

Abstract
The A19L open reading frame of vaccinia virus encodes a 9-kDa protein that is conserved in all sequenced chordopoxviruses, yet until now it has not been specifically characterized in any species. We appended an epitope tag after the start codon of the A19L open reading frame without compromising infectivity. The protein was synthesized after viral DNA replication and was phosphorylated independently of the vaccinia virus F10 kinase. The A19 protein was present in purified virions and was largely resistant to nonionic detergent extraction, suggesting a location within the core. A conditional lethal mutant virus was constructed by placing the A19 open reading frame under the control of the Escherichia coli lac repressor system. A19 synthesis and infectious virus formation were dependent on inducer. In the absence of inducer, virion morphogenesis was interrupted, and spherical dense particles that had greatly reduced amounts of the D13 scaffold accumulated in place of barrel-shaped mature virions. The infectivity of purified A19-deficient particles was more than 2 log units less than that of A19-containing virions. Nevertheless, the A19-deficient particles contained DNA, and except for the absence of A19 and decreased core protein processing, they appeared to have a similar protein composition as A19-containing virions. Thus, the A19 protein participates in the maturation of immature vaccinia virus virions to infectious particles.
INTRODUCTION
The Poxviridae family is a family of complex viruses that infect invertebrate or vertebrate hosts (1). Despite their linear, double-stranded DNA genomes, which range from 130 to 300 kbp, all poxviruses replicate entirely in the cytoplasm. Vaccinia virus (VACV), the best-characterized member of the family, has a genome of nearly 200 kbp encoding approximately 200 proteins. Of these proteins, 90 are conserved in all Chordopoxvirinae (2, 3). The conserved genes have been studied to various degrees, and most are thought to be essential for virus replication. Their functions include transcription, genome replication, virion assembly, morphogenesis, and virus entry. Although the A19L open reading frame (ORF) is conserved in all chordopoxviruses, it has not been specifically analyzed in any species. However, there have been a few references to the A19 protein as part of large screening studies. For example, a VACV genome-wide yeast two-hybrid analysis revealed an interaction of the A12 virion protein with A19 (4). One mass spectrometry study suggested that the A19 protein is a minor component of purified virions (5), which contain about 80 proteins, although A19 was not detected in two other such studies (6, 7). A genome-wide analysis indicated that the A19 ORF is transcribed at the intermediate and late stages of VACV replication (8). In view of its conservation, an important role for the A19 protein in poxvirus replication is predicted. We now describe combined genetic, biochemical, and microscopic studies demonstrating that the A19 protein is required for a late step in virion morphogenesis.
VACV morphogenesis is a complex process that remains to be fully elucidated (9). The first recognizable step is the formation of a crescent-shaped membrane structure stabilized by trimers of the D13 protein, which forms a honeycomb lattice on the cytoplasmic side of the membrane (10, 11). The crescents engulf core proteins and enlarge to become spherical immature virions (IVs) containing the DNA genome. The subsequent transition to barrel-shaped infectious mature virions (MVs) involves the disruption of the D13 scaffold, proteolytic processing of certain membrane and core proteins, and the formation of intramolecular disulfide bonds (12–14). Here we show that spherical, electron-dense particles with little or no infectivity are formed when expression of the A19L ORF is repressed, indicating a role for this protein in the transition from IVs to MVs.
MATERIALS AND METHODS
Cells and viruses.
African green monkey kidney epithelial BS-C-1 cells (ATCC CCL-26) were grown in minimum essential medium with Earle's salts supplemented with 10% fetal bovine serum, 100 units of penicillin, and 100 μg of streptomycin per ml (Quality Biologicals, Gaithersburg, MD). The recombinant viruses vT7LacOI (15) and vF10-V5i (16) have been described. Virus particles were purified by centrifugation through a 36% sucrose cushion and banding on 25 to 40% sucrose density gradient (17).
Construction of recombinant viruses.
A recombinant virus expressing the A19 protein with the fused FLAG- and streptavidin-binding (FS) tandem affinity peptide tag at the N terminus (vFS-A19) was constructed by homologous recombination as follows. The FS-A19 ORF under the control of the natural promoter was inserted into the endogenous locus along with the enhanced green fluorescent protein (GFP) ORF regulated by the VACV P11 late promoter to enable fluorescent selection of recombinant virus plaques. A modified two-step version of the pVOTE system (18) was used to construct an A19L-inducible virus. The FS-A19 ORF under the control of a T7 RNA polymerase promoter and encephalomyocarditis virus cap-independent translation enhancer element was cloned into the pVOTE-DsRED plasmid, which was modified from pVOTE.1 by replacing the Escherichia coli gpt gene with DsRED. In the first recombination event, T7-FS-A19 DNA was inserted into parental vT7LacOI virus genome at the A56L locus. The intermediate virus vT7LacOI-FS-A19i-DsRED was plaque purified, followed by replacement of the original A19 gene with the P11-GFP gene. The final inducible A19 virus, named vFS-A19i for short, was clonally purified, and the relevant regions were confirmed by DNA sequencing. The vFS-A11 virus, used as a control in this study, expressed the A11 ORF with the FS tandem affinity tag and was also constructed by homologous recombination.
Western blotting.
Proteins were resolved by electrophoresis in 4 to 12% NuPAGE gels and blotted onto a nitrocellulose membrane (iBlot; Life Technologies). The nitrocellulose membrane was blocked with 5% nonfat milk in phosphate-buffered saline containing 0.05% Tween 20 and then incubated with a primary antibody for 1 h at room temperature or overnight at 4°C. Unbound antibodies were removed by washing twice with phosphate-buffered saline containing Tween 20 followed by phosphate-buffered saline without detergent. Secondary donkey anti-mouse IRDye 800 and donkey anti-rabbit IRDye 680 antibodies were used at 1:10,000 dilution and analyzed with a LI-COR Odyssey infrared imager (LI-COR Biosciences, Lincoln, NE). Antibodies used for Western blotting included the following: mouse monoclonal antibodies (MAbs) M2 to FLAG (Sigma-Aldrich, St. Louis, MO) and to glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (Covance, Princeton, NJ) and rabbit polyclonal antibodies against A3 (R. Doms and B. Moss, unpublished results), A17 (19), L1 (20), F9 (21), D13 (22), and I7 (23).
Pulse-chase analysis.
