Mutagenesis experiments have confirmed that these loops form a “kissing loops” tertiary interaction (32), and their low SHAPE reactivity is entirely consistent with that structure (Fig. 1E, dashed lines).
The three instances of unprotected nucleotides within predicted stems occur in the 5′- CL (Fig. 1B), 2C-CRE (Fig. 1D), and RNase L ciRNA (Fig. 1E). In the first two cases, the SHAPE data are consistent with the top portion of the accepted structure, but the 5′ side of the bottom stem is unprotected, which is inconsistent with a base-pairing interaction. We speculate that these structures may be only partially formed in the virion RNA, perhaps requiring the stabilizing presence of additional viral or cellular factors. Alternatively, in the case of 5′-CL, high reactivity of nt 4 to 8 may be an artifact of their proximity to the 5′ end of the genome, where the large signal from the full-length transcript tends to artificially increase the signal from adjacent nucleotides. In the case of 2C-CRE, we note that the 14-nt “core” sequence and upper stem, which are the portions shown to be minimally required for 2C-CRE function (33–35), do exhibit SHAPE reactivities consistent with the accepted structure and that this shorter region is the only portion of 2C-CRE which is evolutionarily conserved according to the pairing probability analysis (Fig. 2C). Similarly, in the RNase L ciRNA, the predicted stem near position 5900 that appears to consist of unprotected nucleotides is in a portion of the structure that was shown to be unnecessary for RNase L inhibition (10). Overall, we found that the SHAPE data were highly consistent with previously described functional RNA structures in the poliovirus genome, validating the use of this approach for a global analysis.
We therefore extended our analysis to globally examine the poliovirus genome (Fig. 2B). First, we characterized the relationship between RNA structure and protein structure. In HIV-1, flexible protein linkers were found to be encoded by more structured RNA segments than the rest of the genome (14). However, in poliovirus, a similar bootstrap analysis did not detect a statistically significant relationship between RNA and protein structure (data not shown). Thus, the presence of RNA structures within the open reading frame that regulate local translation kinetics does not appear to be employed by poliovirus in a significant way. Next, we attempted to predict a global RNA secondary structure model for the poliovirus genome similar to that described for HIV-1 (14), using our SHAPE reactivity data as an additional thermodynamic constraint in the folding algorithm RNA structure (19, 36). We found the structure folding and prediction process to be highly user and parameter dependent, producing several alternative structures with similarly low free energies. Ultimately, we were unable to predict a single model with high confidence and so chose to study local, functional RNA structures.
Identification of novel RNA structures in the poliovirus genome.
To identify local RNA structures, the global SHAPE data were examined using a sliding 75-nt moving window (14) over the full length of the poliovirus genome (Fig. 2B). Regions where the local median dips below the global median (0.38, solid red line in Fig. 2B) and reaches a minimum value of at least half the global median (0.19) were considered likely to represent an RNA structure. Thirteen such regions were identified (Table 1 and dashed red lines in Fig. 2B). Five of the thirteen regions represent the two largest previously characterized RNA structures in the PV genome, the IRES (Fig. 1B) and the RNase L ciRNA (Fig. 1E).
Table 1.
Putative local RNA structures
| Low SHAPE reactivity region (nt) | Median pairing probability | Gene (known structure) |
|---|---|---|
| 135–473 | 9.92E−01 | 5′ UTR (IRES) |
| 482–537 | 9.93E−01 | 5′ UTR (IRES) |
| 544–687 | 9.76E−01 | 5′ UTR (IRES) |
| 694–801 | 1.01E−03 | 5′ UTR-VP4 |
| 1779–1872 | 2.93E−03 | VP3 |
| 2327–2470 | 7.10E−04 | VP3 |
| 5276–5396 | 4.50E−05 | 3A-3B |
| 5715–5828 | 6.36E−03 | 3C (RNase L ciRNA) |
| 5900–5993 | 2.11E−01 | 3C (RNase L ciRNA) |
| 6180–6341 | 6.34E−06 | 3D |
| 6354–6477 | 7.56E−07 | 3D |
| 6929–7060 | 8.99E−01 | 3D |
| 7132–7284 | 3.85E−03 | 3D |
A low local median SHAPE reactivity indicates the existence of local RNA structure, but it does not reveal whether the structure is functional or merely represents a thermodynamically stable conformation. We reasoned that functional RNA structures may be evolutionarily conserved, particularly those that are integral to the viral life cycle. We thus employed an evolutionary conservation analysis to probe the relevance of these novel poliovirus RNA structures. This orthogonal analysis takes into account the rate and pattern of nucleotide changes in an alignment of poliovirus sequences and predicts the likelihood that a particular nucleotide is involved in a base pair interaction (pairing probability [Fig. 2C]).
We examined the median pairing probability for each of the 13 putative structured regions that we identified based on SHAPE reactivity (Table 1). As expected, the regions corresponding to the IRES had extremely high levels of conservation. The evolutionary conservation of the RNase L ciRNA structure was lower but was well above the global median (1.9 × 10−4). Of the remaining eight regions, three have pairing probabilities below the global median, suggesting that they do not represent a structure with a conserved function in the polioviruses. The other five exhibit moderate to extremely high conservation. By far the highest median pairing probability was that of the region centered around position 7000, within the portion of the genome encoding the viral RNA-dependent RNA polymerase, 3Dpol. We refer to this structure as 3D-7000. A structure (α) in the same region and exhibiting substantial overlap with 3D-7000, as well as a second structure (β) with putative functional redundancy and corresponding to the structure centered around position 7200 (Table 1), were independently identified by Song et al. (12).
Mutagenesis of the 3D-7000 element.
Thermodynamic and evolutionary folding predictions for the 3D-7000 element, encompassing nt 6902 to 7117, generated several possible secondary structures. Further analysis of these predictions led to the structure proposed in Fig. 3A. This structure is highly conserved both within the polioviruses (Fig. 3B and C) and across the human enterovirus C group (Fig. 3C). To examine its role in the poliovirus life cycle, we subdivided the region into 6 smaller regions of 36 nt each. Within each region, we introduced the maximum possible number of synonymous mutations. We changed nucleotides that would result in a maximal disruption of the predicted structure (Fig. 3A) and/or are highly conserved (Fig. 3B). Six initial mutants were generated with between 11 and 16 synonymous mutations each (Fig. 4A).
