

Abstract
Controlled regulation of genomic DNA synthesis is a universally conserved process for all herpesviruses, including human cytomegalovirus (HCMV), and plays a key role in viral pathogenesis, such as persistent infections. HCMV UL105 is believed to encode the helicase of the DNA replication machinery that needs to localize in the nuclei, the site of viral DNA synthesis. No host factors that interact with UL105 have been identified. In this study, we show that UL105 specifically interacts with Snapin, a human protein that is predominantly localized in the cytoplasm and associated with cellular vesicles. UL105 was found to interact with Snapin in both the yeast two-hybrid screen and coimmunoprecipitation experiments in HCMV-infected cells. The nuclear and cytoplasmic levels of UL105 were decreased and increased in cells overexpressing Snapin, respectively, while the levels of UL105 in the nuclei and cytoplasm were increased and decreased in cells in which the expression of Snapin was downregulated with anti-Snapin small interfering RNA (siRNA) molecules, respectively. Furthermore, viral DNA synthesis and progeny production were decreased in cells overexpressing Snapin and increased in the anti-Snapin siRNA-treated cells, respectively. Our results provide the first direct evidence to suggest that Snapin interacts with UL105 and alters its cellular distribution, leading to modulation of viral DNA synthesis and progeny production. Our study further suggests that modulation of the cellular distribution of viral helicase by Snapin may represent a possible mechanism for regulating HCMV genomic DNA synthesis, a key step during herpesvirus lytic and persistent infections.
INTRODUCTION
Human cytomegalovirus (HCMV) is a member of the herpesvirus family, which includes herpes simplex virus 1 (HSV-1), Epstein-Barr virus (EBV), and Kaposi's sarcoma-associated herpesvirus (KSHV) (1). This virus causes mild or subclinical diseases in immunocompetent adults but may lead to severe morbidity or mortality in neonates and immunocompromised individuals (1, 2). HCMV can infect a wide range of tissues and cells, such as neuronal cells, and like all other herpesviruses, it can establish lytic, persistent, and latent infections in many of these tissues (3, 4). During lytic productive infection, HCMV gene products are expressed temporally, and their expression consists of three sequential phases, described as the immediate early (IE), early (E), and late (L) phases (1). The process of viral DNA synthesis, which occurs in the nucleus of infected cells (5), is highly conserved among all herpesviruses and is the target for most of the current FDA-approved antiherpesvirus therapeutic agents (6).
Lytic DNA replication of herpesviruses is believed to be a complex and highly regulated event. The viral DNA replication complex contains at least six essential proteins that are conserved in all herpesviruses (1, 7). The HCMV replication factors consist of a two-subunit DNA polymerase encoded by UL54 and UL44, a single-stranded DNA-binding protein encoded by UL57, a primase encoded by UL70, a helicase encoded by UL105, and the primase-helicase-associated factor encoded by UL102. DNA replication in HCMV is considered to be analogous to that in HSV and other herpesviruses, with the function of several HCMV proteins (e.g., UL70 and UL105) being predicted on the basis of sequence and functional homology with their counterparts in HSV (1, 5).
HCMV UL105 is believed to encode the viral helicase because this protein contains six motifs (I to VI) typical of the superfamily 1 class of helicase proteins that are highly conserved not only among all sequenced HCMV isolates but also among 32 host helicase homologues (7). HCMV helicase and its herpesviral homologues form a tight helicase-primase complex, which consists of UL105, UL70, and UL102 in HCMV (7–10). It is believed that the helicase UL105 tracks along the lagging strand and unwinds the DNA in front of the replication fork, while the UL70-encoded primase synthesizes short RNA primers for single-stranded DNA which the DNA polymerase extends via deoxynucleoside triphosphate polymerization (1, 5). Although the precise role of each subunit needs further investigation, it would be expected, by analogy with observations in other herpesviruses (e.g., HSV-1), that an assembled subcomplex containing UL105 and UL70 subunits retains the enzymatic activities, while the UL102 subunit modulates these activities (8, 11–13).
As genomic DNA replication of herpesviruses occurs in the nuclei, all HCMV replication proteins, such as UL105, need to be imported into the nuclei (14). Studies have been carried out to identify human proteins that potentially interact with HCMV proteins and modulate their transport to the nuclei (1). For example, many HCMV proteins, including DNA replication core proteins UL44, UL54, and UL57, which possess nuclear localization signal sequences (NLSs), have been found to interact with human cellular importin to facilitate their import into the nucleus through the nuclear pore complex (15–20). More recently, our laboratory (21) has shown that UL70 interacts with cellular Snapin, a cellular vesicle-associated protein that is predominantly localized in the cytoplasm (22–24). Modulation of the expression of Snapin affects the nuclear and cytoplasmic distribution of UL70 and the levels of HCMV DNA genomic replication and progeny production in cultured cells (21). These results suggest that control of nuclear import and the availability of the HCMV replication proteins may represent a key step in regulating viral DNA replication. However, it has not been reported how the UL105-encoded essential helicase is imported into the nuclear compartment where viral DNA replication occurs (25). Equally elusive is how the cellular distribution of UL105 is modulated. Currently, there is no report of interactions of UL105 with any human proteins.
In this study, we performed a yeast two-hybrid screen to identify human proteins that potentially interact with UL105. Our results provide the first direct evidence that UL105 interacts with Snapin at a domain that is different from that bound by UL70. Furthermore, our results suggest that by altering the cellular localization of UL105, Snapin may play a role in modulating viral DNA synthesis and lytic productive infection and may represent a possible means of regulating viral persistent infection during HCMV pathogenesis.
MATERIALS AND METHODS
Antibodies, viruses, and cells.