BS-C-1 cells were infected with 3 PFU of vT7LacOI or vFS-A19i virus per cell and incubated in regular medium at 37°C. Eight hours postinfection, the medium was replaced with cysteine- and methionine-free medium for 30 min at 37°C and pulse-labeled for 15 min with 100 μCi per ml of a mixture of [35S]methionine and [35S]cysteine (PerkinElmer, Waltham, MA). The cells were harvested immediately after pulse-labeling or washed and chased for an additional 16 h with unlabeled medium and harvested. The cells were lysed directly in 1× NuPAGE sample buffer containing 1× reducing agent (Life Technologies, Grand Island, NY) by heating at 100°C for 15 min. The proteins were separated in 4 to 12% Novex NuPAGE acrylamide gels (Life Technologies), dried, and imaged by exposure of phosphorimager film (Typhoon scanner; GE Healthcare, Waukesha, WI).
Protein phosphorylation.
BS-C-1 cells were infected with 3 PFU of virus per cell and incubated for 7 h at 37°C in regular medium. The cells were then washed three times and then incubated in phosphate-free medium containing 100 μCi of 32Pi per ml. At 24 h after infection, the cells were harvested, lysed in buffer containing 50 mM Tris (pH 8.0), 200 mM NaCl, 1% Triton X-100, and 1 mM sodium orthovanadate, a general phosphatase inhibitor, for 30 min at 4°C. Soluble extracts obtained by low-speed centrifugation were allowed to bind to streptavidin-agarose beads (Millipore, Billerica, MA) for 3 h at 4°C. The beads were washed three times with the same buffer, and bound proteins were eluted with buffer containing d-biotin (1 mg/ml). The proteins were resolved by electrophoresis in 4 to 12% NuPAGE gels and either dried and exposed to phosphorimager film or blotted onto a nitrocellulose membrane (iBlot; Life Technologies) for Western blotting as described above.
Electron microscopy.
BS-C-1 cells in 60-mm-diameter dishes were infected with 3 PFU of vFS-A19i virus per cell in the presence or absence of 100 μM isopropyl-β-d-thiogalactopyranoside (IPTG). After 9 or 18 h, the cells were fixed with 2% glutaraldehyde and embedded in EmBed-182 resin (Electron Microscopy Sciences, Hatfield, PA), or cryosections were immunogold labeled as described previously (24). The sections were viewed with a FEI Tecnai Spirit transmission electron microscope (FEI, Hillboro, OR).
Virus counter.
Purified virus samples were analyzed using the virus counter 2100 (Virocyt, Denver, CO) per the manufacturer's instructions. Briefly, purified virus samples were diluted in sample buffer at 1:20 dilution and subsequent 10-fold dilutions. The diluted sample (200 μl) was transferred to a Virocyt vial and incubated with 100 μl of Combo dye, which stains nucleic acids and protein, for 30 min at room temperature in the dark. Nucleic acid and protein peaks were recorded, and coincident peaks were defined as virus particles using the Virocyt software.
RESULTS
Expression and localization of the A19 protein.
The transcriptional program of A19 was classified as postreplicative intermediate or intermediate/late based on global screening of VACV promoters by a transfection protocol (8). To confirm protein expression during virus infection and affinity purify A19 in later experiments, we generated a recombinant VACV expressing A19 with an N-terminal fused FLAG- and streptavidin-binding (FS) epitope tag regulated by its natural promoter (Fig. 1A). The tagged virus formed normal-size plaques, indicating that the additional amino acids did not adversely affect virus replication. A protein of approximately the size predicted for the 14.6-kDa FS-A19 fusion protein was detected by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and Western blotting with anti-FLAG MAb between 6 and 9 h after infection with vFS-A19, coincident with synthesis of the late A3 protein, which appears as a doublet due to proteolytic processing (Fig. 1B). At 9 and 12 h, there appeared to be a minor band migrating slightly slower than the major FS-A19 band. Inhibition of A19 synthesis by the DNA replication inhibitor cytosine arabinoside (AraC) confirmed postreplicative expression (Fig. 1B).
Fig 1.
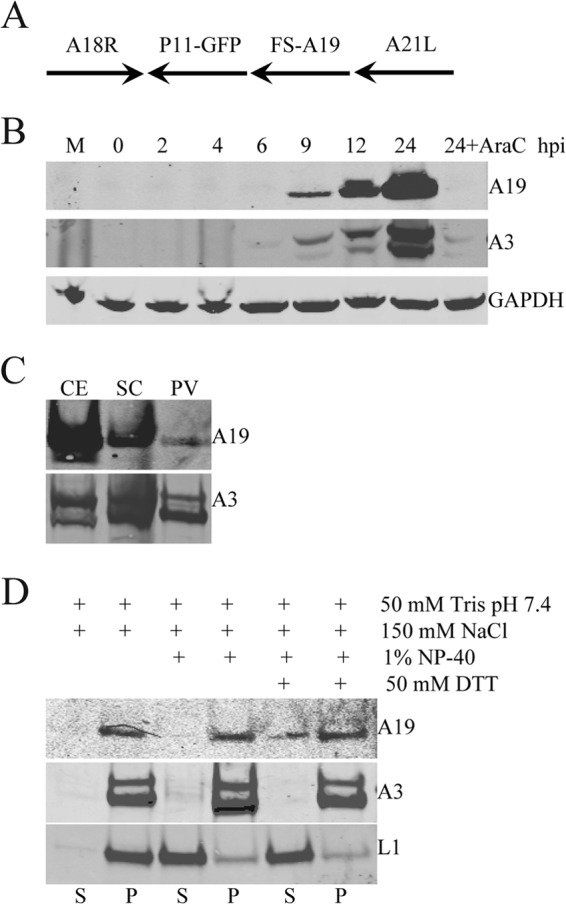
Expression and characterization of the A19 protein. (A) Schematic representation of the DNA construct used for generating recombinant vFS-A19. The FLAG- and streptavidin-binding peptide tag fused at the N terminus of the A19 ORF (FS) is indicated. The arrows denote the direction of transcription. (B) A19 expression. BS-C-1 cells were infected with 3 PFU of vFS-A19 per cell in the absence or presence of AraC for 24 h. Whole-cell lysates were analyzed by SDS-PAGE followed by transfer to a nitrocellulose membrane and Western blotting. Mouse MAbs were used to detect FLAG fused to A19 and GAPDH, and a rabbit polyclonal antibody to A3 was used to detect the latter protein. The time after infection (in hours postinfection [hpi]) is shown above the lanes. M, mock infected. (C) Virion association of A19. BS-C-1 cells were infected with vFS-A19, and virions were purified from cell extracts (CE) by centrifugation through a cushion of sucrose (SC) and sucrose density gradient centrifugation to yield purified virus particles (PV). Fractions from successive stages of purification were analyzed by SDS-PAGE and Western blotting as described above for panel B. (D) Detergent extraction of purified virions. Purified vFS-A19 virions were incubated with buffers containing (+) the components (DTT, dithiothreitol) indicated, and soluble (S) and pellet (P) fractions were analyzed by SDS-PAGE and Western blotting with antibodies to A19 (FLAG), the core protein A3, and the membrane protein L1.
