

Abstract
Expression of cell-intrinsic antiviral factors suppresses HIV-1 replication. We hypothesized that cellular activation modulates host restriction and susceptibility to HIV-1 infection. We measured the gene expression of 34 antiviral factors in healthy peripheral blood mononuclear cells (PBMC). Cellular activation induced expression of interferon-stimulated gene 15 (ISG15), tripartite motif 5α (TRIM5α), bone marrow stromal cell antigen 2 (BST-2)/tetherin, and certain apolipoprotein B mRNA editing enzyme 3 (APOBEC3) family members. Expression of RTF1, RNA polymerase II-associated factor 1 (PAF1), TRIM11, TRIM26, and BST-2/tetherin correlated with decreased HIV-1 infectivity. This report demonstrates synchronous effects of activation-induced antiviral genes on HIV-1 infectivity, providing candidates for pharmacological manipulation.
TEXT
Restriction factors are potent antiviral host proteins that confer protection against retroviral infections (1). Prototypical examples of such proteins are APOBEC3G and APOBEC3F, which inhibit replication of HIV-1 variants lacking a functional vif gene. APOBEC3G is incorporated into HIV-1 virions and deaminates cytosines to uracils in replication intermediates, providing templates for lethal guanine-to-adenine mutations (2). The TRIM family members have also received much attention and are known to target the viral capsid and to inhibit viral transcription (3). Recently, other anti-HIV-1 factors have been described and studied in detail. SAMHD1 was recently shown to inhibit HIV and SIV infection in quiescent CD4+ T cells (4, 5) and myeloid cells (6, 7) by eliminating the intracellular pool of deoxynucleoside triphosphates (dNTPs) required for viral cDNA production during reverse transcription.
Evaluating the overall anti-HIV-1 repertoire in primary cells is critical to identifying host cell-intrinsic defenses against the virus that may be targeted by small molecules, cytokines, or gene therapeutic strategies. Most studies to date have focused on the expression of individual restriction factors or restriction factor gene families, such as APOBEC3 (8–10) or TRIM. In this study, we hypothesized that cellular activation modulates a broad spectrum of host restriction mechanisms and that variations in restriction factor gene expression profiles between activated donor cells influence HIV-1 infectivity in vitro.
Abbreviations.
APOBEC, apolipoprotein B mRNA editing enzyme; BST-2, bone marrow stromal cell antigen 2; SAMHD1, sterile alpha motif (SAM) domain- and HD domain-containing protein 1; TRIM, tripartite motif; ISG, interferon-stimulated gene; CDKN1A, cyclin-dependent kinase inhibitor 1A; PAF1, RNA polymerase II-associated factor 1; DMSO, dimethyl sulfoxide; FCS, fetal calf serum; IL-2, interleukin-2; PHA-P, phytohemagglutinin-P; CT, threshold cycle; TLDA, TaqMan low-density array; IFN-α, alpha interferon; RLU, relative light units; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; TCID50, 50% tissue culture infective dose; PBMC, peripheral blood mononuclear cells; EIF2AK2, eukaryotic translation initiation factor 2-alpha kinase 2; HERC5, HECT domain and RLD domain containing E3 ubiquitin protein ligase 5; IFITM, interferon-induced transmembrane; MOV10, Moloney leukemia virus 10 homolog; SLFN11, Schlafen family member 11.
We first investigated the expression profile of host anti-HIV-1 factors in PBMC before and after cellular activation in vitro. Human primary PBMC were isolated by the use of a Ficoll gradient from buffy coats obtained from 43 anonymous HIV-1-seronegative individuals from the Stanford blood bank. After processing, PBMC were stored and frozen in 10% DMSO–FCS prior to subsequent analyses. Cryopreserved PBMC were quickly thawed and used for RNA extraction. Alternatively, cryopreserved PBMC from the same blood donor were thawed and placed in a T-25 culture flask containing 30 ml of growth medium (RPMI 1640, 2 mM l-glutamine, 25 mM HEPES, 20% heat-inactivated FCS, 100 U/ml IL-2, and 50 μg gentamicin/ml) and PHA-P (5 μg/ml) for 1 day at 37°C. PHA was used for stimulation of PBMC as it provides a strong mitogenic activation of cells by binding nonspecifically to the cell membrane glycoproteins, cross-linking the T cell receptors in a donor-independent manner, yielding rates of proliferation and upregulation of CD25 and CD69 comparable to the rates seen with anti-CD3/CD28 costimulation (11). Total RNA was extracted from whole PBMC using TRIzol followed by DNase treatment. DNase-treated RNA was transcribed into cDNA using random primers. Quantitative real-time PCR utilizing a custom-made TLDA and thermal cycling was performed using an ABI Prism 7900HT sequence detection system. A panel of 6 housekeeping genes (GAPDH, 18S, ACTB, PPIA, RPLP0, and UBC) was included in the TLDA plates, and PPIA was identified as the most stably expressed gene using the GeNorm algorithm (12). Fold induction was determined using the comparative CT method. All gene cards (Applied Biosystems) used for quantification are depicted in Table S1 in the supplemental material.
