

Abstract
To successfully complete their replication cycles, picornaviruses modify several host proteins to alter the cellular environment to favor virus production. One such target of viral proteinase cleavage is AU-rich binding factor 1 (AUF1), a cellular protein that binds to AU-rich elements, or AREs, in the 3′ noncoding regions (NCRs) of mRNAs to affect the stability of the RNA. Previous studies found that, during poliovirus or human rhinovirus infection, AUF1 is cleaved by the viral proteinase 3CD and that AUF1 can interact with the long 5′ NCR of these viruses in vitro. Here, we expand on these initial findings to demonstrate that all four isoforms of AUF1 bind directly to stem-loop IV of the poliovirus 5′ NCR, an interaction that is inhibited through proteolytic cleavage of AUF1 by the viral proteinase 3CD. Endogenous AUF1 was observed to relocalize to the cytoplasm of infected cells in a viral protein 2A-driven manner and to partially colocalize with the viral protein 3CD. We identify a negative role for AUF1 in poliovirus infection, as AUF1 inhibited viral translation and, ultimately, overall viral titers. Our findings also demonstrate that AUF1 functions as an antiviral factor during infection by coxsackievirus or human rhinovirus, suggesting a common mechanism that targets these related picornaviruses.
INTRODUCTION
The replication cycle of cytoplasmic RNA viruses involves a complex interplay between cellular and viral factors to alter the cellular environment for the production of progeny virions. For picornaviruses, this involves the shutdown of many host cell functions, including cellular transcription and cap-dependent translation, as well as the disruption of innate immune signaling and nucleocytoplasmic transport. This overhaul of cellular processes is accomplished mainly through the cleavage of key host proteins by the virally encoded proteinases. For enteroviruses, including poliovirus, coxsackievirus, and human rhinovirus (HRV), these proteinases include 2A and 3C/3CD. For poliovirus, cleavage of nucleoporin proteins in the nuclear membrane by the 2A proteinase disrupts nucleocytoplasmic trafficking, causing redistribution of several cellular proteins such as SRp20, hnRNP C, and Sam68 (1–3; for a review, see reference 4). Polypeptide 3CD is the precursor to the virally encoded RNA-dependent RNA polymerase 3Dpol and the proteinase 3Cpro. The poliovirus 3CD lacks polymerase activity but is proteolytically active and responsible for cleavage of the viral polyprotein as well as its own autocatalytic cleavage. Picornavirus 3C/3CD proteinases also have several known cellular targets, cleavage of which either directly aids in the viral replication cycle or disrupts cellular processes to the advantage of the virus. For example, cleavage of cellular proteins poly(rC) binding protein 2 (PCBP2) and polypyrimidine tract-binding protein (PTB) may play a role in viral RNA template switching from translation to RNA replication (for a review, see reference 5). Additionally, cleavage of other factors, such as RIG-I, MDA-5, and NF-κB, disrupts the cellular innate immune response (6–8; for a review, see reference 9).
We recently described another target of 3C/3CD cleavage, AU-rich binding factor 1 (AUF1), a cellular protein that binds to AU-rich elements, or AREs, in the 3′ noncoding regions (3′ NCRs) of mRNAs to affect the stability of the RNA (10). Through its interaction with AREs in specific cellular mRNAs, AUF1 (also known as hnRNP D) can target the RNAs for degradation via ARE-mediated decay. AUF1 binds specific proto-oncogene and cytokine mRNAs containing AREs, leading to degradation of these mRNAs (for a review, see reference 11). There are also a few examples of AUF1 acting to stabilize transcripts, for example, parathyroid hormone and estrogen receptor α mRNAs (12, 13). Cellular AUF1 exists in four isoforms, each resulting from alternative splicing of the same mRNA and named for its molecular mass, ranging from 37 to 45 kDa (Fig. 1) (14). All isoforms contain tandem RNA recognition motifs (RRMs), both of which are required for efficient RNA binding. The N terminus of all four isoforms of AUF1 contains a dimerization domain that is thought to contribute to oligomeric binding of AUF1 on ARE-containing RNAs (for a review, see reference 11).
Fig 1.

Schematic of human AUF1 isoforms and poliovirus/human rhinovirus 3CD cleavage site. AUF1 exists in four isoforms (p37, p40, p42, and p45), each resulting from alternative splicing of the same pre-mRNA with the inclusion or omission of exon 2 or exon 7. All isoforms contain a dimerization domain in exon 1 and tandem RNA recognition motifs (RRM1 and RRM2) required for efficient RNA binding. At the N terminus of all isoforms is an ideal recognition site for poliovirus or human rhinovirus 3C/3CD proteinase cleavage at the Q-G position, with an alanine in the P4 position, as shown in the top line displaying amino acid residues 29 to 42. To render this site uncleavable, the P1 and P1′ amino acids were mutatagenized to I and D, respectively, as previously described by Rozovics et al. (10).
For enteroviruses in the family Picornaviridae, the 5′ noncoding region (5′ NCR) of genomic RNAs contains a binding site(s) for AUF1 (10). These 5′ NCRs harbor both a viral replication element at the 5′ end (stem-loop I) and an internal ribosome entry site (IRES) encompassing stem-loops II to VI. Several other cellular proteins are known to bind to enterovirus 5′ NCRs to regulate translation or replication of the viral RNA. Most known binding factors, including PTB, La autoantigen, SRp20, unr, and PCBP2, act as positive regulators of translation (1, 15–21). PCBP2, a cellular RNA binding protein normally involved in cellular mRNA stability and translation regulation, binds to the poliovirus 5′ NCR and plays a role in translation and RNA replication initiation. In addition, PCBP2 has been shown to enhance the stability of poliovirus mRNAs (22). AUF1 was initially identified as another player in picornavirus replication via an RNA affinity screen for proteins interacting with the 5′ NCR of poliovirus RNA. Subsequently, endogenous AUF1 was observed to relocalize to the cytoplasm of poliovirus-infected cells, where it colocalized with the viral nonstructural protein 2B but was excluded from viral protein 3A-containing cytoplasmic regions. Near the N terminus of all isoforms is an optimal recognition site for poliovirus 3C/3CD proteinase cleavage (see Fig. 1). Cleavage of AUF1 at this site was confirmed for poliovirus and human rhinovirus 3CD proteinases both during infection and in vitro using recombinant proteins (10).
In the present report, we further define the interaction of AUF1 with the poliovirus 5′ NCR and find that the cleavage of AUF1 by poliovirus proteinase 3CD inhibits this interaction. While AUF1 and 3CD partially colocalize by 4 h after poliovirus infection, relocalization occurs in a 2A-driven manner. Using both in vitro assays and cells genetically ablated for AUF1, we discovered an inhibitory role for AUF1 during poliovirus infection, as the absence of AUF1 increases viral translation and viral titer. Additionally, higher titers for the related enterovirus, coxsackievirus B3 (CVB3), as well as human rhinovirus 1a (HRV1a), are produced in cells lacking AUF1. Taken together, these results suggest that AUF1 acts as a restriction factor during enterovirus/rhinovirus infections and that viral proteolytic cleavage of AUF1 functions as a mechanism to evade such a host cell antiviral response.
MATERIALS AND METHODS
Cell culture and DNA constructs.
MEF-AUF1+/+ and MEF-AUF1−/− mouse embryonic fibroblast (MEF) cell lines were a generous gift from Robert Schneider (New York University School of Medicine) (23). HeLa cells or MEF cells were grown as monolayers in Dulbecco's modified Eagle's medium (DMEM) supplemented with 8% newborn calf serum (NCS) or 10% fetal bovine serum (FBS), respectively. For MEF cells, plates were coated with 0.1% gelatin. Plasmids for purification of recombinant AUF1 isoforms were kindly provided by Robert Schneider. Expression plasmids for FLAG-tagged poliovirus 3CD and 2A proteinases are described elsewhere (24).
Generation of 3CD polyclonal antiserum.
Purification of 3CD(μ10) was performed as previously described (10). Purified fractions were pooled and separated on a 12.5% polyacrylamide gel. Side strips of the gel were removed and stained using colloidal blue (Life Technologies). Strips were then aligned, and portions of the unstained gel containing 3CD protein were excised and cut into small slices. Gel slices were emulsified in a 2-ml microcentrifuge tube with a 1-ml syringe plunger. Protein was passively eluted overnight at 30°C in 1 ml of elution buffer (50 mM Tris-HCl [pH 7.5], 150 mM NaCl, 0.1 mM EDTA). Samples were centrifuged at 7,500 RCF for 10 min, and supernatant was collected for antiserum generation. This purified protein was used to induce antiserum production in rabbits (Bethyl).
Immunofluorescence.
