

Abstract
In the canonical STAT3 signaling pathway, binding of agonist to receptors activates Janus kinases that phosphorylate cytoplasmic STAT3 at tyrosine 705 (Y705). Phosphorylated STAT3 dimers accumulate in the nucleus and drive the expression of genes involved in inflammation, angiogenesis, invasion, and proliferation. Here, we demonstrate that human cytomegalovirus (HCMV) infection rapidly promotes nuclear localization of STAT3 in the absence of robust phosphorylation at Y705. Furthermore, infection disrupts interleukin-6 (IL-6)-induced phosphorylation of STAT3 and expression of a subset of IL-6-induced STAT3-regulated genes, including SOCS3. We show that the HCMV 72-kDa immediate-early 1 (IE1) protein associates with STAT3 and is necessary to localize STAT3 to the nucleus during infection. Furthermore, expression of IE1 is sufficient to disrupt IL-6-induced phosphorylation of STAT3, binding of STAT3 to the SOCS3 promoter, and SOCS3 gene expression. Finally, inhibition of STAT3 nuclear localization or STAT3 expression during infection is linked to diminished HCMV genome replication. Viral gene expression is also disrupted, with the greatest impact seen following viral DNA synthesis. Our study identifies IE1 as a new regulator of STAT3 intracellular localization and IL-6 signaling and points to an unanticipated role of STAT3 in HCMV infection.
INTRODUCTION
Human cytomegalovirus (HCMV) is a human herpesvirus that infects the majority of the world population. Primary exposure results in a lifelong infection. HCMV is an opportunistic pathogen that causes serious disease in immunocompromised patients and is a leading cause of congenital birth defects (1, 2). The current FDA-approved antiviral compounds inhibit viral DNA replication and have significantly improved the management of HCMV-associated diseases. Although the use of antivirals usually resolves viremia, the compounds fail to remove the latent reservoirs of HCMV within the body. Moreover, their use is limited due to toxicity, poor oral bioavailability, and the selection of antiviral-resistant variants (3–5). Efforts are under way to identify additional antiviral compounds to increase treatment options.
The 72-kDa immediate-early 1 (IE1) protein of HCMV is a key regulatory phosphoprotein conditionally required for viral early gene expression and replication in fibroblasts (6–8). IE1 localizes to the host cell nucleus, targeting both interchromatin compartments termed nuclear domain 10 (ND10) (9–11) and chromatin (12). Our work and a consecutive study by Huh et al. have demonstrated that IE1 forms physical complexes with STAT1 and STAT2 in the nuclei of infected cells, prevents association of STAT1, STAT2, and interferon (IFN) regulatory factor 9 with promoters of type I IFN-stimulated genes, and inhibits IFN-α-induced transcription (13–15). Consequently, IE1 disrupts type I IFN-dependent STAT signaling, endowing the virus with partial resistance to the antiviral effects of IFN-α and IFN-β (13–15). Notably, this activity was subsequently shown to be conserved across IE1 homologs of the human betaherpesvirus subfamily (16). Conversely, following ectopic expression in an inducible cell model (TetR/TetR-IE1), IE1 elicited a transcriptional response dominated by the upregulation of proinflammatory and immune-modulatory genes normally induced by IFN-γ (17). Although IE1-mediated gene expression proved to be independent of IFN-γ, it requires the tyrosine-phosphorylated form of STAT1. Accordingly, STAT1 accumulates in the nucleus and becomes associated with IE1 target genes upon expression of the viral protein (17).
Another member of the STAT protein family, STAT3 is involved in regulating diverse responses. In total, four isoforms of STAT3 have been identified: full-length STAT3α and truncated STAT3β, STAT3γ, and STAT3δ (for a review, see reference 18). Although the functions of the truncated isoforms are unclear, studies are beginning to suggest that they have distinct cellular activities from STAT3α (19–21). STAT3 is activated by a variety of different stimuli, including interleukin-6 (IL-6) and other cytokines and growth factors (18, 22). In the canonical STAT3 signaling pathway, binding of agonist to receptors activates Janus kinases (JAKs), which phosphorylate cytoplasmic STAT3 at tyrosine 705 (Y705). Phosphorylated STAT3 dimers accumulate in the nucleus and drive the expression of genes involved in inflammation, angiogenesis, invasion, and proliferation (18, 22). Nuclear translocation is mediated by the importin-α and -β1 heterodimer complex (23, 24). Furthermore, phosphorylation at serine 727 (S727) is necessary for maximal STAT3 transcriptional activity (25, 26). Recent studies have demonstrated that STAT3 when unphosphorylated at Y705 shuttles between the cytoplasm and the nucleus and is also transcriptionally active (27–30).
In this study, we determined a mechanism used by HCMV to regulate STAT3 during infection. We demonstrate that HCMV IE1 is both necessary and sufficient to promote early nuclear localization of STAT3, which is predominately unphosphorylated at Y705. One functional consequence is the IE1-mediated disruption of STAT3-mediated IL-6 signaling. In addition, we found that inhibition of STAT3 nuclear localization is linked to reduced viral DNA replication and late gene expression.
MATERIALS AND METHODS
Biological reagents.
MRC-5 fibroblasts, ARPE19 epithelial cells, and U373 astrocytoma cells were propagated in Dulbecco's modified Eagle's medium (DMEM) supplemented with 7% fetal bovine serum (FBS; Life Technologies, Carlsbad, CA) and 1% penicillin-streptomycin (Life Technologies). Unless otherwise stated, cells were grown until confluent, serum starved in 0.5% fetal bovine serum for 2 days, and then infected at a multiplicity of infection ranging from 0.25 to 5 infectious units (IU) per cell in DMEM supplemented with 0.5% FBS. In several experiments, cells were treated 24 h prior to infection with inhibitors: 15 μM curcumin (Sigma-Aldrich, St. Louis, MO), 30 to 150 μM S3i-201 (NSC 74859; ThermoFisher Scientific, Waltham, MA), 5 μM Stattic (Santa Cruz Biotechnology), 4 μM WP1066 (Santa Cruz Biotechnology), or dimethyl sulfoxide (DMSO; Sigma). Compounds were replaced every 24 h. As a control for pSTAT3- and STAT3-regulatable gene expression, U373 cells were treated with 183 ng/ml of carrier-free recombinant human IL-6 (BioLegend, San Diego, CA) for 15 min for Western blot analysis or for 45 min for gene expression studies. Studies using MRC-5 cells also included recombinant human IL-6 receptor alpha (IL6Rα; R&D Systems, Minneapolis, MN). TetR-IE1 and TetR cells have been previously described (17) and were treated with doxycycline (Dox) for 0 to 72 h at a final concentration of 1 μg/ml. Viability and total cell numbers were determined with Viacount (EMD Millipore, Billerica, MA) and a Gauva EasyCyte miniflow cytometer (Millipore). Control small interfering RNAs (siRNAs; Cell Signaling Technology), siRNA targeting importin-β1 (Life Technologies), or siRNA targeting STAT3 (On Target Smart pool; ThermoFisher) were transfected using Lipofectamine 2000 (Life Technologies).
Bacterial artificial chromosome (BAC)-derived HCMV strains AD169 (ADwt) (31), ADin27F (32), and Towne wild type (wt) (33) were propagated in primary fibroblasts, and Towne dlIE1 virus (14) was propagated in TetR-IE1 cells. BAC-derived HCMV clinical virus TB40/E (34) was propagated in ARPE19 epithelial cells. Viral titers were determined in an infectious units assay (35) or a standard plaque assay.
Analysis of protein, DNA, and gene expression.
Preparation of cell extracts, immunoprecipitation, Western blot analysis, and immunofluorescence microscopy were completed as previously described (14, 32, 36). The antibodies used are listed below. Immunofluorescence was observed with a 63× lens in a Leica DMRX inverted microscope (Leica Microsystems, Wetzlar, Germany) equipped with a Retiga-SRV digital camera and Image-Pro Plus 6.2 software (Q-Imaging, Surrey, Canada) (see Fig. 3 and 5, below; see also Fig. S2 in the supplemental material). Alternatively, a 60× lens in an Eclipse Ti-U inverted microscope (Nikon, Melville, NY) equipped with a CoolSNAP ES2 charge-coupled-device camera (Photometrics), multifluorescent Sedat Quad ET filter set (multichroic splitter; Chroma), and NIS-Elements software (Nikon) were used for image analysis (see Fig. 1, 2, 6, and 8, below; see also Fig. S1 in the supplemental material). The mean fluorescent intensities of STAT3 within the nucleus and cytoplasm were obtained from an average of 20 to 30 cells and from at least two replicate experiments unless otherwise noted. The data are presented as the nuclear-to-cytoplasmic ratio (N/C) ± the standard error of the mean (SEM). Viral DNA content and RNA expression from infected cells were determined using quantitative or quantitative reverse transcriptase PCR (qPCR or qRT-PCR), respectively, as previously described (14, 32) with the primers listed below. Quantities for unknown samples were defined relative to an arbitrary standard curve consisting of 10-fold serial dilutions of one sample and completed for each primer pair. Chromatin immunoprecipitation (ChIP) coupled to qPCR was performed as described previously (17) using 10 μg anti-STAT3 C-20 or normal rabbit IgG and primers specific to the human SOCS3 promoter or transcribed region (see below). For flow cytometry, U373 cells were dissociated from culture plates by using enzyme-free cell dissociation buffer (Millipore). Cells were resuspended in phosphate-buffered saline (PBS) with 2% bovine serum albumin (BSA) containing antibody conjugate (Alexa Fluor 647) and incubated for 30 min at 4°C. Cells were washed three times using PBS with 2% BSA and then fixed with 2% paraformaldehyde. Data acquisition was performed on a BD LSR II apparatus and analyzed using FlowJo software (Treestar, Ashland, OR).