Human astrocytoma U373MG and glioblastoma U251 cells were obtained from Sigma, Inc. (St. Louis, MO), and the American Type Culture Collection (ATCC; Manassas, VA), respectively. Human foreskin fibroblasts (HFFs) and U251, HeLa, U373MG, and 293T cells were maintained in Dulbecco's modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum (26, 27), and HCMV (strains Towne and TowneBAC) was propagated in these cells as described previously (28). The antibodies against human actin and Snapin were purchased from Sigma, Inc. (St. Louis, MO), and Santa Cruz Biotech, Inc. (Santa Cruz, CA), respectively. The monoclonal antibodies that react with HCMV proteins UL44, IE1, and gH have been described previously (29, 30).
To express antigens for generation of the anti-UL70 and anti-UL105 monoclonal antibodies, the UL70- and UL105-coding sequences were cloned into expression vector pET-28a(+) (Novagen, Madison, WI). His-tagged UL105 was expressed in the Escherichia coli BL21(DE3) strain, purified by gel electrophoresis, and then used to immunize BALB/c mice. Spleen cells were isolated from immunized mice and fused with myeloma cells. Positive fused cells were selected by the use of hypoxanthine-aminopterin-thymidine culture medium and enzyme-linked immunosorbent assay screening. The resulting monoclonal antibodies were collected from the supernatants of the culture medium of positive hybridoma cell cultures and kept at −20°C. We further characterized the reactivity of the antibodies using Western blot analyses, coimmunoprecipitation (co-IP), and indirect immunofluorescence assays.
To generate the U251-S, U251-SΔH1, U251-SΔH2, and U251-C cell lines, DNAs of constructs pCMV-Snapin/UL105, pCMV-SΔH1, pCMV-SΔH2, and the empty vector [pCDNA3.1(+)], respectively, were cotransfected into U251 cells with DNA of the LXSN vector (26, 29). At 48 to 72 h postinfection, neomycin was added to the culture medium at a final concentration of 600 μg/ml. Cells were subsequently selected in the presence of neomycin for 2 weeks, and neomycin-resistant cells were cloned (31, 32). The levels of Snapin and its mutants in individual cell clones were determined by Western analysis.
Plasmid construction.
The plasmid constructs generated in this study are listed in Table 1. The construct pUL105 was derived from the DNA of TowneBAC (28), and the construct pCMV-XLS-Snapin containing the coding sequence of Snapin was purchased from Origene, Inc. (Rockville, MD). The construct pGBKT7-UL105, which was used for the yeast two-hybrid screen, was generated by cloning the HCMV UL105-coding sequence into NdeI/BamHI-digested pGBKT7 (Clontech, Mountain View, CA). The construct pCMV-Myc-UL105, which was used for expression in human cells, was generated by cloning the UL105-coding sequence into SalI/KpnI-digested pCMV-Myc (Clontech).
Table 1.
Plasmid constructs used in the study
| Plasmid | Description | Source |
|---|---|---|
| pGADT7 | Cloning vector for protein expression fused with the GAL4 activation domain in yeast | Clontech |
| pGBKT7 | Cloning vector for protein expression fused with the GAL4 DNA-binding domain in yeast | Clontech |
| pCMV-Myc | Cloning vector for protein expression fused with c-Myc tag in mammalian cell | Clontech |
| pCDNA3.1(+) | Cloning vector for protein expression without a tag in mammalian cells | Clontech |
| pCMV6-XL5-Snapin | pCMV6-XL5 containing Snapin cDNA full-length sequence | OriGene |
| pGBKT7-UL105 | pGBKT7 containing HCMV UL105 full-length sequence | This study |
| pACT2-SNAPIN/UL105 | pACT2 containing full-length human Snapin cDNA sequence | This study |
| pCMV-UL105 | pCDNA3.1(+) containing full-length HCMV UL105 sequence | This study |
| pCMV-Myc-UL105 | pCMV-Myc containing HCMV UL105 full-length sequence | This study |
| pRK11-FLAG-UL83 | pRK11-FLAG containing full-length HCMV UL83 sequence | This study |
| pGADT7-Snapin/UL105 | pGADT7 containing full-length human Snapin cDNA sequence | This study |
| pGADT7-SΔHD(1-20aa) | pGADT7 containing a truncated Snapin cDNA sequence with deletion of the sequence coding for the region from amino acid residues 1 to 20 | This study |
| pGADT7-SΔL1(20-36aa) | pGADT7 containing a truncated Snapin cDNA sequence with deletion of the sequence coding for the region from amino acid residues 20 to 36 | This study |
| pGADT7-SΔH1(37-65aa) | pGADT7 containing a truncated Snapin cDNA sequence with deletion of the sequence coding for the region from amino acid residues 37 to 65 | This study |
| pGADT7-SΔL2(66-80aa) | pGADT7 containing a truncated Snapin cDNA sequence with deletion of the sequence coding for the region from amino acid residues 66 to 80 | This study |
| pGADT7-SΔH2(81-126aa) | pGADT7 containing a truncated Snapin cDNA sequence with deletion of the sequence coding for the region from amino acid residues 81 to 126 | This study |
| pGADT7-SΔCD(127-136aa) | pGADT7 containing a truncated Snapin cDNA sequence with deletion of the sequence coding for the region from amino acid residues 127 to 136 | This study |
| pRK11-FLAG-Snapin | pRK11-FLAG containing full-length human Snapin cDNA sequence | This study |
| pRK11-FLAG-SΔHD(1-20aa) | pRK11-FLAG containing a truncated Snapin cDNA sequence with deletion of the sequence coding for the region from amino acid residues 1 to 20 | This study |
| pRK11-FLAG-SΔL1(20-36aa) | pRK11-FLAG containing a truncated Snapin cDNA sequence with deletion of the sequence coding for the region from amino acid residues 20 to 36 | This study |
| pRK11-FLAG-SΔH1(37-65aa) | pRK11-FLAG containing a truncated Snapin cDNA sequence with deletion of the sequence coding for the region from amino acid residues 37 to 65 | This study |
| pRK11-FLAG-SΔL2(66-80aa) | pRK11-FLAG containing a truncated Snapin cDNA sequence with deletion of the sequence coding for the region from amino acid residues 66 to 80 | This study |
| pRK11-FLAG-SΔH2(81-126aa) | pRK11-FLAG containing a truncated Snapin cDNA sequence with deletion of the sequence coding for the region from amino acid residues 81 to 126 | This study |