Immunofluorescence of N-terminal FLAG-tagged 2A or 3CD was performed as previously described (24). Briefly, HeLa cells seeded on coverslips were transfected with FLAG-tagged 2A or 3CD and fixed with 3.7% formaldehyde at different times posttransfection. Cells were incubated with anti-FLAG mouse monoclonal (Agilent; 1:1,000) or anti-AUF1 rabbit polyclonal (Millipore; 1:100) antibodies, followed by Alexa Fluor 594 goat anti-rabbit IgG and Alexa Fluor 488 goat anti-mouse IgG (Molecular Probes; 1:1,000). Nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI). Proteins were visualized with a Zeiss LSM700 laser scanning confocal microscope, and images were processed with Zen software.
For visualization of colocalization of endogenous AUF1 and 3CD, HeLa cells were seeded onto coverslips and either mock infected or infected with poliovirus at a multiplicity of infection (MOI) of 20 at 37°C after adsorption for 30 min at room temperature. Samples were fixed in formaldehyde for 15 min at different times postinfection and permeabilized in 0.5% NP-40–phosphate-buffered saline (PBS) for 5 min. Coverslips were washed three times in 1% NCS–PBS, blocked in 3% milk–1× PBS, and incubated with anti-AUF1 for 1 h. Cells were washed, and then Alexa Fluor 594 goat anti-rabbit IgG was added for 1 h to label anti-AUF1 antibody. Samples were washed and blocked with normal donkey serum (500 μg/ml) for 30 min followed by a second blocking step with unconjugated Fab fragments (Jackson ImmunoResearch) (7 μg/ml). A second formaldehyde fixation was performed, followed by incubation with anti-3CD488 (1:10) for 1 h. Anti-3CD antibody (generated as described above) was conjugated to Alexa Fluor 488 using an Apex antibody labeling kit (Molecular Probes) to generate anti-3CD488. Nuclei were labeled by DAPI staining, and coverslips were then mounted using mounting media (EMS), dried overnight, and imaged as described above. All steps were performed at room temperature unless otherwise indicated.
RNA affinity, electrophoretic mobility shift, and UV-cross-linking assays.
Preparation of NP-40 lysates from mock- or poliovirus-infected HeLa cells at 4 h postinfection was performed as described previously (10). For generation of RNA templates, pT7-5′NCR (25) was linearized with EcoRI (5′ NCR) or DdeI (stem-loop I). pT220-460 was linearized with HindIII to generate a transcription template corresponding to stem-loop IV (26). RNA transcription was performed using a MEGAshortscript T7 transcription kit (Ambion) in the presence of biotinylated CTP. Purification of RNA was performed using RNeasy columns (Qiagen). RNA affinity assays were performed as previously described (10). Briefly, precleared NP-40 cytoplasmic lysates from mock- or poliovirus-infected cells were incubated with biotinylated RNA-streptavidin resin for 2 h at 4°C in 50 mM KCl buffer (50 mM KCl, 5% glycerol, 1 mM dithiothreitol [DTT], 0.5 mM EDTA, 25 μM E. coli tRNA). Following three washes in 100 mM KCl buffer, complexes were resuspended in Laemmli sample buffer (LSB) and subjected to SDS-PAGE and Western blot analysis using anti-AUF1 rabbit polyclonal antibody (Millipore) or anti-PCBP2 mouse monoclonal antibody (27). When indicated, membranes were stripped using mild stripping buffer (25 mM glycine [pH 2.5], 1% SDS, 1% Tween 20). Membranes were washed twice in mild stripping buffer for 10 min each time at room temperature, washed four times with PBST (1× PBS, 0.1% Tween 20), and blocked with 3% milk–PBST for 1 h at room temperature prior to reprobing.
Preparation of recombinant AUF1 and poliovirus 3CD was performed as described elsewhere (10, 28); purified recombinant AUF1 isoforms were dialyzed into AUF1 dialysis buffer (20 mM Tris-HCl [pH 7.5], 1 mM DTT, 5% glycerol). Electrophoretic mobility shift assays were performed as previously described, using [32P]CTP (PerkinElmer) to label RNA probes during transcription (29). Recombinant purified AUF1 with or without recombinant purified 3CD was incubated in RNA-binding buffer (5 mM HEPES [pH 7.4], 2.5 mM MgCl2, 20 mM dithiothreitol, 3.8% glycerol, 1 mg/ml tRNA, 8 U of RNasin [Promega], 0.5 mg/ml bovine serum albumin) for 10 min at 30°C. Radiolabeled poliovirus stem-loop I or stem-loop IV RNA probe was then added to reach a final concentration of 5 nM for 10 min at 30°C. Glycerol was added to reach a 10% concentration, and complexes were resolved on a native 4% polyacrylamide gel at 4°C. Gels were dried for 2 h and imaged using a phosphorimager (Bio-Rad).
RNA probes for UV cross-linking assays were generated as described elsewhere (29), except that the RNA probes were transcribed in the presence of [32P]UTP (PerkinElmer). Conditions for UV cross-linking assays were the same as for the electrophoretic mobility shift assays except that after incubation at 30°C, samples were treated with 30 U of RNase T1 and 0.5 μg of RNase A for 30 min at 37°C. Samples were then exposed to 254-nm UV light for 1 min using the Stratalinker (Stratagene). LSB was added, samples were boiled, and products were then analyzed by SDS-PAGE. Poliovirus lysates labeled with [35S]methionine were used as molecular mass markers.
In vitro translation reactions.
HeLa S10 cytoplasmic extracts were generated as described elsewhere (29). HeLa S10 cytoplasmic extract (50% of final total volume) was incubated with DNA oligonucleotides, recombinant AUF1, or buffer alone in 80% of the final volume for 20 min at 30°C. DNA oligonucleotides (Operon) were reconstituted in RNase-free water. For addition of recombinant AUF1 isoforms, AUF1 dialysis buffer was used as the buffer-only control and for dilution of recombinant proteins. After a 20-min incubation, 250 ng poliovirus virion RNA, [35S]methionine (PerkinElmer), and all four buffer (1 mM ATP, 0.25 mM GTP, 0.25 mM UTP, 0.25 mM CTP, 60 mM potassium acetate, 30 mM creatine phosphate, 0.4 mg/ml creatine kinase, 15.5 mM HEPES-KOH [pH 7.4]) were added and translation was allowed to proceed for 4 h at 30°C. Samples were boiled with LSB and subjected to electrophoresis on an SDS-containing 12.5% polyacrylamide gel. Proteins were visualized by phosphorimaging following fluorography.
Transfection of poliovirus RNA.
Poliovirus virion RNA was generated as previously described (30). Transcript RNA for transfection into mouse cells was generated using MEGAscript T7 kits (Ambion) and RNeasy columns (Qiagen), with EcoRI-digested pT7PV1 plasmid (31) as a template. Poliovirus RNA was incubated with TS buffer (137 mM NaCl, 4.4 mM KCl, 0.7 mM Na2HPO4, 0.5 mM MgCl2, 0.68 mM CaCl2, 25 mM Tris [pH 7.5]) and 1 mg/ml DEAE-dextran. MEF monolayers were washed twice with 1× PBS, and then 125 μl of transfection mixture per 10-cm2 plate of cell monolayer was added to the cells in a dropwise manner. After 30 min incubation at room temperature, DMEM–10% FBS was added and cells were incubated at 37°C. Cells and supernatant were collected at indicated times after transfection and subjected to five freeze-thaw cycles, and plaque assays were performed on HeLa cells to determine the titer.
Virus stocks and infections.
Virus stock of HRV1a was kindly provided by Yury Bochkov (University of Wisconsin—Madison). Virus was expanded by serial passage through HeLa cell monolayers at 34°C. Cells were infected at an MOI of 5 for 1 h at room temperature with HRV1a. Cells were overlaid with liquid media, incubated at 34°C, and harvested at the indicated times postinfection. Titers were determined by plaque assay on HeLa cells and are reported as PFU per cell.
For one-step growth analysis, MEF cell monolayers were washed twice in 1× PBS and infected with CVB3 at an MOI of 100 for 45 min at room temperature. After adsorption, cells were washed twice in 1× PBS, overlaid with DMEM–10% FBS, and incubated at 37°C. Cells were scraped and collected with supernatant and subjected to five freeze-thaw cycles, and virus yields were determined on HeLa cells by plaque assay.
RESULTS
AUF1 is relocalized to the cytoplasm in a poliovirus 2A-dependent manner.
