

Abstract
Like all other positive-strand RNA viruses, hepatitis C virus (HCV) induces rearrangements of intracellular membranes that are thought to serve as a scaffold for the assembly of the viral replicase machinery. The most prominent membranous structures present in HCV-infected cells are double-membrane vesicles (DMVs). However, their composition and role in the HCV replication cycle are poorly understood. To gain further insights into the biochemcial properties of HCV-induced membrane alterations, we generated a functional replicon containing a hemagglutinin (HA) affinity tag in nonstructural protein 4B (NS4B), the supposed scaffold protein of the viral replication complex. By using HA-specific affinity purification we isolated NS4B-containing membranes from stable replicon cells. Complementing biochemical and electron microscopy analyses of purified membranes revealed predominantly DMVs, which contained viral proteins NS3 and NS5A as well as enzymatically active viral replicase capable of de novo synthesis of HCV RNA. In addition to viral factors, co-opted cellular proteins, such as vesicle-associated membrane protein-associated protein A (VAP-A) and VAP-B, that are crucial for viral RNA replication, as well as cholesterol, a major structural lipid of detergent-resistant membranes, are highly enriched in DMVs. Here we describe the first isolation and biochemical characterization of HCV-induced DMVs. The results obtained underline their central role in the HCV replication cycle and suggest that DMVs are sites of viral RNA replication. The experimental approach described here is a powerful tool to more precisely define the molecular composition of membranous replication factories induced by other positive-strand RNA viruses, such as picorna-, arteri- and coronaviruses.
INTRODUCTION
Hepatitis C virus (HCV) is a major human pathogen persistently infecting 130 to 170 million individuals worldwide, thereby increasing the risk for chronic liver diseases, including steatosis, fibrosis, liver cirrhosis, and hepatocellular carcinoma (1). Despite recent advances in the development of promising HCV-specific drugs (2), current therapies suffer from the occurrence of severe side effects and the risk of therapy resistance (3). Thus, more-efficient therapeutic treatments, for which a better understanding of the fundamental principles governing the viral replication cycle is necessary, are required.
HCV is the only member of the genus Hepacivirus within the family Flaviviridae (4). Owing to its high genetic variability, HCV is classified into 7 genotypes and more than 100 subtypes (5). An ∼9.6-kb single-strand uncapped RNA molecule of positive polarity constitutes the HCV genome, which contains a single long open reading frame (ORF) that is flanked by 5′ and 3′ untranslated regions (UTRs). Both UTRs are highly structured and are implicated in viral RNA replication, while an internal ribosome entry site (IRES) contained in the 5′ UTR mediates translation of the positive-strand RNA viral genome (reviewed in reference 6). Upon translation of the ORF, at least 10 HCV proteins are generated from a polyprotein precursor that is co- and posttranslationally cleaved by cellular and viral proteases (6). The resulting cleavage products are three structural proteins (core, envelope protein 1 [E1], and E2), the viroporin p7, and six nonstructural (NS) proteins (NS2, NS3, NS4A, NS4B, NS5A, and NS5B). While p7 and NS2 are required for virus assembly and release (7, 8, 9), NS3 to -5B constitute the minimal viral replicase machinery (10, 11). Indeed, HCV “minigenomes” (termed subgenomic replicons) comprising both UTRs and encoding NS3 to -5B autonomously replicate in cell culture (10, 11). They have been used extensively to study basic principles of HCV replication and to develop directly acting antivirals (DAAs) (12).
Like for all other positive-strand RNA viruses, HCV RNA replication is thought to occur in tight association with remodeled cytoplasmic host cell membranes, which form distinct organelle-like structures designated the membranous web in the case of HCV (13, 14, 15) and “viral replication factories” for many other viruses (reviewed in references 16, 17, and 18). Recent electron tomography studies of infected cells revealed that HCV-induced membrane rearrangements are predominantly vesicular double-membrane protrusions of the endoplasmic reticulum (ER) (19). Such double-membrane vesicles (DMVs) have also been observed in cells containing subgenomic HCV replicon RNA (15, 19, 20). DMV formation is induced by a concerted action of several replicase proteins, with NS4B playing a key role (13, 14, 15, 19, 20). NS4B is a highly hydrophobic protein and is thought to remodel intracellular membranes by self-oligomerization (13, 14, 15; reviewed in reference 21). Notably, replication-impaired NS4B mutants exhibit an altered DMV morphology, suggesting the presence of this viral replicase factor in DMV membranes (15).
A major limitation in our understanding of HCV RNA replication is the lack of knowledge about the molecular composition of the membranous replication compartment. In this study we developed an affinity purification method and present a detailed characterization of HCV replicase-containing membranes. We demonstrate that DMVs are associated with replicase activity and represent distinct virus-induced membranous compartments. Our method overcomes a major restriction and likely is applicable to the study of the membranous replication compartments of other positive-strand RNA viruses.
MATERIALS AND METHODS
Antibodies.
Mouse monoclonal antibody against NS3 of the JFH1 isolate (2E3) was from BioFront Technologies (Tallahassee, FL). Mouse monoclonal antibody 9E10 recognizing NS5A domain III of the HCV isolates Con1 and JFH1 was a kind gift from Charles M. Rice. Rabbit polyclonal antibody raised against NS4B of Con1 and cross-reacting with NS4B of JFH1 was generated by the immunization of rabbits with recombinant hexahistidine-tagged NS4B produced in a baculovirus expression system in insect cells and affinity purified via Ni2+-nitrilotriacetic acid spin columns (Qiagen, Hilden, Germany) as described elsewhere (22). Rabbit polyclonal antibody against human calnexin (CANX; ADI-SPA-865) was obtained from Enzo Life Sciences (Farmingdale, NY), mouse monoclonal antibody recognizing human GAPDH (glyceraldehyde-3-phosphate dehydrogenase) was obtained from Santa Cruz Biotechnology (Santa Cruz, CA), J2 mouse monoclonal antibody recognizing double-stranded RNA (dsRNA) was purchased from English & Scientific Consulting (Szirák, Hungary), and rat antibromodesoxyuridine (BrdU) antibody (M01269M), which cross-reacts with bromouridine (BrU), was obtained from Meridian Life Science (Memphis, TN). Rabbit polyclonal antibodies against human vesicle-associated membrane protein-associated protein A (VAP-A) and VAP-B were purchased from Sigma-Aldrich (St. Louis, MO). Antibiotin rabbit polyclonal antiserum was obtained from Rockland Immunochemicals, Inc. (Gilbertsville, PA). Primary antibodies against β-actin (A5441), the hemagglutinin (HA) tag (H3663), and HA-specific agarose beads (A2095), as well as secondary horseradish peroxidase-conjugated antibodies, were purchased from Sigma-Aldrich.
Cell culture.
Huh7-Lunet cells (23) and Huh7.5 cells (24) were maintained in Dulbecco's modified Eagle medium (DMEM) (Invitrogen, Karlsruhe, Germany) supplemented with 2 mM l-glutamine, nonessential amino acids, 100 U/ml penicillin, 100 μg/ml streptomycin, and 10% fetal calf serum (DMEM cplt). G418 (Geneticin; Invitrogen) was added at a final concentration of 500 μg/ml in case of replicon-harboring cell lines. Huh7-Lunet cells stably overexpressing calnexin that was C-terminally fused to the HA tag (calnexinHA [CANXHA]) were generated by lentiviral gene transfer as described previously (25) and cultured in the presence of 10 μg/ml blasticidin (Invitrogen).
Plasmid constructs.
All nucleotide and amino acid numbers refer to the genome of the JFH1 isolate (GenBank accession no. AB047639). Plasmids carrying the subgenomic replicons pFK_i389LucNS3-3′_JFH_δg, the nonreplicative mutant pFK_i389LucNS3-3′_NS5BΔGDD_JFH_δg, and the full-length genomes of pFK_Jc1_δg and pFK_Jc1ΔE1E2_δg have been described previously (26, 27). The HA-coding sequence (YPYDVPDYA) or FLAG-coding sequence (DYKDDDDK) was introduced in-frame after codon 38, 131, 140, or 258 in the NS4B-coding region by using overlap PCR. Amplicons were inserted into pBSK_SacI_JFH (15) and subsequently transferred into pFK_i389LucNS3-3′_JFH_δg as described recently (15). Selectable replicons were generated by replacing the first cistron encoding firefly luciferase with the gene encoding neomycin phosphotransferase by using flanking AgeI and KpnI restriction sites. The full-length Jc1 genome containing the HA tag insertion after codon 38 and the pseudoreversion Q31R in NS4B were created by transferring an NsiI/MluI fragment from pFK_i389LucNS3-3′-NS4BHA31R_JFH_δg into pFK_Jc1_δg. The human CANX-coding sequence (GenBank accession no. NM_001746.3) was fused at the 3′ end with the HA-coding sequence (YPYDVPDYA), and the resulting product was inserted into the lentiviral self-inactivating vector pWPI-BSD (25) encoding blasticidin S deaminase. The sequence integrity of all constructs was confirmed by DNA sequencing.
Immunoblot analysis.
Samples were denatured in 2× protein sample buffer (200 mM Tris [pH 8.8], 5 mM EDTA, 0.1% bromophenol blue, 10% sucrose, 3% SDS, 2% β-mercaptoethanol) and incubated for 5 min at 95°C. Proteins were separated by SDS-polyacrylamide gel electrophoresis and electro-transferred onto polyvinylidene difluoride membranes. After blocking of the membranes with 5% nonfat milk, they were incubated overnight at 4°C with primary antibodies (NS3 2E3, 1:2,000; NS4B, 1:1,000; NS5A 9E10, 1:10,000; β-actin, 1:5,000; CANX, 1:2,000; GAPDH, 1:1,000; HA, 1:1,000) and after they were washed with phosphate-buffered saline (PBS)-0.5% Tween 20, with secondary horseradish peroxidase-conjugated antibodies. Membranes were developed by using Western Lightning Plus-ECL reagent (PerkinElmer, Waltham, MA), and bands were visualized on Amersham Hyperfilm ECL (GE Healthcare Life Sciences, Uppsala, Sweden).
Immunofluorescence analysis and confocal microscopy.