Fig 3.
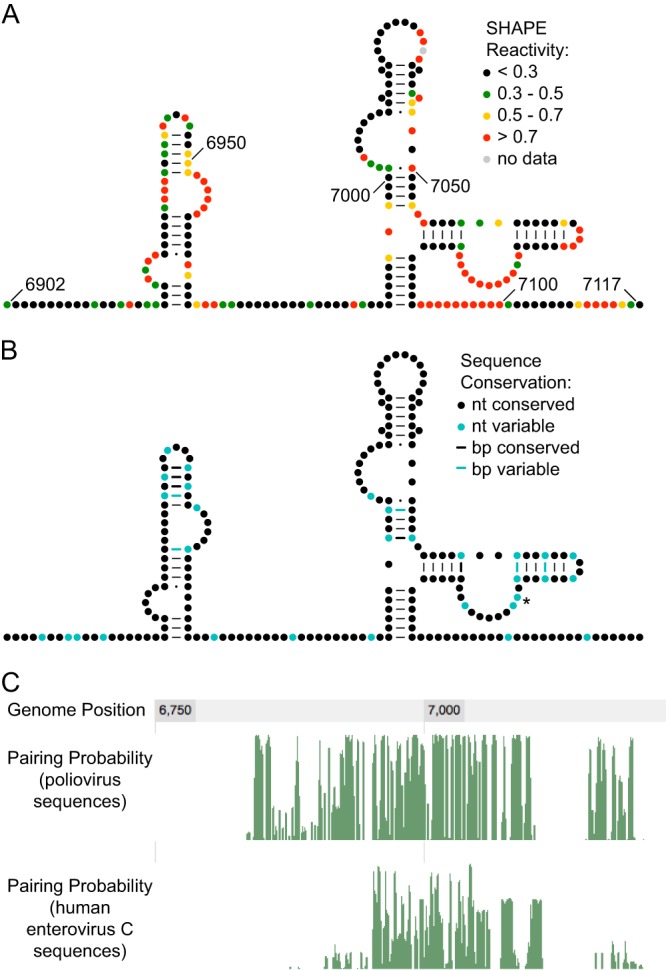
SHAPE reactivity and conservation of 3D-7000 element. (A) SHAPE reactivity of 3D-7000 element. The predicted RNA secondary structure model for nt 6902 to 7117 of the poliovirus 1 (Mahoney) genome is colored by SHAPE reactivity (constrained nucleotides are black, moderately reactive nucleotides are green or yellow, and flexible nucleotides are red). A-U and C-G base pairing is indicated by lines, and G-U (wobble) base pairing is indicated by dots. (B) Conservation of the 3D-7000 element. Nucleotide variability was determined by examining selected other poliovirus genomes: Mahoney (PV1; GenBank NC_002058), Sabin 1 (see note below) (GenBank AY184219), Lansing/MEF-1 (PV2; GenBank M12197/AY238473; identical for this region), Sabin 2 (AY184220), Saukett (PV3; C. P. Burrill, E. F. Goldstein, and R. Andino, unpublished data), and Sabin 3/Leon (GenBank AY184221/K01392; identical for this region). (C7071U encodes the T362I substitution in 3Dpol in the attenuated Sabin 1 vaccine strain; this is the only nonsynonymous change found in this region in any of the poliovirus sequences examined.) (C) Pairing probability of region containing the 3D-7000 element. Green lines represent the pairing probability for each nucleotide obtained by analyzing an alignment of poliovirus (top) or human enterovirus C (bottom) sequences.
Fig 4.

Mutagenesis of the 3D-7000 element and characterization of initial mutants. (A) Mutants. On the schematic, color indicates nucleotides targeted in each mutant. The actual nucleotide changes and corresponding wild-type sequence and nucleotide positions for each mutant are listed below. For WT structure, A-U and C-G base pairing is indicated by lines and G-U (wobble) base pairing by dots. (B) PLuc assay. Mutant (colored) or wild-type (black) replicon RNA was transfected into HeLa S3 cells at 37°C. Cells were incubated in the presence (dashed lines) or absence (solid lines) of 2 mM GdnHCl. Luciferase activity (relative light units) corresponding to ∼5.3 × 105 cells was measured every hour. Data are plotted as the means ± standard deviations of three or four independent transfections. Replication delays at 3 h posttransfection are significant compared to results for the wild type in 3D2 (P = 0.026), 3D3 (P = 0.003), and 3D4 (P = 0.015). (C) Plaque assays with plaque size measurement. Clonal stocks were used to infect HeLa S3 cells at 37°C. Each box plot indicates median plaque area in pixels, 25% and 75% quartiles, and 1.5× interquartile range; outliers are shown as open circles. 3D2 (P = 8.4E−09), 3D3 (P < 2.2E−16), and 3D4 (P < 2.2E−16) plaques are significantly smaller than the wild type. (D) Frequency of A7006C mutation in large and small plaques derived from 3D3 stock. Plaques were picked at 72 (large; n = 45) or 96 (small; n = 46) hours postinfection and amplified. RT-PCR products representing the region in question were sequenced.
Mutations within 3D-7000 reduce viral fitness.
Plaque morphology provides a useful read-out of the phenotype of each of the mutant populations, and we measured the area of individual plaques to quantify the effect of the synonymous mutations introduced in the region surrounding the 3D-7000 element. Several of the mutants exhibited a highly significant small or minute plaque phenotype (Fig. 4C). The most severe phenotypes were found in the mutants 3D3 (nt 6974 to 7009) and 3D4 (nucleotides 7010 to 7045). Next, we employed a poliovirus replicon, PLuc, which carries the firefly luciferase reporter gene in lieu of the capsid coding sequence P1 (17). Since PLuc translates and replicates like wild-type virus, luciferase activity provides a quantitative measure of viral translation and RNA replication (37). Mutants 3D2, 3D3, and 3D4 exhibited an RNA replication delay at 3 h posttransfection; however, this defect was transient, and by 5 h, all mutants reached wild-type levels of RNA production (Fig. 4B).