Although AUF1 is a shuttling protein, in uninfected cells it is predominantly nuclear. Previously published work determined that AUF1 relocalizes from the cytoplasm to the nucleus during poliovirus or human rhinovirus 16 (HRV16) infection (10, 32). The proteinase activity of poliovirus 2A has been shown to inhibit transportin nuclear import pathways (2). All four isoforms of AUF1 contain a transportin 1 binding site; therefore, the disruption of nuclear-cytoplasmic trafficking by poliovirus 2A during infection could contribute to the redistribution of AUF1. Alternatively, we have previously shown that the viral proteinase 3CD cleavage of AUF1 releases the N-terminal dimerization domain of all four AUF1 isoforms (10). Hetero-oligomerization of AUF1 isoforms has been hypothesized to contribute to their localization (33). 3CD cleavage of the dimerization domain likely inhibits AUF1 oligomerization and may, in turn, alter AUF1 localization. To confirm that AUF1 relocalization is the result of viral protein expression, we examined the localization of AUF1 after expression of either poliovirus proteinase 2A or 3CD (Fig. 2). HeLa cells were transfected with constructs encoding N-terminal FLAG-tagged 3CD or FLAG-tagged 2A proteins, and cells were fixed at several time points after transfection. We then examined the expression of FLAG-tagged proteinases and localization of endogenous AUF1 by immunofluorescence. Comparing mock-transfected cells (Fig. 2A) to cells expressing FLAG-2A (seen in green; Fig. 2C), endogenous AUF1 (red) was partially relocalized to the cytoplasm by 12 h posttransfection. This relocalization of AUF1 was seen as soon as 6 h posttransfection (data not shown). In contrast, no relocalization of AUF1 was observed over 24 h posttransfection in cells expressing FLAG-3CD (Fig. 2B). Wild-type FLAG-3CD protein was expressed, thereby producing both FLAG-3CD proteinase and its FLAG-3C cleavage product by the autocatalytic activity of 3C. Additionally, FLAG-3CD, which is incapable of autocleavage into 3C and 3D due to a mutation at the 3C/3D junction but retains catalytic activity (24, 34), was unable to effect relocalization of AUF1 in transfected cells (data not shown). To determine the requirement for proteinase activity in the redistribution of AUF1 by 2A, a previously described mutated 2A protein lacking proteinase activity (2A-L98P) was expressed, and localization of AUF1 was examined (24, 35, 36). When proteinase activity is abolished, no relocalization of AUF1 is detected (Fig. 2D). These results demonstrate that poliovirus 2A expression alone is sufficient to cause redistribution of AUF1 to the cytoplasm and that the proteinase activity of 2A is required for this activity.
Fig 2.

Expression of poliovirus 2A proteinase is sufficient to cause AUF1 relocalization. HeLa cells seeded on coverslips were mock transfected (A) or transfected with FLAG-3CD (B), FLAG-2A (C), or FLAG-2A(98) (D). At 12 (C) or 24 (A, B, and D) h posttransfection, cells were fixed and incubated with anti-FLAG (green) and anti-AUF1 (red) antibodies, followed by Alexa Fluor secondary antibodies. Nuclei were stained with DAPI (blue), and localization was determined using confocal microscopy.
AUF1 partially colocalizes with 3CD in the cytoplasm of poliovirus-infected cells.
In poliovirus-infected cells, AUF1 displays a distinct localization pattern, redistributing to the cytoplasm in regions around the periphery of the nucleus where it partially colocalizes with viral protein 2B; however, by 4 h postinfection, AUF1 is excluded from cytoplasmic regions containing the viral protein 3A (10). Protein 2B is proposed to have roles in the rearrangement of cellular membranes during infection, while protein 3A is thought to take part in anchoring the viral replication complex to cytoplasmic membrane structures. Viral protein 3CD acts in viral replication as well as in the shutdown of host cell processes (for a review, see reference 37). To determine if relocalized AUF1 is associated with poliovirus 3CD, we analyzed the localization of AUF1 and 3CD during poliovirus infection. As previously established, in mock-infected cells AUF1 displays a predominantly nuclear localization (Fig. 3, top panels). Virally expressed 3CD displays a pattern similar to the 2B pattern, with localization throughout the cytoplasm. Relocalized AUF1 partially colocalizes with 3CD in the periphery of the cytoplasm (white arrowheads, bottom panels); however, AUF1 remains absent from a distinct 3CD-containing region of the cytoplasm. These results expand on previously published results, demonstrating that AUF1 partially colocalizes with viral proteins 3CD and 2B in the cytoplasm but is distinctly absent from 3CD-, 2B-, and 3A-containing perinuclear regions (Fig. 3, bottom panel, and reference 10).
Fig 3.
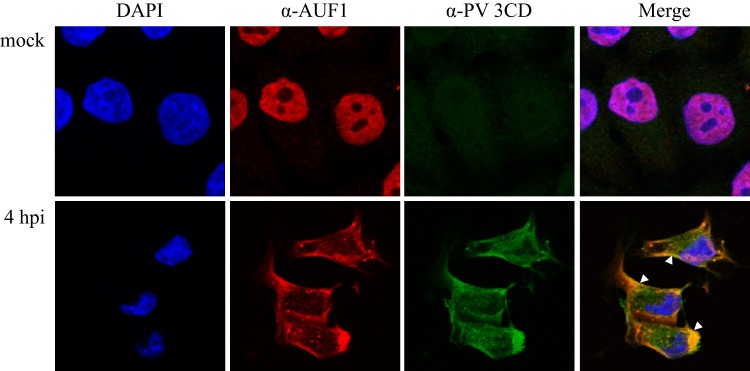
AUF1 partially colocalizes with 3CD in poliovirus-infected cells and is excluded from distinct perinuclear regions. HeLa cells were seeded on coverslips and either mock infected or infected with poliovirus. Cells were fixed at 4 h postinfection, and localization of AUF1 in relation to viral protein 3CD, and 3CD containing precursors, was examined by confocal microscopy with antibodies against AUF1 (red) and 3CD (green). DAPI (blue) was used to visualize nuclei. Colocalization of 3CD and AUF1 is indicated by white arrowheads.
AUF1 interacts directly with stem-loop IV of the poliovirus 5′ NCR.
The 5′ NCR of poliovirus contains multiple secondary structures: (i) the IRES, important for viral translation via a cap-independent mechanism, and (ii) a cloverleaf-like structure located at the most 5′ end of the positive strand (called stem-loop I), important in RNA replication initiation. It was previously shown that AUF1 interacts with the 5′ NCR of both poliovirus and HRV16 RNAs (10); however, the location of this interaction within the 5′ NCR was not defined. To initially determine if AUF1 was binding to the 5′ stem-loop I replication element or to the IRES, an RNA affinity assay was performed using RNA transcripts corresponding to poliovirus stem-loop I or stem-loop IV (Fig. 4A, top panel). In Fig. 4A, lanes 1 and 2 show input protein extracts from either mock- or poliovirus-infected cells; the faster migration of AUF1 isoforms in lane 2 confirms the cleavage of AUF1 in poliovirus-infected cells (10). Using NP-40 lysates from mock-infected or poliovirus-infected HeLa cells, we confirmed the interaction of AUF1 with the 5′ NCR and found that AUF1 can copurify with either stem-loop I or stem-loop IV in a manner independent of the cleavage state (lanes 5 to 10). There appears to be a slight increase in AUF1 bound to the 5′ NCR in lysates from infected cells (e.g., compare lane 5 to lane 6 or lane 7 to lane 8); however, this could be a result of the accumulation of AUF1 in the cytoplasm following infection. Reactions were also examined for PCBP2 binding, as a positive control. As expected, PCBP2 bound to the full-length 5′ NCR, as well as both stem-loop I and stem-loop IV (Fig. 4A, bottom panel). It should be noted that in several different experiments, the interaction of AUF1 with stem-loop I was variable, whereas AUF1 was consistently observed to interact with the full-length 5′ NCR and stem-loop IV.
Fig 4.

AUF1 binds directly to stem-loop IV of the poliovirus 5′ NCR. (A) Streptavidin-bound biotinylated transcripts corresponding to the entire 5′ NCR (lanes 5 and 6), stem-loop I (lanes 7 and 8), or stem-loop IV (lanes 9 and 10) of poliovirus RNA were incubated with either mock-infected (M, odd-numbered lanes) or poliovirus-infected (PV, even-numbered lanes) lysates in an excess of tRNA. As a negative control, tRNA alone was incubated with lysates and streptavidin resin (lanes 3 and 4). Bound complexes were analyzed by Western blotting with anti-AUF1 antibody (top panel). Membranes were stripped and reprobed with anti-PCBP2 monoclonal antibodies as a control for RNA binding (bottom panel) (27). Lanes 1 and 2 represent 20% of the experimental input extract. (B) An RNA electrophoretic mobility shift assay was performed with radiolabeled stem-loop IV and increasing concentrations of recombinant purified AUF1 isoforms ranging from 250 nM (lanes 2, 6, 10, and 14) to 500 nM (lanes 3, 7, 11, and 15) and 750 nM (lanes 4, 8, 12, and 16). Following incubation, complexes were resolved on a native 4% polyacrylamide gel. Lanes 1, 5, 9, and 13 included stem-loop IV probe alone. Electrophoretic mobilities of free stem-loop IV probe and stem-loop IV/AUF1 complexes are indicated on the left by arrows. (C) UV-cross-linking assays were performed with radiolabeled stem-loop IV and increasing concentrations of recombinant AUF1 ranging from 1.5 μM (lanes 3, 6, 9, and 12) to 3 μM (lanes 4, 7, 10, and 13) and 6 μM (lanes 5, 8, 11, and 14). Lane 1 included [35S]-methionine-labeled poliovirus cytoplasmic extract, used as a size marker. Lane 2 included stem-loop IV alone, incubated without added protein.