Cells grown on glass coverslips in a 24-well plate were washed 3 times in PBS, fixed with 4% paraformaldehyde (PFA) in PBS for 10 min at room temperature (RT), and permeabilized as specified in the figure legends with digitonin (50 μg/ml in PBS) for 20 min at RT or with 0.5% Triton X-100 in PBS for 5 min at RT. After blocking with 3% bovine serum albumin (BSA) in PBS for 30 min at RT, cells were incubated with primary antibodies (NS4B and HA, 1:200; CANX, 1:100) diluted in 1% BSA-PBS. After 1 h, cells were washed 3 times for 10 min with PBS and incubated with secondary antibodies conjugated to Alexa 488 or Alexa 568 (goat anti-mouse and goat anti-rabbit for replicon-transfected cells or the other way around for CANXHA-overexpressing cells) diluted 1:1,000 in 1% BSA-PBS. After 45 min of incubation in the dark, cells were washed, and nuclei were stained with Hoechst (Sigma-Aldrich). Coverslips were mounted on glass slides with Fluoromount-G (Southern Biotech, Birmingham, AL), and samples were examined using a Leica SP2 confocal laser scanning microscope (Leica, Heidelberg, Germany). Pictures were edited and merged by using the ImageJ software package (NIH).
In vitro replicase assays.
An HCV in vitro replicase activity assay was carried out in a reaction mixture containing 20 mM Tris-HCl [pH 8], 10 mM MgCl2, 5 mM dithiothreitol, 5 mM KCl, 40 μg/ml actinomycin D, 16 mM creatine phosphate, 40 ng/μl creatine kinase (Roche, Mannheim, Germany), 1 U RNasin, and a 25-μl sample fraction in a total volume of 30 μl at 30°C for 60 min. Nucleotide concentrations were as follows for different experimental setups: 1 mM (each) ATP, CTP, and UTP and 5 mM GTP for total HCV RNA quantification; 20 μCi of [α-32P]CTP, 10 μM CTP, 1 mM (each) ATP and UTP, and 5 mM GTP for in vitro replicase activity assays and measurement by autoradiography; and 1 mM (each) ATP, CTP, and bromouridine-triphosphate (BrUTP) and 5 mM GTP for immunolabeling. In the case of the latter, reaction products were directly subjected to immunolabeling as described. Reaction products were isolated using a NucleoSpin RNAII kit (Macherey-Nagel, Düren, Germany) for total HCV RNA quantification. In the case of radioactively labeled RNA, reaction products were purified by phenol-chloroform extraction and isopropanol precipitation and then analyzed by denaturing glyoxal agarose gel electrophoresis prior to autoradiography.
In vitro transcription and RNA transfection.
In vitro transcripts from given pFK plasmids were generated and purified and transfected into human hepatoma cells as described previously (15).
Nuclease and protease protection assay.
In order to determine nuclease protection of HCV RNA in purified membranous material, we subjected equal aliquots to nuclease (1 U/μl benzonase), protease (8 mg/ml proteinase K), and detergent (1% Triton X-100) treatment for 1 h at 25°C. HCV RNA was extracted and quantified as described below.
Purification of HCV-remodeled and control ER membranes.
Huh7-Lunet cells (7.5 × 107) containing subgenomic replicon RNA encoding either wild-type (wt) or HA-tagged NS4B and control Huh7-Lunet cells stably overexpressing CANXHA were washed 3 times, scraped into PBS, and pelleted by centrifugation at 800 × g for 10 min at RT. Cells were resuspended in 2 ml hypotonic buffer (20 mM Tris [pH 8], 1.5 mM MgCl2, 10 mM Na-acetate) and incubated on ice for 30 min. Cells were lysed by 50 strokes with a dounce homogenizer, and nuclei and unbroken cells were removed by centrifugation at 800 × g for 10 min at 4°C. Postnuclear supernatants were layered on top of a discontinuous (70% to 30%) sucrose gradient and centrifuged for 4 h at 130,000 × g using a SW40 rotor (Beckman Coulter, Fullerton, CA). Thirteen fractions (1 ml each) were collected from the bottom and analyzed for density and for protein and HCV RNA content. For HA affinity capture, equal amounts (∼1 mg protein) contained in pooled fractions 7 to 11 were equilibrated to 150 mM NaCl. Incubation with HA-agarose beads (Sigma-Aldrich, St. Louis, MO) was carried out for 4 h by continuously inverting tubes at 4°C. Beads were washed 5 times with 40 volumes of IP buffer (20 mM Tris [pH 8], 1.5 mM MgCl2, 10 mM Na-acetate, 150 mM NaCl). Bound material was eluted by incubation with 100 μl IP buffer containing 250 μg/ml HA peptide (Sigma-Aldrich) 2 times, each for 15 min at RT. Elution fractions were further analyzed as described in the Results section.
Quantification of HCV RNA.
Prior to quantification, RNA was extracted using a NucleoSpin RNA II kit (Macherey-Nagel, Düren, Germany) according to the instructions of the manufacturer. Five microliters of purified RNA was used for quantitative reverse transcription-PCR analysis using an ABI Prism 7000 sequence detector system (Applied Biosystems, Foster City, CA). HCV JFH1-specific reverse transcription-PCRs were conducted in triplicate using OneStep RT-PCR kits (Qiagen, Hilden, Germany), primers S-146 (5′-TCTGCGGAACCGGTGAGT-3′) and A-219 (5′-GGGCATAGTGTGGGTTTATCC-3′), and the probe 5′-6-carboxyfluorescein (6FAM)-AAAGGACCCAGTCTTCCCGGCAATT-6-carboxytetramethylrhodamine (TAMRA)-3′ (Sigma-Aldrich). The total volume of the reaction mixture was 15 μl, and it contained the following components: 4 mM MgCl2, 0.66 mM deoxynucleoside triphosphates (dNTPs), 0.266 μM HCV probe, 1 μM each primer, and 0.6 μl enzyme mix. The amount of HCV RNA was calculated by comparison with serially diluted in vitro transcripts (1 × 108, 1 × 107, 1 × 106, 1 × 105, 1 × 104, and 1 × 103 copies/PCR). Reactions were performed in three stages by using the following conditions: reverse transcription for 60 min at 50°C, heat inactivation of reverse transcription and activation of Taq polymerase for 15 min at 95°C, and 40 amplification cycles for 15 s at 95°C and 1 min at 60°C.
Sample preparation and ultrastructural analysis by transmission electron microscopy (TEM).
Flat embedding of cultured cells was performed as described recently (15). For examination of purified membranous material, 9-μl droplets of HA-captured material were adsorbed to carbon- and pioloform-coated 300-mesh copper grids (Science Services GmbH, Munich, Germany) for 10 min at RT, and the remaining liquid was drained using a Whatman paper. Structures were negatively stained as described below or subjected to immunolabeling as described in the Results section. For immunolabeling, all incubation steps were performed by floating the grids on top of drops. Prior to immunogold labeling, grids were incubated with blocking solution (0.8% BSA [Roth, Karlsruhe, Germany], 0.1% fish skin gelatin [Sigma-Aldrich], 50 mM glycine in PBS). For immunolabeling, primary antibodies were diluted (NS3 2E3, 1:120; NS5A 9E10, 1:5,000; dsRNA J2, 1:4; BrdU, 1:50; VAP-A and VAP-B, 1:20) and incubated for 20 min in blocking solution. After five 2-min washes with PBS, grids were incubated with rabbit anti-mouse- or anti-rat-bridging antibody (Dako Cytomation, Hamburg, Germany) diluted 1:150 in blocking solution for 30 min. After five additional washing steps in PBS, bound antibodies were labeled with protein A coupled to 10-nm gold particles (Cell Microscopy Center, Utrecht, The Netherlands) diluted 1:60 in blocking reagent for 30 min. Grids were washed five times with PBS and fixed with 1% glutaraldehyde (Science Services GmbH, Munich, Germany) in PBS for 5 min. In the case of double-labeling experiments, membranes were first stained for NS5A with 15-nm gold particles and after fixation with 1% glutaraldehyde, stained for VAP-A or VAP-B using 10-nm colloidal gold particles as described above. For cholesterol-labeling experiments, membranes were incubated with biotinylated perfringolysin O (P-O) (1:150) (28), which was detected with rabbit antibiotin antibody (1:10,000) and protein A–10-nm colloidal gold particles. After fixation, membranes were additionally labeled for NS5A using 15-nm colloidal gold particles as described above. After the last fixation step of the immunolabeling assays, grids were washed 5 times with PBS and in 7 additional washing steps, each for 2 min, with water. Structures were negatively stained using a mixture of 3% aqueous uranylacetate and methyl-cellulose (60:40, vol/vol) for 8 min on ice and examined by using a Zeiss EM-10 transmission electron microscope (Zeiss, Goettingen, Germany) with a built-in MegaView camera (Olympus, Tokyo, Japan).
Selection for pseudorevertants, amplification of HCV RNA by reverse transcription-PCR, and sequence analysis of cloned amplicons.
Huh7-Lunet cells transfected with a selectable replicon RNA were resuspended in 20 ml DMEM cplt and seeded onto two 10-cm-diameter culture dishes. Twenty-four hours later, G418 was added to a final concentration of 500 μg/ml. Medium was exchanged at least twice a week until single-cell clones became visible. Two cell clones per 10-cm-diameter dish were isolated and expanded for further analyses. The total RNA of each cell clone was obtained by using a NucleoSpin RNA II kit (Macherey-Nagel) according to the instructions of the manufacturer. Five micrograms of total RNA served as the template for cDNA synthesis using oligonucleotide A/2A/9842 (5′-GGAACAGTTAGCTATGGAGTGTACC-3′) and SuperScript III (Invitrogen) as specified by the manufacturer. A region spanning NS3 to NS5A was amplified with oligo(f_Ns3_NsiI) (5′-TACCAATGAGGTCACCCTCACACAC-3′) and oligo(r_NS5A_RsrII) (5′-AGGCGGAACCTGTCTCTGAGG-3′) using the Expand long template PCR system (Roche, Mannheim, Germany). After digestion with NsiI and RsrII, amplicons were inserted into pFK_i389LucNS3-3′_JFH_δg and at least two DNA clones per cell clone were sequenced to confirm the presence of the inserted affinity tag and to identify conserved pseudoreversions.
Statistical analysis.
