

Abstract
Middle East respiratory syndrome coronavirus (MERS-CoV) is associated with an outbreak of more than 90 cases of severe pneumonia with high mortality (greater than 50%). To date, there are no antiviral drugs or specific therapies to treat MERS-CoV. To rapidly identify potential inhibitors of MERS-CoV replication, we expressed the papain-like protease (PLpro) and the 3-chymotrypsin-like protease (3CLpro) from MERS-CoV and developed luciferase-based biosensors to monitor protease activity in cells. We show that the expressed MERS-CoV PLpro recognizes and processes the canonical CoV-PLpro cleavage site RLKGG in the biosensor. However, existing CoV PLpro inhibitors were unable to block MERS-CoV PLpro activity, likely due to the divergence of the amino acid sequence in the drug binding site. To investigate MERS-CoV 3CLpro activity, we expressed the protease in context with flanking nonstructural protein 4 (nsp4) and the amino-terminal portion of nsp6 and detected processing of the luciferase-based biosensors containing the canonical 3CLpro cleavage site VRLQS. Importantly, we found that a small-molecule inhibitor that blocks replication of severe acute respiratory syndrome (SARS) CoV and murine CoV also inhibits the activity of MERS-CoV 3CLpro. Overall, the protease expression and biosensor assays developed here allow for rapid evaluation of viral protease activity and the identification of protease inhibitors. These biosensor assays can now be used to screen for MERS-CoV-specific or broad-spectrum coronavirus PLpro and 3CLpro inhibitors.
TEXT
The novel coronavirus Middle East respiratory syndrome coronavirus (MERS-CoV; previously known as London1, novel CoV, and human CoV-EMC) was first identified in 2012 in patients suffering from severe respiratory infection that led to pneumonia and 50% mortality (1–5). MERS-CoV replicates in cell culture, and the viral RNA can be detected by reverse transcription-PCR (RT-PCR) using pan-coronavirus primers that recognize conserved CoV sequences or primers that distinguish MERS-CoV from other CoVs (6, 7). Deep sequencing and bioinformatics analysis identified MERS-CoV as belonging to the genus Betacoronavirus within the subfamily Coronavirinae, most closely related to bat coronaviruses HKU4 and HKU5 (1, 8). The taxonomy supports the hypothesis that MERS-CoV emerged in the Middle East from an animal reservoir, with bats as a likely host. Currently, it is unclear if there exists an intermediate host, such as the masked palm civets implicated as the intermediate host for the transmission of severe acute respiratory syndrome CoV (SARS-CoV) in 2002 to 2003 (9, 10). As of 30 July 2013, there have been 91 infections with MERS-CoV, resulting in 46 fatalities, and chronic diseases, such as diabetes and kidney disease, may more frequently be associated with a fatal outcome (11). Limited human-to-human transmission of MERS-CoV resulting from contact with patients who traveled from the Middle East to England, France, and Italy has been documented, with clusters of infections in Saudi Arabia and Jordan (3–5, 12, 13). This evidence of human-to-human transmission of MERS-CoV indicates that the virus may pose a significant threat to human health and that the screening and development of antiviral drugs that either specifically inhibit MERS-CoV or broadly inhibit CoV replication is a research priority.
When considering antiviral drug development, an important issue is whether prophylactic or immediate treatment with antivirals would reduce the viral spread and mortality associated with MERS-CoV. Analysis of viral load in the respiratory tracts of SARS patients revealed that peak viral load occurred at 10 days after onset of fever (14). These findings suggest that there may be a window of opportunity for administration of antiviral therapy within the first 10 days of illness, potentially reducing viral load and mortality. Indeed, over the last 10 years, there has been considerable effort aimed at generating effective antiviral therapies for SARS-CoV, resulting in over 3,500 publications on SARS-CoV-inhibitory compounds (15). Much of this work has focused on inhibiting the viral proteases that are required for viral replication. These proteases, the papain-like protease (PLpro) and 3-chymotrypsin-like protease (3CLpro; also termed the main protease, Mpro), are critical for processing the viral replicase polyprotein produced upon translation of the CoV positive-sense RNA genome after infection (16). High-throughput screening and structure-activity relationship refinement of leads has identified inhibitors directed against SARS-CoV PLpro and 3CLpro (17–24). Currently, it is unclear whether any of these inhibitors designed to block SARS-CoV proteases also block MERS-CoV proteases. Our goal was to develop a rapid, sensitive, cell-based assay that could be performed in a biosafety level 2 environment to evaluate existing and new drugs for their ability to inhibit CoV protease activity. These novel screening methods could be used to identify inhibitors specific to MERS-CoV or pan-coronaviral inhibitors that could block proteases from MERS-CoV, SARS-CoV, or other potentially emerging coronaviruses.
Here we report the expression and activities of MERS-CoV PLpro and 3CLpro in cell-based assays. We show that coexpression of a MERS-CoV protease domain with a cleavage-activated luciferase substrate allowed for both endpoint evaluation and live-cell imaging profiles of protease activity. We found that a characterized SARS-CoV PLpro inhibitor has limited efficacy on MERS-CoV PLpro activity. However, a previously identified 3CLpro inhibitor of SARS-CoV and murine coronavirus replication also effectively blocks the activity of MERS-CoV 3CLpro. This study describes approaches that can be used to identify CoV protease inhibitors and identifies a compound, CE-5, that can be used to help characterize the active site of MERS-CoV 3CLpro.
MATERIALS AND METHODS
Cells and transfections.
HEK293T cells were cultured in Dulbecco's modified Eagle's medium (DMEM) containing 10% fetal calf serum (FCS) and 2% glutamine. For transfection experiments, cells were plated in CellBIND plates (Corning), and when cells were approximately 70% confluent, they were subjected to transfection using TransIT-LT1 transfecting reagent (Mirus) according to the manufacturer's suggested protocol.
Protease expression plasmids.