Fig 3.

HCMV IE1 interacts with STAT3 and promotes STAT3 nuclear accumulation. (A) Growth-arrested MRC-5 cells were mock infected or infected with wt or dlIE1 at 3 PFU/cell in 10% serum. Samples were fixed at the indicated times, incubated with antibodies against STAT3 (green) and HCMV IE2 (red), and counterstained for DNA using DAPI (blue). Bar, 10 μm. The mean fluorescent intensities of STAT3 within the nucleus and cytoplasm were obtained from an average of 100 cells. The data are presented as the mean nuclear-to-cytoplasmic ratio ± the standard deviation. (B) Cells were infected with wt or dlIE1 at 3 PFU/cell. Samples were fixed at 48 hpi and stained as described for panel A. STAT3 staining of IE2-positive mitotic cells (left panels) or STAT3 colocalization with IE1 at mitotic chromatin (right panels) is shown. Bars, 10 μm. (C) Cells were infected as described for panel B, and extracts were isolated at 24 hpi. Samples were subjected to immunoprecipitation (IP) using antibodies to IE1 or IE2, and Western blot analysis was completed on lysate and IP samples using the indicated antibodies. (D) Cells were infected as described for panel B, and extracts were isolated at 6 to 72 hpi. Samples were subjected to immunoprecipitation using an antibody to STAT3 or normal rabbit IgG, and Western blot analysis was completed on lysates and IP samples with the indicated antibodies.
Fig 5.

IE1 is sufficient to alter STAT3 localization and to inhibit STAT3 phosphorylation, DNA binding, and target gene expression. (A) TetR-IE1 and TetR cells were treated with Dox for 0 to 72 h or 72 h, respectively. Samples were fixed, incubated with antibodies against IE1 (green) and STAT3 (red), and counterstained for DNA using DAPI (blue). Bar, 10 μm. (B) The percentage of positive cell nuclei was determined from 100 randomly selected cells per sample (IE1 -, no IE1 staining above background; IE1 +, weak, mostly punctate IE1 staining; IE1 ++, strong, diffuse IE1 staining; STAT3 -, STAT3 staining mostly cytoplasmic; STAT3 +, STAT3 staining cytoplasmic and nuclear; STAT3 ++, STAT3 staining mostly nuclear). (C) TetR-IE1 and TetR cells were treated with Dox. Samples were fixed at 48 hpi and stained as described for panel A. Representative mitotic cells are shown. Bar, 10 μm. (D) TetR-IE1 cells were treated with Dox for 0 to 72 h, and Western blot analysis was completed using the indicated antibodies. (E) TetR-IE1 and TetR cells were treated with Dox for 0 to 72 h. Relative SOCS3 mRNA levels were determined by qRT-PCR and are presented relative to TUBB expression and TetR at time zero. Data represent two biological and two technical replicates, and values are given as the mean ± standard deviation. (F) TetR-IE1 and TetR cells were treated with Dox for 72 h and with IL-6 and IL6Rα or solvent. Western blot analysis was completed using the indicated antibodies. (G) TetR-IE1 and TetR cells were treated with Dox for 72 h and with IL-6 and IL6Rα or solvent for 30 min. Samples were subjected to ChIP with an antibody to STAT3 or normal rabbit IgG and primers specific for sequences in the SOCS3 promoter or transcribed region. The percentage of output versus input DNA was calculated and is presented as the difference between STAT3 and normal IgG ChIPs. Data represent two biological and two technical replicates, and values are given as the mean ± standard deviation. *, below detection limit. (H) TetR and TetR-IE1 cells were treated with Dox for 72 h and with IL-6 and IL6Rα or solvent. Relative SOCS3 mRNA levels were determined by qRT-PCR and are presented relative to TUBB expression and TetR. Data represent three biological and two technical replicates, and values are given as the mean ± standard deviation.
Fig 1.

Infection increases the levels of unphosphorylated STAT3 in the nucleus and inhibits IL-6-stimulated gene expression. (A) Serum-starved U373 cells were mock infected or infected with ADwt at 5 IU/cell in 0.5% serum. Samples were fixed at the indicated times, incubated with antibodies against STAT3 (green) and pSTAT3 at Y705 (red), and counterstained for DNA with DAPI (blue). Where indicated, mock samples were treated with IL-6 at 183 ng/ml. The mean fluorescent intensities of STAT3 within the nucleus and cytoplasm were obtained from an average of 20 to 30 cells and from at least two replicate experiments. The data are presented as the nuclear-to-cytoplasmic ratio ± the SEM. (B) Cells were infected as described for panel A and treated with IL-6 or DMSO for 15 min prior to Western blot analysis with the indicated antibodies. The α- and β-STAT3 isoforms were evident upon sufficient electrophoretic separation. (C) The above-described experiment was repeated using the clinical isolate TB40/E. (D) Cells were infected at 1 IU/cell and treated with IL-6 or DMSO for 45 min just prior to harvest at 24 and 48 hpi. Levels of the indicated mRNAs were quantified by qRT-PCR and are presented relative to GAPDH. Data represent two biological replicate experiments and are presented as the means ± SEM. *, P < 0.05. (E) Serum-starved U373 cells were infected as described for panel A. Cells were fixed, stained with anti-IL6Rα antibody conjugated to Alexa Fluor 647, and analyzed using flow cytometry. As a control, cells were treated with trypsin solution for 15 min prior to antibody staining (gray), and the values represent the mean fluorescence intensity from two biological experiments.
Fig 2.

Nuclear accumulation of STAT3 reduces IL-6-induced phosphorylation and is dependent on viral gene expression. (A) Serum-starved U373 cells were transfected with control siRNAs or an siRNA targeting importin-β1 24 h prior to infection. Cells were mock infected or infected with ADwt virus at 5 IU/cell and treated with IL-6 for 15 min prior to harvest at 24 hpi. Western blot analysis was completed using the indicated antibodies. (B) Cells were transfected, infected, and treated with IL-6 as described above. At 24 hpi, cells were processed for immunofluorescence analysis using anti-STAT3 (green) and DAPI (blue). The mean fluorescent intensities of STAT3 within the nucleus and cytoplasm were obtained from an average of 20 to 30 cells and from at least two replicate experiments. The data are presented as the nuclear-to-cytoplasmic ratio ± the SEM. (C) U373 cells were mock infected or infected at 5 IU/cell with either untreated or UV-irradiated ADwt virus. At 5 hpi, cells were processed for immunofluorescence analysis using anti-STAT3 (green) and DAPI (blue). The mean fluorescent intensities of STAT3 within the nucleus and cytoplasm were determined as described above. (D) U373 cells were mock infected or infected at 5 IU/cell with either untreated or UV-irradiated ADwt virus. At 5 hpi, cells were harvested and levels of IE1 mRNA were quantified by qRT-PCR. Data presented are relative to results with GAPDH. Data represent two biological replicate experiments and are presented as the means ± SEM.
Fig 6.

Chemical disruption of STAT3 inhibits HCMV replication. (A) U373 cells were treated with increasing concentrations of S3i-201, curcumin, Stattic, or WP1066. At 72 h, cell viability was quantified. The data represent two biological replicate experiments and are presented as the mean ± SEM. (B) Cells were pretreated with DMSO, S3i-201, curcumin, Stattic, or WP1066. After 24 h, cells were infected at 5 IU/cell with ADwt and processed for immunofluorescence analysis using an antibody to STAT3 (green) and the DNA stain DAPI (blue). The mean fluorescent intensities of STAT3 within the nucleus and cytoplasm were obtained from an average of 20 to 30 cells and from at least two biological replicate experiments. The data are presented as the nuclear-to-cytoplasmic ratio ± the SEM. (C) Cells were pretreated with drug as described above. After 24 h, cells were infected at 0.25 IU/cell with ADwt. Viral genomes were quantified at 72 hpi by qPCR and normalized to cellular DNA. Data represent two biological replicates, and values are given as the mean ± SEM. *, P < 0.05.
Fig 8.