The A19 protein was detected in purified virions (Fig. 1C), confirming results obtained by mass spectrometry (5). To evaluate the relative degree of A19 incorporation into virions, we compared the amounts of A19 and A3 (a major core protein) in cell extracts and two successive purification steps. The relative amount of A19 recovered in purified virions compared to cell extract was much less than for A3, suggesting that A19 is a less abundant component. The virion-associated A19 largely remained in the pellet fraction after extraction with nonionic detergent, suggesting an internal location, although some release occurred with detergent and reducing agent (Fig. 1D).
A19 is phosphorylated.
Several VACV proteins are phosphorylated by virus-encoded or cellular kinases, and A19 has multiple serine and threonine residues. Evidence that A19 is phosphorylated was obtained by incubating vFS-A19-infected cells with 32Pi. The cells were lysed and incubated with streptavidin beads in order to capture the A19 protein. Since A11 is known to be phosphorylated (25), vFS-A11-infected cells were labeled and analyzed in parallel as a positive control. Western blot analysis demonstrated binding of the two proteins to streptavidin beads (Fig. 2A). When the samples were analyzed by autoradiography, the presence of radioactively labeled bands of the expected sizes indicated that A19 and A11 were phosphorylated, although the latter was more intense (Fig. 2B).
Fig 2.
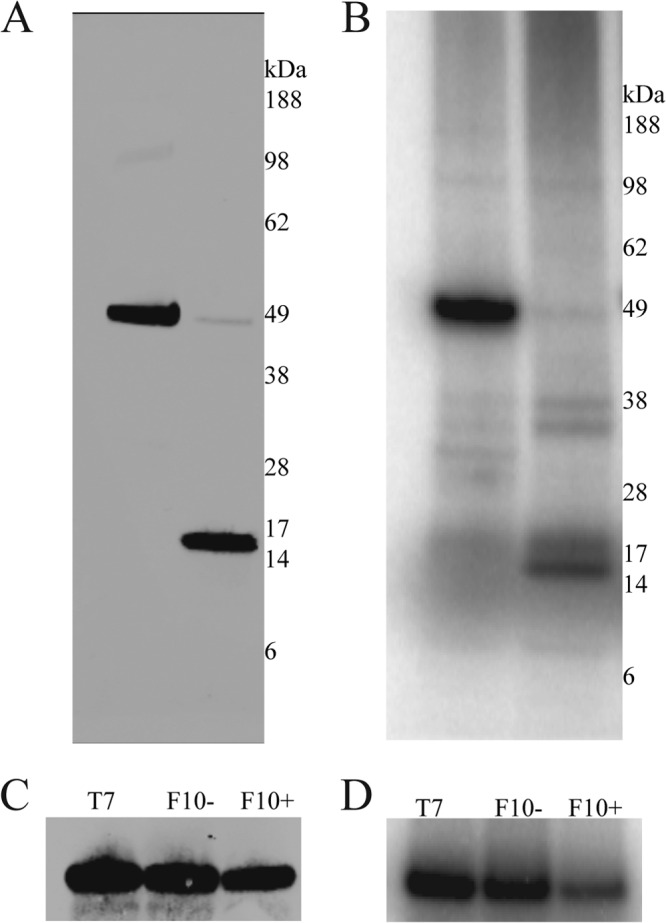
A19 phosphorylation. (A) Western blot analysis. BS-C-1 cells were infected with vFS-A11 and vFS-A19 in the presence of 100 μCi/ml 32Pi. After 18 h, the cells were lysed, and the soluble extract was bound to streptavidin-agarose beads. Bound proteins were eluted with biotin and analyzed by SDS-PAGE and Western blotting using anti-FLAG antibodies. The positions and masses (in kilodaltons) of marker proteins are shown to the right of the gel. (B) Same as panel A except that the gels were dried and exposed to a phosphorimager screen. (C and D) Repression of F10 kinase. BS-C-1 cells were infected with vT7LacOI (T7) or vF10-V5i in the absence (F10−) and presence (F10+) of IPTG. After 1 h of adsorption, the cells were transfected with an FS-A19-expressing plasmid for an additional 18 h. Cells were lysed, and FS-A19 was affinity purified using streptavidin-agarose beads. The A19 protein and phosphorylation were detected by Western blotting (C) and phosphorimager analysis (D), respectively.
An F10-inducible virus was employed to determine whether the VACV-encoded F10 kinase was responsible for A19 phosphorylation. In preliminary experiments, we confirmed the previously reported (26) IPTG dependence of F10 expression by this recombinant virus. Cells were infected with the inducible virus in the presence or absence of IPTG and transfected with an FS-A19 expression plasmid. Upon labeling with 32Pi, A19 was captured with streptavidin-agarose beads and analyzed by SDS-PAGE. The autoradiograph revealed that the phosphorylation of A19 was independent of F10 expression (Fig. 2C and D).
A19 is essential for VACV replication.
To determine whether A19 is required for replication, we modified the pVOTE system, which enables a gene to be placed under the stringent control of the Escherichia coli lac operator (18). The parent VACV for this system is vT7LacOI, which expresses the bacteriophage T7 RNA polymerase regulated by a VACV late promoter and the E. coli lac operator and constitutively expresses the E. coli lac repressor. The FS-A19 gene under the control of a T7 promoter and lac operator was inserted into a modified pVOTE vector that contained the DsRED ORF regulated by the VACV P11 late promoter flanked by segments of the A56R gene. The plasmid was transfected into cells that had been infected with vT7LacOI virus. Recombinant virus plaques were identified and isolated by screening for red fluorescence. In the next step, homologous recombination was used to replace the original A19L ORF with GFP regulated by the P11 promoter using a similar screening procedure (Fig. 3A). The final virus vFS-A19i, containing only an inducible copy of A19, was clonally purified in the presence of the inducer IPTG.
Fig 3.