Since AUF1 is known to interact with PCBP2 in uninfected cells (38), the interaction of AUF1 with stem-loop I or stem-loop IV could be indirect. To determine if AUF1 binds to the 5′ NCR directly, we performed electrophoretic mobility shift assays with purified recombinant AUF1 and radiolabeled stem-loop I (data not shown) or stem-loop IV (Fig. 4B). When recombinant AUF1 isoforms were incubated with stem-loop I, there was a low-level shift in signal of the radiolabeled probe; however, this shift was not dose dependent and may represent a weak, nonspecific interaction (data not shown). In contrast, recombinant AUF1 isoforms formed a shifted RNP complex with radiolabeled stem-loop IV in a dose-dependent manner (Fig. 4B, lanes 2 to 4, 6 to 8, and 14 to 16). Three of the four isoforms of AUF1 bound stem-loop IV directly by a electrophoretic mobility shift assay; however, isoform AUF1p42 displayed markedly decreased binding activity (lanes 10 to 12). These data confirm that AUF1 can interact directly with stem-loop IV of the poliovirus 5′ NCR in vitro.
To investigate whether the decreased binding of AUF1 isoform p42 to poliovirus stem-loop IV was an authentic observation or a result of experimental conditions of the electrophoretic mobility shift assay, we performed UV cross-linking assays with recombinant AUF1 isoforms and radiolabeled stem-loop IV (Fig. 4C). All four isoforms of AUF1 bound to stem-loop IV in a dose-dependent manner, with isoform p42 displaying a weaker interaction (lanes 3 to 8 and 12 to 14 compared to lanes 9 to 11). It is known that the AUF1 isoforms display different affinities for RNA sequences (14). While the weak interaction of isoform p42 and p42mut with stem-loop IV might be attributable to different efficiencies of expression and purification of recombinant AUF1 isoforms leading to differences in the specific activity of recombinant protein preparations, we suggest that it represents an authentic difference in affinity for stem-loop IV RNA. In experiments not shown here, each of our purified AUF1 isoforms, including isoform p42, bound a radiolabeled probe containing both the poliovirus 3′ NCR and a poly(A) tract in a dose-dependent manner, confirming the binding activity of recombinant p42. These data suggest that while all four isoforms can interact with poliovirus stem-loop IV, they may display different affinities for the RNA.
Cleavage of AUF1 by 3CD reduces binding to stem-loop IV.
Cleavage of AUF1 by 3CD releases the most N-terminal 35 amino acids, which are required for dimerization of AUF1 (10, 14). Since AUF1 has been shown to oligomerize on RNA (39–41), we next investigated the effects of cleavage of AUF1 on its affinity for poliovirus stem-loop IV RNA (Fig. 5A). A fixed concentration of recombinant AUF1 was incubated with increasing concentrations of poliovirus 3CD. For each isoform of AUF1, preincubation with proteolytically active 3CD appeared to result in a reduction of RNP complex formation (compare lanes 2, 8, 14, and 20 to lanes 5, 11, 17, and 23). To confirm that the proteolytic activity of 3CD was responsible for the reduction in levels of bound complexes, AUF1 isoforms were incubated with the highest concentration of 3CD harboring an active site mutation [3CD(C147A)] (lanes 6, 12, 18, and 24). Proteolytically inactive 3CD was not effective in decreasing the levels of AUF1-RNA complexes, indicating that the proteolytic activity of 3CD is required to inhibit AUF1 binding to the viral RNA. To further determine if the cleavage of AUF1 prevents its interaction with poliovirus stem-loop IV in the presence of 3CD, we performed the same experiment using recombinant AUF1 proteins with a mutation in the 3CD cleavage site (see Fig. 1). Data showing that mutation of the Q-G amino acids at the N-terminal cleavage site to I-D abolishes poliovirus or human rhinovirus 3CD cleavage of AUF1 have been previously published (10). As seen in Fig. 5B, incubation of uncleavable AUF1 isoforms with proteolytically active 3CD did not markedly decrease RNP complex formation (lanes 4, 9, and 19). These results demonstrate that 3CD cleavage may destabilize the interaction between AUF1 and stem-loop IV RNA in vitro. As discussed above and as seen in Fig. 4B and C, AUF1 isoforms p42 or p42mut do not form a reproducibly stable complex with stem-loop IV (Fig. 5A, lanes 14 to 18; Fig. 5B, lanes 12 to 15).
Fig 5.

Poliovirus 3CD cleavage of AUF1 inhibits binding to stem-loop IV. (A) RNA mobility shifts were performed with 500 nM AUF1 preincubated with or without a proteolytically active form of recombinant 3CD that is incapable of self-cleavage at the 3C-3D junction [3CD(μ10)] (34) or a proteolytically inactive 3CD(C147A) (74, 75). Recombinant AUF1 was incubated with increasing concentrations of 3CD ranging from 250 nM to 500 nM and 750 nM (lanes 3 to 5, 9 to 11, 15 to 17, and 21 to 23). The concentration of 3CD(C147A) was fixed at 1,000 nM (lanes 6, 12, 18, and 24). After incubation with radiolabeled stem-loop IV, RNP complexes were resolved on a native 4% polyacrylamide gel. The levels of shifted RNP complexes were quantified using Quantity One (Bio-Rad). Levels of shifted RNP complexes when incubated in the absence of 3CD (lanes 2, 8, 14, and 20) were set at 100%. When AUF1 was preincubated with the highest concentration of proteolytically active 3CD, the proportions of shifted probe compared to AUF1 incubated without 3CD were 45%, 32%, and 72% for p37, p40, and p45, respectively. The proportions of shifted stem-loop IV were 84%, 83%, and 100% for p37, p40, and p45, respectively, when incubated with 3CD(C147A). These numbers are representative of three separate sets of experiments. (B) RNA electrophoretic mobility shift assays were performed as described for panel A with a mutated version of AUF1. Recombinant AUF1 isoforms were mutated at the P1-P1′ positions, rendering them uncleavable by 3CD (refer to Fig. 1), denoted by a superscript “mut.” Mutated recombinant AUF1 protein was incubated alone (lanes 2, 7, 12, and 17), with 3CD at 500 nM and 1,000 nM (lanes 3 and 4, 8 and 9, 13 and 14, and 18 and 19), or with 3CD(C147A) at 1,000 nM (lanes 5, 10, 15, and 20). The proportions of the shifted complex were calculated as described for panel A and were 117%, 97%, and 144% for p37mut, p40mut, and p45mut, respectively, incubated with the highest concentration of proteolytically active 3CD and 85%, 85%, and 90% for p37mut, p40mut, and p45mut, respectively, incubated with 3CD(C147A). An asterisk indicates twice as much recombinant AUF1 (p42 or p42mut) was used to visualize binding (1,000 nM).
Poliovirus translation is inhibited by AUF1 in vitro.
To determine the effect of the interaction between AUF1 and stem-loop IV, an important RNA element of the IRES, we first examined the potential role of AUF1 in poliovirus translation. Based on the high-affinity interaction of AUF1 with telomeric sequences, DNA oligomers corresponding to G-rich telomeric sequences [(TTAGG)4] have previously been used to sequester AUF1 in cytoplasmic extracts (42). For the experiment whose results are shown in Fig. 6A, cytoplasmic extracts from HeLa cells were preincubated with increasing concentrations of (TTAGG)4 oligodeoxynucleotides to sequester AUF1 prior to in vitro translation. Poliovirus virion RNA was then added with [35S]methionine to label proteins, and translation was allowed to proceed for 4 h. The addition of telomeric DNA oligomers enhanced poliovirus translation in a dose-dependent manner (Fig. 6A, lanes 1 and 3 compared to lane 7). Over five experimental samples, levels of stimulation ranged from 10% above the level seen with the buffer-only control for the lowest concentration of oligomers added (for example, see Fig. 6A, lane 1 compared to lane 3) to 4-fold stimulation for the highest concentrations (for example, see lane 7 of Fig. 6A). Electrophoretic mobility shift assays with recombinant AUF1 and radiolabeled stem-loop IV were performed with increasing amounts of oligomers to confirm binding activity. Unlabeled (TTAGG)4 DNA oligomers were able to successfully decrease stem-loop IV–AUF1 complex formation but had no effect on stem-loop IV–PCBP2 complex formation (data not shown).
Fig 6.