We then examined the genetic stability of the mutations introduced at the 3D-7000 structural element. We observed a rapid selection at position 7006 in the 3D3 virus: adenosine in the mutant was replaced by cytidine in the virus population after one round of replication. We further confirmed the strong negative selection against adenosine at position 7006 by isolating large and small plaques from the initial 3D3 virus stock and sequencing the region surrounding 3D-7000. We found that more than half (53.3%) of “large” plaques (those visible at 72 h) contained the A7006C mutation, while less than a fifth (19.6%) of “small” plaques (those invisible until 96 h) contained this substitution (Fig. 4D). We conclude that the nucleotide at position 7006 is critical for virus fitness.
Role of the 3D-7000 element in viral replication and infectivity.
To further define the critical features of the 3D-7000 element, we subdivided the critical 3D4 region into two parts. Thus, we designed two additional mutants, 3D4A and 3D4B (Fig. 5A). 3D4A contains the 10 mutations in the first 18 nt of 3D4, and 3D4B contains the remaining 6 mutations in the last 18 nt. We characterized these mutants in terms of plaque size (Fig. 5C) and PLuc phenotype (Fig. 5B). In both assays, we found that the 3D4A mutant closely recapitulated the 3D4 phenotype, while the 3D4B phenotype resembled that of the wild type.
Fig 5.

Subdivision and further characterization of the 3D4 mutant. (A) Mutants. On the schematic, color indicates nucleotides targeted in each mutant. The actual nucleotide changes and corresponding wild-type sequence and nucleotide positions for each mutant are listed below. For WT structure, A-U and C-G base pairing is indicated by lines and G-U (wobble) base pairing by dots. (B) PLuc assay. Mutant (colored) or wild-type (black) replicon RNA was transfected into HeLa S3 cells at 37°C. Cells were incubated in the presence (dashed lines) or absence (solid lines) of 2 mM GdnHCl. Luciferase activity (relative light units) corresponding to ∼3.1 × 105 cells was measured every 30 min. Data are plotted as the means ± standard deviations for three independent transfections. Replication delays at 2.0 to 3.5 h posttransfection are significant compared to results for the wild type in 3D4 and 3D4A (P < 0.05). (C) Plaque assays with plaque size measurement. Clonal stocks were used to infect HeLa S3 cells at 37°C. Box plots indicate the median plaque area in pixels, 25% and 75% quartiles, and 1.5× interquartile range; outliers are shown as open circles. 3D4 (P < 2.2E−16), 3D4A (P < 2.2E−16), and SL IV 298 (P = 5.6E−05) plaques are significantly smaller than the wild type. (D) Western blots. P1 virus stocks were used to infect HeLa S3 cells (MOI of ∼20). Total protein was harvested at 5 h postinfection. Five microliters of cell lysate was run for each sample and blotted with anti-3D or anti-2C. Membranes were stripped and reblotted with anti-GAPDH. (E) qPCR. P1 virus stocks were used to infect HeLa S3 cells (MOI of ∼25). Total RNA was harvested at 5 h postinfection, and positive-sense RNA was measured by strand-specific qPCR. Data are normalized to the average wild-type value and are plotted as the means ± standard deviations for three replicates.
Based on the replicon phenotype, we suspected a defect in translation and/or RNA synthesis. We therefore examined the abilities of 3D4, 3D4A, and 3D4B to produce viral proteins. Production of both 2CATPase, which is upstream of the 3D-7000 element, and 3Dpol (as well as its proteolytic precursor, 3CDpro, and the alternative proteolysis product 3D′ [38]) was analyzed by Western blotting at 5 h postinfection (Fig. 5D). We found that 3D4 and 3D4A do produce slightly less viral protein than 3D4B and the wild type. Because the processes of viral translation and RNA replication are dependent on each other, we also measured the number of RNA genomes present at 5 h postinfection and found reduced levels in the 3D4A mutant (Fig. 5E). It is thus likely that the slight decrease in protein levels (Fig. 5D) in the 3D-7000 mutants is attributable to the decrease in the amount of viral RNA available for translation (Fig. 5E). In contrast, a mutant with a genuine translation defect, SL IV 298 (24), produced more viral RNA than the 3D4A mutant (Fig. 5E) but less viral protein (Fig. 5D). Regardless, a decrease in translation cannot explain the severe plaque phenotype of the 3D-7000 mutants, since SL IV 298 exhibited only a small reduction in plaque size (Fig. 5C).
To reconcile the small effect of 3D-7000 mutations on RNA production with the very dramatic effects of these mutations on plaque size, we next considered whether 3D-7000 may have an additional function in the viral life cycle. To this end, we measured both RNA synthesis and infectious virus production during a single replication cycle. Using a strand-specific quantitative RT-PCR (qRT-PCR) assay similar to one previously described (23), we found that synthesis of both positive and negative strands was delayed in 3D4A compared to that in the wild type (Fig. 6C) but reached wild-type levels by 6 h postinfection. This finding was consistent with the results obtained using the PLuc replicon system (Fig. 4B and 5B). Analysis of virus production in this mutant showed that the final virus titer was dramatically reduced (Fig. 6B). Thus, while the 3D4A mutant can eventually produce almost wild-type levels of viral RNA and proteins, its production of infectious viral particles is impaired.
Fig 6.