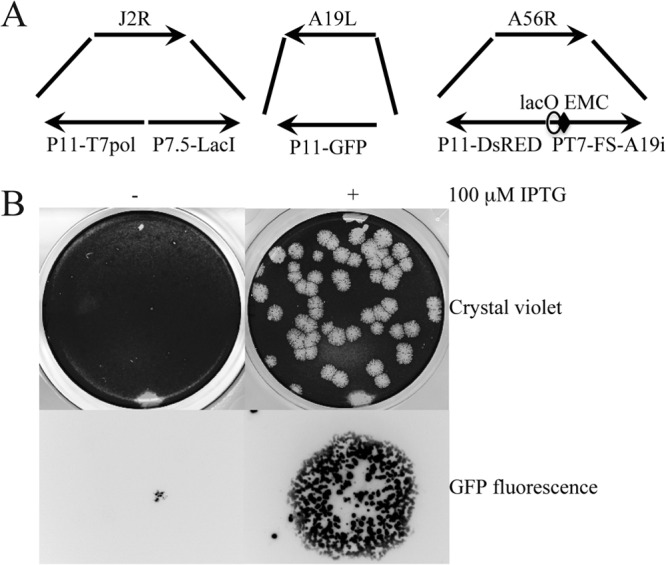
Construction of A19-inducible virus. (A) Schematic representations of portions of the VACV genome in vFS-A19i. Abbreviations: T7pol, bacteriophage T7 RNA polymerase ORF; LacI, E. coli lac repressor ORF; GFP, green fluorescent protein ORF; DsRED, red fluorescent protein ORF; P11, VACV late promoter; P7.5, VACV early/late promoter; PT7, bacteriophage T7 promoter; lacO, lac operator, EMC, encephalomyocarditis virus cap-independent translation enhancer element; FS, fused FLAG and streptavidin epitope tags. J2R, A19L, and A56R refer to the ORFs that were deleted or interrupted by insertion of DNA. The arrows show the direction of transcription. (B) Plaque formation of A19-inducible virus. BS-C-1 cells were infected with vFS-A19i in the absence (−) and presence (+) of 100 μM IPTG for 48 h. The plaques were visualized by staining with crystal violet or by GFP fluorescence. A single fluorescent plaque is shown at high magnification.
The requirement of A19 for VACV replication was ascertained by infecting BS-C-1 cells with vFS-A19i in the presence and absence of 100 μM IPTG. Plaques similar in size to the parental virus formed in the presence of the inducer, whereas plaques were pinpoint in size in the absence of inducer (Fig. 3B). A defect in plaque formation could be due to failure of virus replication or spread. To distinguish between these steps, virus yields were determined under synchronous infection conditions. BS-C-1 cells were infected with vFS-A19i in medium containing a range of IPTG concentrations, since in some cases excess inducer can have a negative effect. The expression of A19 was determined by Western blotting, and virus yields were measured by determining plaque numbers in BS-C-1 cells in the presence of 100 μM IPTG. A19 was undetectable in the absence of IPTG and increased between 20 and 80 μM (Fig. 4A). From 0 to 20 μM IPTG, the virus yield increased by nearly 2 log units and then plateaued with higher IPTG concentrations (Fig. 4A). Virus replication increased between 6 and 24 h after infection under one-step growth conditions in the presence of 100 μM IPTG (Fig. 4B). Following the eclipse period, only a slight increase in virus yield occurred in the absence of added IPTG (Fig. 4B). Thus, repressions of A19 synthesis and virus replication were IPTG dependent.
Fig 4.

Dependence of virus replication on A19 synthesis. (A) IPTG dependence. BS-C-1 cells were infected with FS-A19i in the presence of the indicated concentration of IPTG. Virus yields were determined after 24 h by titration on BS-C-1 cells in the presence of 100 μM IPTG (top). In a parallel experiment, A19 synthesis was determined by Western blotting with anti-FLAG antibodies (bottom). (B) One-step growth curve. BS-C-1 cells were infected with 3 PFU per cell of vFS-A19i in the absence or presence of 100 μM IPTG. Virus yields were determined as described above for panel A at the indicated times after infection. (C) trans-Complementation. BS-C-1 cells were infected with vFS-A19i in the absence of IPTG and transfected with plasmids expressing FS-H7 or FS-A19 from their natural promoters. Complementation efficiency was determined by plaque assay as described above for panel A. (D) Western blot analysis. Samples used in panel C were analyzed for protein by Western blotting with anti-FLAG antibody.
A trans-complementation experiment was carried out to confirm the specific role of A19 and rule out neighboring gene effects. Cells were infected with vFS-A19i in the absence of IPTG and transfected with a plasmid containing the FS-A19 ORF with its natural promoter or a control plasmid FS-H7, encoding a protein needed for viral membrane formation, with its natural promoter. An increase in virus yield was obtained with FS-A19, but it did not reach the level of the control with IPTG (Fig. 4C) as usual in such experiments. Transfection of the FS-H7 plasmid had no effect on virus yield (Fig. 4C), although FS-H7 was expressed (Fig. 4D).
Viral protein synthesis and processing.
Because of the shutdown of host protein synthesis, VACV proteins can be detected by radioactive amino acid pulse-labeling at 6 or more hours after infection. Pulse-labeling at 8 h after infection indicated that viral protein synthesis was similar in cells infected with vFS-A19i in the presence and absence of inducer (Fig. 5A). The labeled cells were resuspended in fresh medium, and incubation was continued to determine whether there was a defect in proteolytic processing of certain proteins, which can reveal a morphogenesis defect (12). For a control, cells were infected with the parental virus vT7LacOI in the absence or presence of rifampin, a drug known to inhibit morphogenesis at an early stage of viral membrane formation (27, 28), and also pulse-labeled. Cell lysates, obtained following the pulse and chase, were analyzed by SDS-PAGE and autoradiography. Inspection of the autoradiographs revealed several alterations in the intensity of bands following the chase, the most noticeable change indicated by the arrow in Fig. 5A corresponded to the processed form of A3. The appearance of this band was largely inhibited in the presence of rifampin and partially reduced in the absence of IPTG compared to the presence of IPTG. Western blotting showed a relatively small effect of A19 repression on A3 processing, compared to the greater effect of rifampin (Fig. 5B). Unprocessed and processed forms of the A17 protein were detected in the presence and absence of IPTG (Fig. 5B). These data suggested that A19 might be involved in core morphogenesis.
Fig 5.