AUF1 inhibits poliovirus translation in vitro. (A) AUF1 was sequestered in HeLa S10 cytoplasmic extract by preincubation with increasing concentrations (0.1 μM, 1 μM, 5 μM, 20 μM, and 40 μM; lanes 3 to 7) of a single-stranded DNA (ssDNA) oligonucleotide (oligo) known to bind strongly to AUF1 (42). After addition of virion RNA, translation was allowed to proceed for 4 h at 30°C. Translation products were labeled with [35S]methionine. In lane 1, translation was allowed to proceed with buffer alone. In lane 2, virion RNA was omitted as a control. Some viral proteins are indicated on the left for reference. (B) HeLa S10 cytoplasmic extracts were incubated with exogenous recombinant AUF1 isoforms in concentrations of 250 nM, 500 nM, and 1,000 nM (lanes 3 to 14) prior to addition of virion RNA. Reaction mixtures were then incubated at 30°C for an additional 4 h, and proteins were labeled with [35S]methionine. (C) Quantity One (Bio-Rad) was used to quantify band intensity of in vitro translation reactions carried out as described for panel B. Levels of VP3 were compared for all reactions as a percentage of the buffer-only control reaction (for an example, see panel B, lane 1) and are displayed as a bar graph, with darker shades reflecting increasing concentrations of exogenous AUF1 or BSA added. Error bars represent standard deviations of the results from three or more experiments.
To confirm the negative effect of AUF1 on poliovirus translation, we performed the converse assay, wherein HeLa cell cytoplasmic extracts were preincubated with exogenous recombinant AUF1 isoforms in increasing concentrations (Fig. 6B and C). Addition of each isoform of AUF1 to in vitro reactions inhibited poliovirus translation in a dose-dependent manner (Fig. 6B [compare lane 1 to lanes 5, 8, 11, and 14]; quantified in Fig. 6C). As a control for any nonspecific effects of adding exogenous protein, bovine serum albumin (BSA) was preincubated with HeLa cytoplasmic extracts prior to viral translation. At the highest concentrations, BSA caused a less than 10% decrease in poliovirus translation compared to an over 50% decrease in parallel experiments when the highest concentrations of AUF1 isoforms were added (Fig. 6C). Additionally, when vRNA was incubated with recombinant isoforms of AUF1 alone and resolved on an agarose gel, no differences in vRNA levels were observed, confirming the absence of RNase contamination in recombinant AUF1 preparations (data not shown). These results suggest that AUF1 negatively regulates poliovirus replication at the level of translation.
AUF1 negatively impacts the enterovirus infectious cycle.
To determine the role of AUF1 in the context of the infected cell, we took advantage of the availability of mouse embryonic cultured fibroblasts lacking AUF1 (MEF-AUF1−/−), as well as the parental wild-type MEF cells (23). Although mice are not naturally infected by poliovirus, transgenic mice expressing the human poliovirus receptor are used as an animal model for poliovirus infection (43, 44). Transcript RNA corresponding to the full-length poliovirus genome was transfected into wild-type (MEF-AUF1+/+) or MEF-AUF1−/− cell monolayers (Fig. 7A). At 8 h posttransfection, cells and supernatants were collected and titers were determined by plaque assay. In cells lacking AUF1, virus titers were increased by between 1 and 2 log10 units, confirming a negative role for AUF1 in the virus life cycle. A one-step growth analysis was performed in MEF-AUF1+/+ or MEF-AUF1−/− cells to examine the kinetics of poliovirus replication in the absence of AUF1 (Fig. 7B). Virion RNA isolated from purified poliovirus particles was used to transfect MEF cells; virus was harvested every 2 h from 0 to 8 h posttransfection. Although viral titers were too low to be detected early after transfection, a difference in titers of wild-type versus MEF-AUF1−/− cells was observed from 4 to 8 h posttransfection. This indicates that the effect of AUF1 on poliovirus growth either occurs early in infection or has a consistent impact throughout the poliovirus replication cycle.
Fig 7.

Deletion of AUF1 enhances poliovirus infection in mouse embryonic fibroblasts. (A) Either 1 μg or 2 μg of poliovirus transcript RNA was transfected into wild-type MEF-AUF1+/+ or MEF-AUF1−/− cell monolayers using DEAE-dextran. Cells and supernatant were harvested at 8 h posttransfection and subjected to sequential freeze-thaw cycles to release virus particles. Virus yield (PFU) was determined by a plaque assay on HeLa cell monolayers and divided by the total cell count. (B) One-step growth analysis was carried out in MEF-AUF1+/+ (dashed line) or MEF-AUF1−/− (solid line) cell monolayers after DEAE-dextran transfection of 0.5 μg of virion RNA. Quantification of virus yield was performed as described for panel A. The error bars indicate standard deviations of the results from triplicate samples.
Given that transfection of viral RNA is a process that is inherently different from infection via cellular receptor binding, we wanted to confirm that the increased titer seen in poliovirus-transfected MEF-AUF1−/− cells was not a result of bypassing the virus-receptor interaction. To accomplish this, we expanded our studies to include enteroviruses that can infect mouse cells: coxsackievirus and human rhinovirus. MEF-AUF1+/+ or MEF-AUF1−/− cells were infected with CVB3 at a high multiplicity of infection to generate a single-cycle growth analysis. As shown in Fig. 8A, a greater than 1 log10 increase in virus production was detected in cells lacking AUF1 at as early as 8 h postinfection. Cleavage of AUF1 in CVB3-infected cells was confirmed by Western blotting of NP-40 lysates (data not shown). Cleavage products were apparent by 7 h postinfection, indicating a common response to AUF1 across enterovirus species (data not shown). Relocalization of AUF1 in CVB3-infected HeLa cells and MEF-AUF1+/+ cells was confirmed by immunofluorescence (data not shown). AUF1 was effectively relocalized, although to a lesser extent than in poliovirus-infected cells. This slower cleavage and relocalization of AUF1 is not surprising, given the slightly delayed kinetics of coxsackievirus infection and the published differences in relocalization seen with host protein SRp20 (24).
Fig 8.

AUF1 negatively affects the enterovirus/rhinovirus infectious cycle. (A) A one-step growth analysis was carried out in MEF-AUF1+/+ (dashed line) or MEF-AUF1−/− (solid line) cell monolayers infected with CVB3 at an MOI of 100. Cells and supernatant were harvested at different times beginning at 0 h until 24 h postinfection. Virus yield was determined as described in the legend to Fig. 7. (B) MEF-AUF1+/+, MEF-AUF1−/−, or HeLa cell monolayers were infected with HRV1a at an MOI of 5 for 20 h. Cells and supernatant were collected, and virus yield was determined as described for Fig. 7. An asterisk (*) indicates statistical significance (Student's t test; P < 0.05); ns, not significant. The error bars indicate standard deviations of the results from triplicate samples.
We have previously shown that AUF1 is cleaved in HRV14- or HRV16-infected HeLa cells (10). In addition, AUF1 relocalizes to the cytoplasm in HRV16-infected cells (32). To perform rhinovirus infections of mouse cells, we used HRV1a, a minor group human rhinovirus (45). Major group HRV14 and HRV16 bind to intercellular adhesion molecule-1 (ICAM-1), a receptor that is lacking on mouse cells, during cell entry (46). In contrast, minor group human rhinoviruses, including HRV1a, utilize the low-density lipoprotein receptor (LDL-R), which is present on both mouse and human cells (47, 48). Based on published data from single-cycle growth analysis in mouse cells (49), MEF or HeLa cells were infected with HRV1a for 20 h, after which cells were harvested and titers were determined by plaque assay (Fig. 8B). As observed for poliovirus or CVB3, HRV1a displayed a marked increase in viral titer in MEF-AUF1−/− cells. Interestingly, this allowed HRV1a to reach titers near the level observed during infection of HeLa cells. Taken together, these experiments reveal that AUF1 acts broadly as a negative regulator of enterovirus/rhinovirus infections.
DISCUSSION
Work described in this article expands on the previous findings identifying AUF1 as a target of viral proteinase cleavage and defines AUF1 as a negative regulator of enterovirus/rhinovirus infection. The relocalization of AUF1 occurs in a viral protein 2A-dependent manner. After this relocalization, AUF1 is found to partially colocalize with 3CD (or precursors or cleavage products thereof) in the periphery of the cytoplasm while being excluded from a perinuclear region containing viral proteins 2B, 3A, and 3CD (this article and reference 10). Given the canonical role of AUF1 in cellular mRNA destabilization, we predict that this cytoplasmic region of exclusion is where viral RNA replication and encapsidation take place. mRNA decay proteins such as AUF1 would be excluded from replication complexes, presumably to protect the RNA from degradation. Relocalization of AUF1 during picornavirus infection may be an indirect consequence of the 2A-driven disruption of the transportin pathway (for a review, see reference 4), as all isoforms of AUF1 contain a nucleocytoplasmic shuttling signal in exon 8 that interacts with the nuclear import receptor transportin (2, 50). Similar localization patterns were recently published for the cellular enzyme 5′-tyrosyl-DNA phosphodiesterase-2 (TDP2), whose proposed role in infection is the removal of the genome-linked protein (VPg) from viral mRNAs to distinguish between replication and translation templates (51). Like AUF1, this protein may be excluded from replication complexes to protect nascent RNA. This suggests a common mechanism to protect viral RNA from cellular proteins via cytoplasmic relocalization.