Statistical analyses were performed as specified in the figure legends, and significance values were calculated by applying the two-tailed, unpaired Student's t test available in the GraphPad Prism 5 software package (GraphPad Software, Inc., La Jolla, CA).
Transient replication and infectivity assays.
Transient HCV RNA replication and the kinetics of viral progeny release were determined by measuring luciferase activity in cell lysates and by limiting dilution assay, respectively, as described recently (15).
RESULTS
Establishment of a functional HCV replicon encoding an affinity-tagged NS4B.
Since NS4B is thought to be the scaffold protein of the HCV replicase complex and a key player in the induction of membrane rearrangements, we wanted to insert an affinity tag into this protein and express it in the context of a functional HCV replicon (Fig. 1A) to allow the isolation of membrane-associated viral replication factories. We inserted the HA or FLAG tag in frame after Ala-38, Gly-131, Leu-140, or Ile-258 of NS4B (Fig. 1B), which correspond to positions that previously have been shown to tolerate the insertion of a short heterologous sequence (29). As shown in Fig. 1C, all chosen insertions into NS4B severely diminished or completely abolished transient replication of a subgenomic JFH1 replicon (Fig. 1C). Only replicons containing the tag after Ala-38 still replicated to a detectable level, whereas the insertions at the other sites completely abrogated HCV RNA replication, as deduced from comparison with the nonreplicative ΔGDD polymerase mutant. To improve the replication competence of these RNAs, we inserted tagged NS4B sequences into a selectable subgenomic replicon and passaged transfected cells under G418 selection for ∼4 weeks (Table 1). In this way, we aimed to select for second-site compensatory mutations (pseudoreversions) enhancing RNA replication. Indeed, we obtained single-cell clones for several NS4B tag insertion constructs (Table 1). By far the highest number of G418-resistant colonies was obtained with the HA tag insertion mutant after Ala-38, whereas no or only very few cell clones were obtained with all the other mutants. To confirm the presence of the inserted affinity tag and to identify pseudoreversions in selected replicons, for each given cell clone we determined the predominant nucleotide sequence of the NS3- to NS5A-coding region of the replicon contained therein. While the FLAG and HA epitopes inserted after Ala-38 and Gly-131, respectively, were deleted, the HA tag-encoding sequence inserted after Ala-38 was retained (Fig. 1C). In the case of the latter, we additionally identified four conserved mutations, all residing in the N-terminal half of NS4B (Table 1). Next we determined whether any of these second-site mutations enhanced the replication capacity of the primary sg4BHA mutant by inserting them individually or in combination into this mutant construct. Single mutations Q31R and Q117R, a combination thereof, and the double mutation Q11R plus D34A all rescued RNA replication of the NS4B alanine 38 HA tag insertion construct (Fig. 1D). Although delayed replication kinetics, which is most visible for the 24-h values, were observed, replication efficiencies at later time points were comparable to that of the wt replicon (Fig. 1D). Owing to its robust replication capacity and the requirement for only one adaptive mutation, the 4B-38HA mutant containing the Q31R pseudoreversion (construct sg4BHA31R) was used for all subsequent experiments. In order to confirm that this mutated NS4B supported the complete HCV replication cycle, we inserted it into the highly assembly-competent Jc1 full-length genome (27) and determined the release kinetics of infectious HCV progeny by limiting-dilution assay (Fig. 1E). The wt genome and a mutant that due to a deletion in the E1/E2-coding region was unable to produce infectious virus particles served as positive and negative controls, respectively. Considerably fewer infectious particles were released from cells transfected with the NS4B mutant at all time points (Fig. 1E), reflecting delayed RNA replication kinetics of Jc1-4BHA31R and, consistent with earlier reports, indicating a role of NS4B in virus production (15, 30). Nevertheless, a 30-fold increase in viral progeny release from transfected cells was detectable between 24 and 48 h after transfection, demonstrating that the HA-tagged version of NS4B supported the complete HCV replication cycle.
Fig 1.

HCV tolerates an HA tag insertion into the N-terminal region of NS4B. (A) Schematic representation of the bicistronic HCV subgenomic reporter replicon. Firefly luciferase (Fluc) or neomycin-phosphotransferase (neoR) is expressed as an N-terminal fusion with 16 amino acids of the N-terminal region of the core protein (black line) and is translated under the control of the HCV IRES contained in the 5′ UTR. The second cistron (NS3 to NS5B) is translated via the IRES of the encephalomyocarditis virus (EMCV-IRES). (B) Predicted NS4B membrane topology. N-terminal amphipathic α-helices AH1 and AH2, the four transmembrane segments (TM1-4), and the C-terminal α-helices H1 and H2 are schematically depicted. Numbers indicate amino acid positions of the JFH1 isolate. Affinity-tag insertion sites are highlighted by black arrows. (C) Huh7-Lunet cells were transfected with the in vitro-transcribed luciferase replicon RNAs specified at the bottom. Cells were lysed 4, 24, 48, and 72 h after transfection, and the luciferase activity in cell lysates was determined. Data were normalized to the 4-h value that reflects transfection efficiency. The background of the assay is determined by the NS5B active-site polymerase mutant (ΔGDD) (dashed line). Mean values of two independent experiments are shown. Error bars indicate standard deviations. (D) Huh7-Lunet cells were transfected with the in vitro-transcribed luciferase replicon RNAs specified at the bottom. Replication efficiency was determined as described for panel C. (E) Release kinetics of infectious HCV particles. Huh7-Lunet cells were transfected with the full-length HCV RNAs specified on the right. Culture supernatants were harvested at the given time points. Infectivity titers were determined by limiting-dilution assay and are expressed as 50% tissue culture infective doses (TCID50)/ml. Mean values of two independent experiments are shown; error bars indicate standard deviations.
Table 1.
Long-term selection assay of NS4B-tagged insertion constructs
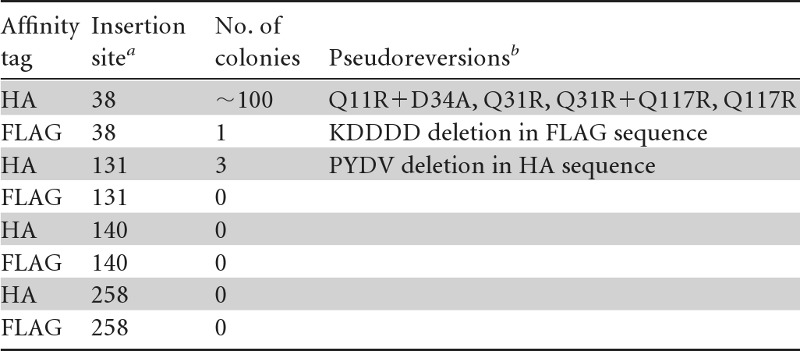
The affinity tag given in the first column was inserted in frame after the given NS4B amino acid residues.
Pseudoreversions detected in replicons that were isolated from individual cell clones were identified by sequence analysis of amplicons spanning the NS3- to NS5A-coding region.
Characterization of HA-tagged NS4B and induced membranous morphotypes.
Next we characterized the 4BHA31R mutant by using Western blotting, immunofluorescence, and electron microscopy (EM) analyses. In lysates of cells transfected with the subgenomic replicon sg4BHA31R and in cells infected with the Jc1-4BHA31R virus, NS4BHA was readily detectable via the HA tag (Fig. 2A and B, respectively). As expected, the HA tag slightly increased the apparent molecular weight of the NS4B protein. In replicon cells, the abundance of 4BHA31R was reduced concomitant with a reduction in NS5A steady-state levels, presumably as a result of delayed replication kinetics (Fig. 2A). Concurrently, in cells infected with Jc1, 4BHA31R and NS5A steady-state protein levels were reduced, reflecting delayed replication kinetics and reduced virus progeny production (Fig. 2B).
Fig 2.

Characterization of HA-tagged NS4B. (A) Huh7-Lunet cells were transfected with the in vitro-transcribed luciferase replicon RNAs specified at the top. Cells were harvested 72 h posttransfection and analyzed by immunoblotting, using the monospecific antibodies indicated on the right. The positions of molecular weight (MW; in thousands) markers are depicted on the left. (B) Huh7.5 cells were infected with the culture supernatants of cells transfected with the Jc1-derived full-length constructs specified at the top. After 72 h, cells were harvested and processed as described for panel A. (C) Huh7-Lunet cells were transfected with the in vitro-transcribed luciferase replicon RNAs specified at the top of each panel. After 48 h, cells were fixed, permeabilized with digitonin, and stained with NS4B- and HA-specific antibodies prior to confocal immunofluorescence microscopy. Only merged images are shown. Scale bars represent 5 μm. Numbers below each panel indicate the mean ± the standard deviation (SD) for NS4B-containing MAF per cell; for each condition, at least 10 different HCV-positive cells were analyzed. (D) Huh7-Lunet cells were transfected with in vitro-transcribed luciferase replicon RNAs as specified at the top of each panel. After 48 h, cells were fixed and flat embedded for TEM analysis. Scale bars represent 100 nm. (E) Naive Huh7-Lunet cells and those overexpressing CANXHA were fixed, permeabilized with Triton X-100, and stained with CANX- and HA-specific antibodies prior to confocal immunofluorescence microscopy. Only merged images are shown. Scale bars represent 5 μm.
Immunofluorescence (IF) analyses of cells transfected with replicon RNA revealed NS4B-induced membrane-associated foci (MAF) (31, 32) that were detected with NS4B- and HA-specific antibodies (Fig. 2C). Their numbers did not differ considerably between wt and NS4BHA-replicating cells. This result confirmed HA tag accessibility and demonstrated that NS4B-induced MAF remained unaltered by insertion of the HA tag. Furthermore, the ability of the sg4BHA31R replicon to induce DMVs, the most prominent membrane alteration observed in cells containing replicating HCV RNA, was verified by TEM analysis (Fig. 2D).
For subsequent membrane purification assays, we additionally established a control cell line stably overexpressing the ER chaperone calnexin that was C-terminally fused to the HA-tag (CANXHA). Calnexin is a type I membrane protein with its ectodomain localizing to the ER lumen and a short cytoplasmic tail to which the HA tag was fused. We chose this protein because HCV-induced membrane alterations are ER derived, thus allowing the purification of control ER membranes. IF analysis confirmed recognition of CANXHA with the HA-specific antibody as well as unaltered ER morphology in cells overexpressing this protein (Fig. 2E).