The sequence of the MERS-CoV PLpro (GenBank accession number AFS88944; ORF1a amino acids 1483 to 1802) was codon optimized, synthesized (GenScript), and cloned into pcDNA3.1-V5/His-B (Invitrogen) (see Fig. S1 in the supplemental material) to generate pMERS-CoV PLpro. A catalytically inactive mutant (in which cysteine 1592 was changed to alanine, designated CA) was generated using site-directed mutagenesis by a two-step overlapping PCR approach using the primers listed in Table S1 in the supplemental material. The MERS-CoV nonstructural protein 4, 5, and N-terminal 6 (nsp4/5/6N) region (ORF1a amino acids 2741 to 3561) was synthesized (GenScript) and cloned into pcDNA3.1-V5/His-B (Invitrogen) (Fig. S2) to generate pMERS-pp3CLpro. Specific mutations in the catalytic residues (C3395 in nsp5) and putative cleavage site of nsp5/6 (Q3553/S3554) were generated by using QuikChange II XL site-directed mutagenesis kit (Stratagene) (Table S1).
Biosensor expression plasmids.
The pGlo-30F vector backbone (Promega) is a circularly permuted Photuris pennsylvanica luciferase optimized for expression in cell culture (25). Oligonucleotides corresponding the amino sequence RLKGG (for PLpro) or VRLQS (for 3CLpro) were ligated into the BamHI and HindIII restriction enzyme cleavage sites (see Table S1 in the supplemental material), and screening for the inserts was performed by restriction enzyme digestion to confirm the presence of engineered AflII (RLKGG) or PstI (VRLQS) sites. The resulting plasmids were designated pGlo-30F-RLKGG and pGlo-30F-VRLQS.
trans-cleavage assay for MERS-CoV PLpro.
HEK293T cells in 12-well plates were transfected using TransIT-LT1 transfection reagent (Mirus) with 25 ng nsp2/3-GFP plasmid and increasing amounts of pcDNA-MERS-CoV PLpro expression plasmids. At 24 h posttransfection, cells were lysed with 300 μl of lysis buffer A, containing 4% SDS, 3% dithiothreitol (DTT), and 0.065 M Tris, pH 6.8. Proteins were separated by SDS-PAGE and transferred to a polyvinylidene difluoride (PVDF) membrane in transfer buffer (0.025 M Tris, 0.192 M glycine, 20% methanol) for 1 h at 65 V at 4°C. The membrane was blocked using 5% dried skim milk in TBST buffer (0.9% NaCl, 10 mM Tris-HCl, pH 7.5, 0.1% Tween 20) overnight at 4°C. The membrane was incubated with polyclonal rabbit anti-green fluorescent protein (anti-GFP) antibody (Life Technologies), followed by incubation with horseradish peroxidase (HRP)-conjugated donkey anti-rabbit secondary antibody (SouthernBiotech). To verify expression of PLpro, the membrane was probed with mouse anti-V5 antibody (Invitrogen). Mouse anticalnexin antibody (Cell Signaling Technology) was used to detect a host cell protein as a loading standard, and HRP-conjugated goat anti-mouse antibody (SouthernBiotech) was used as the secondary antibody. Secondary antibody was detected using Western Lighting Plus chemiluminescence reagent (PerkinElmer) and visualized using a FluorChem E imager.
Biosensor endpoint assay.
HEK293T cells were transfected with 150 ng pGlo-30F-RLKGG or pGlo-30F-VRLQS, 25 ng pRL-TK (Promega), and increasing amounts of protease expression plasmid. Transfections were equalized to 500 ng per well with an empty pcDNA3.1-V5/His-B vector. At 20 h posttransfection, cells were lysed with 1× passive lysis buffer (Promega) and 25 μl of lysate was assayed for luciferase activity using 96-well white-bottom assay plates (Corning) and dual-luciferase-activating reagents (Promega).
Biosensor live-cell assay.
HEK293T cells (96-well format, 0.4 μl TranIT-LT1 per well) were transfected with 37.5 ng pGlo-30F, pGlo-30F-RLKGG, or pGlo-VRLQS, 2 ng pRL-TK (Promega), and either 37.5 ng of SARS-CoV PLpro or 25 ng MERS-CoV 3CLpro DNA. Transfections were equalized to 125 ng with the empty vector pcDNA3.1-V5/His-B. At 13 h posttransfection, GloSensor (Promega) reagent (diluted 1:50 in culture medium) was added to each well. Plates were imaged using a luminometer (Veritas) every hour for 2 h. Cells were then treated with the concentrations of drug indicated in the legend to Fig. 5, with dimethyl sulfoxide (DMSO) at an equivalent volume, and imaged every hour for 4 h.
Fig 5.
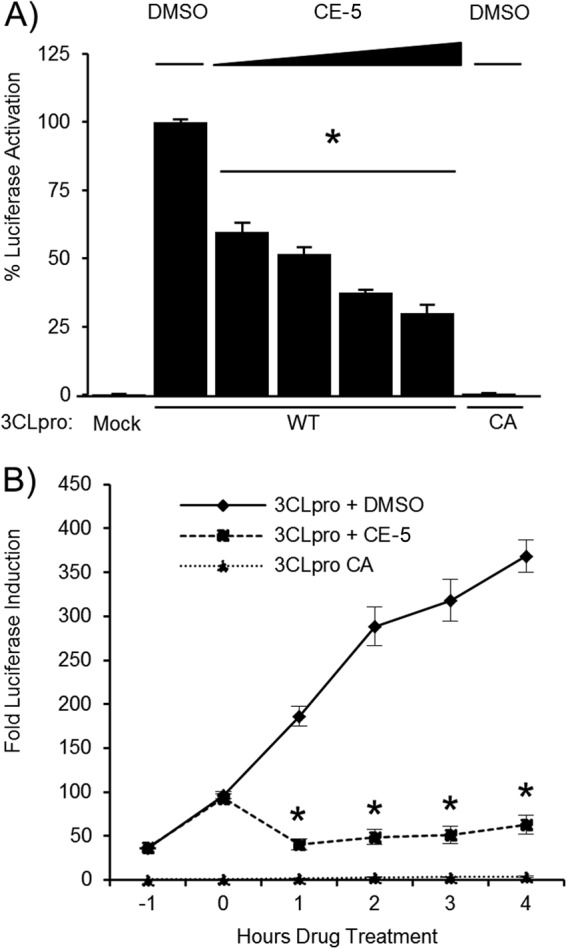
The CoV 3CLpro inhibitor CE-5 blocks MERS-CoV 3CLpro activity. (A) MERS-CoV 3CLpro activity is inhibited by CE-5. HEK293T cells were transfected with wild-type (WT) or catalytic-mutant (CA) pMERS-pp3CLpro and pGlo-VRLQS DNA, incubated for 14 h, and then treated with 6.25, 12.5, 25, or 50 μM CE-5 or DMSO for 6 h. Cells were lysed and assayed for dual-luciferase activity. The experiment was performed in triplicate, with error bars representing the standard deviations of the means. *, P < 0.005, as determined with Student's t test between DMSO- and drug-treated cells. (B) MERS-CoV 3CLpro activity was inhibited by CE-5 in the live-cell assay. HEK293T cells were transfected with wild-type (WT) or catalytic-mutant (CA) pMERS-pp3CLpro and pGlo-VRLQS for 13 h, incubated with GloSensor reagent for 1 h, and then treated with 50 μM CE-5 or DMSO. Luciferase activity was assayed in live cells every hour using a luminometer. The experiment was performed in triplicate, with error bars representing the standard deviations of the means. *, P < 0.005, as determined with Student's t test between DMSO- and drug-treated cells.