| pRK11-FLAG-SΔCD(127-136aa) | pRK11-FLAG containing a truncated Snapin cDNA sequence with deletion of the sequence coding for the region from amino acid residues 127 to 136 | This study |
| pCMV-Snapin/UL105 | pCDNA3.1(+) containing Snapin full-length sequence (amino acid residues 1 to 136) | This study |
| pCMV-SΔH1 | pCDNA3.1(+) containing a truncated Snapin cDNA sequence with deletion of the sequence coding for the region from amino acid residues 37 to 65 | This study |
| pCMV-SΔH2 | pCDNA3.1(+) containing a truncated Snapin cDNA sequence with deletion of the sequence coding for the region from amino acid residues 81 to 126 | This study |
To generate the constructs pGADT7-Snapin/UL105 and pRK11-FLAG-Snapin used for expression in Saccharomyces cerevisiae strain AH109 and in human cells, the coding sequence of Snapin was amplified by PCR using the primers SNAPIN-F (5′-ATGGATCCTAATGGCGGGGGCTGGTT-3′) and SNAPIN-R (5′-ATACTCGAGAATTCTTATTTGCCTGGGGAGCCA-3′) and then inserted into BamHI/XhoI-digested pGADT7 and BamHI/EcoRI-digested pRK11-FLAG, respectively. The constructs that contained the sequences encoding different Snapin deletion mutants were generated by PCR using pGBKT7-Snapin as the template, followed by insertion of the amplified PCR products into NdeI/BamHI-digested pGBKT7 for yeast two-hybrid analysis and SalI/KpnI-digested pCMV-Myc and pCDNA3.1(+) for expression in human cells. The resultant constructs were confirmed by restriction digestion profile and sequencing. The construct containing the HCMV UL83 sequence was generated following the procedures described previously (27).
Yeast two-hybrid analysis.
We obtained a human fetal brain cDNA library and the related yeast two-hybrid screen reagents (e.g., Saccharomyces cerevisiae strain AH109 and control vector pGADT7) from Clontech (Mountain View, CA). We transformed the yeast strain containing pGBKT7-UL105 with the library, selected positive clones on synthetic dropout (SD) medium lacking four nutrients, tryptophan, leucine, adenine, and histidine (SD-minus Trp/Leu/Ade/His; quadruple dropout [QDO]), and tested the positive clones for β-galactosidase activity by a colony-lift filter assay following the manufacturer's recommended procedures, as described previously (27). We then extracted the DNA constructs that contained the sequence coding for the interaction partners of UL105 (designated pACT2-cDNA) from the positive colonies, and the human gene sequences in the pACT2-cDNA constructs from these positive colonies were determined with the sequencing primer 5′-AATACCACTACAATGGAT-3′ (27).
Coimmunoprecipitation and Western blot analysis.
We cotransfected cells with plasmid DNAs with the aid of the Lipofectamine reagent (Invitrogen, Carlsbad, CA). At 48 h posttransfection, we harvested cell lysates and carried out coimmunoprecipitation experiments using the ProFound mammalian FLAG tag IP/co-IP and c-Myc tag IP/co-IP kits following the manufacturer's protocol (Pierce, Rockford, IL) (27). In Western analysis experiments, the denatured polypeptides from cell lysates or co-IP were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred electrically to nitrocellulose membranes. Membranes were blocked in 5% dried milk in phosphate-buffered saline (PBS) plus 0.2% Tween 20, stained with different antibodies, and reacted in an enzyme-linked immunosorbent assay with 1:2,000-diluted horse anti-mouse IgG antibody conjugated with horseradish peroxidase (Vector Laboratories, Burlingame, CA). The membranes were subsequently stained with a chemiluminescent substrate with the aid of enhanced chemiluminescence Western blotting detection reagents kits (GE Healthcare) and quantitated either by densitometry of the films exposed to the samples or with a STORM840 phosphorimager or a gel documentation station (Bio-Rad, Hercules, CA) (26, 29). We carried out the experiments in duplicate and repeated them three times. Quantitation was performed in the linear range of protein detection.
Transfection of siRNA into cells.
Cells (n = 1 × 105) were seeded in 12-well plates and transfected with chemically synthetic anti-Snapin H2-small interfering RNA (siRNA) and control C-siRNA molecules. The H2-siRNA molecules were a pool of three siRNAs (H2-1, H2-2, and H2-3) that targeted different regions of the Snapin mRNA coding for the H2 domain. The oligonucleotides were designed and chemically synthesized by Ribobio Co. Ltd. (Guangzhou, China) and Santa Cruz Biotech, Inc. (Santa Cruz, CA). In each well, 3 μl Lipofectamine (Invitrogen) and 3 μl 20 mM siRNA were diluted in 12 μl Opti-MEM and 50 μl Opti-MEM (Invitrogen), respectively. We then combined both solutions at room temperature after 5 min. After 20 min, we added 400 μl prewarmed Opti-MEM to each transfection reaction mixture, which was subsequently added to the cells. At 10 h posttransfection, we removed the siRNA-containing medium, washed the cells, and then incubated the cells with complete DMEM supplemented with 10% fetal bovine serum. The transfection was repeated once, and at 48 h after the second round of transfection, cells were either infected or prepared for analysis by immunoblotting.
Viral infection and assays for viral gene expression and growth.
Cells (n = 1 × 106) were either mock infected or infected with HCMV at a multiplicity of infection (MOI) of 0.5 to 5 following the experimental procedures described previously (26, 29). To assay viral gene expression, we isolated protein samples from infected cells at 12 to 72 h postinfection, and the protein samples were analyzed by Western blotting experiments, as described previously (29, 30). To assay viral growth, we infected cells (n = 1 × 105) with HCMV at an MOI of 1. We collected the cells and medium at 5 days postinfection and prepared viral stocks by adding an equal volume of 10% (vol/vol) skim milk, followed by sonication. We determined the titers of the viral stocks by infecting 1 × 105 human foreskin fibroblasts and counting the number of plaques at 10 to 14 days after infection. The values obtained were averages from triplicate experiments.