Purification of NS4B-associated membranes from replicon-containing cells.
Having established a cell line with the functional sg4BHA31R replicon and the CANXHA control cell line, we isolated intracellular membranes by using a combination of membrane enrichment via density gradient centrifugation and subsequent HA-affinity capture as schematically depicted in Fig. 3A. Cells containing an analogous subgenomic replicon without the HA tag served as an additional specificity control. First, we analyzed fractions after ultracentrifugation for the presence of HCV proteins and RNA. The majority of viral RNA (Fig. 3B) and proteins (Fig. 3C) was broadly distributed in fractions 7 to 11 (corresponding to densities 1.18 to 1.06 g/cm3), which also contained the ER marker calnexin. These fractions were well-separated from soluble cytosolic proteins, as exemplified with GAPDH. Moreover, gradient distribution of calnexin remained unaltered in the case of cells overexpressing CANXHA. Thus, we pooled fractions 7 to 11 of replicon or CANXHA control lysates and subjected them to HA-affinity capture. After elution under native conditions with the HA peptide, eluates were examined for HCV protein and RNA content. Western blot analysis revealed specific copurification of NS3 and NS5A along with HA-tagged NS4B (Fig. 3D). We also detected CANX in the NS4BHA-specific pull-down assay, corroborating the ER origin of HCV protein-containing membranes. Importantly, HCV RNA was significantly enriched in NS4BHA-captured material compared to that in the untagged negative control, even though slightly higher HCV RNA amounts, presumably reflecting a higher replication efficiency, were detected in the input for the untagged negative control (Fig. 3E).
Fig 3.

Purification and biochemical characterization of NS4BHA-associated membranes. (A) Schematic overview of HCV membrane preparation. Cells containing a stably replicating wild-type replicon (HCV wt), the replicon sg4BHA31R (HCV 4BHA), and control cells stably overexpressing CANXHA were broken by hypotonic lysis. Postnuclear cytosolic supernatants were separated by discontinuous sucrose gradient ultracentrifugation (UC). Subsequently, membrane fractions were pooled and subjected to affinity capture using HA beads. After elution with the HA peptide, purified material was used for further analyses. (B) Distribution of HCV RNA (right y axis, solid lines) and corresponding density of each fraction (left y axis, dashed lines) along the gradient. Mean values and error bars indicating the standard deviations of three independent measurements are depicted. (C) Fractions analyzed for their protein content are shown for HCV NS4BHA. Proteins were separated by SDS-PAGE, and monospecific antibodies were used to detect calnexin (CANX), glyceraldehyde-3-phosphate dehydrogenase (GAPDH), NS3, NS5A, and NS4B, as indicated on the right. The positions of molecular weight (MW) markers are depicted on the left. (D) Protein content after HA-specific affinity capture. Samples were analyzed as described for panel C. (E) HCV RNA content in purified material. Mean values with standard deviations, as indicated by the error bars, from at least three independent experiments, with three measurements each, are given. No HCV RNA was detected in CANXHA samples, as indicated by the inverted triangles (▼). ***, P < 0.0001.
Morphological characterization of purified membranes.
Next we examined the ultrastructures of purified membranes by using negative staining and TEM. Numerous big (>500 nm in length) and partially collapsed membrane structures were detected in control eluates of CANXHA-expressing cells (Fig. 4Aa and b, C, and Da to c). By contrast, a lower number of membranous structures, corresponding predominantly to spherical vesicles with a diameter of ∼50 to 250 nm, were present in the NS4BHA-purified fraction (Fig. 4Ac and d, C, and 4Dd to f). Two mostly concentric membranes, reminiscent in size and morphology of DMVs detected in HCV-replicating cells (Fig. 2D), were clearly discernible for many of these structures. The numbers of membranous structures in NS4BHA preparations were lower than those in CANXHA control samples (Fig. 4B), but this was due to large amounts of overexpressed CANXHA (data not shown). Importantly, quantification of total membranous structures per area revealed significant enrichment in the NS4BHA-containing preparation compared to that in the untagged wt NS4B control (Fig. 4B). Moreover, classification of membrane structures according to their morphology and size revealed distinct differences for CANXHA- and NS4BHA-captured membranes (Fig. 4C), suggesting that for the latter mainly HCV-remodeled membranes had been purified.
Fig 4.

Morphological characterization of purified HCV-remodeled membranes. (A) HA-captured material was negatively stained with uranylacetate and examined by TEM. Representative membranous structures from NS4BHA- and control CANXHA-captured material are shown. Scale bars represent 100 nm. (B) Number of membrane structures per area of 10 randomly chosen hexagons (area/hexagon, ∼2,913 μm2) are given. Horizontal lines indicate mean values. ***, P < 0.0005. (C) Quantification of membrane morphotypes. More than 100 structures per sample were classified as specified at the top and are represented as relative values. (D) HA affinity-purified membranes of CANXHA (a through c) and HCV NS4BHA cells (d through f) were immunolabeled with antibodies recognizing NS3 (a and d), NS5A (b and e), and dsRNA (c and f). Samples were negatively stained with uranylacetate and examined by TEM. The scale bar in each panel corresponds to 100 nm. Percentages of labeled DMVs (n > 75) are given below panels d through f. (E) Quantification of immunolabeling. At least 100 gold particles were counted, and fractions of gold particles associated with membranes per area of the grid are depicted.
In order to investigate the viral constituents of DMVs in more detail, we performed immunolabeling experiments for NS3 and NS5A as well as for dsRNA, which is a commonly accepted marker for viral RNA replication intermediates. Owing to the low number of membranous structures present after immunoprecipitation (IP) of the untagged wt NS4B control (Fig. 4B), we used membranes that were isolated by HA-specific affinity purification from HCV-naive CANXHA-expressing cells as the negative control for this and subsequent assays. A major portion of DMVs in the NS4BHA elution fraction was found to be decorated with gold particles after immunolabeling (Fig. 4Dd to f), and 48%, 59%, and 63% of DMVs were positive for NS3, NS5A, and dsRNA, respectively. Only background labeling was detected on naive ER membranes (Fig. 4Da to c). The specificity of immunolabeling was confirmed by quantification of gold particles on membranes, which revealed a considerably higher level of labeling for all tested antibodies on NS4BHA-containing membranes (Fig. 4E). Taken together, these results demonstrate that besides NS4BHA, NS3 and NS5A, as well dsRNA, are associated with HCV-induced DMVs, suggesting that DMVs might harbor viral replicase complexes.
HCV RNA in NS4BHA preparations resides in a nuclease-protected environment and is replication competent.
With the aim to more precisely determine the localization of replicating HCV RNA, we exploited the possibility of isolating HCV-induced membranes by NS4BHA-specific affinity purification. Viral replicon RNA copurified with the membranes was resistant to nuclease and protease treatment but was destabilized upon the addition of detergent (Fig. 5). The combination of nuclease and detergent treatments led to complete degradation of HCV RNA, arguing for its localization in a membrane-protected environment. This observation is in good agreement with previous publications describing protection of HCV RNA in enriched membrane fractions that were isolated from cells containing a stably replicating genotype 1 subgenomic RNA (33, 34, 35). Likewise, the small amounts of unspecifically captured HCV RNA present in the preparation with the nontagged NS4B were also nuclease resistant (Fig. 5).
Fig 5.
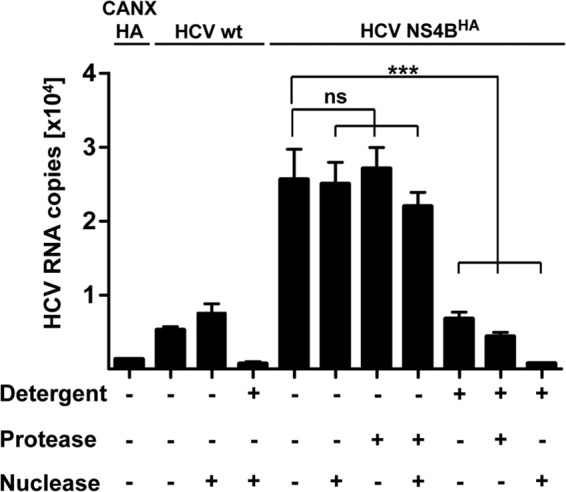
HCV RNA associated with affinity-purified membranes is protected against nuclease. Equal aliquots of NS4BHA-purified and unspecifically bound (NS4B wt) material were subjected to treatment with nuclease (1 U/μl Benzonase), protease (8 mg/ml proteinase K), and detergent (1% Triton X-100) for 1 h at 25°C as indicated at the bottom. Subsequently, HCV RNA was extracted and quantified by quantitative reverse transcription-PCR. Mean values and error bars indicating the standard deviations for at least three independent experiments are depicted. ns, P > 0.4; ***, P < 0.0003.
Next we determined in vitro replicase activity in NS4BHA-purified membranes. Equal aliquots of elution fractions were incubated in the absence or presence of ribonucleotides (rNTPs), and HCV RNA was quantified by quantitative reverse transcription-PCR. A small but significant increase in the HCV RNA copy number was detected when NS4BHA-purified membranes were incubated with rNTPs, arguing for in vitro replicase activity (Fig. 6A). To corroborate this assumption, we performed metabolic radiolabeling of the viral subgenomic RNA (Fig. 6B). The incorporation of [α-32P]CTP into nascent viral RNA confirmed that isolated NS4BHA-associated membranes contained functional HCV replicase. This signal was specific, because it was not detected in reactions using CANXHA membrane preparations that were processed in parallel.
Fig 6.