We developed a heat map illustrating the relative fold changes in antiviral gene expression in all 43 donors following in vitro activation (Fig. 1A). Interestingly, we found that APOBEC3A and APOBEC3F clustered together and were both significantly downregulated after activation. APOBEC3F showed an expression pattern substantially different from that of APOBEC3G, which was upregulated in most donors, as were the antiviral factors TRIM5α, BST-2/tetherin, and ISG15.
Fig 1.

Host antiviral gene expression analysis in resting and activated PBMC. PBMC (n = 43) from the same blood donor were left untreated or were PHA-P (5 μg/ml) activated for 1 day. RNA was extracted using TRIzol followed by cDNA synthesis. Quantitative real-time PCR was performed using custom-made TaqMan low-density arrays (TLDA). A panel of 6 housekeeping genes was included in the TLDA plates, and PPIA was identified as the most stably expressed gene and was used for normalization of results by the comparative CT method. All gene cards (Applied Biosystems) used for quantification are depicted in Table S1 in the supplemental material. (A) Heat map depicting fold induction of antiviral gene expression following activation. (B) Ascending fold changes in gene expression. Bars represent means + standard errors of the means (SEM) of antiviral gene expression determinations for a total of 43 donors. (C) Comparative analysis of antiviral gene expression in resting and activated donor cells. Statistical analysis was performed using the Mann-Whitney test (***, P < 0.001).
A summary of the mean fold changes (downregulation or upregulation after stimulation) of all antiviral factors in the study population is shown in Fig. 1B. APOBEC3F and APOBEC3A were downregulated nearly 2-fold; all other antiviral factors analyzed were upregulated. BST-2/tetherin, TRIM5α, APOBEC3G, and ISG15 were highly (6-fold to over 25-fold) upregulated.
To better depict the relative levels of gene expression of all the antiviral factors screened at the resting state and after activation, we generated Fig. 1C. All changes in gene expression were statistically significant (P < 0.001). APOBEC3A and APOBEC3C were the most abundant genes in resting cells, and BST-2/tetherin, APOBEC3G, and ISG15 were the most highly expressed genes in activated PBMC.
Taken together, our data demonstrate that levels of antiviral factor gene expression are significantly different in resting and activated PBMC and that the differential levels of expression in the resting and activated states could have implications for control of HIV-1 replication.
We then extended the panel of host antiviral factors and included a total of 34 different anti-HIV-1 genes. All anti-HIV-1 genes chosen for this screen are substantiated by at least one publication, demonstrating effective, cell-autonomous anti-HIV-1 activity in vitro (Table 1).
Table 1.
List of the 34 anti-HIV-1 host factors measured
| Factor name | NCBI Entrez gene description | Key anti-HIV-1 role (s) | Reference(s) |
|---|---|---|---|
| APOBEC3A to -H | Apolipoprotein B mRNA editing enzyme, catalytic polypeptide-like 3 | Hypermutation; lethal mutations in viral DNA; inhibition of reverse transcription; inhibition of integration | 2, 13, 14, 15, 16, 17, 18, 19, 20 |
| TRIM family (11 members) | Tripartite motif family | Targeting of viral capsid; inhibition of viral transcription | 21, 22, 23, 24, 25 |
| BST-2/tetherin | Bone marrow stromal cell antigen 2 | Blocks release of enveloped viruses | 26, 27 |
| CDKN1A (P21) | Cyclin-dependent kinase inhibitor 1A (p21, Cip1) | Blocks reverse transcription; blocks RNA transcription by reducing activity of CDK9 | 28 |
| PAF1 | Paf1, RNA polymerase II-associated factor 1 | Inhibits early events of viral life cycle from reverse transcription to integration | 29 |
| CTR9 | Ctr9, Paf1/RNA polymerase II complex component | Inhibits early events of viral life cycle from reverse transcription to integration | 29 |