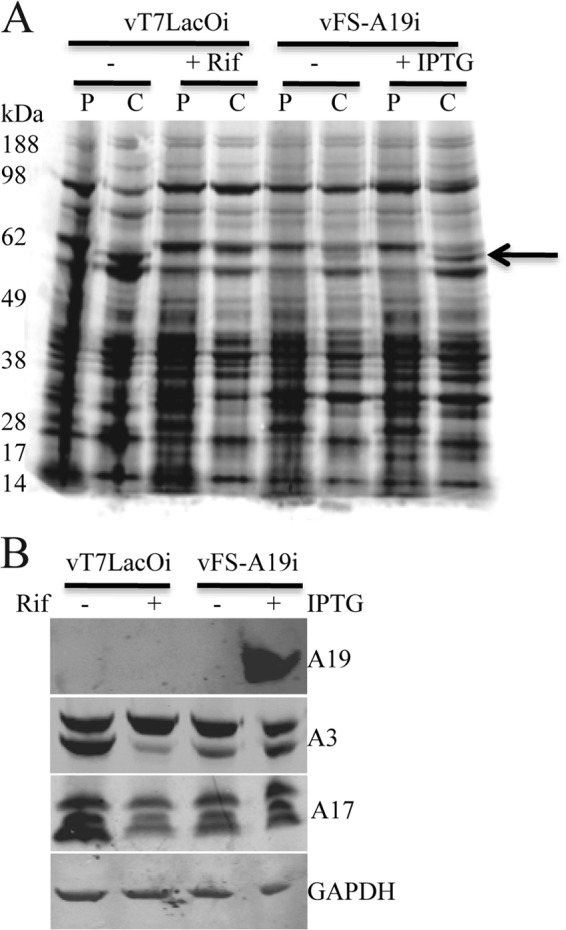
Viral protein synthesis and processing. (A) Pulse-chase analysis. BS-C-1 cells were infected with parental vT7LacOI in the absence (−) or presence (+) of 100 μg/ml rifampin (Rif) or vFS-A19i virus in the absence (−) or presence (+) of 100 μM IPTG for 8 h. The cells were pulse-labeled with [35S]methionine and [35S]cysteine for 15 min and either harvested immediately or chased for an additional 16 h. Proteins in the whole-cell lysates were resolved by SDS-PAGE, and the dried gels were exposed to phosphorimager screens. P and C denote pulse and chase, respectively. The arrow points to the processed A3 protein. (B) Western blot analysis. BS-C-1 cells were infected with vT7LacOI or vFS-A19i for 18 h. Lysates were analyzed by SDS-PAGE and Western blotting with antibodies to FLAG (A19), A3, A17, and GAPDH.
A19 is required for the transition of IVs to infectious MVs.
To determine whether morphogenesis is dependent on A19 expression, vFS-A19i-infected cells were analyzed by transmission electron microscopy. Low- and high-magnification images are shown in Fig. 6. Cells infected with vFS-A19i for 9 h in the presence of IPTG had all morphological forms that are normally observed with wild-type virus (Fig. 6A). Cells infected in the absence of IPTG had crescent-shaped membranes and IVs. Although nucleoids could be discerned in some IVs, in about half of the cases they appeared abnormal looking and were surrounded by low electron density. Most striking, however, was the absence of the barrel-shaped MVs and the presence of numerous dense spherical particles (Fig. 6B). The failure to form fully mature particles in the absence of IPTG was not merely due to a delay since the differences between the characteristics seen in the presence and absence of IPTG persisted even when samples obtained at 18 h after infection were analyzed (Fig. 6C and D). Occasional dense spherical particles were found wrapped in membranes and exocytosed, similar to that of normal extracellular enveloped virions (EVs) (Fig. 6B, inset, and D). The numbers and types of different particles present at 9 and 18 h after infection in the presence and absence of IPTG were enumerated (Table 1).
Fig 6.

Electron micrographs of vFS-A19i-infected cells. (A to D) BS-C-1 cells were infected with vFS-A19i at 3 PFU per cell in the presence (A and C) or absence (B and D) of IPTG. After 9 h (A and B) or 18 h (C and D), the cells were fixed and embedded, and thin sections were prepared for transmission electron microscopy. Abbreviations: IV, immature virion; nu, nucleoid; MV, mature virion; WV, wrapped virion; EV, extracellular enveloped virion; D, dense spherical particle; c, crescent; DEV, dense EV-like particle. The inset in panel B shows examples of EV-like dense, spherical particles protruding from the cell surface. Magnification is indicated by the bar at the bottom left-hand corner of each panel.
Table 1.
Structures in the presence and absence of A19 proteina
| Virus structure | No. of viral structures at the indicated time after infection: |
|||
|---|---|---|---|---|
| 9 h |
18 h |
|||
| +IPTG | −IPTG | +IPTG | −IPTG | |
| Crescent-shaped membrane structure | 203 | 155 | 600 | 473 |
| Immature virion | 394 | 420 | 1,383 | 883 |
| Immature virion + nucleoid | 103 | 51 | 55 | 44 |
| Mature virion | 47 | 0 | 55 | 0 |
| Dense particle | 1 | 8 | 5 | 99 |
| Wrapped mature virion | 46 | 0 | 593 | 0 |
| Wrapped dense particle | 0 | 2 | 0 | 16 |
| Extracellular mature virion | 62 | 0 | 190 | 4 |
| Extracellular dense particle | 0 | 3 | 0 | 95 |
HeLa cells were infected with 3 PFU/cell of vA19i in the presence (+) or absence (−) of IPTG for 9 and 18 h. Samples were prepared for transmission electron microscopy, and a thin section of 50 cells of each sample was examined. The numbers and types of structures are indicated.
Removal of D13 from spherical, dense particles.
The spherical shape of IVs is maintained by a honeycomb lattice, comprised of D13 trimers (11, 29). Removal of the scaffold is coincident with processing of the A17 membrane protein by the I7 protease, preceding further steps in morphogenesis (14). Immunoelectron microscopy was carried out to determine whether the spherical, dense A19-deficient (A19−) particles retained D13. Cells infected with vFS-A19i in the presence or absence of IPTG were cryosectioned and probed with antibody to D13 followed by gold spheres conjugated to protein A. In the presence of IPTG, the gold spheres heavily decorated the IVs (Fig. 7A), but few grains were observed on MVs (Fig. 7B). Gold spheres also decorated the IVs formed in the absence of IPTG (Fig. 7C) but were largely absent from the dense, spherical particles (Fig. 7D). This result was consistent with processing of the A17 membrane protein and disruption of the D13 scaffold.
Fig 7.