Western blot detection of MERS-pp3CLpro cleavage products.
To determine the catalytic activity of MERS-pp3CLpro, HEK293T cells in 24-well CellBIND plates were transfected with increasing amounts of pcDNA-pp3CLpro expression plasmid DNA. At 20 h posttransfection, cells were lysed in 100 μl of lysis buffer A, followed by Western blotting as described above. The protein level of pp3CLpro and its cleaved products were detected using mouse anti-V5 antibody (Invitrogen). After being probed with anti-V5, the membrane was treated with stripping buffer (62.5 mM Tris-Cl, pH 6.8, 2% SDS, 100 mM 2-beta-mercaptoethanol) and reblotted using a mouse monoclonal antibody to beta-actin (Ambion). HRP-conjugated goat anti-mouse (SouthernBiotech) was used as the secondary antibody.
RESULTS AND DISCUSSION
Evaluating MERS-CoV papain-like protease activity.
To determine if the predicted papain-like protease domain of MERS-CoV can be expressed in trans as a functional protease, the MERS-CoV PLpro domain was codon optimized and cryptic splice sites were removed, synthesized, and cloned into pcDNA3.1 for transient-transfection studies (Fig. 1A). The synthetic MERS-CoV PLpro extends from amino acids 1485 to 1802 of ORF1a, with the addition of 2 amino acids at the N terminus to allow efficient translation (methionine and alanine) and a V5 epitope tag on the C terminus (see Fig. S1 in the supplemental material for the modified nucleotide sequence). A catalytic-mutant MERS-CoV PLpro was generated by mutating the catalytic cysteine residue (amino acid 1594) to an alanine (oligonucleotides are listed in Table S1 in the supplemental material). To evaluate protease activity in cell culture, plasmid DNA expressing the wild-type or catalytic-mutant form of MERS-CoV PLpro was transfected into HEK293T cells along with a plasmid DNA expressing the SARS-CoV nsp2/3-GFP substrate. The nsp2/3-GFP substrate is commonly used to assess the cleavage ability of transiently expressed CoV papain-like proteases (26). We detected evidence of processing of the nsp2/3-GFP substrate in the presence of the catalytically active form of MERS-CoV PLpro but not in the presence of the catalytic-mutant MERS-CoV PLpro (Fig. 1B). These data confirm that the putative papain-like protease domain located within nsp3 of the MERS-CoV genome indeed functions as a papain-like protease capable of cleaving LXGG-containing polyprotein substrates. Alignment of the PLpro domain of MERS-CoV with the PLpro domains of other betacoronaviruses, including SARS-CoV, bat coronaviruses HKU-4 and HKU-5, and the murine coronavirus mouse hepatitis virus (MHV) revealed the conservation of the catalytic triad (Cys-1594, His-1759, and Asp-1774) and the four cysteine residues that comprise the zinc-binding finger domain (Cys-1672, -1675, -1707, and -1709). However, the overall sequence identity between MERS-CoV PLpro and SARS-CoV PLpro is low, at only 30% amino acid identify (Fig. 1C). MERS-CoV PLpro also contains a canonical RLKGG site located between the catalytic histidine at position 1759 and the catalytic aspartic acid at position 1774. This cleavage site is unique to MERS-CoV, but due to the proximity to the catalytic residues, it is unlikely that this cleavage site is accessible for PLpro cleavage. However, further experiments must be done to address the functionality of this putative cleavage site.
Fig 1.

Activity of MERS-CoV PLpro. (A) Schematic diagram of MERS-CoV ORF1a/b, with predicted PLpro cleavage sites indicated; pMERS-PLpro corresponds to amino acid residues (aa) 1485 to 1802. ADRP, ADP-ribose-1″-monophosphatase; PLpro, papain-like protease; 3CL, 3-chymotrypsin-like protease; Prm, primase; RdRp, RNA-dependent RNA polymerase; ZBD, zinc-binding domain; Hel, helicase; ExoN, exoribonuclease; NMT, N7 methyltransferase; NendoU, endoribonuclease; OMT, 2′ O-methyltransferase. (B) trans-cleavage activity of MERS-PLpro. pMERS-PLpro and plasmid DNA expressing the SARS-CoV nsp2/3-GFP substrate were transfected into HEK293T cells, lysates were harvested at 24 h posttransfection, and protein expression was analyzed by Western blotting with the indicated antibodies. WT, wild type. Numbers at the right are molecular masses (in kilodaltons). (C) Alignment of the PLpro domains from selected betacoronaviruses using the ViPR software MUSCLE alignment algorithm; identical residues are highlighted. *, catalytic residues; #, zinc-binding cysteines. Underlined residues in the SARS PLpro indicate a flexible loop that binds specific inhibitors. Accession numbers are as follows: for MERS-CoV amino acids 1484 to 1802, JX869059; for bat CoV-HKU4 amino acids 1526 to 1844, NC_009019; for bat CoV-HKU5 amino acids 1560 to 1878, NC_009020; for SARS-CoV amino acids 1541 to 1855, AY278741; and for MHV amino acids 1609 to 1911, NC_001846.
Exploiting a biosensor assay to evaluate MERS-CoV PLpro activity.