Phenotype and location of second site suppressor mutations. (A) Plaque assays with plaque size measurement. Clonal stocks were used to infect HeLa S3 cells at 37°C. Each box plot indicates median plaque area in pixels, 25% and 75% quartiles, and 1.5× interquartile range; outliers are shown as open circles. Suppressor mutants (3D3-A7006C, 3D3-3CA11G, and 3D4A-3CY113F) are significantly different from both the relevant RNA structure mutant (3D3 or 3D4A) and the WT (P < 2.2E−16 in all cases except 3D3-A7006C versus WT: P = 7.1E−05). (B) One-step growth curves. P1 stocks of 3D4A (pink), 3D4A-3CY113F (magenta), or wild-type (black) virus were used to infect HeLa S3 cells (MOI of ∼25). Samples were taken every hour, and titers were determined by TCID50. Data are plotted as means ± standard deviations for three replicates. 3D4A is significantly (P < 0.05) lower than the wild type from 4 to 8 h postinfection. 3D4A-3CY113F is significantly (P < 0.05) higher than 3D4A at 4 to 5 h postinfection and lower than the wild type at 4 h postinfection. (C) Strand-specific qPCR. P1 stocks of 3D4A (pink), 3D4A-3CY113F (magenta), or wild-type (black) virus were used to infect HeLa S3 cells (MOI of ∼25). Samples were taken every hour, and the number of positive-sense (solid lines) or negative-sense (dashed lines) viral genomes was determined by strand-specific qRT-PCR. Data are plotted as means ± standard deviations for three replicates. For both strands, 3D4A is significantly (P < 0.05) lower than the wild type from 0 to 5 h postinfection. 3D4A-3CY113F is significantly (P < 0.05) higher than 3D4A at 4 to 6 h postinfection and significantly (P = 0.004) lower than the wild type at 4 h postinfection. (D and D) RNA packaging (ratio of packaged genomes to total genomes) (D) and virion infectivity (ratio of infectious units to packaged genomes) (E). After titer determination, the 3D4A (pink), 3D4A-3CY113F (magenta), and wild-type (white) 8-h replicates from the one-step growth curves (A) were subjected to virion immunoprecipitation, and the number of packaged genomes was assayed by strand-specific RT-qPCR. This number was compared to either the average number of total viral genomes at 8 h from panel B or the 8-h replicate viral titer from panel A. Data are plotted as means ± standard deviations for three replicates. Background levels from a negative control (boiled viral lysate) were more than 1,000-fold lower than the lowest reported experimental values. (F) PV 3Cpro. A11 is shown in red, and Y113 is shown in pink. The catalytic triad (H40, E71, and C147) are in blue, and the residues known to bind to the 5′CL (KFRDIR 82 to 87) are in green.
A simple interpretation of these results is that the 3D-7000 element is involved in RNA packaging. According to this view, even if 3D4A produces wild-type levels of RNA, it makes a lower number of infectious particles. To examine this possibility directly, we purified virus particles by immunoprecipitation and determined the proportion of encapsidated viral RNA. Interestingly, while the 3D4A and wild-type viruses packaged similar amounts of RNA into virions (Fig. 6D), fewer virions with the 3D4A mutation were infectious, resulting in significant reduction in the ratio of infectious units to packaged RNA (Fig. 6E). Thus, 3D-7000 mutants do not exhibit a gross defect in RNA packaging but produce viruses of lower infectivity.
Mutations in 3C/3CDpro partially suppress the 3D-7000 mutant phenotype.
To further characterize the mechanism underlying the replication and infectivity defects of the 3D-7000 mutants, we looked for second site suppressors of our mutations using a two-pronged approach. First, we examined the viruses derived from the “large” plaques (those visible at 72 h) of the 3D3 virus (Fig. 4D). Of the 45 large plaques initially sequenced in the 3D-7000 region, 24 contained the A7006C mutation and were not considered further. Of note, our sequence analysis included the region identified as β by Song et al. (12), and no second site mutations were found in that region. The 21 remaining plaques were tested to confirm the presence of a genetically stable large-plaque phenotype. Most did not exhibit larger plaques than the parental 3D3 mutant; however, three clones exhibited a reproducible large-plaque phenotype (data not shown). The entire viral genome of each of these clones was sequenced. Two of the three contained a mutation in the region encoding the viral protease, 3Cpro: C5469G, which encodes an amino acid change, A11G, in 3Cpro. (The third clone contained a mutation in the region encoding the viral protease, 2Apro: G3826A, which encodes an amino acid change, M147I, in 2Apro. Since this mutation was not identified in multiple clones, its effects were not further analyzed.) No other mutations were found in these clones.
We also examined the genetic stability of the 3D4A mutant. We observed that the severity of the small-plaque phenotype (Fig. 5C) was exacerbated at 39°C compared to that at 37°C (data not shown). We therefore reasoned that while 3D4A was genetically stable upon passage at 37°C, it might not be so at 39°C. Indeed, after two passages at 39°C, a single point mutation was fixed in the population in seven out of seven clones examined. That mutation was in the region encoding the viral protease, 3Cpro: A5775U, which encodes an amino acid change, Y113F, in 3Cpro. The entire viral genome was sequenced in all seven clones, and no other mutations were observed at the consensus level.
Further characterization of the two mutations in 3Cpro shows that they do indeed partially suppress the small-plaque phenotype exhibited by the 3D3 and 3D4A mutants (Fig. 6A). We also found that the 3CY113F mutation could partially suppress the RNA synthesis and infectivity defects of the 3D4A mutant (Fig. 6C and E). This suppression could be due either to a long-range RNA-RNA interaction or to interaction of the 3D-7000 RNA structure with the 3Cpro protein. This is difficult to test directly, because in both cases the nucleotide change is in the second position in the codon, and there are no synonymous changes possible that could distinguish between these possibilities. However, several factors argue in favor of interaction with the 3Cpro protein or one of its precursors. Nucleotide 5775 is within the RNase L ciRNA structure (Fig. 1E) and has a high SHAPE reactivity, arguing strongly against any kind of base-pairing interaction with the 3D-7000 structure. Nucleotide 5469 does not fall in a region predicted by our global RNA structure analysis to be structured (Fig. 2B and Table 1). However, within the three-dimensional structure of the 3Cpro protein, the amino acids A11 and Y113 are in close physical proximity (Fig. 6F), and an analysis of contacts of structural units (39), which calculates the surface area of every atom and its intersection with other atoms, predicts a noncovalent interaction between these residues based on the structure of 3C (Protein Data Bank [PDB]: 1L1N). Attempts to demonstrate a direct interaction between the 3D-7000 RNA and the viral protein 3Cpro (or its precursor, 3CDpro) have thus far been unsuccessful, perhaps because the 3D-7000 structure does not fold properly in vitro or requires long-range interaction with another RNA structure, such as 5′-CL, to bind to 3Cpro. Nonetheless, these results suggest that the role of the 3D-7000 RNA structure in viral replication and infectivity is mediated by interaction with the viral protein 3Cpro, since mutations in a particular interface of the viral protein can partially suppress defects caused by mutations in the RNA structure.