Assaying intracellular viral DNA level.
We performed quantitative real-time PCR (qPCR) analysis of viral DNA as described previously (33). Briefly, we isolated DNA samples from mock-infected or HCMV-infected cells using a DNeasy tissue kit (Qiagen) according to the manufacturer's instructions. The levels of intracellular viral DNAs were quantified with primers P53 (5′-GTCAGCGTTCGTGTTTCCCA-3′) and P33 (5′-GGGACACAACACCGTAAAGC-3′) and a probe for amplifying the HCMV UL83 sequence. The TaqMan probe (5′-FAM-CCCGCAACCCGCAACCCTTCATG-TAMRA-3′, where FAM is 6-carboxyfluorescein and TAMRA is 6-carboxytetramethylrhodamine) was purchased from Applied Biosystems Inc. (Foster City, CA) and was labeled at the 5′ end with FAM and at the 3′ end with the fluorescent quencher TAMRA. We normalized the number of viral genomes to the number of cellular copies of β-actin with a previously described set of primers and probe (33). Unknown sample values were determined on the basis of a standard curve of known copy numbers of UL83 (TowneBAC) and β-actin (pβ-actin). We carried out the PCR amplification in a 50-μl reaction mixture (which contained 21 μl of DNA extract, 25 μl of 2× TaqMan Universal PCR master mix [Applied Biosystems Inc.], 1 μl of each primer at 10 μM, 2 μl of the fluorogenic probe at 5 μM) using either an ABI 7500 device (Applied Biosystems Inc., Foster City, CA) or an iCycler real-time PCR detection system (Bio-Rad, Hercules, CA). Thermal cycling conditions were as follows: 50°C for 2 min, 95°C for 15 min, and 45 cycles of 95°C for 30 s and 60°C for 1 min. The PCR results were derived from three independent experiments.
Preparation of nuclear and cytoplasmic extracts.
We resuspended the cells in buffer A (10 mM HEPES [pH 7.4], 10 mM KCl, 1 mM dithiothreitol, 0.6% NP-40) at 4°C for 10 min. After centrifugation at 1,000 × g for 5 min, the supernatant was collected as the cytoplasmic fraction and the nuclear fraction was prepared by resuspending the remaining pellet in buffer B (20 mM HEPES [pH 7.4], 150 mM NaCl, 1 mM dithiothreitol). We stained the nuclear and cytoplasmic extracts for the presence of UL70, UL105, actin, and histone H1 by immunoblotting.
Immunofluorescence microscopy analysis.
Cells (grown on glass coverslips) were cotransfected with DNAs of the different constructs and at 48 h posttransfection were either mock infected or infected with HCMV at an MOI of 1 to 5 (34). At different time points postinfection, cells were fixed with 4% formaldehyde, blocked with 0.1% bovine serum albumin and 0.1% sodium azide in PBS, and stained with primary antibodies, followed by incubation with secondary antibody Alexa Fluor 555 anti-mouse IgG or Alexa Fluor 488 anti-rabbit IgG (Invitrogen). DAPI (4′,6-diamidino-2-phenylindole) was used for nuclear staining (Invitrogen). We collected confocal images using an Olympus confocal FV1000 microscope in separate channels and analyzed the images using Olympus Fluoview software. We determined the percentages of cytoplasmic and nuclear UL105 from at least three independent experiments. In some of the experiments, we also captured the images using a Nikon Eclipse TE300 microscope using a SPOT RT Slider camera and imaging software (Diagnostic Instruments, Inc., Sterling Heights, MI) (34). The digital images were subsequently merged by the use of SimplePCI software (Compix Inc., Sewickley, PA). For some experiments, we also manually counted cells exhibiting different patterns of UL105 localization and determined the percentages of the numbers of cells from at least three independent experiments.
RESULTS
Interaction of Snapin with UL105 revealed by yeast two-hybrid analysis.
To identify cellular factors that potentially interact with UL105, we carried out the yeast two-hybrid screen by transforming S. cerevisiae AH109 containing pGBKT7-UL105, which contained the full-length UL105 coding sequence, with a cDNA library derived from human fetal brain. Those yeast cells that grew on synthetic dropout (SD) medium lacking four nutrients, tryptophan, leucine, adenine, and histidine (SD-minus Trp/Leu/Ade/His), and yielded blue signals in a colony-lift filter test for β-galactosidase activity were identified (27). One of the identified constructs, pACT2-SNAPIN/UL105, which contained the full-length coding sequence of Snapin, was consistently found to interact with UL105 in our yeast two-hybrid screens (data not shown). Of nearly 1.5 × 107 independent cDNA clones tested, 27 yeast colonies yielded positive results, and 13 of these contained plasmid constructs with the full-length coding sequence of Snapin.
Interaction of Snapin with UL105 in human cells revealed by co-IP.
In our co-IP assays, we first cloned the sequences coding for Snapin and UL105 into mammalian expression vectors pRK11-FLAG and pCMV-Myc to generate constructs pRK11-FLAG-Snapin and pCMV-Myc-UL105, respectively, in which each open reading frame (ORF) was expressed as a fusion protein with an amino-terminal FLAG tag and Myc epitope tag, respectively (22–24, 28). Protein lysates from human glioblastoma U251 cells transfected with these constructs were first immunoprecipitated with either anti-FLAG or anti-Myc and then immunoblotted with antibodies against the FLAG and Myc epitope tags. We observed coprecipitation of the Myc-tagged UL105 with the FLAG-tagged Snapin (Fig. 1, lanes 4 and 11). In contrast, in control experiments we detected no significant binding or coprecipitation between the Myc-tagged UL105 and the FLAG-tagged UL83 of HCMV, which is not known to interact with UL105 and serves as a negative control (lanes 1 to 3 and 7 to 9) (27). We also observed similar results in HeLa cells, 293T cells, astrocytoma U373MG cells, and HCMV-infected U251 cells transfected with the constructs expressing FLAG-tagged Snapin and the Myc-tagged UL105 proteins (data not shown). These observations confirmed the specificity of the co-IP assay and suggested that the UL105-Snapin interaction may occur in human cells.