Developing assays to rapidly detect the activity of coronavirus proteases is a key step toward the goal of evaluating specific and pan-coronavirus protease inhibitors. Therefore, we developed a luciferase-based biosensor assay to measure protease activity within transfected cells. The system takes advantage of an inverted, circularly permuted luciferase construct (pGlo-30F) separated by an engineered site corresponding to the canonical coronavirus PLpro cleavage recognition sequence, RLKGG (Fig. 2A). The protease recognition site is limited in size, and because coronaviral proteases recognize short consensus sequences within the viral polyprotein, only 5 amino acids were engineered into the construct. The 5-amino-acid cleavage site contains the consensus LXGG motif necessary for recognition by coronaviral papain-like proteases, so this substrate should be recognized by MERS-CoV PLpro and other coronavirus PLpros (27–29). The inactive form of luciferase is expressed within transfected cells, and upon cleavage by a viral protease recognizing the engineered cleavage site, there is a conformational change into an active form of luciferase. Protease activity is measured quantitatively by endpoint lysis of the cells and incubation with luciferase substrate reagents or by a live-cell approach to examine the kinetics of protease activity. This system is based on the pGloSensor caspase 3/7 system, which was used previously to evaluate protease activity in live cells and animal models, and on similar systems that were used to evaluate viral protease activity in an in vitro translation system (25, 30–32).
Fig 2.
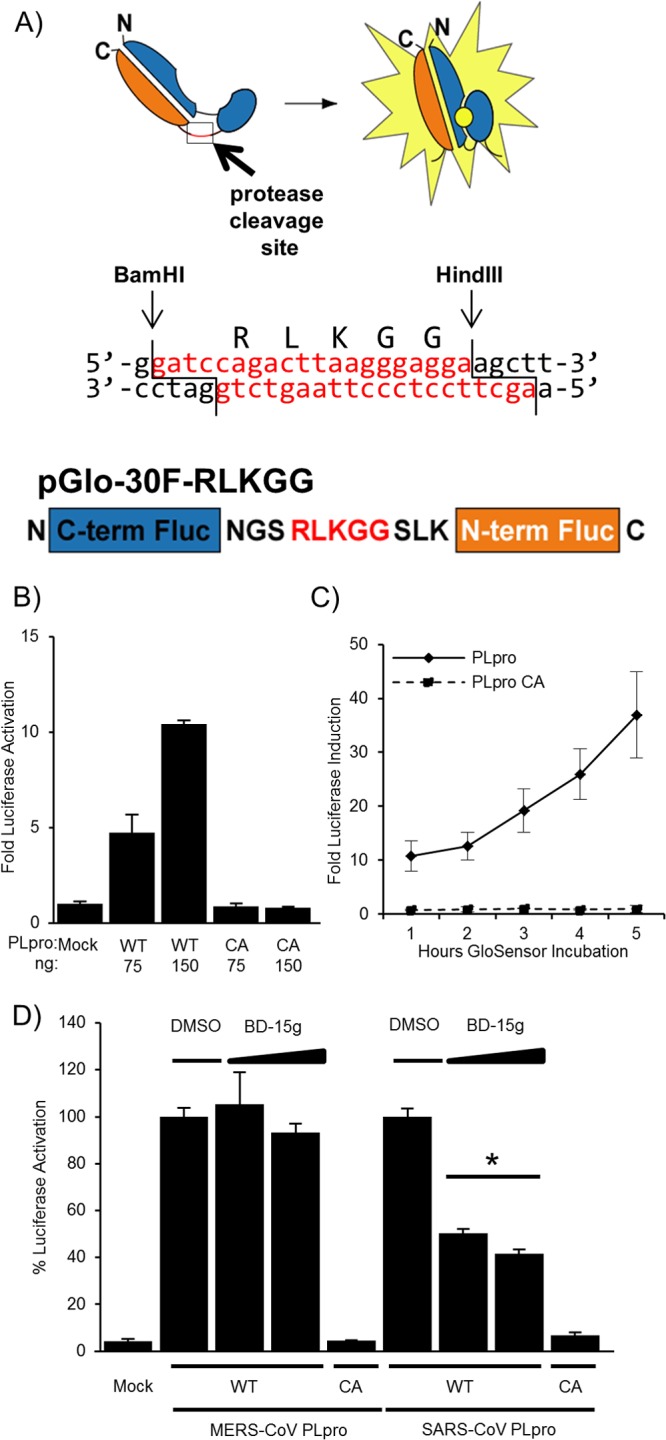
Biosensor assay detecting MERS-CoV PLpro activity in cells. (A) Diagram depicting the circularly permuted luciferase construct linked by the protease cleavage site RLKGG for assessing CoV PLpro activity. (B) Expression of MERS-CoV PLpro activates the biosensor. HEK293T cells are cotransfected with pGlo-30F-RLKGG and either pMERS-PLpro or pMERS-PLpro-CA. At 20 h posttransfection, cells were lysed and assayed using the dual-luciferase assay. The experiment was performed in triplicate, and error bars represent the standard deviations of the means. (C) Live-cell assay of MERS-PLpro activity. Cells were cotransfected with pGlo-30F-RLKGG and either pMERS-PLpro or pMERS-PLpro-CA. At 14 h posttransfection, cells were incubated with GloSensor reagent, and luminescence was read hourly. The experiment was performed in triplicate, and error bars represent the standard deviations of the means. (D) A previously identified SARS-CoV inhibitor does not inhibit MERS-CoV PLpro. HEK293T cells were transfected with the wild-type (WT) or catalytic-mutant (CA) SARS-CoV PLpro or MERS-CoV PLpro and pGlo-RLKGG constructs for 13 h and then treated with 12.5 or 6.25 μM BD-15g for 3 h or with the equivalent volume of DMSO diluted in medium. Cells were then lysed and assayed for dual-luciferase activity. The experiment was performed in triplicate, with error bars representing the standard deviations of the averages. *, P < 0.005, as determined with Student's t test between DMSO- and drug-treated cells.