siRNA targeting STAT3 attenuates viral DNA replication. (A) Serum-starved U373 cells were transfected with control siRNA or siRNA targeting STAT3. After 24 h, Western blot analysis was completed using the indicated antibodies. (B) Cells were transfected as described above. After 24 h, cells were processed for immunofluorescence analysis using anti-STAT3 (green) and DAPI (blue). The mean fluorescent intensities of STAT3 within the nucleus and cytoplasm were obtained from an average of 20 to 30 cells and from at least two replicate experiments. The data are presented as the nuclear-to-cytoplasmic ratio ± SEM. (C) Cells were transfected as described above. After 24 h, cells were infected with ADwt virus at 0.25 IU/cell. Viral genomes were quantified at 72 hpi by qPCR and normalized to cellular DNA. Data represent two biological replicates, and values are given as the mean ± SEM. *, P < 0.05.
Antibodies.
The following antibodies were used in these studies: normal rabbit IgG (Sigma-Aldrich), mouse anti-FLAG M2 (Sigma-Aldrich), mouse anti-glyceraldehyde 3-phosphate dehydrogenase (GAPDH) clone 0411 (Santa Cruz Biotechnology), rabbit anti-GAPDH ab9485 (Abcam), mouse anti-hemagglutinin (HA) clone HA-7 (Sigma-Aldrich), rabbit anti-STAT1 E-23 (Santa Cruz Biotechnology), rabbit anti-STAT2 H-190 (Santa Cruz Biotechnology), mouse anti-STAT3 clone 124H6 (Cell Signaling Technology), rabbit anti-STAT3 C-20 (Santa Cruz Biotechnology), rabbit anti-pSTAT3 (Y705) (Cell Signaling Technology), rabbit anti-pSTAT3 (S727) (Cell Signaling Technology), rabbit anti-H2A ab15653 (Abcam), and rabbit anti-importin β1 (Cell Signaling Technology). The antibodies against HCMV proteins were mouse anti-pUL123 clone 1B12, mouse anti-pUL122 clone 3A9, mouse anti-pTRS1, mouse anti-pUL99, mouse anti-pUL37 clone 2A1D, mouse anti-pUL38 clone 3D12 (generously provided by Tom Shenk, Princeton University), and mouse anti-pUL44 (Virusys). Secondary antibodies included goat anti-mouse–horseradish peroxidase (HRP) and donkey anti-rabbit–HRP (Jackson ImmunoResearch) conjugates for Western blot analysis and anti-mouse–Alexa Fluor 488, anti-mouse–Alexa Fluor 568, anti-mouse–Alexa Fluor 594, anti-rabbit–Alexa Fluor 488 (Life Technologies) conjugates, and Alexa Fluor 647 conjugated to anti-CD126 (IL-6Rα; Biolegend, San Diego, CA) for immunofluorescence. Cellular DNA was stained with 4′,6-diamidino-2-phenylindole (DAPI; Life Technologies).
PCR oligonucleotides.
The following oligonucleotide pairs were used in these studies: IL-6 (5′-AGCCACTCACCTCTTCAGAACGAA-3′ and 5′-AGTGCCTCTTTGCTGCTTTCACAC-3′), SOCS3 (5′-ATTCGCCTTAAATGCTCCCTGTCC-3′ and 5′-TGGCCAATACTTACTGGGCTGACA-3′) and (5′-GGCCACTCTTCAGCATCTC-3′ and 5′-ATCGTACTGGTCCAGGAACTC-3′), SOCS3 promoter (5′-AGCCTTTCTCTGCTGCGAGT-3′ and 5′-CCCGATTCCTGGAACTGC-3′), β-tubulin (TUBB) (5′-TATCAGCAGTACCAGGATGC-3′ and 5′-TGAGAAGCCTGAGGTGATG-3′), GAPDH (5′-ACCCACTCCTCCACCTTTGAC-3′ and 5′-CTGTTGCTGTAGCCAAATTCGT-3′), UL123 (5′-GCCTTCCCTAAGACCACCAAT-3′ and 5′-ATTTTCTGGGCATAAGCCATAATC-3′), UL122 (5′-CCAGTATGCACCAGGTGTTAG-3′ and 5′-CTGGATGCCCTTGTTGTTC-3′), UL38 (5′-ACGGTGCTATCGTGCTGGAGTATT-3′ and 5′-AAGACCATCACCAGGTCGTCCATA-3′), UL99 (5′-TTCACAAGGTCCACCCACC-3′ and 5′-GTGTCCCATTCCGACTCG-3′), and UL83 (5′-TGAGCATCTCAGGTAACCTGTTG-3′ and 5′-CAGCCACGGGATCGTACTG-3′).
Statistical analysis.
The data are representative of at least two independent experiments, and values are given as the mean of replicate experiments ± the SEM unless otherwise stated. For all experiments where we used the Student t test, we determined that the variances were not different by using an F-test prior to using the t test. A significant P value (P < 0.05) is indicated by an asterisk in the figures.
RESULTS
HCMV infection localizes unphosphorylated STAT3 to the nucleus and disrupts IL-6-induced gene expression.
Limited information is available on the impact of HCMV infection on the cellular transcription factor STAT3. To determine whether HCMV influences STAT3, we evaluated subcellular localization at 2 and 24 h postinfection (hpi). We infected U373 astrocytoma cells by using HCMV strain AD169 (ADwt) at a multiplicity of infection (MOI) of 5 IU per cell in 0.5% serum. As a control, uninfected cells were treated with or without human IL-6 to stimulate phosphorylation of STAT3. Reduced serum conditions allowed for more robust IL-6-stimlulated responses. Cells were fixed and stained using antibodies against STAT3 and pSTAT3 at Y705. To quantify changes in localization, we determined the N/C ratio of mean fluorescent intensities between nuclear and cytosolic staining. We detected increased STAT3 within the nuclei of infected cells at 2 hpi (N/C, 2.95 ± 0.05) and 24 hpi (N/C, 6.71 ± 0.68) compared to uninfected cells (N/C, 1.25 ± 0.16) (Fig. 1A). As expected, we observed increased nuclear localization of STAT3 (N/C, 3.42 ± 0.35) and pSTAT3 in mock-infected and IL-6-treated cells (Fig. 1A). To our surprise, we detected little to no pSTAT3 within the nuclei of infected cells (Fig. 1A). These data suggest that HCMV infection rapidly promotes nuclear localization of STAT3 in the absence of robust phosphorylation at Y705.
In general, phosphorylation of cytoplasmic STAT3 at Y705 occurs following cytokine and growth factor stimulation and results in STAT3 nuclear accumulation and DNA binding (18, 22). We next investigated whether HCMV influences STAT3 phosphorylation during both infection and cytokine stimulation. U373 cells were infected at an MOI of 5 IU/cell, and cultures were treated at the indicated times postinfection with or without IL-6 (Fig. 1B). We evaluated steady-state protein levels by using Western blot analysis on whole-cell lysates isolated from a population of cells. Compared to mock-infected cells treated with IL-6, HCMV infection suppressed IL-6-induced phosphorylation of STAT3 at Y705 by 2 hpi and continued through 48 hpi (Fig. 1B). In contrast, phosphorylation at S727 occurred regardless of infection and was independent of IL-6 stimulation (Fig. 1B). At a lower MOI, we observed via Western blotting increased STAT3 phosphorylation at Y705, which likely occurs in uninfected cells in the population, as determined by immunofluorescence analysis (see Fig. S1 in the supplemental material). To determine whether the response also occurs when using a clinically relevant HCMV strain, we completed the experiment using the TB40/E virus (34). Similar to AD169, TB40/E infection suppressed IL-6-induced phosphorylation at Y705 but not S727 (Fig. 1C). These data demonstrate that HCMV infection disrupts IL-6-induced phosphorylation of STAT3 at Y705.
We evaluated the impact of infection on the expression of two genes known to be regulated by STAT3, those for IL-6 and SOCS3 (18, 37). We infected U373 cells at 1 IU/cell using ADwt virus with or without IL-6 at the indicated times postinfection and determined changes in gene expression by using qRT-PCR relative to GAPDH RNA levels. Compared to mock controls, infection significantly decreased gene expression of IL-6 and SOCS3 following IL-6 stimulation (Fig. 1D). These data support the conclusion that HCMV infection disrupts expression of two IL-6-induced STAT3-regulated genes. To exclude the possibility that disruption of IL-6 signaling is a consequence of decreased IL-6 receptor cell surface expression, we measured the impact of infection on IL6Rα levels. We infected U373 cells at 3 IU/cell, and surface levels of IL6Rα were determined by flow cytometry. Compared to mock infection, we observed similar levels of IL6Rα during HCMV infection at 6 and 24 hpi (Fig. 1E). These data rule out the possibility of HCMV-mediated loss of endogenous IL6Rα surface expression during the time of altered gene expression.
To test whether HCMV-mediated inhibition of IL-6 signaling depends on STAT3 nuclear localization, we evaluated the effects on STAT3 phosphorylation after disruption of nuclear import. Nuclear translocation of STAT3 is mediated by the importin-α and -β1 heterodimer complex (23, 24). We transfected U373 cells with either a control siRNA or an siRNA targeting importin-β1 and observed a reduction in importin-β1 levels (Fig. 2A). Under these conditions, reduced importin-β1 resulted in increased pSTAT3 in IL-6-treated HCMV-infected cells (Fig. 2A). Furthermore, disruption of importin-β1 reduced IL-6-induced STAT3 nuclear accumulation in both mock-infected cells (N/C, 1.27 ± 0.11) and HCMV-infected cells (N/C, 1.55 ± 0.12) compared to control siRNA-treated mock-infected (N/C, 2.19 ± 0.03) and infected (N/C, 4.15 ± 0.10) cells (Fig. 2B). These data suggest that HCMV promotes nuclear accumulation of STAT3 early during infection, thereby moving STAT3 away from the cytosolic regulators.