In agreement with a proposed role for AUF1 as an antiviral factor, we found that sequestering AUF1 in vitro enhanced viral translation, while exogenous AUF1 added to in vitro translation reactions had the opposite effect. In MEF cells lacking AUF1, poliovirus, CVB3, or HRV1a reached higher titers, confirming a negative role for AUF1 in enterovirus/rhinovirus infection. Additionally, we demonstrated that the cleavage of AUF1 by 3CD destabilizes its interaction with poliovirus stem-loop IV RNA, suggesting that 3CD cleavage may act as a viral defense against AUF1. While we have determined that AUF1 has an overall inhibitory effect on enterovirus/rhinovirus infection, the precise mechanism of this negative regulation is unknown. Defining the mechanism of AUF1 regulation of enterovirus/rhinovirus infection may be challenging due to the multiple, and contradictory, known roles of AUF1. Depending on the cellular or viral RNA, AUF1 can act to increase or decrease stability (e.g., parathyroid hormone or granulocyte macrophage colony-stimulating factor [GM-CSF], respectively), alter translation levels (e.g., c-myc), or regulate transcription (e.g., CD-21 and Epstein-Barr virus latency C promoter) (12, 52–55). As a consequence, AUF1 affects RNAs involved in multiple cellular functions, including the immune response (e.g., tumor necrosis factor alpha [TNF-α]), apoptosis (e.g., Bcl2), cell cycle regulation (e.g., cyclin D1), and differentiation (e.g., c-fos) (56–59; for a review, see reference 11). Notably, AUF1 interacts with multiple players reported to participate in the poliovirus replication cycle, including PCBP2, nucleolin, and PABP (38, 60, 61). The interaction of AUF1 with these proteins during viral infection could decrease their availability for use in viral translation and RNA replication. As such, the cleavage and cellular sequestration of AUF1 may act to disrupt protein-protein interactions and release its cellular binding partners, such as PCBP2 and PABP, allowing them to be used in the viral life cycle.
Another potential mechanism for negative regulation of viral infection by AUF1 is that the binding of AUF1 to the 5′ NCR of enterovirus RNA may work directly to recruit mRNA decay factors, thereby contributing to the degradation of the viral RNA. AUF1 is an ARE-binding protein (AUBP) involved in the recruitment of deadenylases for ARE-mediated decay (AMD). After AUBP-dependent recruitment of AMD proteins, mRNAs undergo deadenylation followed by recruitment of exo- or endonucleases and degradation of the RNA (for a review, see reference 11). The disruption of the AUF1-stem-loop IV interaction by 3CD cleavage could act as a viral defense against host mRNA decay machinery. Evasion and modification of mRNA decay machinery during virus infections are not exclusive to AUF1 (for a review, see reference 62). For example, the 5′ exonuclease Xrn1, the decapping enzyme Dcp1a, and the deadenylase complex component Pan3 were found to be degraded in poliovirus-infected cells; the functional consequences of this degradation are unknown, but they may aid in the disruption of processing bodies (complexes of stalled mRNAs) seen during poliovirus infection (63). Enterovirus 71 (EV71) has been shown to induce cleavage of a DNA binding protein, far upstream element-binding protein 2 (FBP2), which, like AUF1, has roles in cellular translation and RNA stability (64).
AUF1 may also have an indirect effect on enterovirus replication by altering specific cellular mRNA levels. AUF1 is linked to the stability of many mRNAs involved in the innate immune response, including c-myc, interleukin-6 (IL-6), and TNF-α (56, 65, 66). Could AUF1-mediated alterations in cytokine mRNA levels alter enterovirus replication? Generally, AUF1 has been found to have a destabilizing effect on specific mRNAs. These mRNA targets include members of both the proinflammatory (e.g., IL-6) and anti-inflammatory (e.g., IL-10) cytokines (65, 67, 68). Previous studies noted a correlation in the timing of AUF1 relocalization and a reduction in the levels of chemokine CXCL10 (gamma interferon [IFN-γ]-inducible protein 10 [IP-10]) during HRV16 infection (32). Alternatively, depending on the cell type, AUF1 can act as a stabilizing factor for TNF-α mRNA (56); this stabilization could potentially play a role in virus-induced apoptosis. Based on the cascade of cytokine signals, deregulation of AUF1 could have either a positive or negative effect on the cellular immune response. AUF1-mediated alteration of mRNA levels linked to the immune response could thus alter the kinetics of enteroviral infection. Additionally, at peak times of poliovirus infection, AUF1 exists in both cleaved and uncleaved forms. Preferential cleavage of AUF1 interacting with the viral genome or viral proteins could allow for differences in the cleavage state of local concentrations of AUF1. This could prevent AUF1 from binding to the viral RNA in complexes associated with viral RNA synthesis while leaving uncleaved AUF1 in the periphery of the cytoplasm to destabilize cytokine or other cellular mRNAs. A similar phenomenon has been reported for the poliovirus proteinase 2A, which preferentially cleaves eIF4G, which is associated with eIF4E (and presumably actively translating ribosomes), to disrupt cap-dependent cellular translation (69). It is unlikely that cytoplasmic lysates used for in vitro translation experiments retain signaling pathways necessary to elicit cytokine production. For this reason, we predict that AUF1-driven alterations of cellular mRNA stability are not the sole cause of its negative role in poliovirus translation, as our in vitro experiments recapitulated the negative effects of AUF1.
A role for AUF1 in viral infection is not without precedent, as a recent paper confirmed our original findings for poliovirus (this article and reference 10) with coxsackievirus B3 (70). Wong and colleagues reported that AUF1 is relocalized and cleaved at the N terminus during CVB3 infection and can interact with the 3′ NCR of coxsackievirus RNA (70). It is not clear whether the 3′ NCR construct used by this group contained a poly(A) tract; however, it is known that AUF1 can interact with poly(A) RNA (71). The functional consequence of AUF1 binding to both ends of the viral RNA is unknown, but such binding may act to further stabilize these RNP complexes, thereby increasing the negative effects of AUF1 on the viral replication cycle. It was also reported that transient knockdown of AUF1 leads to an increase in accumulation of cellular mRNAs that contain an IRES (e.g., c-myc) during CVB3 infection, possibly leading to an increase in cytokine levels (70). Whether an increase in c-myc mRNA levels affects CVB3 titers or the increase in mRNAs correlated to c-myc protein levels was not addressed. Further investigation on the impact, if any, of c-myc on virus infection is necessary to assess the relevance of this observation. In contrast to its negative role in poliovirus translation, AUF1 has been shown to be required for translation of another positive-strand RNA virus, hepatitis C virus (HCV). Interestingly, AUF1 has a positive role in the translation of HCV but a negative role in overall viral RNA accumulation (42). Similarly, during HIV infection, AUF1 is postulated to play a role in HIV Gag and Env synthesis either through affecting RNA stability or by export or alteration of the RNP complex involved in export (72). In addition to roles in RNA virus infection, AUF1 has been shown to bind to the Epstein-Barr virus F promoter, the latency C promoter, and the EBER1 noncoding RNAs (52, 61, 73). These findings, along with work described in this paper, further highlight the diverse and context-dependent functions of AUF1.
Overall, we found that AUF1 negatively regulates replication of three related picornaviruses in mammalian cells. These viruses appear to have overcome this inhibitory effect, in part, by promoting the relocalization of AUF1 in infected cells and by proteolytic cleavage of AUF1. These results provide new insights into the potential roles of picornavirus proteinase cleavage of cellular proteins that act as restriction factors to viral infections. They also highlight the intricate interplay between cellular and viral functions that ultimately determines the outcome of a viral infection. Further studies of the mechanism by which AUF1 acts as a negative regulator are necessary to understand this complicated virus-host interaction.
ACKNOWLEDGMENTS
We are grateful to Amanda Chase, Eric Baggs, and Ilya Belalov for critical comments on the manuscript. We are indebted to Robert Schnieder for generously providing MEF-AUF1+/+ and MEF-AUF1−/− cell lines and Yury Bochkov for HRV1a virus stock. We thank Hung Nguyen and MyPhuong Tran for their expert technical assistance. We also thank Adeeyla Syed at the UCI Optical Biology Core for confocal microscopy training. Confocal microscopy images were generated at the UCI Optical Biology Core facility, which is supported by the Comprehensive Cancer Center award P30CA062203 from the National Cancer Institute.
J.M.R. was supported by a postdoctoral fellowship from the George E. Hewitt Foundation for Medical Research. This research was supported by Public Health Service grant AI026765 from the National Institutes of Health and by the California Center for Antiviral Drug Discovery (a Multicampus Research Program Initiative from the University of California).