| RTF1 | Rtf1, Paf1/RNA polymerase II complex component | Inhibits early events of viral life cycle from reverse transcription to integration | 29 |
| EIF2AK2 (PKR) | Eukaryotic translation initiation factor 2-alpha kinase 2 | Inhibits viral protein translation by protein phosphorylation; promotes innate immune signaling | 30 |
| HERC5 | HERC5, HECT domain and RLD domain containing E3 ubiquitin protein ligase 5 | Blocks early stage of retroviral particle assembly | 31 |
| IFITM family (3 members) | Interferon-induced transmembrane protein | Inhibits cytosolic entry | 46 |
| ISG15 | ISG15 ubiquitin-like modifier | Blocks interaction between HIV-1 Gag and Tsg101 (ESCRT-I) required for efficient budding of HIV-1 | 32, 33 |
| MOV10 | Mov10, Moloney leukemia virus 10, homolog | Inhibits proteolytic processing of Gag and reverse transcription | 34, 35, 36 |
| RNASEL | RNase L (2′,5′-oligoisoadenylate synthetase dependent) | Cleaves single-stranded RNA in U-rich sequences; activates antiviral innate immunity | 37 |
| RSAD2 (viperin) | Radical S-adenosyl methionine domain containing 2 | Inhibits viral production | 38 |
| SAMHD1 | SAM domain and HD domain 1 | Inhibits HIV replication in quiescent CD4+ T cells and myeloid cells by regulating cellular dNTP supply | 4, 5, 6, 7 |
| SLFN11 | Schlafen family member 11 | Inhibits viral protein synthesis | 39 |
We activated PBMC cultures for 1 day with PHA-P (5 μg/ml), as described above, extracted RNA, and measured the overall expression of anti-HIV-1 genes using our custom-made TLDA. The gene expression levels are intercomparable, allowing the inference of relative expression levels across the surveyed genes. All gene cards (Applied Biosystems) used for quantification are depicted in Table S1 in the supplemental material.
We found that the expression levels of IFITM1, viperin, and TRIM22 were highest among all 34 genes screened (Fig. 2). APOBEC3B, APOBEC3F, and APOBEC3H were the least abundant restriction factors, exhibiting a more than 1-log reduction in expression compared to the other genes. The expression levels of TRIM15 and TRIM31 were below the level of detection (Fig. 2).
Fig 2.
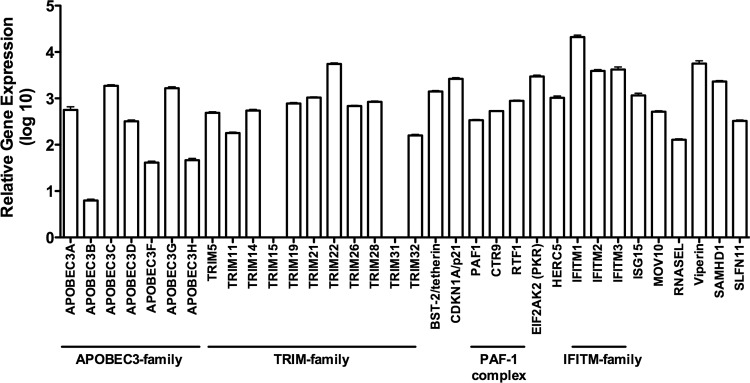
Antiviral profile of activated PBMC. (A) Extended antiviral gene expression analysis in activated PBMC. PBMC (n = 43) were PHA-P (5 μg/ml) activated for 1 day. RNA was extracted using TRIzol followed by cDNA synthesis. Quantitative real-time PCR was performed using custom-made TaqMan low-density arrays (TLDA). A panel of 6 housekeeping genes was included in the TLDA plates, and PPIA was identified as the most stably expressed gene and was used for normalization of results by the comparative CT method. All gene cards (Applied Biosystems) used for quantification are depicted in Table S1 in the supplemental material.
To complement our observation that host anti-HIV-1 gene expression is strongly induced by cellular activation, we next examined how the differential levels of gene expression observed between activated donor cells influenced HIV-1 infectivity ex vivo. To test whether the relative levels of expression of anti-HIV-1 factors correlated with HIV-1 infectivity, we performed ex vivo standardized HIV-1 infection assays.
We used the WITO4160 CCR5-tropic transmitted/founder strain of subtype B HIV-1 (40) with a Tat-regulated Renilla luciferase reporter gene (41) to quantify viral infectivity. Virus stocks were generated by transfection of 293T cells. PBMC were IL-2/PHA-P activated for 1 day, as described above, washed, and infected with 5-fold serial dilutions of virus made in quadruplicate for a total of 11 dilutions, in 96-well round-bottom tissue-culture plates. RLU were measured after 6 days of infection (this is sufficient time for multiple rounds of replication to occur) using a ViviRen Renilla luciferase kit (Promega), and the TCID50 was calculated according to the method described in reference 42. Cell viability was determined by trypan blue staining using a Countess cell counter.