DISCUSSION
In this study, we combine global chemical probing (SHAPE) of a viral genome with evolutionary analysis to identify several RNA structures in the poliovirus genome that may be functionally relevant to viral replication (Table 1). This combinatorial analysis proved a powerful tool to assess potentially functional regions, since these may be conserved across the human enterovirus C group. Indeed, two such highly conserved structures were independently identified by others (12), validating this approach as a means of identifying novel, functional RNA structures. Site-directed mutagenesis of one of these structures revealed a functional RNA element that plays an important role in viral replication and infectivity. Mutants in which the structure is disrupted exhibit delays in RNA synthesis that are observable in both the PLuc replicon system (Fig. 4B and 5B) and the virus (Fig. 6C), as well as a striking reduction in particle infectivity (Fig. 6E). These defects can be partially suppressed by mutations in the viral protease 3Cpro (Fig. 6). These results suggest that both RNA synthesis and the infectivity of the virus are directly modulated by this RNA structure, likely via interaction with the viral protein 3Cpro or its precursor, 3CDpro. This finding raises intriguing questions about the potential for interaction of 3D-7000 with the other RNA structural elements involved in replication, such as 5′-CL and 2C-CRE, both of which are known to interact with 3Cpro and/or 3CDpro (24, 35). Whether these interactions reflect a single RNP complex or a dynamic series of complexes will be an interesting question for future study.
SHAPE is a powerful tool for RNA secondary structure analysis. Its many benefits and limitations have been thoroughly reviewed elsewhere (40, 41). It has been used to analyze the structure of the full-length HIV-1 genomic RNA (14) and to analyze portions of the HIV-1 genome (42) and other retroviral genomes (43–45; reviewed in reference 46) and more recently the SIV (15) and SMTV (16) genomes. The identification of several novel putative structures in the open reading frame (Fig. 2 and Table 1) suggests that even a virus as well studied as poliovirus can reveal surprises when analyzed by unbiased, global methods and argues that SHAPE could be used to great effect to identify functional RNA structures in the genomes of less well studied RNA viruses. Recent advances could allow such studies to be performed even without the large amounts of purified virions used in this study (45).
We have identified several regions where a low median SHAPE reactivity indicates the presence of an RNA structure (Fig. 2B and Table 1). Of the eight identified regions that do not represent previously described structures, five have moderate to high median pairing probabilities (Fig. 2C and Table 1). This analysis provides an independent line of evidence supporting the presence of a functionally conserved RNA structure. Two of the five putative structure fall in the 3Dpol-coding region and correspond roughly to the α and β structures described by Song et al. (12). Interestingly, the other three are partially or totally within the P1 portion of the genome, which encodes the structural (capsid) proteins. A number of codon shuffling and deoptimization experiments have targeted the P1 region (47–50). The results of those experiments indicate that the strongly attenuated phenotypes are due to systematic alterations in codon usage and/or dinucleotide abundance, rather than specific local disruption of an RNA structural element. However, in light of the RNA structures identified here, it is difficult to exclude the possibility that one or more of the structures in this region play an important role in the viral life cycle; the nature of synonymous mutation means that ∼2/3 of nucleotides remain unchanged, which can suffice to maintain a structure well enough to preserve its function.
Within the region of the genome encoding 3Dpol, we identified a highly conserved and functional structure, which we call 3D-7000 and which corresponds roughly to the α element described by Song et al. (12). The phenotypes of our six initial mutants indicated that the central portion of this region (6974 to 7045), mutated in the 3D3 and 3D4 mutants, was critical for viral replication. The strong selection for the A7006C mutation in the 3D3 mutant (Fig. 4D) and our ability to localize the severe 3D4 phenotype to the first 18 nt (7010 to 7027; 3D4A mutant in Fig. 5) further narrowed the region of functional importance. We note that the sequence at nt 7004 to 7044 is completely conserved in all the strains of poliovirus that we examined (Fig. 3B). Moreover, nut 7005 to 7026 exhibit extremely low SHAPE reactivity despite being predicted to include parts of two different loops (Fig. 3A). As we noted in our analysis of previously identified RNA structures in the poliovirus genome, unreactive loops can often indicate a tertiary interaction, such as a pseudoknot or kissing loops, and we speculate that one or both of these loops may be involved in a longer-range interaction of this type.
Of interest is whether phenotypes previously attributed to 3Dpol amino acid changes might in fact result from defects in this RNA element. According to the comprehensive list of known 3Dpol mutations compiled by Tellez et al. (51), only two 3Dpol mutations in amino acids 330 to 353, corresponding to nucleotides 6974 to 7045, have been published. These are D339A/S331A/D349A (52, 53) and L342A (54). These amino acid residues are thought to be involved in 3Dpol oligomeric interfaces, and in vitro biochemical work with the polymerase mutants supports that view (52, 54). However, the viral phenotypes described include an RNA synthesis defect (52) and small plaques (52, 54), with the latter phenotype exhibiting a marked temperature sensitivity (53, 54). All of these phenotypes are also exhibited by our synonymous 3D-7000 mutants.