Footnotes
Published ahead of print 31 July 2013
REFERENCES
- 1.Fitzgerald KD, Semler BL. 2011. Re-localization of cellular protein SRp20 during poliovirus infection: bridging a viral IRES to the host cell translation apparatus. PLoS Pathog. 7:e1002127. 10.1371/journal.ppat.1002127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gustin KE, Sarnow P. 2001. Effects of poliovirus infection on nucleo-cytoplasmic trafficking and nuclear pore complex composition. EMBO J. 20:240–249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.McBride AE, Schlegel A, Kirkegaard K. 1996. Human protein Sam68 relocalization and interaction with poliovirus RNA polymerase in infected cells. Proc. Natl. Acad. Sci. U. S. A. 93:2296–2301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Castelló A, Alvarez E, Carrasco L. 2011. The multifaceted poliovirus 2A protease: regulation of gene expression by picornavirus proteases. J. Biomed. Biotechnol. 2011:369648. 10.1155/2011/369648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chase AJ, Semler BL. 2012. Viral subversion of host functions for picornavirus translation and RNA replication. Future Virol. 7:179–191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Barral PM, Sarkar D, Fisher PB, Racaniello VR. 2009. RIG-I is cleaved during picornavirus infection. Virology 391:171–176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Drahos J, Racaniello VR. 2009. Cleavage of IPS-1 in cells infected with human rhinovirus. J. Virol. 83:11581–11587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Neznanov N, Kondratova A, Chumakov KM, Angres B, Zhumabayeva B, Agol VI, Gudkov AV. 2001. Poliovirus protein 3A inhibits tumor necrosis factor (TNF)-induced apoptosis by eliminating the TNF receptor from the cell surface. J. Virol. 75:10409–10420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Racaniello VR. 2010. Innate immune responses, p 287–302 In Ehrenfeld E, Domingo E, Roos RP. (ed), The picornaviruses. ASM Press, Washington, DC [Google Scholar]
- 10.Rozovics JM, Chase AJ, Cathcart AL, Chou W, Gershon PD, Palusa S, Wilusz J, Semler BL. 2012. Picornavirus modification of a host mRNA decay protein. mBio 3:e00431–12. 10.1128/mBio.00431-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gratacós FM, Brewer G. 2010. The role of AUF1 in regulated mRNA decay. Wiley Interdiscip. Rev. RNA 1:457–473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sela-Brown A, Silver J, Brewer G, Naveh-Many T. 2000. Identification of AUF1 as a parathyroid hormone mRNA 3′-untranslated region-binding protein that determines parathyroid hormone mRNA stability. J. Biol. Chem. 275:7424–7429 [DOI] [PubMed] [Google Scholar]
- 13.Ing NH, Massuto DA, Jaeger LA. 2008. Estradiol up-regulates AUF1p45 binding to stabilizing regions within the 3′-untranslated region of estrogen receptor alpha mRNA. J. Biol. Chem. 283:1764–1772 [DOI] [PubMed] [Google Scholar]
- 14.Wagner BJ, DeMaria CT, Sun Y, Wilson GM, Brewer G. 1998. Structure and genomic organization of the human AUF1 gene: alternative pre-mRNA splicing generates four protein isoforms. Genomics 48:195–202 [DOI] [PubMed] [Google Scholar]
- 15.Bedard KM, Daijogo S, Semler BL. 2007. A nucleo-cytoplasmic SR protein functions in viral IRES-mediated translation initiation. EMBO J. 26:459–467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hellen CU, Witherell GW, Schmid M, Shin SH, Pestova TV, Gil A, Wimmer E. 1993. A cytoplasmic 57-kDa protein that is required for translation of picornavirus RNA by internal ribosomal entry is identical to the nuclear pyrimidine tract-binding protein. Proc. Natl. Acad. Sci. U. S. A. 90:7642–7646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Meerovitch K, Svitkin YV, Lee HS, Lejbkowicz F, Kenan DJ, Chan EK, Agol VI, Keene JD, Sonenberg N. 1993. La autoantigen enhances and corrects aberrant translation of poliovirus RNA in reticulocyte lysate. J. Virol. 67:3798–3807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Costa-Mattioli M, Svitkin Y, Sonenberg N. 2004. La autoantigen is necessary for optimal function of the poliovirus and hepatitis C virus internal ribosome entry site in vivo and in vitro. Mol. Cell. Biol. 24:6861–6870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hunt SL, Hsuan JJ, Totty N, Jackson RJ. 1999. unr, a cellular cytoplasmic RNA-binding protein with five cold-shock domains, is required for internal initiation of translation of human rhinovirus RNA. Genes Dev. 13:437–448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fitzgerald KD, Semler BL. 2009. Bridging IRES elements in mRNAs to the eukaryotic translation apparatus. Biochim. Biophys. Acta 1789:518–528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Borman A, Howell MT, Patton JG, Jackson RJ. 1993. The involvement of a spliceosome component in internal initiation of human rhinovirus RNA translation. J. Gen. Virol. 74:1775–1788 [DOI] [PubMed] [Google Scholar]
- 22.Murray KE, Roberts AW, Barton DJ. 2001. Poly(rC) binding proteins mediate poliovirus mRNA stability. RNA 7:1126–1141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lee C, Gyorgy A, Maric D, Sadri N, Schneider RJ, Barker JL, Lawson M, Agoston DV. 2008. Members of the NuRD chromatin remodeling complex interact with AUF1 in developing cortical neurons. Cereb. Cortex 18:2909–2919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fitzgerald KD, Chase AJ, Cathcart AL, Tran GP, Semler BL. 2013. Viral proteinase requirements for the nucleocytoplasmic relocalization of cellular splicing factor SRp20 during picornavirus infections. J. Virol. 87:2390–2400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dildine SL, Semler BL. 1992. Conservation of RNA-protein interactions among picornaviruses. J. Virol. 66:4364–4376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dildine SL, Semler BL. 1989. The deletion of 41 proximal nucleotides reverts a poliovirus mutant containing a temperature-sensitive lesion in the 5′ noncoding region of genomic RNA. J. Virol. 63:847–862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sean P, Nguyen JH, Semler BL. 2008. The linker domain of poly(rC) binding protein 2 is a major determinant in poliovirus cap-independent translation. Virology 378:243–253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wilson GM, Brewer G. 1999. Identification and characterization of proteins binding A + U-rich elements. Methods 17:74–83 [DOI] [PubMed] [Google Scholar]
- 29.Walter BL, Parsley TB, Ehrenfeld E, Semler BL. 2002. Distinct poly(rC) binding protein KH domain determinants for poliovirus translation initiation and viral RNA replication. J. Virol. 76:12008–12022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dewalt PG, Semler BL. 1987. Site-directed mutagenesis of proteinase 3C results in a poliovirus deficient in synthesis of viral RNA polymerase. J. Virol. 61:2162–2170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Haller AA, Semler BL. 1992. Linker scanning mutagenesis of the internal ribosome entry site of poliovirus RNA. J. Virol. 66:5075–5086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Spurrell JC, Wiehler S, Zaheer RS, Sanders SP, Proud D. 2005. Human airway epithelial cells produce IP-10 (CXCL10) in vitro and in vivo upon rhinovirus infection. Am. J. Physiol. Lung Cell. Mol. Physiol. 289:L85–L95 [DOI] [PubMed] [Google Scholar]
- 33.Sarkar B, Lu JY, Schneider RJ. 2003. Nuclear import and export functions in the different isoforms of the AUF1/heterogeneous nuclear ribonucleoprotein protein family. J. Biol. Chem. 278:20700–20707 [DOI] [PubMed] [Google Scholar]
- 34.Semler BL, Johnson VH, Dewalt PG, Ypma-Wong MF. 1987. Site-specific mutagenesis of cDNA clones expressing a poliovirus proteinase. J. Cell. Biochem. 33:39–51 [DOI] [PubMed] [Google Scholar]
- 35.Crowder S, Kirkegaard K. 2005. Trans-dominant inhibition of RNA viral replication can slow growth of drug-resistant viruses. Nat. Genet. 37:701–709 [DOI] [PubMed] [Google Scholar]
- 36.Yu SF, Lloyd RE. 1991. Identification of essential amino acid residues in the functional activity of poliovirus 2A protease. Virology 182:615–625 [DOI] [PubMed] [Google Scholar]
- 37.Rozovics JM, Semler BL. 2010. Genome replication I: the players, p 107–125 In Ehrenfeld E, Domingo E, Roos RP. (ed), The picornaviruses. ASM Press, Washington, DC [Google Scholar]
- 38.Kiledjian M, DeMaria CT, Brewer G, Novick K. 1997. Identification of AUF1 (heterogeneous nuclear ribonucleoprotein D) as a component of the alpha-globin mRNA stability complex. Mol. Cell. Biol. 17:4870–4876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.DeMaria CT, Sun Y, Long L, Wagner BJ, Brewer G. 1997. Structural determinants in AUF1 required for high affinity binding to A + U-rich elements. J. Biol. Chem. 272:27635–27643 [DOI] [PubMed] [Google Scholar]