We found statistically significant negative correlations between HIV-1 infectivity and the expression of PAF1 (R = −0.3708, P = 0.0144), RTF1 (R = −0.3629, P = 0.0168) (Fig. 3A), TRIM11 (R = −0.4203, P = 0.0050) (Fig. 3B), TRIM26 (R = −0.3784, P = 0.0124) (Fig. 3B), and BST-2/tetherin (R = −0.3016, P = 0.0494) (Fig. 3C). None of the 34 surveyed antiviral genes exhibited positive correlations with HIV-1 infectivity ex vivo.
Fig 3.
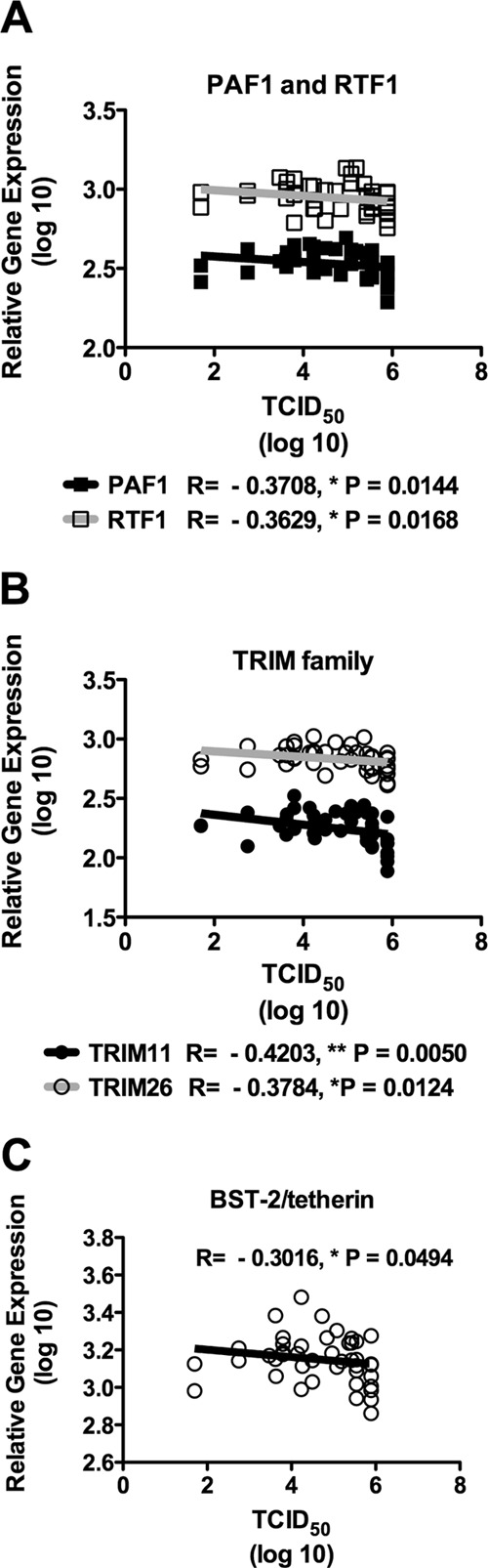
Host antiviral gene expression negatively correlates with ex vivo HIV-1 infectivity in activated PBMC. Individual donor PBMC (n = 43) were assessed for their susceptibility to infection by an CCR5-tropic transmitted/founder strain of subtype B HIV-1 using Tat-regulated Renilla luciferase reporter gene expression to quantify infection. The virus used was an infectious molecular clone that contained the entire ectodomain of HIV-1 WITO4160 and also contained a Renilla luciferase reporter gene. PBMC (n = 43) were activated for 1 day with PHA-P (5 μg/ml), washed, and placed in fresh growth medium without PHA-P. Five-fold serial dilutions of virus were made in quadruplicate for a total of 11 dilutions in 96-well round-bottom tissue-culture plates. PBMC were added and incubated for 6 days. Luminescence was measured using a ViviRen Renilla luciferase kit, and the TCID50 was calculated according to the method described in reference 42. Cell viability was determined by trypan blue staining using a Countess cell counter. Significant negative Spearman correlations were observed between PAF1 and RTF1 (A), TRIM family members (TRIM11 and TRIM26) (B), and BST-2/tetherin (C). P < 0.05 was considered significant. Solid lines depict linear regressions.