To determine if this system can be used to examine MERS-CoV PLpro activity, the MERS-CoV PLpro expression plasmid, in addition to pGlo-RLKGG and renilla luciferase plasmids, was transfected into HEK293T cells. The cells were lysed 20 h posttransfection, and a dual-luciferase assay was performed on the lysates, with assessment of the firefly luciferase activity generated by cleavage of the pGlo-RLKGG substrate and the renilla luciferase activity to control for transfection efficiency and toxicity. Wild-type MERS-CoV PLpro recognized and cleaved the pGlo-RLKGG substrate, resulting in luciferase induction 10-fold above that in the mock experiment (Fig. 2B). A dose response was evident, with increasing amounts of protease expression leading to higher levels of luciferase activity. The catalytic-mutant PLpro did not cleave the substrate, and there was no detectable increase in luciferase activity above background. To evaluate MERS-CoV PLpro activity in real time, we exploited an assay based on detecting luciferase activity using a live-cell readout. The firefly luciferase encoded in the pGlo reporter can be detected in live cells by using a cell-permeable luciferase activator substrate, GloSensor, added to tissue culture media during incubation (30). HEK293T cells in a 96-well format were transfected with MERS-CoV PLpro (wild type or catalytic mutant) and the pGlo-RLKGG-expressing substrate. At 15 h posttransfection, the cells were incubated in GloSensor reagent and monitored for luciferase activity using a luminometer. Cells expressing MERS-CoV PLpro showed a 10-fold increase over levels in the mock experiment as early as 1 h after GloSensor incubation, and luciferase activity continued to increase over the duration of the experiment (Fig. 2C). The catalytic-mutant MERS-CoV PLpro-expressing cells show no increase over levels in the mock transfection cell background. The mock-transfected cells gave an extremely low background in the live-cell assay (only ∼15 luciferase units), allowing for excellent sensitivity of protease activity. Initial screening of the existing SARS-CoV PLpro inhibitor, a benzodioxolane derivative termed 15 g (BD-15g) against MERS-CoV PLpro, revealed no significant inhibition (Fig. 2D) (17). One possible explanation for the specificity of BD-15g for SARS-CoV PLpro over MERS-CoV PLpro is that the inhibitor associates with a flexible loop that differs between SARS-CoV PLpro and MERS-CoV PLpro (underlined in Fig. 1C and shown in reference 20). This loop has been shown to close upon BD-15g binding to SARS-CoV PLpro, and the loop closure prevents the substrate from accessing the active site. Because there are differences in the amino acid sequences of the flexible loop, it is possible that BD-15g cannot bind the loop of MERS-CoV PLpro, leaving the active site accessible to the substrate. Overall, these results validate the use of the biosensor assay, with a known positive control, for evaluating the specificity of small-molecule inhibitors directed against CoV PLpros. Further work is needed to identify inhibitors that block MERS-CoV PLpro or are broadly inhibitory to CoV PLpros.
Cloning and expression of MERS-CoV 3CLpro.
To evaluate MERS-CoV 3CLpro activity, an nsp4/5/6N-V5 codon-optimized DNA encoding amino acids 2741 (N terminus of nsp4) to 3561 (within nsp6) with its splice site removed was synthesized and cloned into the pcDNA3.1 expression vector and designated pMERS-pp3CLpro (for polyprotein containing the nsp5/3CLpro domain). The 3CLpro cleavage site between nsp5 and -6 was either retained, resulting in expression of MERS-3CLpro, which cleaves between nsp4/5 and nsp5/6 and is untagged, or mutated (pMERS-pp3CLpro-AA), resulting in a protease that cleaves the nsp4/5 site but should retain the V5 epitope tag at the nsp5/6N-V5 C terminus (Fig. 3A). Currently, there are no antibodies available to detect MERS-CoV 3CLpro; therefore, we relied on detection of cleavage intermediates and epitope-tagged products to assess expression and activity. To determine if MERS-pp3CLpro expresses an active protease, we transfected plasmid DNA into HEK293T cells and harvested cell lysates at 20 h posttransfection for Western blot evaluation. As expected, the wild-type pp3CLpro protease cleaved the polyprotein, generating an nsp5/6N-V5 product that can be detected by Western blotting (Fig. 3B). As expected, this product is more abundant when the nsp5/6 cleavage site is mutated (Fig. 3B, compare the WT lanes [with QS] to the WT-AA lanes). The nsp6-V5 product is predicted to be only 5.6 kDa and was undetectable by Western blotting in the wild-type sample. The catalytic-mutant pMERS-pp3CLpro-CA does not generate the cleavage product, instead generating a polyprotein of the expected size and an aggregate that does not efficiently enter the SDS-PAGE gel, presumably because of the trans-membrane domain that remains in the polyprotein (Fig. 3C, asterisk). These results demonstrate the activity of the MERS-CoV 3CLpro and the requirement for the catalytic cysteine residue Cys-3395.
Fig 3.

MERS-CoV 3CLpro activity. (A) Schematic diagram of the 3CLpro domain and predicted cleavage sites in ORF1a/b. The region corresponding to the amino acid residues 2741 to 3561 spanning nsp4/5/6 was synthesized, cloned, and designated pMERS-pp3CLpro. The catalytic cysteine was changed to alanine, and the nsp5/6 cleavage site QS was changed to AA. (B) Expression and activity of 3CLpro. pMERS-pp3CLpro was transfected into HEK293T cells. Lysates were prepared at 20 h posttransfection and protein products analyzed by Western blotting. Anti-V5 detects polyprotein and processed products, and anti-beta-actin was used to monitor protein loading. *, protein aggregates. Numbers at the right are molecular masses (in kilodaltons). (C) Alignment of 3CLpro regions from selected betacoronaviruses. Alignments were performed using ViPR software MUSCLE alignment algorithms. Sequence identity is indicated with shading. Catalytic residues are boldface and marked with an asterisk. Accession numbers are listed in the legend to Fig. 1.