Changes in STAT3 phosphorylation and localization are detectable as early as 2 hpi, indicating a role for HCMV virions or newly expressed proteins in manipulating STAT3. To identify the source of the activity, U373 cells were infected with either untreated ADwt or UV-irradiated virus and evaluated by immunofluorescence microscopy. STAT3 nuclear localization occurred following infection with untreated (N/C, 3.27 ± 0.15) but not UV-irradiated (N/C, 1.09 ± 0.02) virus (Fig. 2C). Under these conditions of UV treatment, we did not detect IE1 RNA expression (Fig. 2D). These data demonstrated that viral gene expression is necessary for relocalization of STAT3.
HCMV IE1 promotes STAT3 nuclear accumulation and disrupts IL-6-induced STAT3 phosphorylation, DNA binding, and target gene expression.
HCMV protein IE1 is known to regulate both STAT1 and STAT2 (13–15, 17). To determine whether IE1 expression could influence STAT3 localization, MRC-5 fibroblasts were mock infected or infected at 3 PFU/cell with either wild-type virus (wt) or an IE1-deficient virus (dlIE1) of the HCMV Towne strain. Using immunofluorescence microscopy, we observed increased staining of STAT3 within the nuclei of IE2-positive infected cells by 6 hpi with wt virus (N/C, 1.73 ± 0.04) but not dlIE1 (N/C, 0.99 ± 0.15) compared to mock-infected cells (N/C, 1.09 ± 0.13) (Fig. 3A). This increase was also observed at both 24 (N/C, 1.73 ± 0.55) and 72 hpi (N/C, 2.13 ± 0.56) (Fig. 3A). At these later times, we did observe a few cells in the dlIE1 infections that had increased nuclear STAT3 staining; however, this was not significant among the population, as indicated by the N/C ratio determined from a random selection of cells (n = 95). These data demonstrated that IE1 is necessary for the efficient nuclear localization of STAT3 during infection. HCMV IE1 has been previously demonstrated to localize to mitotic chromatin (14). In cells undergoing mitosis in the infected population, we observed colocalization of STAT3 with DAPI-stained chromatin (Fig. 3B, left and right panels). Moreover, STAT3 and IE1 colocalized in wt- but not dlIE1-infected cells (Fig. 3B, right panel). To assess a possible physical interaction between IE1 and STAT3, we isolated whole-cell lysates at 24 hpi and immunoprecipitated protein complexes by using an antibody against IE1. Following immunoprecipitation, we detected the slower-migrating STAT3α but not the smaller STAT3 isoform by Western blotting from Towne strain wt-infected cells (Fig. 3C). We did not observe an interaction when we used an antibody against IE2 or following infection by the dlIE1 virus (Fig. 3C). IE1 was also specifically detected throughout the viral infectious cycle (6 to 72 h) in protein complexes isolated by immunoprecipitation with a STAT3-directed antibody (Fig. 3D). These data demonstrate that HCMV IE1 associates with at least one STAT3 isoform during infection.
Finally, we evaluated the functional impact of IE1 on STAT3 following IL-6 stimulation. These studies were completed by adding both IL-6 and soluble IL-6 receptor alpha (IL-6/IL6Rα) to the culture medium, because MRC-5 cells are largely unresponsive to IL-6 alone. Following the addition of exogenous IL-6/IL6Rα, infection by wt but not dlIE1 virus resulted in reduced levels of Y705-phosphorylated STAT3 at 16 hpi compared to the mock group (Fig. 4A). Under these conditions in mock-infected cells, IL-6/IL6Rα triggered robust STAT3 DNA binding at the SOCS3 promoter and little binding at the SOCS3 transcribed region, as determined in a chromatin immunoprecipitation (ChIP) assay (Fig. 4B). During infection, IL-6/IL6Rα-induced STAT3 DNA binding was substantially diminished when we used wt virus but not with the dlIE1 virus (Fig. 4B). Concordantly, SOCS3 gene induction was significantly reduced during wt infection (Fig. 4C). Infection by the dlIE1 virus altered SOCS3 expression, but to a lesser degree than wt virus (Fig. 4C). Expression of HCMV IE2 was unaltered between the different conditions of infection (Fig. 4C). Our results support the conclusion that HCMV IE1 binds to STAT3α and is necessary to localize STAT3 to the nucleus at early times of infection. Furthermore, expression of IE1 disrupts IL-6-induced phosphorylation of STAT3, DNA association by STAT3, and SOCS3 gene induction during infection.
Fig 4.
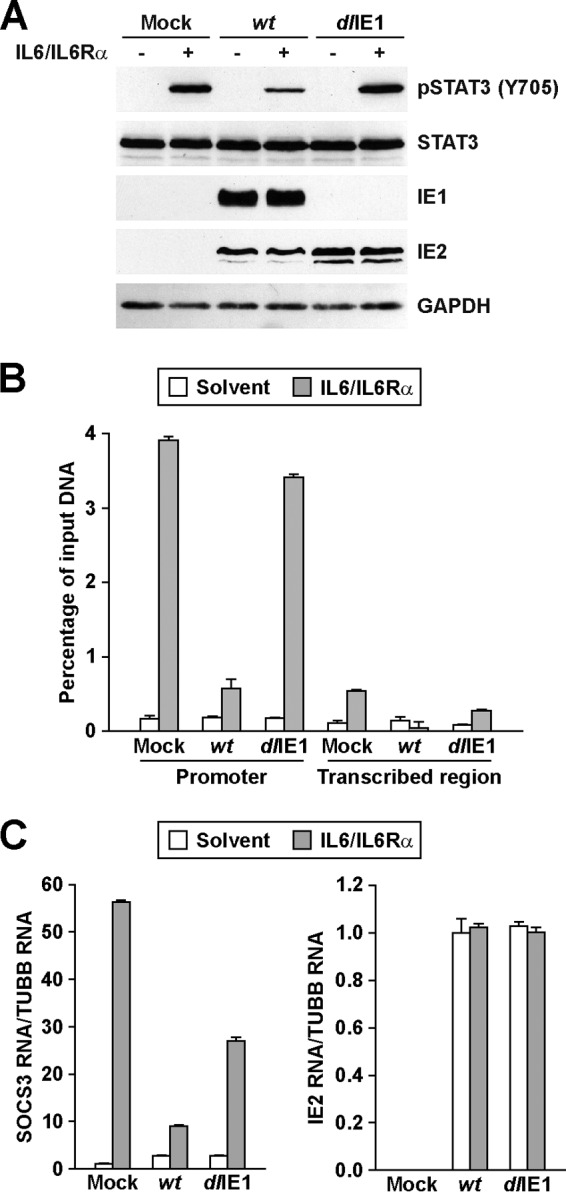
HCMV IE1 inhibits STAT3 phosphorylation, DNA binding, and target gene expression. (A) Serum-starved MRC-5 cells were infected with wt or dlIE1 at 3 PFU/cell and treated with IL-6 and IL6Rα or solvent. Samples from 16 hpi were subjected to Western blot analysis using the indicated antibodies. (B) Serum-starved cells were infected as described for panel A and treated with IL-6 and IL6Rα or solvent for 30 min. Samples from 16 hpi were subjected to ChIP with an antibody to STAT3 or normal rabbit IgG and primers specific for sequences in the SOCS3 promoter or transcribed region. The percentage of output versus input DNA was calculated and is presented as the difference between STAT3 and normal IgG ChIPs. Data represent two biological and two technical replicates, and values are given as the mean ± standard deviation. (C) Serum-starved cells were infected and treated as described for panel A. Relative SOCS3 and IE2 mRNA levels at 16 hpi were determined by qRT-PCR and are presented relative to TUBB expression. Data represent three biological and two technical replicates, and values are given as the mean ± standard deviation.