HCV-induced double-membrane vesicles are sites of RNA replication. (A) Equal fractions of NS4BHA-purified material were incubated without (−) or with (+) exogenously added ribonucleotides in an in vitro replicase assay as specified at the bottom. Total HCV RNA was quantified before and after the in vitro replicase assay and is represented as the fold increase. Mean values and error bars representing standard deviations from six replicates in two independent experiments are shown. **, P < 0.001. (B) Equal amounts of control CANXHA and NS4BHA-purified membranes were subjected to an in vitro replicase assay in the absence (−) or presence (+) of radioactively labeled [α-32P]CTP. After RNA purification, samples were analyzed by denaturing glyoxal agarose gel electrophoresis and autoradiography. (C) Membranes from the NS4BHA purification were used for in vitro replicase assay in the absence (−) or presence (+) of BrUTP. After immunolabeling with a BrdU-specific antibody and subsequent negative staining, samples were examined by TEM. White arrowheads show the locations of gold particles. The number below each panel indicates the percentage of non-specifically gold-labeled DMVs (a) or of DMVs with gold labeling on the exterior (b) or apparent in the interior (c) (n > 75). The scale bar in each panel corresponds to 100 nm. (D) Quantification of immunolabeling. At least 100 gold particles were counted, and the fraction of gold particles with respect to their location per area is depicted.
Taking advantage of purified HCV-induced membranes containing functional viral replicase, we next identified the sites of HCV RNA replication. NS4BHA-associated membranes were subjected to in vitro replicase assay using 5-bromouridine-5′-triphosphate (BrUTP), which was subsequently detected by immunogold labeling and TEM. Only background labeling of membranes was detected with CANXHA control ER membranes (data not shown) and membranes subjected to reaction mixtures lacking BrUTP (Fig. 6Ca). By contrast, ∼50% of DMVs were immunolabeled after in vitro replicase reaction with BrUTP (Fig. 6Cb and c). The gold label was detected on the outsides of DMVs (27%) (Fig. 6Cb) and also in their interiors (23%) (Fig. 6Cc). In the case of the latter, DMVs appeared to be open toward the exterior. However, we cannot determine whether these DMVs correspond to not yet fully closed structures as was recently described for HCV-infected cells (19) or whether membrane discontinuities have been introduced by purification or subsequent immunolabeling procedures. Quantification of the relative distribution of BrU incorporated into nascent HCV RNA revealed that ∼50% of the label appeared to reside on membranes and ∼25% in the DMV interior (Fig. 6D). In summary, these results demonstrate that DMVs contain active HCV replicase. Our data suggest that DMVs are the sites of HCV RNA replication, which might occur at least in part in the vesicle lumen, in agreement with the nuclease protection of the viral RNA.
HCV–co-opted cellular proteins VAP-A and -B are associated with DMVs.
As obligate intracellular parasites, viruses exploit host cell functions to a large extent. Indeed, the subversion of cellular proteins by positive-strand RNA viruses, including HCV, has been a major focus of research in recent years (reviewed in references 18 and 36). However, their localization with respect to HCV-remodeled membranes remains largely unknown. Hence, we wondered whether purified DMVs were suitable for identification of host cell proteins associated with these membranes. Among HCV–co-opted host factors, VAP-A and VAP-B have been identified as NS5A and NS5B interactors and are required for viral RNA replication (37, 38, 39). Thus, as a proof of concept, we investigated whether these host proteins are associated with DMVs by using immunogold labeling of purified HCV-induced membranes and TEM. Membrane structures were double labeled for the viral replicase protein NS5A in order to unambiguously identify DMVs and additionally for VAP-A or VAP-B. While only a low level of the background label of NS5A (15-nm colloidal gold) was observed on ER control membranes (Fig. 7Aa and b), cellular proteins VAP-A and -B (10-nm colloidal gold) were readily detected. Importantly, HCV-induced DMVs were labeled intensively with NS5A and with VAP-A and VAP-B (Fig. 7Ac and d). Interestingly, 10- and 15-nm colloidal gold, corresponding to VAP-A or -B and NS5A, respectively, were frequently found next to each other (Fig. 7Ac and d), supporting earlier reports that these proteins interact with each other (37, 38, 39). Quantification of gold particles on membranes (Fig. 7B) confirmed the specificity of detection of NS5A and excluded cross-reactivity of used antibodies. Moreover, quantitative analysis of differently sized gold particles on membranes revealed that most NS5A-positive membranous structures from the NS4BHA capture also contained VAP-A or -B, while ER control membranes almost exclusively exhibited single labeling for VAP-A or -B (Fig. 7C). Finally, we calculated the number of 10-nm gold particles per micrometer of membrane of NS5SA-positive structures in order to explore whether VAP-A and -B would be locally concentrated on HCV-remodeled membranes. The results revealed an ∼2-fold enrichment of both cellular proteins on HCV-induced DMVs compared to that on ER control membranes (Fig. 7D).
Fig 7.

HCV–co-opted host proteins VAP-A and VAP-B are enriched in DMVs. (A) Purified ER control (a and b) and HCV-remodeled membranes (c and d) were consecutively labeled for NS5A (15-nm gold) and for VAP-A (a and c) or VAP-B (b and d) (10-nm gold). After negative staining, they were examined by TEM. White and black arrowheads highlight 10- and 15-nm gold particles, respectively. Scale bars represent 100 nm. (B) Quantification of gold particles on membranes. At least 100 gold particles were counted, and the number of gold particles associated with membranes per area of the grid is depicted. (C) Number of single- and double-labeled structures containing either NS5A or VAP-A/B, or both viral and cellular protein, respectively, per area on the grid. At least 50 membranous structures per condition were analyzed. (D) Amount of 10-nm colloidal gold particles per micrometer of membrane length. Only membranes positive for NS5A were considered for the analysis of NS4BHA-captured membranes. The analysis is based on ∼50 membranous structures with a total membrane length of more than 50 μm for each condition.
Cholesterol is highly enriched in HCV-induced DMVs.
Apart from cellular proteins, specific host cell lipids likely contribute to HCV RNA replication and remodeling of intracellular membranes (reviewed in reference 40). For instance, HCV nonstructural proteins appear to reside in detergent-resistant membranes (DRMs) (41). Although by analogy to lipid rafts one might assume that HCV-induced DMVs are enriched for sphingolipids and cholesterol, their lipid composition is unknown. Hence we sought to determine if cholesterol is present in DMV membranes, resembling described DRM properties, by using NS4BHA-purified material. For this we employed biotinylated and noncytolytic P-O that has been developed to specifically detect cholesterol in cellular membranes by EM (28). Purified membranes were double labeled for cholesterol using P-O and for NS5A as a marker of DMVs, and membranous structures were examined by TEM. CANXHA-purified ER control membranes were only sporadically decorated with gold particles after P-O immunolabeling, indicating an overall low abundance of cholesterol in ER membranes (Fig. 8Aa). Interestingly, some mostly vesicular structures in the CANXHA preparation showed higher labeling densities, possibly representing cholesterol-rich domains (Fig. 8Ab). By contrast, NS5A-positive DMVs were heavily labeled for cholesterol, arguing for massive accumulation of this lipid in DMV membranes (Fig. 8Ac and d). Quantification of 10- and 15-nm colloidal gold particles present on membranes per area of the grid showed explicit tagging of cholesterol and NS5A and excluded cross-reactivity of used antibodies (Fig. 8B). Determination of cholesterol label density revealed a significant, ∼9-fold enrichment of cholesterol in HCV-remodeled membranes compared to that in control ER membranes, with 3.5 ± 1.1 and 29.6 ± 2.6 (means ± standard error of the means [SEMs]) gold particles per micrometer of ER control and HCV DMV membranes, respectively (Fig. 8C). Labeling density was significantly reduced upon cholesterol depletion using methyl-β-cyclodextrin (MβCD) treatment, thus confirming the specificity of our labeling method (Fig. 8D and E).
Fig 8.

DMV membranes contain large amounts of cholesterol. (A) Purified ER control (a and b) and HCV-remodeled membranes (c and d) were consecutively labeled for cholesterol using biotinylated perfringolysin O (10-nm gold) and for NS5A (15-nm gold). After negative staining, they were examined by TEM. Scale bars correspond to 100 nm. (B) Quantification of gold particles on membranes. At least 100 gold particles were counted, and the number of gold particles associated with membranes per area of the grid is depicted. (C) Amount of 10-nm colloidal gold particles per micrometer of membrane length of a given structure. Only membranes also positive for NS5A were considered for the analysis of NS4BHA-captured membranes. The analysis is based on at least 75 membranous structures with a total membrane length of more than 60 μm for each condition. Horizontal lines indicate mean values. ***, P < 0.0001. (D) Purified ER control (CANXHA) and HCV-remodeled membranes (NS4BHA) were treated with 10 mM MβCD for 1 h at 25°C as indicated, subsequently labeled for cholesterol using biotinylated perfringolysin O, and after negative staining examined by TEM. Scale bars correspond to 100 nm. (E) Amount of 10-nm colloidal gold particles per micrometer of membrane length of a given structure. The analysis is based on at least 50 membranous structures with a total membrane length of more than 50 μm for each condition. Horizontal lines indicate mean values. ***, P < 0.0001. (F) DMV diameters after MβCD treatment. Membranes from the NS4BHA capture were MβCD treated as described for panel D, and DMV diameters were measured. Horizontal lines indicate mean values. n > 100; ***, P < 0.0001. (G) Equal aliquots of NS4BHA-purified material were subjected to treatment with methyl-β-cyclodextrin (MβCD; 10 mM), nuclease (1 U/μl Benzonase), and/or detergent (1% Triton X-100) for 1 h at 25°C as indicated at the bottom. HCV RNA was extracted and quantified by reverse transcription-quantitative PCR. Mean values and error bars indicating the standard deviations for three independent experiments are depicted. #, P > 0.05; ***, P < 0.0001.
In order to investigate a functional role of cholesterol in HCV-remodeled membranes, we depleted it from DMV membranes by MβCD treatment and quantified HCV RNA copies as a measure of DMV integrity. The HCV RNA amount was reduced ∼2-fold (Fig. 8G), indicating a structural function of cholesterol for DMV and thus for HCV RNA stability. However, nuclease treatment of cholesterol-depleted membranes did not further decrease HCV RNA amounts, as seen in the case of detergent-only or detergent-plus-nuclease treatment (Fig. 8G), thus excluding a general membrane solubilization by MβCD. Indeed, when examined by TEM, MβCD-treated NS4BHA-captured membranes still exhibited a vesicular morphology (Fig. 8D). However, vesicle size was significantly decreased for cholesterol-depleted membranes, with a mean diameter of 121 ± 5.1 nm compared to 183 ± 5.5 nm (means ± SEMs) in the untreated control (Fig. 8F). These results emphasize the role of cholesterol as a structural component of DMV membranes and demonstrate a distinct lipid composition for HCV-induced DMVs.