To generate a biosensor 3CLpro substrate, we evaluated the predicted 3CLpro cleavage sites in the MERS-CoV ORF1a/b polyprotein and noted that the predicted cleavage sites for MERS-CoV 3CLpro conform to the consensus cleavage sites for previously characterized CoV 3CLpros (33). Oligonucleotides encoding the consensus cleavage site (VRLQ↓S) were synthesized and ligated into pGlo-30F to generate the pGlo-30F-VRLQS biosensor for CoV 3CLpro activity (Fig. 4A). Transfecting cells with plasmid DNA encoding pMERS-pp3CLpro, pGlo-VRLQS, and renilla luciferase reporters and lysing the cells after 20 h allowed for the evaluation of MERS 3CLpro activity. Recognition of the pGlo-VRQLS cleavage site engineered into the luciferase biosensor resulted in a 250-fold increase in luciferase activity above the level detected in mock-transfected cells. We also noted a dose response of the reporter relative to the increasing expression of 3CLpro (Fig. 4B). The catalytic-mutant 3CLpro did not generate significant luciferase activity above background. To determine if we could detect MERS 3CLpro activity in the live-cell kinetic assay, we transfected HEK293T cells with the protease and substrate plasmid DNAs and evaluated activity using the GloSensor assay as described above. The wild-type 3CLpro construct led to a rapid activation of luciferase, with a 50-fold increase detectable only 1 h after incubation with the substrate reagent (Fig. 4C). This activation increased to levels of >300-fold above levels in the mock experiment after 5 h of incubation. The catalytic-mutant 3CLpro was not capable of cleaving the luciferase biosensor, and there was no increase over levels in the mock live-cell assay.
Fig 4.
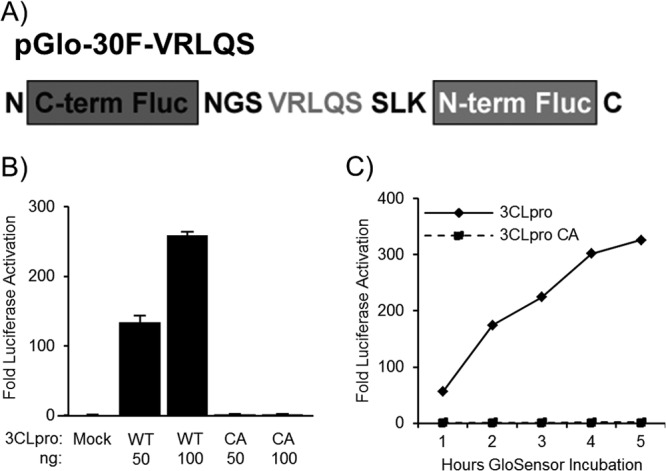
Biosensor assay detecting MERS-3CLpro activity in cell culture. (A) Diagram depicting the circularly permuted luciferase construct linked by the protease cleavage site VRLQS for assessing CoV 3CLpro activity. (B) Expression of MERS-pp3CLpro activates the biosensor. HEK293T cells were cotransfected with pGlo-VRLQS and pMERS-pp3CLpro expressing either wild-type or catalytic-mutant (CA) 3CLpro. At 20 h after transfection, cells were lysed and assayed for luciferase activity. The experiment was performed in triplicate, with error bars representing the standard deviations of the means. (C) MERS-CoV 3CLpro activity is detected in a live-cell assay. Cells were transfected as described above, and at 14 h, they were incubated with GloSensor reagent. Luciferase activity was assayed in live cells every hour using a luminometer. The experiment was performed in triplicate, with error bars representing the standard deviations of the means.
Identification of a SARS-CoV 3CLpro inhibitor that blocks MERS-CoV 3CLpro activity.
The structure for the MERS-CoV 3CLpro in complex with a peptidomimetic inhibitor termed N3 was recently solved, and significant structural similarities between SARS-CoV and MERS-CoV 3CLpros were noted (34). These results suggest that broadly reactive 3CLpro inhibitors may block MERS 3CLpro activity. To test this hypothesis, the efficacy of antiviral drugs optimized for inhibition of SARS-CoV 3CLpro that were also shown to block murine coronavirus replication were tested for the ability to block MERS-CoV 3CLpro activity. The antiviral inhibitor tested is a chloropyridine ester, CE-5 (35). Structural studies by Verschueren and colleagues showed that this class of benzotriazole esters acts as suicide inhibitors by covalently modifying the catalytic cysteine residue necessary for protease activity (36). This drug inhibits SARS-CoV 3CLpro activity in vitro in addition to inhibiting viral replication in SARS-CoV-infected VeroE6 cells and murine coronavirus replication in DBT cells. Because of the structural similarity between SARS-CoV and MERS-CoV 3CLpros, it is possible that 3CLpro inhibitors like CE-5 will be cross-reactive and have inhibitory properties against MERS-CoV 3CLpro.
The efficacy of the SARS-CoV 3CLpro inhibitor CE-5 was tested against MERS-CoV 3CLpro using the pGlo-VRLQS biosensor assay. To determine if CE-5 inhibited MERS-CoV 3CLpro, cells were transfected with the pGlo-VRLQS construct, renilla luciferase plasmid, and MERS-CoV 3CLpro wild-type and catalytic-mutant expression constructs. After 14 h of transfection, CE-5 was added to the media at final concentrations of 50 μM, 25 μM, 12.5 μM, and 6.25 μM, and incubation continued for 6 h. The cells were lysed and assayed for luciferase activity to measure the amount of reporter activity (Fig. 5A). The luciferase levels measured for the MERS-CoV 3CLpro wild type were set to 100%. The drug treatment inhibited the activity of MERS-CoV 3CLpro to 30% of that of DMSO-treated cells at a maximum dose of 50 μM. The endpoint evaluation of CE-5 indicated a 50% effective concentration (EC50) in cell culture of ∼12.5 μM.
To evaluate the real-time effects of CE-5 inhibition of MERS-CoV 3CLpro, the live-cell protease cleavage assay described earlier was used. HEK293T cells transfected with pGlo-VRLQS, renilla luciferase plasmids, and MERS-CoV 3CLpro were incubated in GloSensor reagent and then measured for luminescence beginning at 14 h posttransfection. We noted the expected increase in luciferase activity in the DMSO-treated group, while the luciferase activity in the CE-5-treated cells declined sharply (Fig. 5B). The efficacy of this previously reported SARS-CoV 3CLpro inhibitor to act on MERS 3CLpro is consistent with broad-spectrum inhibition. Further studies are needed to identify whether other compounds with activity inhibitory to SARS-CoV proteases may have effects inhibitory to MERS-CoV.
Conclusion.
In summary, the successful expression of MERS-CoV PLpro and 3CLpro and the development of the luciferase-based biosensor for protease activity will facilitate screening and identification of effective small-molecule inhibitors for MERS-CoV and future emerging coronaviruses.