To determine whether IE1 expression is sufficient to mediate these changes, we induced IE1 in the absence of infection by using a tetracycline repressor-regulated expression system in human fibroblasts (TetR/TetR-IE1) (17). Following induction of IE1 expression using doxycycline, we observed strong accumulation of STAT3 within the nuclei of TetR-IE1 cells (Fig. 5A). STAT3 relocalization did not occur in control TetR cells (Fig. 5A). By 24 to 72 h poststimulation, greater than 90% of the IE1-positive cells contained STAT3 within the nucleus (Fig. 5B), but these nuclei did not exhibit detectable pSTAT3 (see Fig. S3 in the supplemental material). Furthermore, STAT3 again colocalized with the viral protein and DAPI-stained mitotic chromatin upon induction of IE1 (Fig. 5C). Consistent with previous studies (17), induction of IE1 expression relocalized only a fraction of STAT1 to the nucleus, with delayed kinetics compared to STAT3 (see Fig. S2A and C in the supplemental material), while not apparently altering the subcellular distribution of STAT2 (see Fig. S2B and C). We also evaluated changes in STAT3 phosphorylation upon IE1 expression. Increased expression of IE1 correlated with decreased levels of phosphorylation at Y705, while the total levels of STAT3 remained constant (Fig. 5D). Under these conditions, IE1 was sufficient to suppress the levels of SOCS3 RNA (Fig. 5E). Finally, induction of IE1 suppressed exogenous IL-6/IL6Rα-stimulated phosphorylation of STAT3 (Fig. 5F; see also Fig. S3), SOCS3 promoter binding by STAT3 (Fig. 5G), and SOCS3 expression (Fig. 5H). These data indicate that expression of IE1 is sufficient to promote the nuclear accumulation of mostly unphosphorylated STAT3, inactive for sequence-specific DNA binding at the SOCS3 promoter, and to alter expression of the STAT3-regulated gene SOCS3.
Disruption of STAT3 relocalization inhibits HCMV DNA replication.
The rapid accumulation of unphosphorylated STAT3 in the nucleus during infection suggests that HCMV might utilize STAT3 for viral replication. To test this hypothesis, we evaluated the impact of chemical inhibitors of STAT3 on HCMV infection. The inhibitors included the following; S3i-201, which inhibits STAT3 dimerization and DNA binding (38); curcumin, a natural plant polyphenol which functions, in part, by inhibiting STAT3 DNA binding (39–42); Stattic, which interferes with STAT3 phosphorylation and dimerization (43); WP1066, which blocks upstream JAK2-mediated phosphorylation (44). Initially, we tested whether the compounds influenced HCMV-mediated localization of STAT3. U373 cells were pretreated with DMSO or noncytotoxic concentrations of each compound (Fig. 6A). We infected cells at a multiplicity of 5 IU/cell and evaluated STAT3 accumulation by immunofluorescence microscopy. Compared to DMSO (N/C, 5.30 ± 0.12), both S3i-201 (N/C, 1.26 ± 0.10) and curcumin (N/C, 1.13 ± 0.40) treatments significantly reduced the accumulation of STAT3 in the nuclei of infected cells (Fig. 6B). Stattic treatment resulted in an intermediate phenotype (N/C, 2.64 ± 0.80), while inhibition of JAK-mediated phosphorylation by using WP1066 failed to block the HCMV-mediated change in STAT3 localization (N/C, 5.45 ± 0.90) (Fig. 6B).
Next, we quantified the impact of inhibiting STAT3 on HCMV replication. U373 cells were pretreated with DMSO or compound and infected at 0.25 IU/cell using ADwt virus. We quantified changes in HCMV viral DNA levels by using qPCR. The addition of S3i-201 resulted in a 99.7% reduction in viral DNA levels, curcumin resulted in a 94.0% reduction, Stattic resulted in an 89.5% reduction, and WP1066 had no effect on DNA replication (Fig. 6C). Interestingly, the percent reduction was proportional to the change in the nuclear/cytosolic ratio for STAT3 (Fig. 6B). With S3i-201, we observed the greatest decrease in DNA replication at ≥120 μM (Fig. 7A). The efficacy of S3i-201 inhibition was influenced by the MOI. Although still inhibiting replication, infection at 3 IU/cell resulted in an approximately 71.2% decrease in viral DNA replication (Fig. 7B). We also evaluated the antiviral efficacy of S3i-201 during infection of primary human foreskin fibroblasts and retinal pigmented epithelial cells. Fibroblasts and U373 cells were infected at 0.25 IU/cell with ADwt virus, while epithelial cells were infected using the clinical isolate TB40/E. Chemical inhibition of STAT3 significantly reduced viral DNA replication at 72 hpi in all of the cell types and viral strains (Fig. 7C). Finally, we determined the impact of STAT3 inhibition on viral titers at 96 hpi from cells treated with either DMSO or S3i-201. We observed that the addition of S3i-201 resulted in an average 2.3-log reduction in viral titers (Fig. 7D). These data suggest that STAT3 nuclear localization is linked to efficient HCMV DNA replication and virus production in multiple cell types.
Fig 7.

S3i-201 inhibits HCMV replication in multiple cell types. (A) Cells were pretreated with increasing concentrations of S3i-201. After 24 h, cells were infected at 0.25 IU/cell with ADwt. Viral genomes were quantified at 72 hpi by qPCR and normalized to cellular DNA. Data represent two biological replicates, and values are given as the mean ± SEM. *, P < 0.05. (B) U373 cells were pretreated with 125 μM S3i-201. After 24 h, cells were infected at 3 IU/cell with ADwt. Viral genomes were quantified as described above. Data represent two biological replicates, and values are given as the mean ± SEM. *, P < 0.05. (C) Different cell types were pretreated with 125 μM S3i-201. U373 and HFF cells were infected at 0.25 IU/cell with ADwt, while ARPE19 cells were infected at 0.25 IU/cell with TB40/E. Viral genomes were quantified as described above. Data represent two biological replicates, and values are given as the mean ± SEM. *, P < 0.05. (D) U373 cells were pretreated with drug as described above. At 24 h, cells were infected at 0.25 IU/cell with ADwt. Viral titers were determined from culture supernatants obtained at 96 hpi. Data represent two biological replicates, and values are given as the mean ± SEM. *, P < 0.05.
To provide evidence that the disruption is STAT3 dependent, we knocked down STAT3 expression by transfecting U373 cells with an siRNA targeting STAT3 or with control siRNA. We analyzed changes in STAT3 protein expression by Western blotting and observed a reduction in STAT3 levels with the specific siRNA compared to the control (Fig. 8A). Albeit reduced, we did observe STAT3 within the nucleus in cells transfected with the specific siRNA (Fig. 8B). To evaluate the impact on HCMV viral DNA replication, siRNA-transfected U373 cells were infected at 0.25 IU/cell, and we quantified changes in viral DNA levels by using qPCR. Compared to the control, we observed a 5-fold reduction in viral DNA replication upon reduced levels of STAT3 (Fig. 8C). These data provide additional evidence that STAT3 is necessary for fully efficient viral DNA replication.
Herpesvirus gene expression is temporally regulated, with kinetic classes defined as immediate early (IE), early (E), early-late (E-L), and late (L). Efficient late gene expression is dependent upon DNA replication (1). To identify the steps in replication that require STAT3, we quantified changes in viral gene expression. U373 cells were pretreated with DMSO or S3i-201 and infected using the ADwt virus at 0.25 IU/cell. Total RNA was harvested, and viral gene expression was quantified relative to GAPDH RNA. We observed similar levels of expression for the IE and E genes UL123 (IE1) and UL38, respectively, between mock and S3i-201 treatments (Fig. 9A). However, beginning around 24 hpi, the addition of S3i-201 significantly decreased expression of UL122 (IE2), UL83, and UL99 (Fig. 9A). We confirmed these changes by Western blot analysis of whole-cell lysates from HCMV-infected U373 cells (Fig. 9B). In addition to changes in the IE2-86kDa (pUL122) protein, we observed decreased expression of the IE2-60kDa late isoform following S3i-201 treatment (Fig. 9B). Furthermore, expression levels of two viral proteins with E-L kinetics, pTRS1 and pUL44, were also inhibited (Fig. 9B) beginning at 24 hpi. These data demonstrated that inhibition of STAT3 disrupts viral gene expression beginning around 24 hpi, with the greatest impact seen after 48 hpi for HCMV IE2, E-L, and L genes.
Fig 9.

STAT3 is necessary for efficient HCMV gene expression and genome replication. (A) U373 cells were pretreated with DMSO or 125 μM S3i-201. After 24 h, cells were infected at 0.25 IU/cell with ADwt. Levels of the indicated RNAs were quantified by qRT-PCR and are presented relative to GAPDH. Data represent two biological replicate experiments, and values are given as the mean ± SEM. *, P < 0.05. (B) U373 cells were pretreated with drug as described above, and after 24 h cells were infected for 2 to 72 h with ADwt at 0.25 IU/cell. Western blot analysis was completed using the indicated antibodies. (C) U373 cells were infected at 0.25 IU/cell with ADwt. Cells were then treated for 24 h with 125 μM S3I-201 at either 2 hpi or 48 hpi. After 72 hpi, viral genomes were quantified by qPCR and normalized to cellular DNA levels. Data represent two biological replicates, and values are given as the mean ± SEM. *, P < 0.05.
The accumulation levels of E-L and L transcripts as well as of UL122 is dependent on viral DNA synthesis (45–47). Furthermore, HCMV IE2, pUL44, and pTRS1 have been demonstrated to contribute to genome replication (33, 48–53). To test the timing of STAT3's contribution to HCMV replication, we treated U373 cells with S3i-201 at different times during infection. Cells were infected at 0.25 IU/cell with ADwt virus and treated with S3i-201 at 2 hpi or at 48 hpi for 24 h. We isolated DNA at 72 hpi and quantified viral genomes by using qPCR. Consistent with previous data, S3i-201 treatment early during infection resulted in a significant decrease in viral DNA levels (Fig. 9C). Conversely, treatment at 48 hpi resulted in no significant difference in viral DNA compared to control infection (Fig. 9C). These data suggest that HCMV relocalizes STAT3 early to regulate early and late events during infection, including efficient viral DNA replication.