Fig 1.
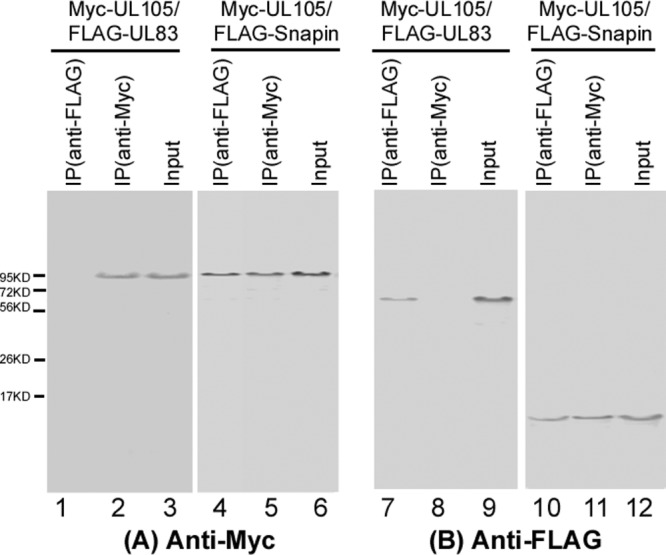
Identification of the interaction between the Myc-tagged UL105 and FLAG-tagged Snapin by co-IP. Human U251 cells were cotransfected with a combination of two plasmids expressing FLAG- and Myc-tagged proteins. Cellular lysates were prepared at 48 h posttransfection. The input protein samples (70 μg; Input, lanes 3, 6, 9, and 12) and samples (10 μg) that were immunoprecipitated with either anti-Myc [IP(anti-Myc), lanes 2, 5, 8, and 11] or anti-FLAG [IP(anti-FLAG), lanes 1, 4, 7, and 10] antibodies were separated on SDS-containing polyacrylamide gels and analyzed with anti-Myc (anti-Myc) and anti-FLAG (anti-FLAG) antibodies, respectively.
To determine if the native (untagged) UL105 specifically interacts with endogenous Snapin in human cells and in the presence of HCMV infection, we expressed UL105 in E. coli and used the expressed protein as the antigen to generate an anti-UL105 monoclonal antibody. To determine if Snapin may nonspecifically interact with other viral proteins in addition to UL105, we also investigated if Snapin is associated with UL44, which encodes the HCMV polymerase processivity factor essential for viral DNA replication (1). Protein lysates from HCMV-infected U251 cells were first immunoprecipitated with either anti-UL105, anti-UL44, or anti-Snapin and then immunoblotted with antibodies against UL105, UL44, and Snapin. UL105 was coprecipitated with the endogenous Snapin (Fig. 2, lanes 7 and 11), while we observed no significant binding or coprecipitation between UL44 and Snapin (Fig. 2, lanes 1 to 6). Similar results were also observed in HFFs infected with HCMV and uninfected HeLa, 293T, and U251 cells that were transfected with pCMV-UL105 and pCMV-UL44 (data not shown). These results suggest that Snapin may specifically interact with UL105 but not UL44 during HCMV infection.
Fig 2.
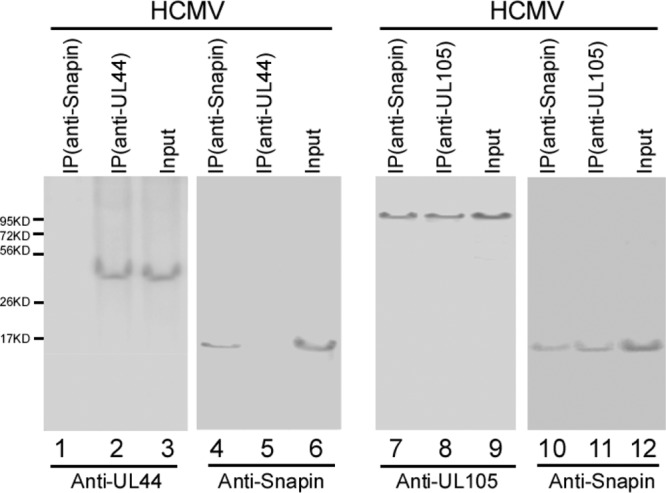
Co-IP of viral proteins and cellular Snapin in HCMV-infected cells. Human U251 cells were infected with HCMV (MOI = 1), and cellular lysates were prepared at 72 h postinfection. The input protein samples (60 μg; Input, lanes 3, 6, 9, and 12) and samples (15 μg) that were immunoprecipitated with anti-UL44 [IP(anti-UL44), lanes 2 and 5], anti-UL105 [IP(anti-UL105), lanes 8 and 11], or anti-Snapin [IP(anti-Snapin), lanes 1, 4, 7, and 10] antibodies were separated on SDS-polyacrylamide gels and analyzed with anti-UL44 (anti-UL44), anti-UL105 (anti-UL105), and anti-Snapin (anti-Snapin) antibodies, respectively.
Critical roles of the conserved coiled-coil H2 domain of Snapin in the interaction with UL105.