Supplementary Material
ACKNOWLEDGMENTS
We thank Frank Fan and Brock Binkowski (Promega) for providing the pGlo-30F vector, Arun Ghosh (Purdue University) for protease inhibitors, and Karina Durso for technical support.
Financial support for this work was provided by the National Institutes of Health (NIAID grant R01AI085089 to S.C.B.). A.K. was supported by an NIH training grant in experimental immunology (NIH grant T32 AI512795).
Footnotes
Published ahead of print 28 August 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JVI.02105-13.
REFERENCES
- 1.Zaki AM, van Boheemen S, Bestebroer TM, Osterhaus AD, Fouchier RA. 2012. Isolation of a novel coronavirus from a man with pneumonia in Saudi Arabia. N. Engl. J. Med. 367:1814–1820 [DOI] [PubMed] [Google Scholar]
- 2.de Groot RJ, Baker SC, Baric RS, Brown CS, Drosten C, Enjuanes L, Fouchier RA, Galiano M, Gorbalenya AE, Memish Z, Perlman S, Poon LL, Snijder EJ, Stephens GM, Woo PC, Zaki AM, Zambon M, Ziebuhr J. 15 May 2013. Middle East respiratory syndrome coronavirus (MERS-CoV); announcement of the Coronavirus Study Group. J. Virol. 10.1128/JVI.01244-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Memish ZA, Zumla AI, Al-Hakeem RF, Al-Rabeeah AA, Stephens GM. 2013. Family cluster of Middle East respiratory syndrome coronavirus infections. N. Engl. J. Med. 368:2487–2494 [DOI] [PubMed] [Google Scholar]
- 4.Guery B, Poissy J, El Mansouf L, Sejourne C, Ettahar N, Lemaire X, Vuotto F, Goffard A, Behillil S, Enouf V, Caro V, Mailles A, Che D, Manuguerra JC, Mathieu D, Fontanet A, van der Werf S, the MERS-CoV Study Group 2013. Clinical features and viral diagnosis of two cases of infection with Middle East respiratory syndrome coronavirus: a report of nosocomial transmission. Lancet 381:2265–2272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Assiri A, McGeer A, Perl TM, Price CS, Al Rabeeah AA, Cummings DAT, Alabdullatif ZN, Assad M, Almulhim A, Makhdoom H, Madani H, Alhakeem R, Al-Tawfiq J, Cotten M, Watson SJ, Kellam P, Zumla AI, Memish ZA. 2013. Hospital outbreak of Middle East respiratory syndrome coronavirus. N. Engl. J. Med. 369:407–416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vijgen L, Moes E, Keyaerts E, Li S, Van Ranst M. 2008. A pancoronavirus RT-PCR assay for detection of all known coronaviruses. Methods Mol. Biol. 454:3–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Corman VM, Muller MA, Costabel U, Timm J, Binger T, Meyer B, Kreher P, Lattwein E, Eschbach-Bludau M, Nitsche A, Bleicker T, Landt O, Schweiger B, Drexler JF, Osterhaus AD, Haagmans BL, Dittmer U, Bonin F, Wolff T, Drosten C. 2012. Assays for laboratory confirmation of novel human coronavirus (hCoV-EMC) infections. Euro Surveill. 17(49):pii=20334 http://www.eurosurveillance.org/ViewArticle.aspx?ArticleId=20334 [DOI] [PubMed] [Google Scholar]
- 8.van Boheemen S, de Graaf M, Lauber C, Bestebroer TM, Raj VS, Zaki AM, Osterhaus AD, Haagmans BL, Gorbalenya AE, Snijder EJ, Fouchier RA. 2012. Genomic characterization of a newly discovered coronavirus associated with acute respiratory distress syndrome in humans. mBio 3(6):e00473–12. 10.1128/mBio.00473-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lau SK, Woo PC, Li KS, Huang Y, Tsoi HW, Wong BH, Wong SS, Leung SY, Chan KH, Yuen KY. 2005. Severe acute respiratory syndrome coronavirus-like virus in Chinese horseshoe bats. Proc. Natl. Acad. Sci. U. S. A. 102:14040–14045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Guan Y, Zheng BJ, He YQ, Liu XL, Zhuang ZX, Cheung CL, Luo SW, Li PH, Zhang LJ, Guan YJ, Butt KM, Wong KL, Chan KW, Lim W, Shortridge KF, Yuen KY, Peiris JS, Poon LL. 2003. Isolation and characterization of viruses related to the SARS coronavirus from animals in southern China. Science 302:276–278 [DOI] [PubMed] [Google Scholar]
- 11.World Health Organization (WHO) 21 July 2013. Global alert and response (GAR). Middle East respiratory syndrome coronavirus (MERS-CoV)—update. WHO, Geneva, Switzerland [Google Scholar]
- 12.Centers for Disease Control and Prevention (CDC) 2013. Update: severe respiratory illness associated with Middle East respiratory syndrome coronavirus (MERS-CoV)—worldwide, 2012–2013. MMWR Morb. Mortal. Wkly. Rep. 62:480–483 [PMC free article] [PubMed] [Google Scholar]
- 13.Gulland A. 2013. Two cases of novel coronavirus are confirmed in France. BMJ 346:f3114. 10.1136/bmj.f3114 [DOI] [PubMed] [Google Scholar]
- 14.Peiris JS, Chu CM, Cheng VC, Chan KS, Hung IF, Poon LL, Law KI, Tang BS, Hon TY, Chan CS, Chan KH, Ng JS, Zheng BJ, Ng WL, Lai RW, Guan Y, Yuen KY, HKU/Study Group UCH SARS 2003. Clinical progression and viral load in a community outbreak of coronavirus-associated SARS pneumonia: a prospective study. Lancet 361:1767–1772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Barnard DL, Kumaki Y. 2011. Recent developments in anti-severe acute respiratory syndrome coronavirus chemotherapy. Future Virol. 6:615–631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Perlman S, Netland J. 2009. Coronaviruses post-SARS: update on replication and pathogenesis. Nat. Rev. Microbiol. 7:439–450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ghosh AK, Takayama J, Rao KV, Ratia K, Chaudhuri R, Mulhearn DC, Lee H, Nichols DB, Baliji S, Baker SC, Johnson ME, Mesecar AD. 2010. Severe acute respiratory syndrome coronavirus papain-like novel protease inhibitors: design, synthesis, protein-ligand X-ray structure and biological evaluation. J. Med. Chem. 53:4968–4979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jacobs J, Grum-Tokars V, Zhou Y, Turlington M, Saldanha SA, Chase P, Eggler A, Dawson ES, Baez-Santos YM, Tomar S, Mielech AM, Baker SC, Lindsley CW, Hodder P, Mesecar A, Stauffer SR. 2013. Discovery, synthesis, and structure-based optimization of a series of N-(tert-butyl)-2-(N-arylamido)-2-(pyridin-3-yl) acetamides (ML188) as potent noncovalent small molecule inhibitors of the severe acute respiratory syndrome coronavirus (SARS-CoV) 3CL protease. J. Med. Chem. 56:534–546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ramajayam R, Tan KP, Liang PH. 2011. Recent development of 3C and 3CL protease inhibitors for anti-coronavirus and anti-picornavirus drug discovery. Biochem. Soc. Trans. 39:1371–1375 [DOI] [PubMed] [Google Scholar]