In this study, we found that host-encoded antiviral genes are differentially expressed in response to cellular activation. As expected, most of the genes studied were significantly upregulated following mitogen stimulation, while a minority of factors were significantly downregulated. For example, ISG15 and APOBEC3G, which were the two most upregulated antiviral genes following activation, may play pivotal roles in restricting HIV-1 replication in activated cells. Conversely, APOBEC3F and APOBEC3A, whose expression levels were strongly repressed after activation, may play a more important role in restricting infection in quiescent, resting cells. It is curious that activation had divergent effects on the expression levels of APOBEC3F and APOBEC3G, which typically fluctuate in tandem and behave similarly in response to other stimuli (e.g., IFN-α). This discordance reveals that there may be discrete regulatory pathways associated with these APOBEC3 family members that were not apparent under other experimental conditions.
Most antiviral factors are triggered by interferon; in particular, APOBEC3G, APOBEC3F, and BST-2/tetherin have been shown to be strongly induced by IFN-α in vivo (43), so it is possible that our observed upregulation of antiviral genes occurs through an IFN-α pathway boosted by cellular activation.
Although it may seem paradoxical that activation induces antiviral restriction factors and simultaneously renders the cell more supportive of HIV-1 replication, we do not believe that these phenomena are in conflict, and our group has generated ample data supporting the positive correlation of cellular activation and the expression of restriction factors both in vitro and in vivo.
Cellular activation is known to induce a wide range of factors that enhance HIV-1 replication. These include several transcription factors that accelerate viral transactivation, transcription, and production (44, 45). The overall cell-intrinsic susceptibility to HIV-1 infection, therefore, is determined by the net balance between the induction of inhibitory factors and the induction of supportive cellular cofactors and mechanisms. There is little doubt that stimulation increases the overall susceptibility of target cells to HIV-1 infection. However, our work is based on the hypothesis that the relative differences in the expression of restriction factors between activated cells account for differences in the extents of viral infectivity and production between donors observed ex vivo.
Finally, using an ex vivo standardized HIV-1 replication assay, we showed that the increased levels of certain, but not all, antiviral genes correlated with impaired viral replication. The Env-IMC-LucR construct used in our replication assays bears a minimal Nef defect, which may affect viral propagation to some degree. It is also worth noting that this clone encodes functional Vpu and Vif proteins and therefore antagonizes restriction by BST-2/tetherin and members of the APOBEC3 family.
Another consideration is the contribution of other unrelated cellular variables to viral production. Factors such as CD4 receptor and chemokine coreceptor surface expression levels facilitate viral entry and likely contribute to the susceptibility of PBMC to HIV-1 infection; these parameters were not controlled or explicitly examined in our experiments. Correlations with HIV-1 infectivity were analyzed at the level of the individual gene using univariate statistics. As experiments come to be performed on a larger scale, involving cells from hundreds of donors, a multivariate approach may be implemented to simultaneously evaluate the relative effects of restriction genes on HIV-1 replication ex vivo to test the hypothesis that groups of antiviral factors could act in concert to promote resistance to HIV-1. Moreover, the expression profiling of isolated cellular subsets (e.g., CD4+ T cells) and lymphoid-resident cellular populations will enhance our understanding of the relationship between host restriction mechanisms and HIV-1 pathogenesis.
Identifying and characterizing potent host-encoded, cell-intrinsic anti-HIV-1 mechanisms are of critical importance to the development of novel prophylactic, therapeutic, and curative strategies for HIV-1 infection.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by grants from the National Institutes of Health (AI093179 to D.F.N. and 1K01DA024654 to S.K.P.), the Peter and Shelagh Godsoe Family Foundation through the AIDS Research Institute at UCSF (to D.F.N.), and the UCSF-Gladstone Institute of Virology & Immunology Center for AIDS Research (P30-AI027763 to R.A.S.R.) and in part by a grant provided by the Academic Senate under the UCSF RAP awards program (to D.F.N.).
We thank Emily Eriksson, Sara Holditch, Rex Cheng, and Peter Kuebler for discussions and Christina Ochsenbauer for supplying the HIV-1 WITO4160 with a Tat-regulated Renilla luciferase reporter gene.
We declare that we have no conflicts of interest.
R.A.S.R., M.A.-M., and M.B. performed experiments; R.A.S.R., M.A.-M., S.K.P., and D.F.N. analyzed results and made the figures; R.A.S.R., D.C.M., S.K.P., and D.F.N. designed the research and wrote the paper.