Snapin contains an N-terminal hydrophobic domain (amino acids [aa] 1 to 20) and two conserved helical coiled-coil domains, H1 (amino acids 37 to 65) and H2 (amino acids 81 to 126) (22, 23). The H2 domain has been shown to be the major binding site for SNAP23, while no proteins have been reported to interact with the H1 domain (22, 23). To map the domains of Snapin required for interacting with UL105, we constructed a series of Snapin truncation mutants for yeast two-hybrid analyses (Fig. 3). Furthermore, we generated a series of mammalian expression constructs (Table 1) which encoded the FLAG-tagged Snapin truncation mutants (Fig. 3). These constructs were then cotransfected with pCMV-UL105 into U251 cells, and the interactions between UL105 and different FLAG-tagged Snapin mutants were examined in coimmunoprecipitation experiments (Fig. 4). The results, summarized in Fig. 3, indicate that deletion of the H2 domain [e.g., in mutant SΔH2(81-126aa)] abolished the ability of Snapin to interact with UL105, while deletion of each of the remaining regions (e.g., the amino-terminal hydrophobic region and the H1 domain) had no effect on the binding of Snapin to UL105 (Fig. 4). Thus, the conserved H2 sequence of Snapin is essential for its interaction with UL105.
Fig 3.

Schematic diagram of Snapin and its deletion mutants that interact with UL105 and UL70, as identified by the yeast two-hybrid (YTH) screen and co-IP in U251 cells. + and −, positive and negative interactions, respectively, in the two-hybrid screen or co-IP.
Fig 4.
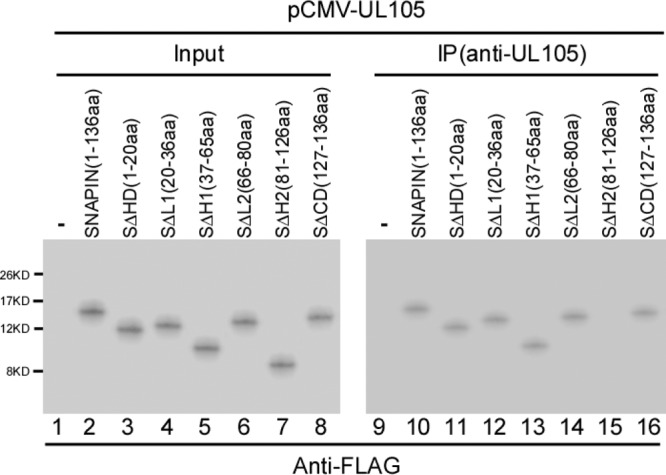
Identification of the interaction between UL105 and different FLAG-tagged Snapin mutants by co-IP. Human U251 cells were cotransfected with pCMV-UL105 and a plasmid expressing FLAG-tagged Snapin protein with different mutations. Cellular lysates were prepared at 48 h posttransfection. The input protein samples (50 μg) (Input, lanes 1 to 8) and samples (10 μg) that were immunoprecipitated with anti-UL105 [IP(anti-UL105), lanes 9 to 16) were separated on SDS-containing polyacrylamide gels and analyzed with anti-FLAG antibodies (anti-FLAG).
Cellular localization of UL105 and Snapin in human cells.
If UL105 is associated with (or binds to) Snapin in cells, these proteins are expected to localize within the same cellular compartments. Microscopy analyses showed that both the Myc-tagged UL105 and FLAG-tagged Snapin were predominantly localized in the cytoplasm in U251 cells transfected with the constructs (Fig. 5a to d). To further confirm these results and investigate the cellular localization of untagged UL105 and endogenous Snapin proteins, cells were transfected with pCMV-UL105, which contained the untagged UL105-coding sequence, and then stained with anti-UL105 and anti-Snapin antibodies. Similar to the results obtained with the tagged proteins (Fig. 5a to d), both UL105 and Snapin were found to be primarily localized in the cytoplasm (Fig. 5e to h). We also observed cytoplasmic localization of these two proteins in HeLa cells and HFFs (data not shown). Anti-UL105 appeared to be specific, as staining with this antibody was observed only in cells transfected with pCMV-UL105 and not in the empty vector, pCDNA3.1(+) (Fig. 5i to l). The localization of UL105 may not completely overlap that of Snapin, possibly because both proteins are dynamically expressed in cells and overexpression of UL105 may lead to the misdistribution of a small amount of UL105 in these cells. Consistent with our results from the two-hybrid screens and co-IP experiments, these results suggest that the tag sequences in Myc-UL105 and FLAG-Snapin do not affect the interaction and colocalization of UL105 with Snapin and that Snapin may specifically interact with UL105. Furthermore, these results provide direct evidence to suggest that UL105 is primarily localized in the cytoplasmic compartment in uninfected cells.
Fig 5.
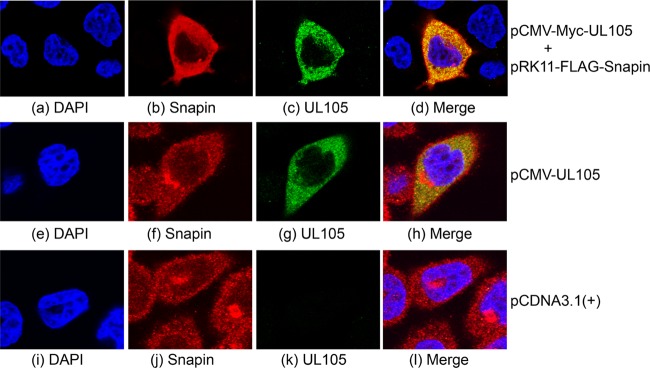
Colocalization of UL105 and Snapin expressed in human cells. Cells were cotransfected with constructs pCMV-Myc-UL105 and pRK11-FLAG-Snapin (a to d), pCMV-UL105 alone (e to h), or pCDNA3.1(+) alone (i to l), fixed at 48 h posttransfection, stained with DAPI (a, e, and i), anti-FLAG (b), anti-Myc (c), anti-Snapin (f and j), and anti-UL105 (g and k) antibodies, and visualized using a confocal microscope (Olympus FV1000). The images of Snapin (b, f and j), UL105 (c, g and k), and the nuclei stained with DAPI (a, e, and i) were used to generate the composite images (d, h, and l). The images show different levels of magnification.
Modulation of the cellular localization of UL105 by up- and downregulation of the expression of Snapin.