Absence of D13 on dense spherical virus particles. (A to D) BS-C-1 cells were infected with 5 PFU of vFS-A19i per cell in the presence (A and B) or absence (C and D) of IPTG for 18 h. Cells were fixed, embedded, and stained with antibodies against D13 protein followed by protein A conjugated to gold spheres. Fields with IVs (A and C) and MVs (B) and dense spherical particles (D) are shown.
Comparison of A19+ and A19− virus particles.
Virus particles were purified from cells infected with vFS-A19i in the presence and absence of IPTG in order to compare their infectivity and protein content. The purified virus particles were characterized and quantified with a virus counter, which is a specialized dual-channel flow cytometer that scores the coincidence of particles containing DNA and protein (Table 2). Analysis of the data indicated that the two preparations had similar numbers of particles per milliliter containing both DNA and protein with similar average peak heights. However, when we compared the specific infectivities of A19− and A19-containing (A19+) virions, the former were more than 100-fold-less infectious (Table 3).
Table 2.
Data from virus counter
| Parametera | Value for parameter |
|
|---|---|---|
| A19− particles | A19+ particles | |
| NA event counts | 11,745 | 9,976 |
| Protein event counts | 8,428 | 6,651 |
| Simultaneous event counts | 8,380 | 6,611 |
| Avg peak ht NA | 2.79 | 3.06 |
| Avg peak ht protein | 1.77 | 1.67 |
| No. of virus particles/ml | 2.1 × 107 | 1.7 × 107 |
NA, nucleic acid.
Table 3.
Specific infectivity of virus particles
| Virus particle | No. of PFU/mla | No. of particles/PFUb |
|---|---|---|
| A19− | 4.37 × 107 | 2.75 × 104 |
| A19+ | 5.84 × 109 | 2.05 × 102 |
Determined by plaque assay.
Determined by optical density.
The A19− and A19+ virus particles were analyzed by SDS-PAGE. The protein patterns visualized by staining with Coomassie blue dye appeared similar (Fig. 8A). Samples from the cell lysate, sucrose cushion, and sucrose gradient centrifugation steps were further analyzed by Western blotting. Virus particles made in the absence of IPTG lacked A19, whereas those made in the presence of IPTG contained A19 as expected (Fig. 8B). We analyzed two additional core proteins, A3 and I7, and two membrane proteins, F9 and A17, as well as the scaffold protein D13. The major core protein A3 was present in virus particles made in the presence and absence of IPTG (Fig. 8B). Both the precursor (top) and cleaved (bottom) form of A3 were present in the A19+ and A19− virus particles, but the relative amounts of precursor were higher in the latter. The I7 protein, which is the protease responsible for A3 cleavage, was detected in both preparations (Fig. 8B). There were similar amounts of unprocessed and processed A17 associated with the A19+ and A19− virus particles (Fig. 8B). A17 is also cleaved by I7 but at an earlier stage of morphogenesis than A3. The F9 membrane protein, involved in cell entry, was also present in A19+ and A19− particles. Similar small amounts of residual D13, which forms a honeycomb lattice on the exterior of immature virions, were detected in the A19+ and A19− particle preparations (Fig. 8B). Thus, protein contents of A19− and A19+ virus particles appeared to be similar.
Fig 8.
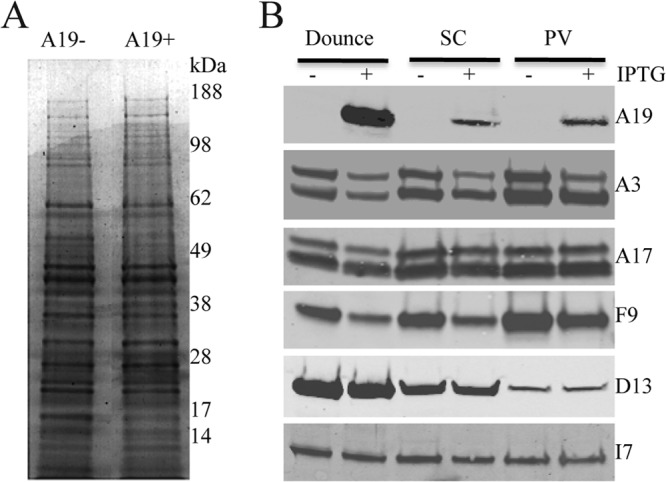
Analysis of proteins associated with purified virus particles. (A) SDS-PAGE. Equal amounts of sucrose gradient-purified A19+ and A19− virus particles were analyzed by SDS-PAGE and stained with Coomassie blue. The positions of molecular mass markers (in kilodaltons) are indicated to the right of the gel. (B) Western blots. Cells infected with vFS-A19i virus in the presence and absence of IPTG were lysed, and virus particles were purified by centrifugation through a sucrose cushion and sucrose gradient. Samples were analyzed by Western blotting with antibodies to A19 (FLAG), A3, A17, F9, D13, and I7. Abbreviations: SC, pellet after centrifugation through a sucrose cushion; PV, purified virus particles following sucrose gradient centrifugation.
DISCUSSION
Except for the A19 protein, the proteins encoded by previously annotated ORFs that are conserved in all chordopoxviruses (2) have been characterized to different degrees. Here, we demonstrated that the A19L ORF encodes a protein that is phosphorylated and expressed following viral DNA replication. A cellular enzyme probably carried out phosphorylation, since this modification was not dependent on the VACV-encoded F10 kinase. Although A19 was detected in purified virions, it was not enriched relative to the amount in the cytoplasm, as was the abundant core protein A3, suggesting that it is a minor component in agreement with a previous mass spectrometry analysis (5).
A requirement of A19 for VACV replication was determined by constructing a recombinant virus with the ORF under stringent E. coli lac operator control. The recombinant virus exhibited a conditional lethal phenotype: A19 synthesis, plaque formation, and production of infectious virus were dependent on the addition of inducer. In the absence of A19, the early stages of morphogenesis appeared normal, but electron-dense spherical particles accumulated instead of barrel-shaped virions. The A19− and A19+ particles were purified by sucrose gradient centrifugation and appeared to have similar ratios of DNA to protein, as determined using a specialized virus flow cytometer. Nevertheless, the specific activity of the A19− particles was more than 2 log units lower than that of A19+ virions. Despite the absence of A19, the virus particles appeared to have a full complement of the major proteins as judged by Coomassie blue staining of SDS-polyacrylamide gels and Western blotting of representative proteins. There was, however, partial impairment in processing of core proteins as found for other morphogenesis mutants and consistent with the electron microscopic images. Nevertheless, the I7 protease, which is responsible for processing of core proteins, was detected by Western blotting in both A19− and A19+ particles. Apparently a proper structure is needed for efficient I7 protease activity on core proteins.