The 3D-7000 element appears to directly affect RNA synthesis. The presence of a phenotype in the PLuc mutants (Fig. 4B and 5B, solid lines) indicates that translation or RNA synthesis must be directly affected, since the replicons do not undergo other viral processes, such as attachment, uncoating, morphogenesis, or virion release. The level of luciferase production in the presence of GdnHCl, a replication inhibitor, suggests that mutations in the 3D-7000 element do not affect the efficiency of translation (Fig. 4B and 5B, dashed lines). 3D4 and 3D4A do show somewhat lower levels of viral proteins (Fig. 5D). However, after comparison to a mutant with a known translation defect, SL IV 298 (24), we attribute that difference to the defect in positive-strand RNA synthesis (Fig. 5E and 6C) (see below), which makes fewer genomes available for translation, rather than any mechanistic defect in translation itself. The defect in RNA synthesis, while significant, is transient. Indeed, by 5 h posttransfection, PLuc mutants exhibit wild-type levels of luciferase activity (Fig. 4B and 5B), and by 6 h postinfection, mutant viruses produced wild-type levels of RNA (Fig. 6C). In addition to the defects in RNA synthesis, 3D-7000 mutants exhibit reduced particle infectivity (Fig. 6E), suggesting that this RNA structure may play multiple roles in the viral life cycle.
As this study was nearing completion, another group independently identified RNA elements that overlap substantially with the 3D-7000 element described here. The authors report that they identified “two unique functionally redundant RNA elements (α and β), each about 75 nt long and separated by 150 nt, in the 3′-terminal coding sequence of RNA polymerase, 3Dpol” (12). The β element (nt 7220 to 7294) corresponds to the most 3′ structure identified in our SHAPE analysis, and its conservation is consistent with our pairing probability analysis of that region as well (Table 1 and Fig. 2). The α element (nt 6995 to 7069) corresponds quite closely with the 3D-7000 element described in this study. The functionally important part of the α element is reported to be a conserved 48-nt stretch (6999 to 7045), which is consistent with the nucleotides identified as functionally important in the current study: 7006, which is subject to rapid selection in the 3D3 mutant, and 7010 to 7027, which is the region altered in the 3D4A mutant (Fig. 4 and 5). While the findings of Song et al. clearly indicate a genetic interaction between the α and β elements, our results argue against a simple functional redundancy. We found that as few as 10 mutations in the α region (3D4A mutant), with no additional mutations in the β region, are sufficient to yield a clear replication defect (Fig. 5 and 6). Clearly the precise mutations engineered in the different experiments differed substantially, despite the fact that both were synonymous with respect to the protein coding capacity of the region. This underscores the difficulty of identifying functionally important RNA structures by mutagenesis while simultaneously maintaining the open reading frame.
The plaque size, replication, and translation phenotypes reported by Song et al. (12) are similar to, though less extensively characterized than, those of the 3D-7000 mutants. However, the most intriguing phenotype identified in the present study is the defect in virion infectivity (Fig. 6E). On a mechanistic level, there are a number of plausible explanations for this phenotype. One possibility is purely biophysical: the lesion on the 3D-7000 RNA structure destabilizes the poliovirus virion, reducing the number of infectious particles. We analyzed the thermostability of 3D4A and WT virions over time and found no significant difference (data not shown), which argues against this possibility, as does the ability of mutations in 3Cpro to partially suppress the phenotype (Fig. 6), since the nonstructural viral proteins are not present in the PV capsid. A second possibility is probabilistic: given that a productive infection represents a series of stochastic events (55), one or more critical events (entry, translation, and replication) could be modulated by the 3D-7000 RNA structure. In mutants, the event(s) would occur less frequently, decreasing the probability of a productive infection. The observed replication defects, as well as the suppressor mutations in 3Cpro, which is involved in many of these events, both argue in favor of this explanation. A third possibility is immunological: the 3D-7000 RNA structure interacts with one or more host antiviral defense mechanisms, in a manner analogous to that of the RNase L ciRNA (10, 11). We cannot exclude this possibility, although the suppressor phenotype suggests that 3Cpro would also have to be involved in counteracting innate antiviral immunity.
In summary, we have used SHAPE to globally interrogate the structure of full-length, native poliovirus virion RNA. Our experimental results, together with orthogonal evolutionary predictions, indicate that there are a number of conserved RNA structures within the poliovirus open reading frame. Our site-directed mutagenesis demonstrated the functional relevance of one such structure, the 3D-7000 element. Disruption of this structure affects viral replication kinetics, delaying both negative- and positive-strand RNA synthesis, as well as the infectivity of the virus particle. Mutations in the viral protein 3Cpro are able to partially suppress the mutant phenotype, suggesting that the 3D-7000 structure interacts, directly or indirectly, with this protein.
ACKNOWLEDGMENTS
We thank members of the Andino laboratory for critical comments on the manuscript.
This work was supported by grants from the NIAID to R.A. (R01 AI36178 and AI40085) and the University of California (CCADD).