Because viral DNA synthesis occurs in the nuclei during HCMV infection, UL105, the essential helicase, needs to be localized in the nuclear compartment. Our results that UL105 interacts with the cytoplasmic Snapin protein and is primarily localized in the cytoplasm in uninfected cells raise the possibility that Snapin may sequester UL105 and affect its cellular localization through binding. If this is the case, UL105 is expected to be accumulated in cells that overexpress Snapin or a Snapin mutant with UL105 binding activity but not a mutant exhibiting no binding to UL105. To study the effect of overexpression of Snapin on the cellular localization of UL105, we constructed a U251 cell line, U251-S, which constitutively expressed full-length Snapin protein, and a control cell line, U251-C, which contained the empty expression vector. Furthermore, we generated two additional cell lines, U251-SΔH1 and U251-SΔH2, which expressed Snapin mutants SΔH1(37-65aa) and SΔH2(81-126aa), respectively. SΔH1(37-65aa) contained a deletion of the conserved helical H1 domain (amino acids 37 to 65) and was able to bind to UL105 in yeast two-hybrid assays and coimmunoprecipitation experiments in U251 cells, while SΔH2(81-126aa) contained a deletion of the conserved helical H2 domain (amino acids 81 to 126) and exhibited no binding to UL105 (Fig. 3). Western blotting of HCMV-infected cell lysates indicated an ∼5-fold elevated level of Snapin protein in cells expressing Snapin and its mutants compared to the levels in parental U251 and control U251-C cells (Fig. 6). The constructed cell lines and the parental U251 cells were indistinguishable in terms of their growth and viability for 3 months (data not shown), suggesting that the overexpression of Snapin did not result in significant cytotoxicity.
Fig 6.
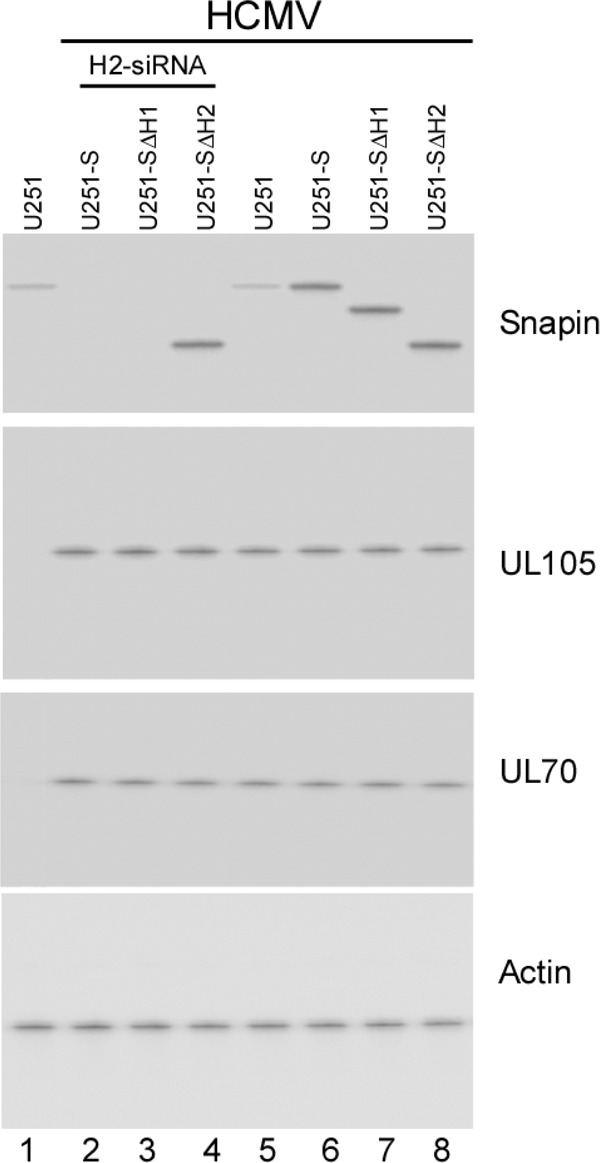
Expression of Snapin and its mutants in cells. Western blot analysis was used to determine the levels of Snapin, UL70, and UL105 in the parental U251 cells (U251) (lanes 1 and 5), cells that expressed full-length Snapin (U251-S; lanes 2 and 6), and Snapin mutants SΔH1(37-65aa) (U251-SΔH1; lanes 3 and 7) and SΔH2(81-126aa) (U251-SΔH2; lanes 4 and 8). Cells were either transfected with anti-Snapin H2-siRNA molecules (lanes 2 to 4) or not transfected with any siRNAs (lanes 1 and 5 to 8) and were either mock infected (lane 1) or infected with HCMV at an MOI of 1 after 48 h of siRNA incubation (lanes 2 to 8). The expression of cellular actin was used as the internal loading control.
To study the localization of UL105 in HCMV-infected cells, we transfected the constructed cell lines with pCMV-Myc-UL105 and then infected these cells with HCMV. We used an MOI of 1 to 5 to ensure that most cells were infected. Indeed, IE1 expression was detected in a majority of the cells when cells were stained with an anti-IE1 antibody (data not shown). HCMV infection appeared to promote nuclear import of UL105 in U251 cells, as we found a substantial amount of the UL105 signal in the nuclei at 48 to 72 h postinfection compared to that in the uninfected cells (compare Fig. 7a to c to Fig. 5). In the presence of HCMV infection, the localization of UL105 in U251-S cells was significantly different from that in the parental U251 cells and the control U251-C cells. At 48 h postinfection, UL105 was exclusively localized in the cytoplasm of U251-S cells (96% cytoplasmic versus 4% nuclear) (Fig. 7d to f; Table 2). In contrast, UL105 was distributed in both the cytoplasm and nuclei of U251 and U251-C cells (60% nuclear versus 40% cytoplasmic) (Fig. 7a to c; Table 2). Similar results were also observed when cells were transfected with pCMV-UL105 instead of pCMV-Myc-UL105 and stained with the anti-UL105 antibody (Table 2), consistent with our observations that the tag sequence does not affect the interaction and colocalization of UL105 with Snapin (Fig. 1, 2, and 5).