DISCUSSION
An important role of HCV-induced DMVs in viral replication has been suggested by several studies (15, 19, 20, 42). However, viral and cellular constituents of DMVs, as well as their precise function in RNA replication, remain enigmatic. Here we report a functional HA-tagged version of NS4B that allows purification of HCV-remodeled membranes and their biochemical and morphological characterization. Our results provide evidence that HCV-induced DMVs harbor active viral replicase complexes and represent specialized membranous structures composed of distinct cellular protein and lipid constituents.
HCV replication is only moderately affected by the insertion of an HA affinity tag in NS4B.
Epitope tagging is a commonly used method to allow purification of proteins with high efficiency. Although it has been used with success in the case of HCV (for examples, see references 43, 44, and 45), the main problem is to identify a site that tolerates the insertion of a heterologous sequence without affecting the functionality of the viral protein. In this respect, NS4B has proven to be very sensitive to sequence alterations, since the exchange of single amino acid residues often exhibits dramatic effects on viral RNA replication (14, 15, 30, 46). Thus, we based our initial analysis on sites within NS4B that previously were shown to tolerate the insertion of a short sequence (29) and inserted the FLAG or HA tag. While most of our insertion constructs were not viable, an NS4B containing an HA tag insertion after Ala-38 still supported HCV RNA replication when at least one additional mutation was present. Interestingly, these pseudoreversions eliminated a negative charge (D34A) or introduced an additional positive charge (Q11R, Q31R, Q117R) (Table 1), indicating compensation of the negative charge resulting from two aspartic acid residues within the HA-coding sequence. This might also explain why four of the five aspartic acid residues within the FLAG sequence were deleted after adaptation of the subgenomic replicon containing the FLAG-coding sequence after Ala-38 (Table 1). Hence, the introduction of negatively charged amino acids by an affinity tag into the NS4B N-terminal domain might interfere with its reported membrane association (47, 48) due to electrostatic repulsion with the negatively charged head groups of membrane phospholipids. Additionally, posttranslational membrane translocation of the NS4B N-terminal domain into the ER lumen (48, 49) could be inhibited. Nonetheless, NS4BHA31R supported all NS4B-associated functions in the viral replication cycle, at least those required in cell culture (Fig. 1D and E and 2A to C). Thus, the advantages of the HA-tagged NS4B for specifically purifying HCV-remodeled membranous structures greatly outweigh the drawback of delayed replication kinetics for at least two reasons: first, because highly specific and efficient HA purification overcomes the inherent limitations of antibodies directly targeting viral proteins and, second, because the possibility of elution of immunocaptured samples with the HA peptide, i.e., under native conditions, is even more important and key to the success of the present study, which would not have been possible with an antibody directly targeting the NS4B protein.
Double-membrane vesicles contain active HCV replicase.
Despite detailed knowledge about the three-dimensional architecture of DMVs in HCV-infected cells (19), determination of their viral and cellular constituents and their exact role in the viral replication cycle is lacking. This is due to difficulties in producing biochemical quantities of highly purified HCV-remodeled membranes. We have overcome this problem and provide conclusive evidence that DMVs contain the viral replicase machinery. First, we showed that replicase proteins NS3 and NS5A, as well as dsRNA, are associated with purified DMVs. Second, we showed that NS4BHA-captured membranes contain active viral replicase complexes. Third, by using metabolic labeling of nascent viral RNA we demonstrated that DMVs are sites of active HCV RNA replication. The latter result is of high importance, as detection of metabolically labeled viral RNA is the only reliable method of identifying viral replication sites. In situ hybridization or detection of viral RNA by dsRNA-specific antibodies does not discriminate between actively replicating RNA and RNA engaged in other reactions, such as translation or virion formation. In fact, the dsRNA-specific antibody can also bind to dsRNA structures present in the genomes of positive-strand RNA viruses, thus excluding specificity for detection of active viral RNA replication sites and explaining the disconnect between the signals of newly synthesized RNA and dsRNA described, e.g., for coronaviruses (50). BrUTP labeling in combination with TEM overcomes these limitations and has also been successfully applied to DMV replication factories of arteriviruses (51) and coronaviruses (52) and to spherule-type replication factories of rubella virus (53), alphaviruses (54), flock house virus (55), and brome mosaic virus (56).
Based on our findings, we propose a model of the membrane-associated HCV replicase complex (Fig. 9). DMVs are formed by an as-yet-unknown mechanism that requires the concerted action of several HCV replicase proteins (15, 19). Our data show that these DMVs contain viral replicase proteins NS3, -4B, and -5A, as well as viral RNA and replicase activity, thus representing a site of viral RNA storage and amplification (Fig. 9). It is not yet clear whether HCV RNA replication occurs primarily on the outer DMV membrane facing the cytoplasm as proposed for picornaviruses (57, 58) or whether it takes place mainly in the DMV lumen as suggested for coronaviruses (51, 52) or both (Fig. 9). Detection of incorporated BrU within the lumen of DMVs requires accessibility to relatively large antibodies and protein A-conjugated 10-nm colloidal gold particles, which might not be possible for the majority of DMVs that exhibit “sealed” inner and outer membranes (19). Nevertheless, a subset of DMVs with apparent membrane discontinuity stained positive for BrU in their interiors, but we cannot determine whether those DMVs correspond to a subset of not yet fully closed structures as observed in HCV-infected cells (19) or whether membrane alterations have been introduced by purification or subsequent immunolabeling procedures. DMV luminal replication sites would best fit theoretical assumptions that consider membranous replication factories to be shielded subcompartments protecting viral replication machinery from nucleases and intracellular RNA sensors (16, 18, 33, 35, 58, 59). This topology requires extensive transport of metabolites and viral RNA between the cytosol and the DMV lumen, which could occur directly for DMVs that are not yet closed and thus linked to the cytosol (19), or might be mediated via a previously unknown transport function of a viral and/or cellular protein (Fig. 9), as was also proposed for coronaviruses (60). In any case, we want to point out that our model does not exclude the possibility that viral RNA replication might also occur on other membranous structures, such as single membrane vesicles, that were either not enriched with our method or not detectable, e.g., because of low replicase activity. Further studies will be required to clarify this possibility.
Fig 9.

Schematic model of an HCV-induced DMV. Cellular lipids, viral RNA, and viral and cellular proteins are depicted as indicated in the panel on the right. The HCV replicase complex is associated with DMV membranes, and the bulk of viral RNA resides in a nuclease-protected environment, likely the DMV lumen. HCV creates a unique membrane environment containing co-opted cellular proteins, exemplified here by VAP-A/B, and also modifies the membrane lipid composition, highlighted by the large quantity of cholesterol. It remains to be determined how transport to and out of the DMV interior might be mediated, as indicated by the question marks. Possible scenarios are not (yet) completely sealed DMVs or a distinct transporter function.
Cellular components of HCV replication factories.
Per their definition as obligate intracellular parasites, all viruses highjack infected host cells in order to replicate their genomes and generate viral progeny. Positive-strand RNA viruses, such as HCV, depend especially heavily on the exploitation of metabolic pathways. Thus, subversion of host cell factors by HCV and other positive-strand RNA viruses has been a major focus of research in recent years (36) and might ultimately lead to the development of host-targeting antiviral drugs (61). Despite detailed knowledge about virus dependency on certain cellular proteins and proven direct interactions between host and viral factors, little is known about their exact localization with respect to viral replication sites. As a proof of principle, we investigated the localization of two well-described HCV host dependency factors, VAP-A and VAP-B (37, 38, 39), which in uninfected cells are implicated in ER homeostasis and vesicular trafficking (62, 63). We verified their presence in DMVs and demonstrated enrichment in the HCV-induced membranous compartment. Hence, we propose that other cellular replicase cofactors are recruited to and thus enriched in HCV-induced DMVs. Thus, determination of the host cell proteome associated with purified DMVs will be the next important task in identifying novel HCV–co-opted host factors.
Beside proteins, lipids constituting membranes of positive-strand RNA virus replication factories are of major interest. Although this group of viruses is known to alter cellular lipid homeostasis and depend on certain lipids for viral propagation (reviewed in reference 18), knowledge about the lipid composition of these specialized membrane compartments is scarce. A fundamental question is whether positive-strand RNA viruses simply reshape preexisting “donor” membranes (e.g., the ER in the case of HCV) without changing their properties or whether they create special membrane compartments with a distinct lipid composition. To address this point, we investigated the cholesterol content of HCV-induced DMVs in comparison to that of ER control membranes, since HCV nonstructural proteins are described to reside in detergent-resistant membranes (41). Strikingly, cholesterol was found to be highly concentrated in DMVs. The ∼9-fold enrichment, as determined by quantitative immunolabeling, indicates a very high relative cholesterol concentration in DMV membranes. Such an enrichment of cholesterol in HCV-induced DMVs would result in local cholesterol concentrations that might be similar to that of endosomes or the plasma membrane (64). Since cholesterol enhances membrane rigidity and stability, it could serve as an important structural component of HCV-remodeled membranes (Fig. 9) and possibly for other DMV-inducing viruses, such as picornaviruses, coronaviruses, and arteriviruses, as well. The reduced DMV size and decreased HCV genome stability induced by cholesterol removal support this assumption.
How could HCV induce this high local concentration of membrane cholesterol? The striking difference between ER and DMV cholesterol contents argues against the simple usage of preexisting cholesterol in ER membranes and favors an active recruitment of cholesterol to DMVs. In spite of low ER cholesterol steady-state levels, this organelle is the site of cholesterol synthesis, implying a high turnover. Hence, ER-localized cholesterol could be recruited and thus concentrated during DMV formation by cholesterol-binding proteins associated with viral replicase proteins. Another appealing hypothesis is that cholesterol enrichment in HCV-remodeled membranes might be mediated via another lipid, phosphatidyl-inositol-4-phosphate (PI4P), and oxysterol-binding proteins (OSBPs). This protein family is implicated in nonvesicular sterol transport between different membrane compartments (65). Characteristic features are the N-terminal pleckstrin homology (PH) domain mediating PI4P binding and the C-terminal sterol-binding domain. OSBPs have been shown to exchange sterols for PI4P between different membranes (66) and thus, in a simplified view, transport cholesterol to PI4P-rich compartments, such as the Golgi compartment and the plasma membrane. However, HCV, as well as picornaviruses, prominently redistributes and induces intracellular PI4P pools by subversion of phosphatidyl-inositol-4-kinase class III alpha and beta (PI4KIII α/β), respectively (67, 68, 69). Furthermore, OSBPs interact via an FFAT motif with VAP-A and VAP-B (62, 70), which are also enriched in HCV-induced DMVs. Hence, virus-induced locally elevated PI4P as well as VAP concentrations might attract OSBPs, delivering cholesterol to viral replication organelles. Further studies are needed to address this unifying hypothesis.