Footnotes
Published ahead of print 21 August 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JVI.02128-13.
REFERENCES
- 1.Harris RS, Hultquist JF, Evans DT. 2012. The restriction factors of human immunodeficiency virus. J. Biol. Chem. 287:40875–40883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bishop KN, Holmes RK, Sheehy AM, Davidson NO, Cho SJ, Malim MH. 2004. Cytidine deamination of retroviral DNA by diverse APOBEC proteins. Curr. Biol. 14:1392–1396 [DOI] [PubMed] [Google Scholar]
- 3.Uchil PD, Quinlan BD, Chan WT, Luna JM, Mothes W. 2008. TRIM E3 ligases interfere with early and late stages of the retroviral life cycle. PLoS Pathog. 4:e16. 10.1371/journal.ppat.0040016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Descours B, Cribier A, Chable-Bessia C, Ayinde D, Rice G, Crow Y, Yatim A, Schwartz O, Laguette N, Benkirane M. 2012. SAMHD1 restricts HIV-1 reverse transcription in quiescent CD4(+) T-cells. Retrovirology 9:87. 10.1186/1742-4690-9-87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Baldauf HM, Pan X, Erikson E, Schmidt S, Daddacha W, Burggraf M, Schenkova K, Ambiel I, Wabnitz G, Gramberg T, Panitz S, Flory E, Landau NR, Sertel S, Rutsch F, Lasitschka F, Kim B, Konig R, Fackler OT, Keppler OT. 2012. SAMHD1 restricts HIV-1 infection in resting CD4(+) T cells. Nat. Med. 18:1682–1687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Laguette N, Sobhian B, Casartelli N, Ringeard M, Chable-Bessia C, Segeral E, Yatim A, Emiliani S, Schwartz O, Benkirane M. 2011. SAMHD1 is the dendritic- and myeloid-cell-specific HIV-1 restriction factor counteracted by Vpx. Nature 474:654–657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hrecka K, Hao C, Gierszewska M, Swanson SK, Kesik-Brodacka M, Srivastava S, Florens L, Washburn MP, Skowronski J. 2011. Vpx relieves inhibition of HIV-1 infection of macrophages mediated by the SAMHD1 protein. Nature 474:658–661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Refsland EW, Stenglein MD, Shindo K, Albin JS, Brown WL, Harris RS. 2010. Quantitative profiling of the full APOBEC3 mRNA repertoire in lymphocytes and tissues: implications for HIV-1 restriction. Nucleic Acids Res. 38:4274–4284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kourteva Y, De Pasquale M, Allos T, McMunn C, D'Aquila RT. 2012. APOBEC3G expression and hypermutation are inversely associated with human immunodeficiency virus type 1 (HIV-1) burden in vivo. Virology 430:1–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Koning FA, Newman EN, Kim EY, Kunstman KJ, Wolinsky SM, Malim MH. 2009. Defining APOBEC3 expression patterns in human tissues and hematopoietic cell subsets. J. Virol. 83:9474–9485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Trickett A, Kwan YL. 2003. T cell stimulation and expansion using anti-CD3/CD28 beads. J. Immunol. Methods 275:251–255 [DOI] [PubMed] [Google Scholar]
- 12.Vandesompele J, De Preter K, Pattyn F, Poppe B, Van Roy N, De Paepe A, Speleman F. 2002. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 3:RESEARCH0034. 10.1186/gb-2002-3-7-research0034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sheehy AM, Gaddis NC, Choi JD, Malim MH. 2002. Isolation of a human gene that inhibits HIV-1 infection and is suppressed by the viral Vif protein. Nature 418:646–650 [DOI] [PubMed] [Google Scholar]
- 14.Yu Q, Chen D, Konig R, Mariani R, Unutmaz D, Landau NR. 2004. APOBEC3B and APOBEC3C are potent inhibitors of simian immunodeficiency virus replication. J. Biol. Chem. 279:53379–53386 [DOI] [PubMed] [Google Scholar]