Footnotes
Published ahead of print 21 August 2013
REFERENCES
- 1.Hogle JM, Chow M, Filman DJ. 1985. Three-dimensional structure of poliovirus at 2.9 A resolution. Science 229:1358–1365 [DOI] [PubMed] [Google Scholar]
- 2.Kew OM, Sutter RW, de Gourville EM, Dowdle WR, Pallansch MA. 2005. Vaccine-derived polioviruses and the endgame strategy for global polio eradication. Annu. Rev. Microbiol. 59:587–635 [DOI] [PubMed] [Google Scholar]
- 3.Pelletier J, Kaplan G, Racaniello VR, Sonenberg N. 1988. Cap-independent translation of poliovirus mRNA is conferred by sequence elements within the 5′ noncoding region. Mol. Cell. Biol. 8:1103–1112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Trono D, Pelletier J, Sonenberg N, Baltimore D. 1988. Translation in mammalian cells of a gene linked to the poliovirus 5′ noncoding region. Science 241:445–448 [DOI] [PubMed] [Google Scholar]
- 5.Andino R, Rieckhof GE, Baltimore D. 1990. A functional ribonucleoprotein complex forms around the 5′ end of poliovirus RNA. Cell 63:369–380 [DOI] [PubMed] [Google Scholar]
- 6.Barton DJ, O'Donnell BJ, Flanegan JB. 2001. 5′ cloverleaf in poliovirus RNA is a cis-acting replication element required for negative-strand synthesis. EMBO J. 20:1439–1448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vogt DA, Andino R. 2010. An RNA element at the 5′-end of the poliovirus genome functions as a general promoter for RNA synthesis. PLoS Pathog. 6:e1000936. 10.1371/journal.ppat.1000936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pilipenko EV, Poperechny KV, Maslova SV, Melchers WJ, Slot HJ, Agol VI. 1996. Cis-element, oriR, involved in the initiation of (-) strand poliovirus RNA: a quasi-globular multi-domain RNA structure maintained by tertiary (‘kissing') interactions. EMBO J. 15:5428–5436 [PMC free article] [PubMed] [Google Scholar]
- 9.Goodfellow I, Chaudhry Y, Richardson A, Meredith J, Almond JW, Barclay W, Evans DJ. 2000. Identification of a cis-acting replication element within the poliovirus coding region. J. Virol. 74:4590–4600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Han JQ, Townsend HL, Jha BK, Paranjape JM, Silverman RH, Barton DJ. 2007. A phylogenetically conserved RNA structure in the poliovirus open reading frame inhibits the antiviral endoribonuclease RNase L. J. Virol. 81:5561–5572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Townsend HL, Jha BK, Han JQ, Maluf NK, Silverman RH, Barton DJ. 2008. A viral RNA competitively inhibits the antiviral endoribonuclease domain of RNase L. RNA 14:1026–1036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Song Y, Liu Y, Ward CB, Mueller S, Futcher B, Skiena S, Paul AV, Wimmer E. 2012. Identification of two functionally redundant RNA elements in the coding sequence of poliovirus using computer-generated design. Proc. Natl. Acad. Sci. U. S. A. 109:14301–14307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wilkinson KA, Merino EJ, Weeks KM. 2006. Selective 2′-hydroxyl acylation analyzed by primer extension (SHAPE): quantitative RNA structure analysis at single nucleotide resolution. Nat. Protoc. 1:1610–1616 [DOI] [PubMed] [Google Scholar]
- 14.Watts JM, Dang KK, Gorelick RJ, Leonard CW, Bess JW, Jr, Swanstrom R, Burch CL, Weeks KM. 2009. Architecture and secondary structure of an entire HIV-1 RNA genome. Nature 460:711–716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pollom E, Dang KK, Potter EL, Gorelick RJ, Burch CL, Weeks KM, Swanstrom R. 2013. Comparison of SIV and HIV-1 genomic RNA structures reveals impact of sequence evolution on conserved and non-conserved structural motifs. PLoS Pathog. 9:e1003294. 10.1371/journal.ppat.1003294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Archer EJ, Simpson MA, Watts NJ, O'Kane R, Wang B, Erie DA, McPherson A, Weeks KM. 2013. Long-range architecture in a viral RNA genome. Biochemistry 52:3182–3190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Herold J, Andino R. 2000. Poliovirus requires a precise 5′ end for efficient positive-strand RNA synthesis. J. Virol. 74:6394–6400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vasa SM, Guex N, Wilkinson KA, Weeks KM, Giddings MC. 2008. ShapeFinder: a software system for high-throughput quantitative analysis of nucleic acid reactivity information resolved by capillary electrophoresis. RNA 14:1979–1990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Deigan KE, Li TW, Mathews DH, Weeks KM. 2009. Accurate SHAPE-directed RNA structure determination. Proc. Natl. Acad. Sci. U. S. A. 106:97–102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.McGinnis JL, Duncan CD, Weeks KM. 2009. High-throughput SHAPE and hydroxyl radical analysis of RNA structure and ribonucleoprotein assembly. Methods Enzymol. 468:67–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Westesson O, Holmes I. 2012. Developing and applying heterogeneous phylogenetic models with XRate. PLoS One 7:e36898. 10.1371/journal.pone.0036898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lauring AS, Acevedo A, Cooper SB, Andino R. 2012. Codon usage determines the mutational robustness, evolutionary capacity, and virulence of an RNA virus. Cell Host Microbe 12:623–632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Plaskon NE, Adelman ZN, Myles KM. 2009. Accurate strand-specific quantification of viral RNA. PLoS One 4:e7468. 10.1371/journal.pone.0007468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gamarnik AV, Andino R. 2000. Interactions of viral protein 3CD and poly(rC) binding protein with the 5′ untranslated region of the poliovirus genome. J. Virol. 74:2219–2226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Beinz K. 1974. Host cell reactions on virus infection (author's transl). Pathol. Microbiol. (Basel) 41:143–150 (In German.) [PubMed] [Google Scholar]