A honeycomb lattice, comprised of D13 trimers, is thought to impose the spherical shape on IVs (10, 11, 30). This scaffold is normally lost during succeeding steps of maturation (22, 31, 32). The I7 protease is responsible for cleaving the A17 membrane protein, and repression of I7 synthesis leads to the formation of dense spherical particles that retain D13 (14). Although repression of A19 also resulted in the accumulation of electron-dense spherical particles, very little D13 was retained. This result was similar to that found for dense spherical particles that form when G1 protein is repressed (14). Thus, disruption of the D13 honeycomb lattice is necessary but not sufficient for the transition of IVs to barrel-shaped MVs. The observation that some aberrant particles were wrapped and exocytosed, which has also been found for other core protein mutants (33), suggests a normal complement of membrane proteins on their surface.
Electron-dense spherical particles, which resemble to different degrees those produced by repression of A19 synthesis, have also been shown to accumulate during infection with other conditional lethal mutants under nonpermissive conditions (Table 4). Unlike the A19 mutant, defective virions resulting from the absence of A32 (34), I6 (35), A22 (36), and G5 (37) lack DNA. In other cases, the aberrant particles caused by mutation or absence of D6 (38), A3 (33), A7 (39), I7 (23, 40, 41), G1 (42, 43), and A12 (44) contain the viral genome. The above proteins have a spectrum of roles with a common feature that they are incorporated into the virus core. An association between A12 and A19 was demonstrated by a genome-wide yeast two-hybrid screen (4). In the accompanying report (45), we describe interactions of A19 with additional proteins.
Table 4.
Mutants that produce spherical, dense staining virus particles
| ORF | Mutation(s)a | DNA packagedb | Role | Reference(s) |
|---|---|---|---|---|
| I7L | I, ts | + | Cysteine protease | 23, 40, 41 |
| G1L | I | + | Putative metalloprotease | 42, 43 |
| A12L | I | + | Not determined | 44 |
| G5R | D | − | Double-strand break repair | 37 |
| A32 | I | − | DNA packaging ATPase | 34 |
| A22R | I | − | HJ resolvase | 36 |
| I6L | ts | − | Telomere binding | 35 |
| D6R | I | + | Early transcription factor | 38 |
| A8R | I | + | Early transcription factor | 39 |
I, inducible; ts, temperature sensitive; D, deletion.
+, DNA packaged; −, DNA not packaged.
ACKNOWLEDGMENTS
We thank Catherine Cotter for help preparing cell cultures.
This research was supported by intramural funds of the National Institute of Allergy and Infectious Diseases, National Institutes of Health.
Footnotes
Published ahead of print 24 July 2013
REFERENCES
- 1.Moss B. 2007. Poxviridae: the viruses and their replication, p 2905–2946 In Knipe DM, Howley PM. (ed), Fields virology, vol 2 Lippincott Williams & Wilkins, Philadelphia, PA [Google Scholar]
- 2.Upton C, Slack S, Hunter AL, Ehlers A, Roper RL. 2003. Poxvirus orthologous clusters: toward defining the minimum essential poxvirus genome. J. Virol. 77:7590–7600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gubser C, Hue S, Kellam P, Smith GL. 2004. Poxvirus genomes: a phylogenetic analysis. J. Gen. Virol. 85:105–117 [DOI] [PubMed] [Google Scholar]
- 4.McCraith S, Holtzman T, Moss B, Fields S. 2000. Genome-wide analysis of vaccinia virus protein-protein interactions. Proc. Natl. Acad. Sci. U. S. A. 97:4879–4884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Resch W, Hixson KK, Moore RJ, Lipton MS, Moss B. 2007. Protein composition of the vaccinia virus mature virion. Virology 358:233–247 [DOI] [PubMed] [Google Scholar]
- 6.Yoder JD, Chen TS, Gagnier CR, Vemulapalli S, Maier CS, Hruby DE. 2006. Pox proteomics: mass spectrometry analysis and identification of vaccinia virion proteins. Virol. J. 3:10. 10.1186/1743-422X-3-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chung CS, Chen CH, Ho MY, Huang CY, Liao CL, Chang W. 2006. Vaccinia virus proteome: identification of proteins in vaccinia virus intracellular mature virion particles. J. Virol. 80:2127–2140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yang Z, Reynolds SE, Martens CA, Bruno DP, Porcella SF, Moss B. 2011. Expression profiling of the intermediate and late stages of poxvirus replication. J. Virol. 85:9899–9908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Condit RC. 2007. Vaccinia, Inc.—probing the functional substructure of poxviral replication factories. Cell Host Microbe 2:205–207 [DOI] [PubMed] [Google Scholar]
- 10.Heuser J. 2005. Deep-etch EM reveals that the early poxvirus envelope is a single membrane bilayer stabilized by a geodetic “honeycomb” surface coat. J. Cell Biol. 169:269–283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Szajner P, Weisberg AS, Lebowitz J, Heuser J, Moss B. 2005. External scaffold of spherical immature poxvirus particles is made of protein trimers, forming a honeycomb lattice. J. Cell Biol. 170:971–981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Katz E, Moss B. 1970. Formation of a vaccinia virus structural polypeptide from a higher molecular weight precursor: inhibition by rifampicin. Proc. Natl. Acad. Sci. U. S. A. 6:677–684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Senkevich TG, White CL, Koonin EV, Moss B. 2002. Complete pathway for protein disulfide bond formation encoded by poxviruses. Proc. Natl. Acad. Sci. U. S. A. 99:6667–6672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bisht H, Weisberg AS, Szajner P, Moss B. 2009. Assembly and disassembly of the capsid-like external scaffold of immature virions during vaccinia virus morphogenesis. J. Virol. 83:9140–9150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Alexander WA, Moss B, Fuerst TR. 1992. Regulated expression of foreign genes in vaccinia virus under the control of bacteriophage T7 RNA polymerase and the Escherichia coli lac repressor. J. Virol. 66:2934–2942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Szajner P, Weisberg AS, Moss B. 2004. Physical and functional interactions between vaccinia virus F10 protein kinase and virion assembly proteins A30 and G7. J. Virol. 78:266–274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Earl PL, Moss B. 1998. Characterization of recombinant vaccinia viruses and their products, p 16.18.1–16.18.11 In Ausubel FM, Brent R, Kingston RE, Moore DD, Seidman JG, Smith JA, Struhl K. (ed), Current protocols in molecular biology, vol 2 Greene Publishing Associates & Wiley Interscience, New York, NY [Google Scholar]