- 15.Bourara K, Liegler TJ, Grant RM. 2007. Target cell APOBEC3C can induce limited G-to-A mutation in HIV-1. PLoS Pathog. 3:e153. 10.1371/journal.ppat.0030153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dang Y, Wang X, Esselman WJ, Zheng YH. 2006. Identification of APOBEC3DE as another antiretroviral factor from the human APOBEC family. J. Virol. 80:10522–10533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Berger G, Durand S, Fargier G, Nguyen XN, Cordeil S, Bouaziz S, Muriaux D, Darlix JL, Cimarelli A. 2011. APOBEC3A is a specific inhibitor of the early phases of HIV-1 infection in myeloid cells. PLoS Pathog. 7:e1002221. 10.1371/journal.ppat.1002221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wiegand HL, Doehle BP, Bogerd HP, Cullen BR. 2004. A second human antiretroviral factor, APOBEC3F, is suppressed by the HIV-1 and HIV-2 Vif proteins. EMBO J. 23:2451–2458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liddament MT, Brown WL, Schumacher AJ, Harris RS. 2004. APOBEC3F properties and hypermutation preferences indicate activity against HIV-1 in vivo. Curr. Biol. 14:1385–1391 [DOI] [PubMed] [Google Scholar]
- 20.OhAinle M, Kerns JA, Malik HS, Emerman M. 2006. Adaptive evolution and antiviral activity of the conserved mammalian cytidine deaminase APOBEC3H. J. Virol. 80:3853–3862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Stremlau M, Owens CM, Perron MJ, Kiessling M, Autissier P, Sodroski J. 2004. The cytoplasmic body component TRIM5alpha restricts HIV-1 infection in Old World monkeys. Nature 427:848–853 [DOI] [PubMed] [Google Scholar]
- 22.Carthagena L, Bergamaschi A, Luna JM, David A, Uchil PD, Margottin-Goguet F, Mothes W, Hazan U, Transy C, Pancino G, Nisole S. 2009. Human TRIM gene expression in response to interferons. PLoS One 4:e4894. 10.1371/journal.pone.0004894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Barr SD, Smiley JR, Bushman FD. 2008. The interferon response inhibits HIV particle production by induction of TRIM22. PLoS Pathog. 4:e1000007. 10.1371/journal.ppat.1000007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li X, Gold B, O'HUigin C, Diaz-Griffero F, Song B, Si Z, Li Y, Yuan W, Stremlau M, Mische C, Javanbakht H, Scally M, Winkler C, Dean M, Sodroski J. 2007. Unique features of TRIM5alpha among closely related human TRIM family members. Virology 360:419–433 [DOI] [PubMed] [Google Scholar]
- 25.Uchil PD, Quinlan BD, Chan WT, Luna JM, Mothes W. 2008. TRIM E3 ligases interfere with early and late stages of the retroviral life cycle. PLoS Pathog. 4:e16. 10.1371/journal.ppat.0040016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Van Damme N, Goff D, Katsura C, Jorgenson RL, Mitchell R, Johnson MC, Stephens EB, Guatelli J. 2008. The interferon-induced protein BST-2 restricts HIV-1 release and is downregulated from the cell surface by the viral Vpu protein. Cell Host Microbe 3:245–252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Neil SJ, Zang T, Bieniasz PD. 2008. Tetherin inhibits retrovirus release and is antagonized by HIV-1 Vpu. Nature 451:425–430 [DOI] [PubMed] [Google Scholar]
- 28.Chen H, Li C, Huang J, Cung T, Seiss K, Beamon J, Carrington MF, Porter LC, Burke PS, Yang Y, Ryan BJ, Liu R, Weiss RH, Pereyra F, Cress WD, Brass AL, Rosenberg ES, Walker BD, Yu XG, Lichterfeld M. 2011. CD4+ T cells from elite controllers resist HIV-1 infection by selective upregulation of p21. J. Clin. Invest. 121:1549–1560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liu L, Oliveira NM, Cheney KM, Pade C, Dreja H, Bergin AM, Borgdorff V, Beach DH, Bishop CL, Dittmar MT, McKnight A. 2011. A whole genome screen for HIV restriction factors. Retrovirology 8:94. 10.1186/1742-4690-8-94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Muto NF, Martinand-Mari C, Adelson ME, Suhadolnik RJ. 1999. Inhibition of replication of reactivated human immunodeficiency virus type 1 (HIV-1) in latently infected U1 cells transduced with an HIV-1 long terminal repeat-driven PKR cDNA construct. J. Virol. 73:9021–9028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Woods MW, Kelly JN, Hattlmann CJ, Tong JG, Xu LS, Coleman MD, Quest GR, Smiley JR, Barr SD. 2011. Human HERC5 restricts an early stage of HIV-1 assembly by a mechanism correlating with the ISGylation of Gag. Retrovirology 8:95. 10.1186/1742-4690-8-95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pincetic A, Kuang Z, Seo EJ, Leis J. 2010. The interferon-induced gene ISG15 blocks retrovirus release from cells late in the budding process. J. Virol. 