- 40.Wilson GM, Sun Y, Lu H, Brewer G. 1999. Assembly of AUF1 oligomers on U-rich RNA targets by sequential dimer association. J. Biol. Chem. 274:33374–33381 [DOI] [PubMed] [Google Scholar]
- 41.David PS, Tanveer R, Port JD. 2007. FRET-detectable interactions between the ARE binding proteins, HuR and p37AUF1. RNA 13:1453–1468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Paek KY, Kim CS, Park SM, Kim JH, Jang SK. 2008. RNA-binding protein hnRNP D modulates internal ribosome entry site-dependent translation of hepatitis C virus RNA. J. Virol. 82:12082–12093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mendelsohn C, Johnson B, Lionetti KA, Nobis P, Wimmer E, Racaniello VR. 1986. Transformation of a human poliovirus receptor gene into mouse cells. Proc. Natl. Acad. Sci. U. S. A. 83:7845–7849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ren RB, Costantini F, Gorgacz EJ, Lee JJ, Racaniello VR. 1990. Transgenic mice expressing a human poliovirus receptor: a new model for poliomyelitis. Cell 63:353–362 [DOI] [PubMed] [Google Scholar]
- 45.Bochkov YA, Palmenberg AC, Lee WM, Rathe JA, Amineva SP, Sun X, Pasic TR, Jarjour NN, Liggett SB, Gern JE. 2011. Molecular modeling, organ culture and reverse genetics for a newly identified human rhinovirus C. Nat. Med. 17:627–632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kolatkar PR, Bella J, Olson NH, Bator CM, Baker TS, Rossmann MG. 1999. Structural studies of two rhinovirus serotypes complexed with fragments of their cellular receptor. EMBO J. 18:6249–6259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hewat EA, Neumann E, Conway JF, Moser R, Ronacher B, Marlovits TC, Blaas D. 2000. The cellular receptor to human rhinovirus 2 binds around the 5-fold axis and not in the canyon: a structural view. EMBO J. 19:6317–6325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Reithmayer M, Reischl A, Snyers L, Blaas D. 2002. Species-specific receptor recognition by a minor-group human rhinovirus (HRV): HRV serotype 1A distinguishes between the murine and the human low-density lipoprotein receptor. J. Virol. 76:6957–6965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rasmussen AL, Racaniello VR. 2011. Selection of rhinovirus 1A variants adapted for growth in mouse lung epithelial cells. Virology 420:82–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chen CY, Xu N, Zhu W, Shyu AB. 2004. Functional dissection of hnRNP D suggests that nuclear import is required before hnRNP D can modulate mRNA turnover in the cytoplasm. RNA 10:669–680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Virgen-Slane R, Rozovics JM, Fitzgerald KD, Ngo T, Chou W, van der Heden van Noort GJ, Filippov DV, Gershon PD, Semler BL. 2012. An RNA virus hijacks an incognito function of a DNA repair enzyme. Proc. Natl. Acad. Sci. U. S. A. 109:14634–14639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Fuentes-Pananá EM, Peng R, Brewer G, Tan J, Ling PD. 2000. Regulation of the Epstein-Barr virus C promoter by AUF1 and the cyclic AMP/protein kinase A signaling pathway. J. Virol. 74:8166–8175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tolnay M, Vereshchagina LA, Tsokos GC. 1999. Heterogeneous nuclear ribonucleoprotein D0B is a sequence-specific DNA-binding protein. Biochem. J. 338(Pt 2):417–425 [PMC free article] [PubMed] [Google Scholar]
- 54.Buzby JS, Lee SM, Van Winkle P, DeMaria CT, Brewer G, Cairo MS. 1996. Increased granulocyte-macrophage colony-stimulating factor mRNA instability in cord versus adult mononuclear cells is translation-dependent and associated with increased levels of A + U-rich element binding factor. Blood 88:2889–2897 [PubMed] [Google Scholar]
- 55.Liao B, Hu Y, Brewer G. 2007. Competitive binding of AUF1 and TIAR to MYC mRNA controls its translation. Nat. Struct. Mol. Biol. 14:511–518 [DOI] [PubMed] [Google Scholar]
- 56.Xu N, Chen CY, Shyu AB. 2001. Versatile role for hnRNP D isoforms in the differential regulation of cytoplasmic mRNA turnover. Mol. Cell. Biol. 21:6960–6971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lal A, Mazan-Mamczarz K, Kawai T, Yang X, Martindale JL, Gorospe M. 2004. Concurrent versus individual binding of HuR and AUF1 to common labile target mRNAs. EMBO J. 23:3092–3102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lapucci A, Donnini M, Papucci L, Witort E, Tempestini A, Bevilacqua A, Nicolin A, Brewer G, Schiavone N, Capaccioli S. 2002. AUF1 Is a bcl-2 A + U-rich element-binding protein involved in bcl-2 mRNA destabilization during apoptosis. J. Biol. Chem. 277:16139–16146 [DOI] [PubMed] [Google Scholar]
- 59.Lin S, Wang W, Wilson GM, Yang X, Brewer G, Holbrook NJ, Gorospe M. 2000. Down-regulation of cyclin D1 expression by prostaglandin A(2) is mediated by enhanced cyclin D1 mRNA turnover. Mol. Cell. Biol. 20:7903–7913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lu JY, Bergman N, Sadri N, Schneider RJ. 2006. Assembly of AUF1 with eIF4G-poly(A) binding protein complex suggests a translation function in AU-rich mRNA decay. RNA 12:883–893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Dempsey LA, Hanakahi LA, Maizels N. 1998. A specific isoform of hnRNP D interacts with DNA in the LR1 heterodimer: canonical RNA binding motifs in a sequence-specific duplex DNA binding protein. J. Biol. Chem. 273:29224–29229 [DOI] [PubMed] [Google Scholar]
- 62.Moon SL, Barnhart MD, Wilusz J. 2012. Inhibition and avoidance of mRNA degradation by RNA viruses. Curr. Opin. Microbiol. 15:500–505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Dougherty JD, White JP, Lloyd RE. 2011. Poliovirus-mediated disruption of cytoplasmic processing bodies. J. Virol. 85:64–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Chen LL, Kung YA, Weng KF, Lin JY, Horng JT, Shih SR. 2013. Enterovirus 71 infection cleaves a negative regulator for viral internal ribosomal entry site-driven translation. J. Virol. 87:3828–3838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Paschoud S, Dogar AM, Kuntz C, Grisoni-Neupert B, Richman L, Kuhn LC. 2006. Destabilization of interleukin-6 mRNA requires a putative RNA stem-loop structure, an AU-rich element, and the RNA-binding protein AUF1. Mol. Cell. Biol. 26:8228–8241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Brewer G. 1991. An A + U-rich element RNA-binding factor regulates c-myc mRNA stability in vitro. Mol. Cell. Biol. 11:2460–2466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Brewer G, Saccani S, Sarkar S, Lewis A, Pestka S. 2003. Increased interleukin-10 mRNA stability in melanoma cells is associated with decreased levels of A + U-rich element binding factor AUF1. J. Interferon Cytokine Res. 23:553–564 [DOI] [PubMed] [Google Scholar]
- 68.Sarkar S, Sinsimer KS, Foster RL, Brewer G, Pestka S. 2008. AUF1 isoform-specific regulation of anti-inflammatory IL10 expression in monocytes. J. Interferon Cytokine Res. 28:679–691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bovee ML, Lamphear BJ, Rhoads RE, Lloyd RE. 1998. Direct cleavage of elF4G by poliovirus 2A protease is inefficient in vitro. Virology 245:241–249 [DOI] [PubMed] [Google Scholar]
- 70.Wong J, Si X, Angeles A, Zhang J, Shi J, Fung G, Jagdeo J, Wang T, Zhong Z, Jan E, Luo H. 2013. Cytoplasmic redistribution and cleavage of AUF1 during coxsackievirus infection enhance the stability of its viral genome. FASEB J. 27:2777–2787 [DOI] [PubMed] [Google Scholar]
- 71.Sagliocco F, Laloo B, Cosson B, Laborde L, Castroviejo M, Rosenbaum J, Ripoche J, Grosset C. 2006. The ARE-associated factor AUF1 binds poly(A) in vitro in competition with PABP. Biochem. J. 400:337–347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Lund N, Milev MP, Wong R, Sanmuganantham T, Woolaway K, Chabot B, Abou Elela S, Mouland AJ, Cochrane A. 2012. Differential effects of hnRNP D/AUF1 isoforms on HIV-1 gene expression. Nucleic Acids Res. 40:3663–3675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Lee N, Pimienta G, Steitz JA. 2012. AUF1/hnRNP D is a novel protein partner of the EBER1 noncoding RNA of Epstein-Barr virus. RNA 18:2073–2082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lawson MA, Semler BL. 1991. Poliovirus thiol proteinase 3C can utilize a serine nucleophile within the putative catalytic triad. Proc. Natl. Acad. Sci. U. S. A. 88:9919–9923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Parsley TB, Cornell CT, Semler BL. 1999. Modulation of the RNA binding and protein processing activities of poliovirus polypeptide 3CD by the viral RNA polymerase domain. J. Biol. Chem. 274:12867–12876 [DOI] [PubMed] [Google Scholar]