DISCUSSION
We have determined that HCMV infection promotes nuclear accumulation of STAT3 that is predominantly or entirely unphosphorylated at Y705. In the canonical pathway from uninfected cells, STAT3 is activated by a variety of different stimuli, including cytokines and growth factors, resulting in phosphorylation of Y705 (pSTAT3) and accumulation in the nucleus (18, 22). Unlike HCMV, herpesviruses that infect cells of lymphoid origin, including Epstein-Barr virus (EBV) (54, 55), Kaposi's sarcoma-associated herpesvirus (KSHV) (56), herpesvirus saimiri (HVS) (57), and varicella-zoster virus (VZV) (58) exploit the survival and oncogenic effects of pSTAT3. Beyond herpesviruses, several oncogenic viruses utilize pSTAT3, while other viruses employ mechanisms to inhibit STAT3 signaling (59, 60). Within our studies, we observed that disrupting the nuclear accumulation of STAT3 or STAT3 expression inhibited HCMV infection at the stage of viral DNA replication. These studies are the first example of a virus that inhibits phosphorylation of STAT3 at Y705 yet still requires its activities for viral replication.
We found that the HCMV IE1 protein is necessary to relocalize STAT3 to the nucleus at early times during infection, and the viral protein is also sufficient to induce nuclear STAT3 accumulation. The effects of IE1 on the subcellular distribution of STAT3 seem to be independent of phosphorylation at Y705 or cytokine stimulation. IE1 (also known as IE1-72kDa, or IE72) is a nuclear regulatory phosphoprotein expressed from the HCMV genome at the start of infection. IE1 has long been known to attach to human chromosomes (12), but it is not considered to bind directly to DNA (61). We observed that IE1 expression promoted the association of STAT3 with mitotic chromatin, yet disrupted STAT3 binding to the SOCS3 promoter. Unphosphorylated STAT3 has been demonstrated to shuttle between the nucleus and the cytosol (27–30), and we speculate that STAT3 nuclear export may be prevented by interactions with IE1 at cellular chromatin and/or other nuclear compartments.
One functional consequence of IE1-mediated nuclear localization of STAT3 is the suppression of IL-6-induced SOCS3 gene expression. When we disrupted nuclear import, we were able to reestablish IL-6-stimulated phosphorylation of STAT3, suggesting that HCMV sequesters STAT3 away from its upstream kinases by nuclear localization. In addition, disruption of JAK signaling by the compound WP1066 failed to prevent HCMV-mediated nuclear localization or disrupt viral DNA replication. However, we cannot exclude the possibility of HCMV modulation of regulatory phosphatases. Previous studies demonstrated that upregulation of IL-6 by HCMV pUS28 resulted in pSTAT3 at Y705 (62). Consistent with these finding and other studies (62, 63), we observed a transient increase in IL-6 expression that occurred only at 2 hpi and was not affected by S3i-201 (data not shown). Furthermore, we did detect increased pSTAT3 (Y705) by Western blotting when we infected most but not all of the cells in culture. Under these conditions, phosphorylation of STAT3 occurred mostly in the uninfected cells within this population. Finally, Le et al. (64) have demonstrated that HCMV infection can disrupt IFN-γ-stimulated STAT3 phosphorylation starting at 24 hpi. Our studies indicate that nuclear STAT3 is predominantly unphosphorylated at Y705 in cells infected by either lab-adapted or clinical strains of HCMV.
Our data indicate that HCMV primarily utilizes unphosphorylated STAT3 to promote, either directly or indirectly, the initiation of HCMV DNA replication. Consistent with this idea, the addition of S3i-201 after 48 hpi had no effect on DNA replication. We found that inhibition of STAT3 severely attenuated viral DNA replication, the expression of numerous viral genes, and consequently, the production of infectious viral progeny. At 24 hpi, we observed similar levels of expression for HCMV IE1, IE2, and pUL38 following STAT3 inhibition. However, we detected a substantial decrease in expression of the viral polymerase subunit pUL44. The decrease in pUL44 levels may be attributed to decreased pTRS1, since pTRS1 functions in cooperation with IE1 and IE2 to stimulate UL44 expression (52, 65). After 24 hpi, the increase in IE2-86kDa, IE2-60kDa, pTRS1, and pUL99 levels failed to occur upon inhibiting STAT3. These changes have been shown to be dependent upon viral DNA replication (66–69). A similar phenotype occurred upon deletion of the HCMV protein pUL21a which, along with pUL97 kinase, negatively regulates the anaphase-promoting complex (45, 67, 70). Disruption of pUL21a resulted in reduced DNA replication and late expression of a subset of proteins, including IE2 and pUL99 (67).
We demonstrated that chemical antagonists of STAT3 significantly inhibited HCMV infection. Similar observations have been made in experiments involving inhibitors of STAT3 and VZV infection (58). Numerous malignancies are characterized by elevated STAT3 expression and activity (71). As a result, STAT3 inhibitors are being developed and are currently entering clinical trials as anticancer agents (71). Several FDA-approved compounds have been shown to inhibit STAT3 activity, such as Celebrex and Sorafenib (72–74), which also inhibit HCMV replication in vitro (75, 76). Overall, our studies indicate that HCMV manipulates STAT3 to promote an environment that supports efficient viral DNA replication, and the findings implicate STAT3 as a possible target for anti-HCMV antiviral research.
Supplementary Material
ACKNOWLEDGMENTS
We thank T. Shenk for the HCMV antibodies. We thank Theresa Knoblach, Benedikt Grandel, Nathalie Czech, and Thomas Harwardt for experimental contributions and Sandra Meinel for technical assistance. We also thank M. Hakki and the members of the Terhune and Nevels labs for their insights and helpful discussions.
This work is supported by NIH grant R01AI083281 to S. Terhune and by Deutsche Forschungsgemeinschaft grant PA851/2-1 to C. Paulus.
Footnotes
Published ahead of print 31 July 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JVI.01197-13.
REFERENCES
- 1.Mocarski E, Shenk T, Pass RF. 2007. Cytomegaloviruses, p 2701–2772 In Knipe DM, Howley PM, Griffin PE, Lamb RA, Martin MA, Roizman B, Straus SE. (ed), Fields virology, 5th ed. Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 2.Nassetta L, Kimberlin D, Whitley R. 2009. Treatment of congenital cytomegalovirus infection: implications for future therapeutic strategies. J. Antimicrob. Chemother. 63:862–867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Avery RK. 2008. Update in management of ganciclovir-resistant cytomegalovirus infection. Curr. Opin. Infect. Dis. 21:433–437 [DOI] [PubMed] [Google Scholar]
- 4.Hakki M, Chou S. 2011. The biology of cytomegalovirus drug resistance. Curr. Opin. Infect. Dis. 24:605–611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lurain NS, Chou S. 2010. Antiviral drug resistance of human cytomegalovirus. Clin. Microbiol. Rev. 23:689–712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Greaves RF, Brown JM, Vieira J, Mocarski ES. 1995. Selectable insertion and deletion mutagenesis of the human cytomegalovirus genome using the Escherichia coli guanosine phosphoribosyl transferase (gpt) gene. J. Gen. Virol. 76:2151–2160 [DOI] [PubMed] [Google Scholar]
- 7.Greaves RF, Mocarski ES. 1998. Defective growth correlates with reduced accumulation of a viral DNA replication protein after low-multiplicity infection by a human cytomegalovirus ie1 mutant. J. Virol. 72:366–379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mocarski ES, Kemble GW, Lyle JM, Greaves RF. 1996. A deletion mutant in the human cytomegalovirus gene encoding IE1(491aa) is replication defective due to a failure in autoregulation. Proc. Natl. Acad. Sci. U. S. A. 93:11321–11326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ahn JH, Hayward GS. 1997. The major immediate-early proteins IE1 and IE2 of human cytomegalovirus colocalize with and disrupt PML-associated nuclear bodies at very early times in infected permissive cells. J. Virol. 71:4599–4613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Korioth F, Maul GG, Plachter B, Stamminger T, Frey J. 1996. The nuclear domain 10 (ND10) is disrupted by the human cytomegalovirus gene product IE1. Exp. Cell Res. 229:155–158 [DOI] [PubMed] [Google Scholar]
- 11.Wilkinson GW, Kelly C, Sinclair JH, Rickards C. 1998. Disruption of PML-associated nuclear bodies mediated by the human cytomegalovirus major immediate early gene product. J. Gen. Virol. 79:1233–1245 [DOI] [PubMed] [Google Scholar]