84:4725–4736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Okumura A, Lu G, Pitha-Rowe I, Pitha PM. 2006. Innate antiviral response targets HIV-1 release by the induction of ubiquitin-like protein ISG15. Proc. Natl. Acad. Sci. U. S. A. 103:1440–1445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Furtak V, Mulky A, Rawlings SA, Kozhaya L, Lee K, Kewalramani VN, Unutmaz D. 2010. Perturbation of the P-body component Mov10 inhibits HIV-1 infectivity. PLoS One 5:e9081. 10.1371/journal.pone.0009081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Burdick R, Smith JL, Chaipan C, Friew Y, Chen J, Venkatachari NJ, Delviks-Frankenberry KA, Hu WS, Pathak VK. 2010. P body-associated protein Mov10 inhibits HIV-1 replication at multiple stages. J. Virol. 84:10241–10253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang X, Han Y, Dang Y, Fu W, Zhou T, Ptak RG, Zheng YH. 2010. Moloney leukemia virus 10 (MOV10) protein inhibits retrovirus replication. J. Biol. Chem. 285:14346–14355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Malathi K, Dong B, Gale M, Jr, Silverman RH. 2007. Small self-RNA generated by RNase L amplifies antiviral innate immunity. Nature 448:816–819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nasr N, Maddocks S, Turville SG, Harman AN, Woolger N, Helbig KJ, Wilkinson J, Bye CR, Wright TK, Rambukwelle D, Donaghy H, Beard MR, Cunningham AL. 2012. HIV-1 infection of human macrophages directly induces viperin which inhibits viral production. Blood 120:778–788 [DOI] [PubMed] [Google Scholar]
- 39.Li M, Kao E, Gao X, Sandig H, Limmer K, Pavon-Eternod M, Jones TE, Landry S, Pan T, Weitzman MD, David M. 2012. Codon-usage-based inhibition of HIV protein synthesis by human schlafen 11. Nature 491:125–128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Salazar-Gonzalez JF, Salazar MG, Keele BF, Learn GH, Giorgi EE, Li H, Decker JM, Wang S, Baalwa J, Kraus MH, Parrish NF, Shaw KS, Guffey MB, Bar KJ, Davis KL, Ochsenbauer-Jambor C, Kappes JC, Saag MS, Cohen MS, Mulenga J, Derdeyn CA, Allen S, Hunter E, Markowitz M, Hraber P, Perelson AS, Bhattacharya T, Haynes BF, Korber BT, Hahn BH, Shaw GM. 2009. Genetic identity, biological phenotype, and evolutionary pathways of transmitted/founder viruses in acute and early HIV-1 infection. J. Exp. Med. 206:1273–1289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Edmonds TG, Ding H, Yuan X, Wei Q, Smith KS, Conway JA, Wieczorek L, Brown B, Polonis V, West JT, Montefiori DC, Kappes JC, Ochsenbauer C. 2010. Replication competent molecular clones of HIV-1 expressing Renilla luciferase facilitate the analysis of antibody inhibition in PBMC. Virology 408:1–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Johnson VA, Byington RE. 1990. Infectivity assay (virus yield assay), p 71–76 In Aldovani A, Walker BD. (ed), Techniques in HIV research. Stockton Press, New York, NY [Google Scholar]
- 43.Pillai SK, Abdel-Mohsen M, Guatelli J, Skasko M, Monto A, Fujimoto K, Yukl S, Greene WC, Kovari H, Rauch A, Fellay J, Battegay M, Hirschel B, Witteck A, Bernasconi E, Ledergerber B, Gunthard HF, Wong JK. 2012. Role of retroviral restriction factors in the interferon-alpha-mediated suppression of HIV-1 in vivo. Proc. Natl. Acad. Sci. U. S. A. 109:3035–3040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tong-Starksen SE, Luciw PA, Peterlin BM. 1987. Human immunodeficiency virus long terminal repeat responds to T-cell activation signals. Proc. Natl. Acad. Sci. U. S. A. 84:6845–6849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Siekevitz M, Josephs SF, Dukovich M, Peffer N, Wong-Staal F, Greene WC. 1987. Activation of the HIV-1 LTR by T cell mitogens and the trans-activator protein of HTLV-I. Science 238:1575–1578 [DOI] [PubMed] [Google Scholar]
- 46.Lu J, Pan Q, Rong L, He W, Liu SL, Liang C. 2011. The IFITM proteins inhibit HIV-1 infection. J. Virol. 85:2126–2137 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.