- 18.Ward GA, Stover CK, Moss B, Fuerst TR. 1995. Stringent chemical and thermal regulation of recombinant gene expression by vaccinia virus vectors in mammalian cells. Proc. Natl. Acad. Sci. U. S. A. 92:6773–6777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wolffe EJ, Moore DM, Peters PJ, Moss B. 1996. Vaccinia virus A17L open reading frame encodes an essential component of nascent viral membranes that is required to initiate morphogenesis. J. Virol. 70:2797–2808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lustig S, Fogg C, Whitbeck JC, Eisenberg RJ, Cohen GH, Moss B. 2005. Combinations of polyclonal or monoclonal antibodies to proteins of the outer membranes of the two infectious forms of vaccinia virus protect mice against a lethal respiratory challenge. J. Virol. 79:13454–13462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Brown E, Senkevich TG, Moss B. 2006. Vaccinia virus F9 virion membrane protein is required for entry but not virus assembly, in contrast to the related L1 protein. J. Virol. 80:9455–9464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sodeik B, Griffiths G, Ericsson M, Moss B, Doms RW. 1994. Assembly of vaccinia virus: effects of rifampin on the intracellular distribution of viral protein p65. J. Virol. 68:1103–1114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ansarah-Sobrinho C, Moss B. 2004. Role of the I7 protein in proteolytic processing of vaccinia virus membrane and core components. J. Virol. 78:6335–6343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Senkevich TG, Wyatt LS, Weisberg AS, Koonin EV, Moss B. 2008. A conserved poxvirus NlpC/P60 superfamily protein contributes to vaccinia virus virulence in mice but not to replication in cell culture. Virology 374:506–514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Resch W, Weisberg AS, Moss B. 2005. Vaccinia virus nonstructural protein encoded by the A11R gene is required for formation of the virion membrane. J. Virol. 79:6598–6609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Szajner P, Weisberg AS, Moss B. 2004. Evidence for an essential catalytic role of the F10 protein kinase in vaccinia virus morphogenesis. J. Virol. 78:257–265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Moss B, Rosenblum EN, Katz E, Grimley PM. 1969. Rifampicin: a specific inhibitor of vaccinia virus assembly. Nature 224:1280–1284 [DOI] [PubMed] [Google Scholar]
- 28.Nagayama A, Pogo BGT, Dales S. 1970. Biogenesis of vaccinia: separation of early stages from maturation by means of rifampicin. Virology 40:1039–1051 [DOI] [PubMed] [Google Scholar]
- 29.Hyun JK, Accurso C, Hijnen M, Schult P, Pettikiriarachchi A, Mitra AK, Coulibaly F. 2011. Membrane remodeling by the double-barrel scaffolding protein of poxvirus. PLoS Pathog. 7:e1002239. 10.1371/journal.ppat.1002239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Unger B, Mercer J, Boyle KA, Traktman P. 2013. Biogenesis of the vaccinia virus membrane: genetic and ultrastructural analysis of the contributions of the A14 and A17 proteins. J. Virol. 87:1083–1097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Essani K, Dugre R, Dales S. 1982. Biogenesis of vaccinia: involvement of spicules of the envelope during virion assembly examined by means of conditional lethal mutants and serology. Virology 118:279–292 [DOI] [PubMed] [Google Scholar]
- 32.Mohandas AR, Dales S. 1995. Involvement of spicules in the formation of vaccinia virus envelopes elucidated by a conditional lethal mutant. Virology 214:494–502 [DOI] [PubMed] [Google Scholar]
- 33.Kato SEM, Strahl AL, Moussatche N, Condit RC. 2004. Temperature-sensitive mutants in the vaccinia virus 4b virion structural protein assemble malformed, transcriptionally inactive intracellular mature virions. Virology 330:127–146 [DOI] [PubMed] [Google Scholar]
- 34.Cassetti MC, Merchlinsky M, Wolffe EJ, Weisberg AS, Moss B. 1998. DNA packaging mutant: repression of the vaccinia virus A32 gene results in noninfectious, DNA-deficient, spherical, enveloped particles. J. Virol. 72:5769–5780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Grubisha O, Traktman P. 2003. Genetic analysis of the vaccinia virus I6 telomere-binding protein uncovers a key role in genome encapsidation. J. Virol. 77:10929–10942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Garcia AD, Moss B. 2001. Repression of vaccinia virus Holliday junction resolvase inhibits processing of viral DNA into unit-length genomes. J. Virol. 75:6460–6471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Senkevich TG, Koonin EV, Moss B. 2009. Predicted poxvirus FEN1-like nuclease required for homologous recombination, double-strand break repair and full-size genome formation. Proc. Natl. Acad. Sci. U. S. A. 106:17921–17926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hu X, Carroll LJ, Wolffe EJ, Moss B. 1996. De novo synthesis of the early transcription factor 70-kDa subunit is required for morphogenesis of vaccinia virions. J. Virol. 70:7669–7677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hu X, Wolffe EJ, Weisberg AS, Carroll LJ, Moss B. 1998. Repression of the A8L gene, encoding the early transcription factor 82-kilodalton subunit, inhibits morphogenesis of vaccinia virions. J. Virol. 72:104–112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kane EM, Shuman S. 1993. Vaccinia virus morphogenesis is blocked by a temperature-sensitive mutation in the I7 gene that encodes a virion component. J. Virol. 67:2689–2698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Byrd CM, Hruby DE. 2005. A conditional-lethal vaccinia virus mutant demonstrates that the I7L gene product is required for virion morphogenesis. Virol. J. 2:4. 10.1186/1743-422X-2-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ansarah-Sobrinho C, Moss B. 2004. Vaccinia virus G1 protein, a predicted metalloprotease, is essential for morphogenesis of infectious virions but not for cleavage of major core proteins. J. Virol. 78:6855–6863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hedengren-Olcott M, Byrd CM, Watson J, Hruby DE. 2004. The vaccinia virus G1L putative metalloproteinase is essential for viral replication in vivo. J. Virol. 78:9947–9953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yang SJ, Hruby DE. 2007. Vaccinia virus A12L protein and its AG/A proteolysis play an important role in viral morphogenic transition. Virol. J. 4:73. 10.1186/1743-422X-4-73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Satheshkumar PS, Olano LR, Hammer CH, Zhao M, Moss B. 2013. Interactions of the vaccinia virus A19 protein. J. Virol. 87:10710–10720 [DOI] [PMC free article] [PubMed] [Google Scholar]