- 12.Lafemina RL, Pizzorno MC, Mosca JD, Hayward GS. 1989. Expression of the acidic nuclear immediate-early protein (IE1) of human cytomegalovirus in stable cell lines and its preferential association with metaphase chromosomes. Virology 172:584–600 [DOI] [PubMed] [Google Scholar]
- 13.Huh YH, Kim YE, Kim ET, Park JJ, Song MJ, Zhu H, Hayward GS, Ahn JH. 2008. Binding STAT2 by the acidic domain of human cytomegalovirus IE1 promotes viral growth and is negatively regulated by SUMO. J. Virol. 82:10444–10454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Krauss S, Kaps J, Czech N, Paulus C, Nevels M. 2009. Physical requirements and functional consequences of complex formation between the cytomegalovirus IE1 protein and human STAT2. J. Virol. 83:12854–12870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Paulus C, Krauss S, Nevels M. 2006. A human cytomegalovirus antagonist of type I IFN-dependent signal transducer and activator of transcription signaling. Proc. Natl. Acad. Sci. U. S. A. 103:3840–3845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jaworska J, Gravel A, Flamand L. 2010. Divergent susceptibilities of human herpesvirus 6 variants to type I interferons. Proc. Natl. Acad. Sci. U. S. A. 107:8369–8374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Knoblach T, Grandel B, Seiler J, Nevels M, Paulus C. 2011. Human cytomegalovirus IE1 protein elicits a type II interferon-like host cell response that depends on activated STAT1 but not interferon-gamma. PLoS Pathog. 7(4):e1002016. 10.1371/journal.ppat.1002016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yu H, Pardoll D, Jove R. 2009. STATs in cancer inflammation and immunity: a leading role for STAT3. Nat. Rev. Cancer 9:798–809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Maritano D, Sugrue ML, Tininini S, Dewilde S, Strobl B, Fu X, Murray-Tait V, Chiarle R, Poli V. 2004. The STAT3 isoforms alpha and beta have unique and specific functions. Nat. Immunol. 5:401–409 [DOI] [PubMed] [Google Scholar]
- 20.Yoo JY, Huso DL, Nathans D, Desiderio S. 2002. Specific ablation of Stat3β distorts the pattern of Stat3-responsive gene expression and impairs recovery from endotoxic shock. Cell 108:331–344 [DOI] [PubMed] [Google Scholar]
- 21.Zammarchi F, de Stanchina E, Bournazou E, Supakorndej T, Martires K, Riedel E, Corben AD, Bromberg JF, Cartegni L. 2011. Antitumorigenic potential of STAT3 alternative splicing modulation. Proc. Natl. Acad. Sci. U. S. A. 108:17779–17784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Aggarwal BB, Kunnumakkara AB, Harikumar KB, Gupta SR, Tharakan ST, Koca C, Dey S, Sung B. 2009. Signal transducer and activator of transcription-3, inflammation, and cancer: how intimate is the relationship? Ann. N. Y. Acad. Sci. 1171:59–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cimica V, Chen HC, Iyer JK, Reich NC. 2011. Dynamics of the STAT3 transcription factor: nuclear import dependent on Ran and importin-beta1. PLoS One 6(5):e20188. 10.1371/journal.pone.0020188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liu L, McBride KM, Reich NC. 2005. STAT3 nuclear import is independent of tyrosine phosphorylation and mediated by importin-α3. Proc. Natl. Acad. Sci. U. S. A. 102:8150–8155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wen Z, Darnell JE., Jr 1997. Mapping of Stat3 serine phosphorylation to a single residue (727) and evidence that serine phosphorylation has no influence on DNA binding of Stat1 and Stat3. Nucleic Acids Res. 25:2062–2067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wen Z, Zhong Z, Darnell JE., Jr 1995. Maximal activation of transcription by Stat1 and Stat3 requires both tyrosine and serine phosphorylation. Cell 82:241–250 [DOI] [PubMed] [Google Scholar]
- 27.Reich NC, Liu L. 2006. Tracking STAT nuclear traffic. Nat. Rev. Immunol. 6:602–612 [DOI] [PubMed] [Google Scholar]
- 28.Timofeeva OA, Chasovskikh S, Lonskaya I, Tarasova NI, Khavrutskii L, Tarasov SG, Zhang X, Korostyshevskiy VR, Cheema A, Zhang L, Dakshanamurthy S, Brown ML, Dritschilo A. 2012. Mechanisms of unphosphorylated STAT3 transcription factor binding to DNA. J. Biol. Chem. 287:14192–14200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yang J, Chatterjee-Kishore M, Staugaitis SM, Nguyen H, Schlessinger K, Levy DE, Stark GR. 2005. Novel roles of unphosphorylated STAT3 in oncogenesis and transcriptional regulation. Cancer Res. 65:939–947 [PubMed] [Google Scholar]
- 30.Yang J, Liao X, Agarwal MK, Barnes L, Auron PE, Stark GR. 2007. Unphosphorylated STAT3 accumulates in response to IL-6 and activates transcription by binding to NFκB. Genes Dev. 21:1396–1408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yu D, Smith GA, Enquist LW, Shenk T. 2002. Construction of a self-excisable bacterial artificial chromosome containing the human cytomegalovirus genome and mutagenesis of the diploid TRL/IRL13 gene. J. Virol. 76:2316–2328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Reitsma JM, Savaryn JP, Faust K, Sato H, Halligan BD, Terhune SS. 2011. Antiviral inhibition targeting the HCMV kinase pUL97 requires pUL27-dependent degradation of Tip60 acetyltransferase and cell-cycle arrest. Cell Host Microbe 9:103–114 [DOI] [PubMed] [Google Scholar]
- 33.Marchini A, Liu H, Zhu H. 2001. Human cytomegalovirus with IE-2 (UL122) deleted fails to express early lytic genes. J. Virol. 75:1870–1878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sinzger C, Hahn G, Digel M, Katona R, Sampaio KL, Messerle M, Hengel H, Koszinowski U, Brune W, Adler B. 2008. Cloning and sequencing of a highly productive, endotheliotropic virus strain derived from human cytomegalovirus TB40/E. J. Gen. Virol. 89:359–368 [DOI] [PubMed] [Google Scholar]
- 35.Mitchell DP, Savaryn JP, Moorman NJ, Shenk T, Terhune SS. 2009. Human cytomegalovirus UL28 and UL29 open reading frames encode a spliced mRNA and stimulate accumulation of immediate-early RNAs. J. Virol. 83:10187–10197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Terhune SS, Moorman NJ, Cristea IM, Savaryn JP, Cuevas-Bennett C, Rout MP, Chait BT, Shenk T. 2010. Human cytomegalovirus UL29/28 protein interacts with components of the NuRD complex which promote accumulation of immediate-early RNA. PLoS Pathog. 6(6):e1000965. 10.1371/journal.ppat.1000965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yu H, Kortylewski M, Pardoll D. 2007. Crosstalk between cancer and immune cells: role of STAT3 in the tumour microenvironment. Nat. Rev. Immunol. 7:41–51 [DOI] [PubMed] [Google Scholar]
- 38.Siddiquee K, Zhang S, Guida WC, Blaskovich MA, Greedy B, Lawrence HR, Yip ML, Jove R, McLaughlin MM, Lawrence NJ, Sebti SM, Turkson J. 2007. Selective chemical probe inhibitor of Stat3, identified through structure-based virtual screening, induces antitumor activity. Proc. Natl. Acad. Sci. U. S. A. 104:7391–7396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Alexandrow MG, Song LJ, Altiok S, Gray J, Haura EB, Kumar NB. 2012. Curcumin: a novel Stat3 pathway inhibitor for chemoprevention of lung cancer. Eur. J. Cancer Prev. 21:407–412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bill MA, Fuchs JR, Li C, Yui J, Bakan C, Benson DM, Jr, Schwartz EB, Abdelhamid D, Lin J, Hoyt DG, Fossey SL, Young GS, Carson WE, III, Li PK, Lesinski GB. 2010. The small molecule curcumin analog FLLL32 induces apoptosis in melanoma cells via STAT3 inhibition and retains the cellular response to cytokines with anti-tumor activity. Mol. Cancer 9:165. 10.1186/1476-4598-9-165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fossey SL, Bear MD, Lin J, Li C, Schwartz EB, Li PK, Fuchs JR, Fenger J, Kisseberth WC, London CA. 2011. The novel curcumin analog FLLL32 decreases STAT3 DNA binding activity and expression, and induces apoptosis in osteosarcoma cell lines. BMC Cancer 11:112. 10.1186/1471-2407-11-112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kumar A, Bora U. 2012. Molecular docking studies on inhibition of Stat3 dimerization by curcumin natural derivatives and its conjugates with amino acids. Bioinformation. 8:988–993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schust J, Sperl B, Hollis A, Mayer TU, Berg T. 2006. Stattic: a small-molecule inhibitor of STAT3 activation and dimerization. Chem. Biol. 13:1235–1242 [DOI] [PubMed] [Google Scholar]
- 44.Ferrajoli A, Faderl S, Van Q, Koch P, Harris D, Liu Z, Hazan-Halevy I, Wang Y, Kantarjian HM, Priebe W, Estrov Z. 2007. WP1066 disrupts Janus kinase-2 and induces caspase-dependent apoptosis in acute myelogenous leukemia cells. Cancer Res. 67:11291–11299 [DOI] [PubMed] [Google Scholar]