In summary, the experimental approach reported here will help to unravel the biochemical properties of HCV replication factories. The methodology should also be applicable to other DMV-inducing viruses, such as picornaviruses, coronaviruses, and arteriviruses. High-throughput quantitative analyses will unravel the protein and lipid constituents of the membranous replication organelles of viruses and thus provide fundamental insights into the cell biology of viral infection.
ACKNOWLEDGMENTS
We thank U. Herian and F. Huschmand for excellent technical and IT assistance, Francois Penin for the image template of HCV protein topology, and members of the Molecular Virology Research Unit for indispensable, stimulating discussions. We thank Wiebke Möbius for biotinylated perfringolysin O, Takaji Wakita for the gift of the JFH-1 clone, and Charles Rice for Huh7.5 cells and the 9E10 antibody. We are grateful to the Electron Microscopy Core Facility of the University of Heidelberg for their continuous support and for providing access to their unit.
This work was supported by a grant from the Deutsche Forschungsgemeinschaft (SFB/TRR 83, Teilprojekt 13).
Footnotes
Published ahead of print 24 July 2013
REFERENCES
- 1.Lavanchy D. 2009. The global burden of hepatitis C. Liver Int. 29(Suppl 1):74–81 [DOI] [PubMed] [Google Scholar]
- 2.Sarrazin C, Hezode C, Zeuzem S, Pawlotsky JM. 2012. Antiviral strategies in hepatitis C virus infection. J. Hepatol. 56(Suppl 1):S88–S100 [DOI] [PubMed] [Google Scholar]
- 3.Vermehren J, Susser S, Lange CM, Forestier N, Karey U, Hughes E, Ralston R, Tong X, Zeuzem S, Sarrazin C. 2012. Mutations selected in the hepatitis C virus NS3 protease domain during sequential treatment with boceprevir with and without pegylated interferon alfa-2b. J. Viral Hepat. 19:120–127 [DOI] [PubMed] [Google Scholar]
- 4.van Regenmortel MHV, Fauquet CM, Bishop DHL, Carstens EB, Estes MK, Lemon SM, Maniloff J, Mayo MA, McGeoch DJ, Pringle CR, Wickner RB. (ed). 2000. Virus taxonomy. Seventh report of the International Committee on Taxonomy of Viruses. Academic Press, San Diego, CA [Google Scholar]
- 5.Simmonds P, Bukh J, Combet C, Deleage G, Enomoto N, Feinstone S, Halfon P, Inchauspe G, Kuiken C, Maertens G, Mizokami M, Murphy DG, Okamoto H, Pawlotsky JM, Penin F, Sablon E, Shin I, Stuyver LJ, Thiel HJ, Viazov S, Weiner AJ, Widell A. 2005. Consensus proposals for a unified system of nomenclature of hepatitis C virus genotypes. Hepatology 42:962–973 [DOI] [PubMed] [Google Scholar]
- 6.Bartenschlager R, Frese M, Pietschmann T. 2004. Novel insights into hepatitis C virus replication and persistence. Adv. Virus Res. 63:71–180 [DOI] [PubMed] [Google Scholar]
- 7.Jones CT, Murray CL, Eastman DK, Tassello J, Rice CM. 2007. Hepatitis C virus p7 and NS2 proteins are essential for production of infectious virus. J. Virol. 81:8374–8383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jirasko V, Montserret R, Appel N, Janvier A, Eustachi L, Brohm C, Steinmann E, Pietschmann T, Penin F, Bartenschlager R. 2008. Structural and functional characterization of nonstructural protein 2 for its role in hepatitis C virus assembly. J. Biol. Chem. 283:28546–28562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Steinmann E, Penin F, Kallis S, Patel AH, Bartenschlager R, Pietschmann T. 2007. Hepatitis C virus p7 protein is crucial for assembly and release of infectious virions. PLoS Pathog. 3:e103. 10.1371/journal.ppat.0030103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lohmann V, Körner F, Koch JO, Herian U, Theilmann L, Bartenschlager R. 1999. Replication of subgenomic hepatitis C virus RNAs in a hepatoma cell line. Science 285:110–113 [DOI] [PubMed] [Google Scholar]
- 11.Blight KJ, Kolykhalov AA, Rice CM. 2000. Efficient initiation of HCV RNA replication in cell culture. Science 290:1972–1974 [DOI] [PubMed] [Google Scholar]
- 12.Horscroft N, Lai VC, Cheney W, Yao N, Wu JZ, Hong Z, Zhong W. 2005. Replicon cell culture system as a valuable tool in antiviral drug discovery against hepatitis C virus. Antivir. Chem. Chemother. 16:1–12 [DOI] [PubMed] [Google Scholar]
- 13.Egger D, Wolk B, Gosert R, Bianchi L, Blum HE, Moradpour D, Bienz K. 2002. Expression of hepatitis C virus proteins induces distinct membrane alterations including a candidate viral replication complex. J. Virol. 76:5974–5984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gouttenoire J, Roingeard P, Penin F, Moradpour D. 2010. Amphipathic α-helix AH2 is a major determinant for the oligomerization of hepatitis C virus nonstructural protein 4B. J. Virol. 84:12529–12537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Paul D, Romero-Brey I, Gouttenoire J, Stoitsova S, Krijnse-Locker J, Moradpour D, Bartenschlager R. 2011. NS4B self-interaction through conserved C-terminal elements is required for the establishment of functional hepatitis C virus replication complexes. J. Virol. 85:6963–6976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.den Boon JA, Ahlquist P. 2010. Organelle-like membrane compartmentalization of positive-strand RNA virus replication factories. Annu. Rev. Microbiol. 64:241–256 [DOI] [PubMed] [Google Scholar]
- 17.Belov GA, van Kuppeveld FJ. 2012. (+)RNA viruses rewire cellular pathways to build replication organelles. Curr. Opin. Virol. 2:740–747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Paul D, Bartenschlager R. 2013. Architecture and biogenesis of plus-strand RNA virus replication factories. World J. Virol. 2:32–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Romero-Brey I, Merz A, Chiramel A, Lee JY, Chlanda P, Haselman U, Santarella-Mellwig R, Habermann A, Hoppe S, Kallis S, Walther P, Antony C, Krijnse-Locker J, Bartenschlager R. 2012. Three-dimensional architecture and biogenesis of membrane structures associated with hepatitis C virus replication. PLoS Pathog. 8:e1003056. 10.1371/journal.ppat.1003056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ferraris P, Blanchard E, Roingeard P. 2010. Ultrastructural and biochemical analyses of hepatitis C virus-associated host cell membranes. J. Gen. Virol. 91:2230–2237 [DOI] [PubMed] [Google Scholar]
- 21.Gouttenoire J, Penin F, Moradpour D. 2010. Hepatitis C virus nonstructural protein 4B: a journey into unexplored territory. Rev. Med. Virol. 20:117–129 [DOI] [PubMed] [Google Scholar]
- 22.Lohmann V, Korner F, Herian U, Bartenschlager R. 1997. Biochemical properties of hepatitis C virus NS5B RNA-dependent RNA polymerase and identification of amino acid sequence motifs essential for enzymatic activity. J. Virol. 71:8416–8428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Friebe P, Boudet J, Simorre JP, Bartenschlager R. 2005. Kissing-loop interaction in the 3′ end of the hepatitis C virus genome essential for RNA replication. J. Virol. 79:380–392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Blight KJ, McKeating JA, Rice CM. 2002. Highly permissive cell lines for subgenomic and genomic hepatitis C virus RNA replication. J. Virol. 76:13001–13014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Steinmann E, Brohm C, Kallis S, Bartenschlager R, Pietschmann T. 2008. Efficient trans-encapsidation of hepatitis C virus RNAs into infectious virus-like particles. J. Virol. 82:7034–7046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schaller T, Appel N, Koutsoudakis G, Kallis S, Lohmann V, Pietschmann T, Bartenschlager R. 2007. Analysis of hepatitis C virus superinfection exclusion by using novel fluorochrome gene-tagged viral genomes. J. Virol. 81:4591–4603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pietschmann T, Kaul A, Koutsoudakis G, Shavinskaya A, Kallis S, Steinmann E, Abid K, Negro F, Dreux M, Cosset FL, Bartenschlager R. 2006. Construction and characterization of infectious intragenotypic and intergenotypic hepatitis C virus chimeras. Proc. Natl. Acad. Sci. U. S. A. 103:7408–7413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Möbius W, Ohno-Iwashita Y, van Donselaar EG, Oorschot VM, Shimada Y, Fujimoto T, Heijnen HF, Geuze HJ, Slot JW. 2002. Immunoelectron microscopic localization of cholesterol using biotinylated and non-cytolytic perfringolysin O. J. Histochem. Cytochem. 50:43–55 [DOI] [PubMed] [Google Scholar]
- 29.Arumugaswami V, Remenyi R, Kanagavel V, Sue EY, Ngoc HT, Liu C, Fontanes V, Dasgupta A, Sun R. 2008. High-resolution functional profiling of hepatitis C virus genome. PLoS Pathog. 4:e1000182. 10.1371/journal.ppat.1000182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jones DM, Patel AH, Targett-Adams P, McLauchlan J. 2009. The hepatitis C virus NS4B protein can trans-complement viral RNA replication and modulates production of infectious virus. J. Virol. 83:2163–2177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lundin M, Monne M, Widell A, Von HG, Persson MA. 2003. Topology of the membrane-associated hepatitis C virus protein NS4B. J. Virol. 77:5428–5438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gretton SN, Taylor AI, McLauchlan J. 2005. Mobility of the hepatitis C virus NS4B protein on the endoplasmic reticulum membrane and membrane-associated foci. J. Gen. Virol. 86:1415–1421 [DOI] [PubMed] [Google Scholar]
- 33.Miyanari Y, Hijikata M, Yamaji M, Hosaka M, Takahashi H, Shimotohno K. 2003. Hepatitis C virus non-structural proteins in the probable membranous compartment function in viral genome replication. J. Biol. Chem. 278:50301–50308 [DOI] [PubMed] [Google Scholar]