- 26.Gamarnik AV, Andino R. 1998. Switch from translation to RNA replication in a positive-stranded RNA virus. Genes Dev. 12:2293–2304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Crotty S, Saleh MC, Gitlin L, Beske O, Andino R. 2004. The poliovirus replication machinery can escape inhibition by an antiviral drug that targets a host cell protein. J. Virol. 78:3378–3386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Burrill CP, Strings VR, Andino R. 2013. Poliovirus: generation, quantification, propagation, purification, and storage. Curr. Protoc. Microbiol. 29:15H.1.1–15H.1.27. 10.1002/9780471729259.mc15h01s29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wilkinson KA, Vasa SM, Deigan KE, Mortimer SA, Giddings MC, Weeks KM. 2009. Influence of nucleotide identity on ribose 2′-hydroxyl reactivity in RNA. RNA 15:1314–1321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Simoes EA, Sarnow P. 1991. An RNA hairpin at the extreme 5′ end of the poliovirus RNA genome modulates viral translation in human cells. J. Virol. 65:913–921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Paul AV, Rieder E, Kim DW, van Boom JH, Wimmer E. 2000. Identification of an RNA hairpin in poliovirus RNA that serves as the primary template in the in vitro uridylylation of VPg. J. Virol. 74:10359–10370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Townsend HL, Jha BK, Silverman RH, Barton DJ. 2008. A putative loop E motif and an H-H kissing loop interaction are conserved and functional features in a group C enterovirus RNA that inhibits ribonuclease L. RNA Biol. 5:263–272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rieder E, Paul AV, Kim DW, van Boom JH, Wimmer E. 2000. Genetic and biochemical studies of poliovirus cis-acting replication element cre in relation to VPg uridylylation. J. Virol. 74:10371–10380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Goodfellow IG, Kerrigan D, Evans DJ. 2003. Structure and function analysis of the poliovirus cis-acting replication element (CRE). RNA 9:124–137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yin J, Paul AV, Wimmer E, Rieder E. 2003. Functional dissection of a poliovirus cis-acting replication element [PV-cre(2C)]: analysis of single- and dual-cre viral genomes and proteins that bind specifically to PV-cre RNA. J. Virol. 77:5152–5166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mathews DH, Disney MD, Childs JL, Schroeder SJ, Zuker M, Turner DH. 2004. Incorporating chemical modification constraints into a dynamic programming algorithm for prediction of RNA secondary structure. Proc. Natl. Acad. Sci. U. S. A. 101:7287–7292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Andino R, Rieckhof GE, Achacoso PL, Baltimore D. 1993. Poliovirus RNA synthesis utilizes an RNP complex formed around the 5′-end of viral RNA. EMBO J. 12:3587–3598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Toyoda H, Nicklin MJ, Murray MG, Anderson CW, Dunn JJ, Studier FW, Wimmer E. 1986. A second virus-encoded proteinase involved in proteolytic processing of poliovirus polyprotein. Cell 45:761–770 [DOI] [PubMed] [Google Scholar]
- 39.Sobolev V, Wade RC, Vriend G, Edelman M. 1996. Molecular docking using surface complementarity. Proteins 25:120–129 [DOI] [PubMed] [Google Scholar]
- 40.Low JT, Weeks KM. 2010. SHAPE-directed RNA secondary structure prediction. Methods 52:150–158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Weeks KM, Mauger DM. 2011. Exploring RNA structural codes with SHAPE chemistry. Acc. Chem. Res. 44:1280–1291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wilkinson KA, Gorelick RJ, Vasa SM, Guex N, Rein A, Mathews DH, Giddings MC, Weeks KM. 2008. High-throughput SHAPE analysis reveals structures in HIV-1 genomic RNA strongly conserved across distinct biological states. PLoS Biol. 6:e96. 10.1371/journal.pbio.0060096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gherghe C, Leonard CW, Gorelick RJ, Weeks KM. 2010. Secondary structure of the mature ex virio Moloney murine leukemia virus genomic RNA dimerization domain. J. Virol. 84:898–906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gherghe C, Lombo T, Leonard CW, Datta SA, Bess JW, Jr, Gorelick RJ, Rein A, Weeks KM. 2010. Definition of a high-affinity Gag recognition structure mediating packaging of a retroviral RNA genome. Proc. Natl. Acad. Sci. U. S. A. 107:19248–19253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Grohman JK, Kottegoda S, Gorelick RJ, Allbritton NL, Weeks KM. 2011. Femtomole SHAPE reveals regulatory structures in the authentic XMRV RNA genome. J. Am. Chem. Soc. 133:20326–20334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sztuba-Solinska J, Le Grice SF.2012. Probing retroviral and retrotransposon genome structures: the “SHAPE” of things to come. Mol. Biol. Int. 2012:530754. 10.1155/2012/530754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Coleman JR, Papamichail D, Skiena S, Futcher B, Wimmer E, Mueller S. 2008. Virus attenuation by genome-scale changes in codon pair bias. Science 320:1784–1787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mueller S, Papamichail D, Coleman JR, Skiena S, Wimmer E. 2006. Reduction of the rate of poliovirus protein synthesis through large-scale codon deoptimization causes attenuation of viral virulence by lowering specific infectivity. J. Virol. 80:9687–9696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Burns CC, Campagnoli R, Shaw J, Vincent A, Jorba J, Kew O. 2009. Genetic inactivation of poliovirus infectivity by increasing the frequencies of CpG and UpA dinucleotides within and across synonymous capsid region codons. J. Virol. 83:9957–9969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Burns CC, Shaw J, Campagnoli R, Jorba J, Vincent A, Quay J, Kew O. 2006. Modulation of poliovirus replicative fitness in HeLa cells by deoptimization of synonymous codon usage in the capsid region. J. Virol. 80:3259–3272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tellez AB, Wang J, Tanner EJ, Spagnolo JF, Kirkegaard K, Bullitt E. 2011. Interstitial contacts in an RNA-dependent RNA polymerase lattice. J. Mol. Biol. 412:737–750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pathak HB, Ghosh SK, Roberts AW, Sharma SD, Yoder JD, Arnold JJ, Gohara DW, Barton DJ, Paul AV, Cameron CE. 2002. Structure-function relationships of the RNA-dependent RNA polymerase from poliovirus (3Dpol). A surface of the primary oligomerization domain functions in capsid precursor processing and VPg uridylylation. J. Biol. Chem. 277:31551–31562 [DOI] [PubMed] [Google Scholar]
- 53.Burgon TB, Jenkins JA, Deitz SB, Spagnolo JF, Kirkegaard K. 2009. Bypass suppression of small-plaque phenotypes by a mutation in poliovirus 2A that enhances apoptosis. J. Virol. 83:10129–10139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hobson SD, Rosenblum ES, Richards OC, Richmond K, Kirkegaard K, Schultz SC. 2001. Oligomeric structures of poliovirus polymerase are important for function. EMBO J. 20:1153–1163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hensel SC, Rawlings JB, Yin J. 2009. Stochastic kinetic modeling of vesicular stomatitis virus intracellular growth. Bull. Math. Biol. 71:1671–1692 [DOI] [PMC free article] [PubMed] [Google Scholar]