- 20.Ratia K, Pegan S, Takayama J, Sleeman K, Coughlin M, Baliji S, Chaudhuri R, Fu W, Prabhakar BS, Johnson ME, Baker SC, Ghosh AK, Mesecar AD. 2008. A noncovalent class of papain-like protease/deubiquitinase inhibitors blocks SARS virus replication. Proc. Natl. Acad. Sci. U. S. A. 105:16119–16124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Anand K, Ziebuhr J, Wadhwani P, Mesters JR, Hilgenfeld R. 2003. Coronavirus main proteinase (3CLpro) structure: basis for design of anti-SARS drugs. Science 300:1763–1767 [DOI] [PubMed] [Google Scholar]
- 22.Gan YR, Huang H, Huang YD, Rao CM, Zhao Y, Liu JS, Wu L, Wei DQ. 2006. Synthesis and activity of an octapeptide inhibitor designed for SARS coronavirus main proteinase. Peptides 27:622–625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Xue X, Yu H, Yang H, Xue F, Wu Z, Shen W, Li J, Zhou Z, Ding Y, Zhao Q, Zhang XC, Liao M, Bartlam M, Rao Z. 2008. Structures of two coronavirus main proteases: implications for substrate binding and antiviral drug design. J. Virol. 82:2515–2527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yang H, Xie W, Xue X, Yang K, Ma J, Liang W, Zhao Q, Zhou Z, Pei D, Ziebuhr J, Hilgenfeld R, Yuen KY, Wong L, Gao G, Chen S, Chen Z, Ma D, Bartlam M, Rao Z. 2005. Design of wide-spectrum inhibitors targeting coronavirus main proteases. PLoS Biol. 3:e324. 10.1371/journal.pbio.0030324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Galban S, Jeon YH, Bowman BM, Stevenson J, Sebolt KA, Sharkey LM, Lafferty M, Hoff BA, Butler BL, Wigdal SS, Binkowski BF, Otto P, Zimmerman K, Vidugiris G, Encell LP, Fan F, Wood KV, Galban CJ, Ross BD, Rehemtulla A. 2013. Imaging proteolytic activity in live cells and animal models. PLoS One 8:e66248. 10.1371/journal.pone.0066248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Frieman M, Ratia K, Johnston RE, Mesecar AD, Baric RS. 2009. Severe acute respiratory syndrome coronavirus papain-like protease ubiquitin-like domain and catalytic domain regulate antagonism of IRF3 and NF-kappaB signaling. J. Virol. 83:6689–6705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kanjanahaluethai A, Jukneliene D, Baker SC. 2003. Identification of the murine coronavirus MP1 cleavage site recognized by papain-like proteinase 2. J. Virol. 77:7376–7382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ratia K, Saikatendu KS, Santarsiero BD, Barretto N, Baker SC, Stevens RC, Mesecar AD. 2006. Severe acute respiratory syndrome coronavirus papain-like protease: structure of a viral deubiquitinating enzyme. Proc. Natl. Acad. Sci. U. S. A. 103:5717–5722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Barretto N, Jukneliene D, Ratia K, Chen Z, Mesecar AD, Baker SC. 2005. The papain-like protease of severe acute respiratory syndrome coronavirus has deubiquitinating activity. J. Virol. 79:15189–15198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Binkowski B, Fan F, Wood K. 2009. Engineered luciferases for molecular sensing in living cells. Curr. Opin. Biotechnol. 20:14–18 [DOI] [PubMed] [Google Scholar]
- 31.Binkowski BF, Butler BL, Stecha PF, Eggers CT, Otto P, Zimmerman K, Vidugiris G, Wood MG, Encell LP, Fan F, Wood KV. 2011. A luminescent biosensor with increased dynamic range for intracellular cAMP. ACS Chem. Biol. 6:1193–1197 [DOI] [PubMed] [Google Scholar]
- 32.Oka T, Takagi H, Tohya Y, Murakami K, Takeda N, Wakita T, Katayama K. 2011. Bioluminescence technologies to detect calicivirus protease activity in cell-free system and in infected cells. Antiviral Res. 90:9–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chuck CP, Chow HF, Wan DC, Wong KB. 2011. Profiling of substrate specificities of 3C-like proteases from group 1, 2a, 2b, and 3 coronaviruses. PLoS One 6:e27228. 10.1371/journal.pone.0027228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ren Z, Yan L, Zhang N, Guo Y, Yang C, Lou Z, Rao Z. 2013. The newly emerged SARS-like coronavirus HCoV-EMC also has an “Achilles' heel”: current effective inhibitor targeting a 3C-like protease. Protein Cell 4:248–250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ghosh AK, Gong G, Grum-Tokars V, Mulhearn DC, Baker SC, Coughlin M, Prabhakar BS, Sleeman K, Johnson ME, Mesecar AD. 2008. Design, synthesis and antiviral efficacy of a series of potent chloropyridyl ester-derived SARS-CoV 3CLpro inhibitors. Bioorg. Med. Chem. Lett. 18:5684–5688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Verschueren KH, Pumpor K, Anemuller S, Chen S, Mesters JR, Hilgenfeld R. 2008. A structural view of the inactivation of the SARS coronavirus main proteinase by benzotriazole esters. Chem. Biol. 15:597–606 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.