- 34.El-Hage N, Luo G. 2003. Replication of hepatitis C virus RNA occurs in a membrane-bound replication complex containing nonstructural viral proteins and RNA. J. Gen. Virol. 84:2761–2769 [DOI] [PubMed] [Google Scholar]
- 35.Quinkert D, Bartenschlager R, Lohmann V. 2005. Quantitative analysis of the hepatitis C virus replication complex. J. Virol. 79:13594–13605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nagy PD, Pogany J. 2012. The dependence of viral RNA replication on co-opted host factors. Nat. Rev. Microbiol. 10:137–149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Evans MJ, Rice CM, Goff SP. 2004. Phosphorylation of hepatitis C virus nonstructural protein 5A modulates its protein interactions and viral RNA replication. Proc. Natl. Acad. Sci. U. S. A. 101:13038–13043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gao L, Aizaki H, He JW, Lai MM. 2004. Interactions between viral nonstructural proteins and host protein hVAP-33 mediate the formation of hepatitis C virus RNA replication complex on lipid raft. J. Virol. 78:3480–3488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hamamoto I, Nishimura Y, Okamoto T, Aizaki H, Liu M, Mori Y, Abe T, Suzuki T, Lai MM, Miyamura T, Moriishi K, Matsuura Y. 2005. Human VAP-B is involved in hepatitis C virus replication through interaction with NS5A and NS5B. J. Virol. 79:13473–13482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Alvisi G, Madan V, Bartenschlager R. 2011. Hepatitis C virus and host cell lipids: an intimate connection. RNA Biol. 8:258–269 [DOI] [PubMed] [Google Scholar]
- 41.Shi ST, Lee KJ, Aizaki H, Hwang SB, Lai MM. 2003. Hepatitis C virus RNA replication occurs on a detergent-resistant membrane that cofractionates with caveolin-2. J. Virol. 77:4160–4168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ferraris P, Beaumont E, Uzbekov R, Brand D, Gaillard J, Blanchard E, Roingeard P. 2013. Sequential biogenesis of host cell membrane rearrangements induced by hepatitis C virus infection. Cell. Mol. Life Sci. 70:1297–1306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Merz A, Long G, Hiet MS, Brugger B, Chlanda P, Andre P, Wieland F, Krijnse-Locker J, Bartenschlager R. 2011. Biochemical and morphological properties of hepatitis C virus particles and determination of their lipidome. J. Biol. Chem. 286:3018–3032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jirasko V, Montserret R, Lee JY, Gouttenoire J, Moradpour D, Penin F, Bartenschlager R. 2010. Structural and functional studies of nonstructural protein 2 of the hepatitis C virus reveal its key role as organizer of virion assembly. PLoS Pathog. 6:e1001233. 10.1371/journal.ppat.1001233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Vieyres G, Brohm C, Friesland M, Gentzsch J, Wolk B, Roingeard P, Steinmann E, Pietschmann T. 2013. Subcellular localization and function of an epitope-tagged p7 viroporin in hepatitis C virus-producing cells. J. Virol. 87:1664–1678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Blight KJ. 2011. Charged residues in hepatitis C virus NS4B are critical for multiple NS4B functions in RNA replication. J. Virol. 85:8158–8171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Elazar M, Liu P, Rice CM, Glenn JS. 2004. An N-terminal amphipathic helix in hepatitis C virus (HCV) NS4B mediates membrane association, correct localization of replication complex proteins, and HCV RNA replication. J. Virol. 78:11393–11400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gouttenoire J, Castet V, Montserret R, Arora N, Raussens V, Ruysschaert JM, Diesis E, Blum HE, Penin F, Moradpour D. 2009. Identification of a novel determinant for membrane association in hepatitis C virus nonstructural protein 4B. J. Virol. 83:6257–6268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lundin M, Lindstrom H, Gronwall C, Persson MA. 2006. Dual topology of the processed hepatitis C virus protein NS4B is influenced by the NS5A protein. J. Gen. Virol. 87:3263–3272 [DOI] [PubMed] [Google Scholar]
- 50.Hagemeijer MC, Vonk AM, Monastyrska I, Rottier PJ, de Haan CA. 2012. Visualizing coronavirus RNA synthesis in time by using click chemistry. J. Virol. 86:5808–5816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pedersen KW, van der Meer Y, Roos N, Snijder EJ. 1999. Open reading frame 1a-encoded subunits of the arterivirus replicase induce endoplasmic reticulum-derived double-membrane vesicles which carry the viral replication complex. J. Virol. 73:2016–2026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gosert R, Kanjanahaluethai A, Egger D, Bienz K, Baker SC. 2002. RNA replication of mouse hepatitis virus takes place at double-membrane vesicles. J. Virol. 76:3697–3708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kujala P, Ahola T, Ehsani N, Auvinen P, Vihinen H, Kaariainen L. 1999. Intracellular distribution of rubella virus nonstructural protein P150. J. Virol. 73:7805–7811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kujala P, Ikaheimonen A, Ehsani N, Vihinen H, Auvinen P, Kaariainen L. 2001. Biogenesis of the Semliki Forest virus RNA replication complex. J. Virol. 75:3873–3884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kopek BG, Perkins G, Miller DJ, Ellisman MH, Ahlquist P. 2007. Three-dimensional analysis of a viral RNA replication complex reveals a virus-induced mini-organelle. PLoS Biol. 5:e220. 10.1371/journal.pbio.0050220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Schwartz M, Chen J, Janda M, Sullivan M, den Boon J, Ahlquist P. 2002. A positive-strand RNA virus replication complex parallels form and function of retrovirus capsids. Mol. Cell 9:505–514 [DOI] [PubMed] [Google Scholar]
- 57.Bienz K, Egger D, Pasamontes L. 1987. Association of polioviral proteins of the P2 genomic region with the viral replication complex and virus-induced membrane synthesis as visualized by electron microscopic immunocytochemistry and autoradiography. Virology 160:220–226 [DOI] [PubMed] [Google Scholar]
- 58.Belov GA, Nair V, Hansen BT, Hoyt FH, Fischer ER, Ehrenfeld E. 2012. Complex dynamic development of poliovirus membranous replication complexes. J. Virol. 86:302–312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kato H, Takahasi K, Fujita T. 2011. RIG-I-like receptors: cytoplasmic sensors for non-self RNA. Immunol. Rev. 243:91–98 [DOI] [PubMed] [Google Scholar]
- 60.Knoops K, Kikkert M, Worm SH, Zevenhoven-Dobbe JC, van der Meer Y, Koster AJ, Mommaas AM, Snijder EJ. 2008. SARS-coronavirus replication is supported by a reticulovesicular network of modified endoplasmic reticulum. PLoS Biol. 6:e226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.von Hahn T, Ciesek S, Manns MP. 2011. Arrest all accessories–inhibition of hepatitis C virus by compounds that target host factors. Discov. Med. 12:237–244 [PubMed] [Google Scholar]
- 62.Wyles JP, McMaster CR, Ridgway ND. 2002. Vesicle-associated membrane protein-associated protein-A (VAP-A) interacts with the oxysterol-binding protein to modify export from the endoplasmic reticulum. J. Biol. Chem. 277:29908–29918 [DOI] [PubMed] [Google Scholar]
- 63.Amarilio R, Ramachandran S, Sabanay H, Lev S. 2005. Differential regulation of endoplasmic reticulum structure through VAP-Nir protein interaction. J. Biol. Chem. 280:5934–5944 [DOI] [PubMed] [Google Scholar]
- 64.van Meer G, Voelker DR, Feigenson GW. 2008. Membrane lipids: where they are and how they behave. Nat. Rev. Mol. Cell Biol. 9:112–124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ngo MH, Colbourne TR, Ridgway ND. 2010. Functional implications of sterol transport by the oxysterol-binding protein gene family. Biochem. J. 429:13–24 [DOI] [PubMed] [Google Scholar]
- 66.de Saint-Jean M, Delfosse V, Douguet D, Chicanne G, Payrastre B, Bourguet W, Antonny B, Drin G. 2011. Osh4p exchanges sterols for phosphatidylinositol 4-phosphate between lipid bilayers. J. Cell Biol. 195:965–978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hsu NY, Ilnytska O, Belov G, Santiana M, Chen YH, Takvorian PM, Pau C, van der Schaar H, Kaushik-Basu N, Balla T, Cameron CE, Ehrenfeld E, van Kuppeveld FJ, Altan-Bonnet N. 2010. Viral reorganization of the secretory pathway generates distinct organelles for RNA replication. Cell 141:799–811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Reiss S, Rebhan I, Backes P, Romero-Brey I, Erfle H, Matula P, Kaderali L, Poenisch M, Blankenburg H, Hiet MS, Longerich T, Diehl S, Ramirez F, Balla T, Rohr K, Kaul A, Buhler S, Pepperkok R, Lengauer T, Albrecht M, Eils R, Schirmacher P, Lohmann V, Bartenschlager R. 2011. Recruitment and activation of a lipid kinase by hepatitis C virus NS5A is essential for integrity of the membranous replication compartment. Cell Host Microbe 9:32–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bianco A, Reghellin V, Donnici L, Fenu S, Alvarez R, Baruffa C, Peri F, Pagani M, Abrignani S, Neddermann P, De Francesco R. 2012. Metabolism of phosphatidylinositol 4-kinase IIIα-dependent PI4P is subverted by HCV and is targeted by a 4-anilino quinazoline with antiviral activity. PLoS Pathog. 8:e1002576. 10.1371/journal.ppat.1002576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Loewen CJ, Roy A, Levine TP. 2003. A conserved ER targeting motif in three families of lipid binding proteins and in Opi1p binds VAP. EMBO J. 22:2025–2035 [DOI] [PMC free article] [PubMed] [Google Scholar]