- 45.Fehr AR, Gualberto NC, Savaryn JP, Terhune SS, Yu D. 2012. Proteasome-dependent disruption of the E3 ubiquitin ligase anaphase-promoting complex by HCMV protein pUL21a. PLoS Pathog. 8(7):e1002789. 10.1371/journal.ppat.1002789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Stenberg RM, Depto AS, Fortney J, Nelson JA. 1989. Regulated expression of early and late RNAs and proteins from the human cytomegalovirus immediate-early gene region. J. Virol. 63:2699–2708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tenney DJ, Colberg-Poley AM. 1991. Human cytomegalovirus UL36-38 and US3 immediate-early genes: temporally regulated expression of nuclear, cytoplasmic, and polysome-associated transcripts during infection. J. Virol. 65:6724–6734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Heider JA, Bresnahan WA, Shenk TE. 2002. Construction of a rationally designed human cytomegalovirus variant encoding a temperature-sensitive immediate-early 2 protein. Proc. Natl. Acad. Sci. U. S. A. 99:3141–3146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Loregian A, Appleton BA, Hogle JM, Coen DM. 2004. Residues of human cytomegalovirus DNA polymerase catalytic subunit UL54 that are necessary and sufficient for interaction with the accessory protein UL44. J. Virol. 78:158–167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Loregian A, Appleton BA, Hogle JM, Coen DM. 2004. Specific residues in the connector loop of the human cytomegalovirus DNA polymerase accessory protein UL44 are crucial for interaction with the UL54 catalytic subunit. J. Virol. 78:9084–9092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sanders RL, Clark CL, Morello CS, Spector DH. 2008. Development of cell lines that provide tightly controlled temporal translation of the human cytomegalovirus IE2 proteins for complementation and functional analyses of growth-impaired and nonviable IE2 mutant viruses. J. Virol. 82:7059–7077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Stasiak PC, Mocarski ES. 1992. Transactivation of the cytomegalovirus ICP36 gene promoter requires the alpha gene product TRS1 in addition to IE1 and IE2. J. Virol. 66:1050–1058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Weiland KL, Oien NL, Homa F, Wathen MW. 1994. Functional analysis of human cytomegalovirus polymerase accessory protein. Virus Res. 34:191–206 [DOI] [PubMed] [Google Scholar]
- 54.Gires O, Kohlhuber F, Kilger E, Baumann M, Kieser A, Kaiser C, Zeidler R, Scheffer B, Ueffing M, Hammerschmidt W. 1999. Latent membrane protein 1 of Epstein-Barr virus interacts with JAK3 and activates STAT proteins. EMBO J. 18:3064–3073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wang Z, Luo F, Li L, Yang L, Hu D, Ma X, Lu Z, Sun L, Cao Y. 2010. STAT3 activation induced by Epstein-Barr virus latent membrane protein1 causes vascular endothelial growth factor expression and cellular invasiveness via JAK3 And ERK signaling. Eur. J. Cancer 46:2996–3006 [DOI] [PubMed] [Google Scholar]
- 56.Punjabi AS, Carroll PA, Chen L, Lagunoff M. 2007. Persistent activation of STAT3 by latent Kaposi's sarcoma-associated herpesvirus infection of endothelial cells. J. Virol. 81:2449–2458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chung YH, Cho NH, Garcia MI, Lee SH, Feng P, Jung JU. 2004. Activation of Stat3 transcription factor by herpesvirus saimiri STP-A oncoprotein. J. Virol. 78:6489–6497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sen N, Che X, Rajamani J, Zerboni L, Sung P, Ptacek J, Arvin AM. 2012. Signal transducer and activator of transcription 3 (STAT3) and survivin induction by varicella-zoster virus promote replication and skin pathogenesis. Proc. Natl. Acad. Sci. U. S. A. 109:600–605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ulane CM, Rodriguez JJ, Parisien JP, Horvath CM. 2003. STAT3 ubiquitylation and degradation by mumps virus suppress cytokine and oncogene signaling. J. Virol. 77:6385–6393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Valmas C, Grosch MN, Schumann M, Olejnik J, Martinez O, Best SM, Krahling V, Basler CF, Muhlberger E. 2010. Marburg virus evades interferon responses by a mechanism distinct from Ebola virus. PLoS Pathog. 6(1):e1000721. 10.1371/journal.ppat.1000721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Munch K, Messerle M, Plachter B, Koszinowski UH. 1992. An acidic region of the 89K murine cytomegalovirus immediate early protein interacts with DNA. J. Gen. Virol. 73:499–506 [DOI] [PubMed] [Google Scholar]
- 62.Slinger E, Maussang D, Schreiber A, Siderius M, Rahbar A, Fraile-Ramos A, Lira SA, Soderberg-Naucler C, Smit MJ. 2010. HCMV-encoded chemokine receptor US28 mediates proliferative signaling through the IL-6-STAT3 axis. Sci. Signal. 3:ra58. 10.1126/scisignal.2001180 [DOI] [PubMed] [Google Scholar]
- 63.Dumortier J, Streblow DN, Moses AV, Jacobs JM, Kreklywich CN, Camp D, Smith RD, Orloff SL, Nelson JA. 2008. Human cytomegalovirus secretome contains factors that induce angiogenesis and wound healing. J. Virol. 82:6524–6535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Le VT, Trilling M, Wilborn M, Hengel H, Zimmermann A. 2008. Human cytomegalovirus interferes with signal transducer and activator of transcription (STAT) 2 protein stability and tyrosine phosphorylation. J. Gen. Virol. 89:2416–2426 [DOI] [PubMed] [Google Scholar]
- 65.Colberg-Poley AM. 1996. Functional roles of immediate early proteins encoded by the human cytomegalovirus UL36-38, UL115-119, TRS1/IRS1 and US3 loci. Intervirology 39:350–360 [DOI] [PubMed] [Google Scholar]
- 66.Alvarez-Castelao B, Martin-Guerrero I, Garcia-Orad A, Castano JG. 2009. Cytomegalovirus promoter up-regulation is the major cause of increased protein levels of unstable reporter proteins after treatment of living cells with proteasome inhibitors. J. Biol. Chem. 284:28253–28262 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 67.Fehr AR, Yu D. 2011. Human cytomegalovirus early protein pUL21a promotes efficient viral DNA synthesis and the late accumulation of immediate-early transcripts. J. Virol. 85:663–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Marshall EE, Bierle CJ, Brune W, Geballe AP. 2009. Essential role for either TRS1 or IRS1 in human cytomegalovirus replication. J. Virol. 83:4112–4120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Nevels M, Brune W, Shenk T. 2004. SUMOylation of the human cytomegalovirus 72-kilodalton IE1 protein facilitates expression of the 86-kilodalton IE2 protein and promotes viral replication. J. Virol. 78:7803–7812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Tran K, Kamil JP, Coen DM, Spector DH. 2010. Inactivation and disassembly of the anaphase-promoting complex during human cytomegalovirus infection is associated with degradation of the APC5 and APC4 subunits and does not require UL97-mediated phosphorylation of Cdh1. J. Virol. 84:10832–10843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Johnston PA, Grandis JR. 2011. STAT3 signaling: anticancer strategies and challenges. Mol. Interv. 11:18–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ghosh N, Chaki R, Mandal V, Mandal SC. 2010. COX-2 as a target for cancer chemotherapy. Pharmacol. Rep. 62:233–244 [DOI] [PubMed] [Google Scholar]
- 73.Liu DB, Hu GY, Long GX, Qiu H, Mei Q, Hu GQ. 2012. Celecoxib induces apoptosis and cell-cycle arrest in nasopharyngeal carcinoma cell lines via inhibition of STAT3 phosphorylation. Acta Pharmacol. Sin. 33:682–690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Yang F, Van Meter TE, Buettner R, Hedvat M, Liang W, Kowolik CM, Mepani N, Mirosevich J, Nam S, Chen MY, Tye G, Kirschbaum M, Jove R. 2008. Sorafenib inhibits signal transducer and activator of transcription 3 signaling associated with growth arrest and apoptosis of medulloblastomas. Mol. Cancer Ther. 7:3519–3526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Baryawno N, Rahbar A, Wolmer-Solberg N, Taher C, Odeberg J, Darabi A, Khan Z, Sveinbjornsson B, FuskevAg OM, Segerstrom L, Nordenskjold M, Siesjo P, Kogner P, Johnsen JI, Soderberg-Naucler C. 2011. Detection of human cytomegalovirus in medulloblastomas reveals a potential therapeutic target. J. Clin. Invest. 121:4043–4055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Michaelis M, Paulus C, Loschmann N, Dauth S, Stange E, Doerr HW, Nevels M, Cinatl J., Jr 2011. The multi-targeted kinase inhibitor sorafenib inhibits human cytomegalovirus replication. Cell. Mol. Life Sci. 68:1079–1090 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.