Fig 7.
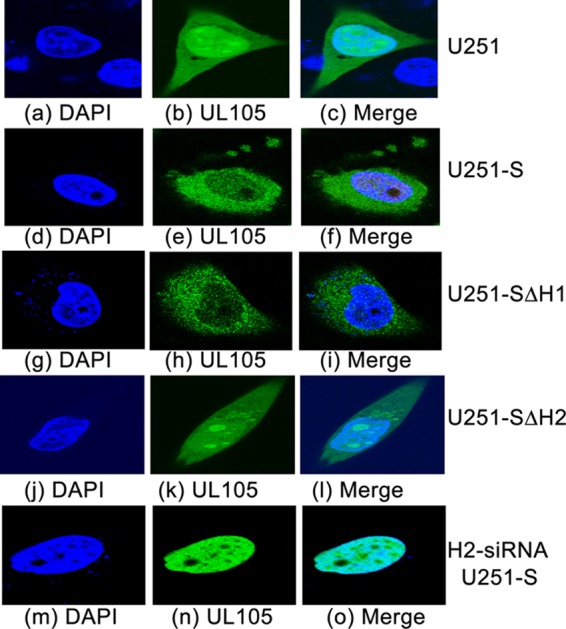
Effect of altered expression of Snapin and its mutants on the cellular distribution of UL105. The cellular localization of UL105 in parental U251 cells (U251; a to c), cells overexpressing full-length Snapin protein (U251-S; d to f), mutants SΔH1(37-65aa) (U251-SΔH1; g to i) and SΔH2(81-126aa) (U251-SΔH2; j to l), or anti-Snapin H2-siRNA-treated U251-S cells (H2-siRNA U251-S; m to o) was examined. Cells were transfected with pCMV-Myc-UL105 in the absence and presence of siRNAs, infected with HCMV (MOI = 5) at 48 h posttransfection, fixed at 48 h postinfection, stained with antibodies, and visualized using a confocal microscope (Olympus FV1000). The images of the DAPI-stained nuclei (blue) (a, d, g, j, m) and Myc-tagged UL105 (green) (b, e, h, k, n) were used to generate the composite images (c, f, i, l, o). The images show different levels of magnification.
Table 2.
Percentages of UL105 found to be localized in the nuclei or cytoplasma
| Expressed protein/cell | % UL105 found at the indicated locations |
|||
|---|---|---|---|---|
| Myc-UL105 |
UL105 |
|||
| Nuclei | Cytoplasm | Nuclei | Cytoplasm | |
| U251 | 60 ± 8 | 40 ± 7 | 62 ± 7 | 38 ± 7 |
| U251-C | 60 ± 7 | 40 ± 6 | 59 ± 7 | 41 ± 8 |
| U251-S | 4 | 96 ± 7 | 4 | 96 ± 8 |
| U251-SΔH1 | 7 | 93 ± 7 | 8 | 92 ± 8 |
| U251-SΔH2 | 60 ± 7 | 40 ± 6 | 60 ± 8 | 40 ± 7 |
| C-siRNA (U251) | 64 ± 6 | 36 ± 6 | 62 ± 7 | 38 ± 7 |
| C-siRNA (U251-C) | 60 ± 7 | 40 ± 6 | 60 ± 7 | 40 ± 7 |
| C-siRNA (U251-S) | 6 | 94 ± 7 | 7 | 93 ± 8 |
| C-siRNA (251-SΔH1) | 9 | 91 ± 8 | 8 | 92 ± 8 |
| C-siRNA (U251-SΔH2) | 60 ± 7 | 40 ± 7 | 61 ± 8 | 39 ± 8 |
| H2-siRNA (U251) | 97 ± 9 | 3 | 96 ± 8 | 4 |
| H2-siRNA (U251-C) | 97 ± 8 | 3 | 97 ± 7 | 3 |
| H2-siRNA (U251-S) | 92 ± 8 | 8 | 93 ± 7 | 7 |
| H2-siRNA (U251-SΔH1) | 92 ± 9 | 8 | 93 ± 8 | 7 |
| H2-siRNA (U251 SΔH2) | 96 ± 7 | 4 | 95 ± 8 | 5 |
Cells were transfected with either pCMV-Myc-UL105 (Myc-UL105) or pCMV-UL105 (UL105) and then infected with HCMV, stained with anti-Myc or anti-UL105, respectively, and visualized using a confocal microscope (Olympus FV1000). The experimental procedures are described in Materials and Methods. The values shown are the means ± standard deviations of three independent experiments. We examined 150 cells in each experiment, with a total of 450 cells examined in the three experiments. The values of standard deviations that were less than 5% are not shown.
The localization of UL105 in U251-SΔH1 cells, which expressed mutant SΔH1(37-65aa) exhibiting UL105 binding activity, was exclusively in the cytoplasm and was similar to that in the U251-S cells (Fig. 7g to i; Table 2). In contrast, the localization of UL105 in U251-SΔH2 cells, which expressed mutant SΔH2(81-126aa) exhibiting no UL105 binding activity, was both cytoplasmic and nuclear and was similar to that in the U251 and U251-C cells (Fig. 7j to l; Table 2). These results suggest that overexpression of Snapin leads to a reduction of nuclear import of UL105 and an accumulation of UL105 in the cytoplasm and that binding of Snapin is required for the observed change of the cellular distribution of UL105.
It is expected that downregulation of Snapin expression may promote nuclear import of UL105, on the basis of our observations that overexpression of Snapin leads to the cytoplasmic accumulation of UL105. To determine whether this is the case, we cotransfected different cells with pCMV-Myc-UL105 and siRNA molecules that were designed either to recognize the region of Snapin mRNA coding the H2 domain (H2-siRNA) or not to recognize any viral or cellular transcripts (control siRNA or C-siRNA) and then either mock infected or infected these cells with HCMV at 48 h posttransfection. Previous studies showed that downregulation of Snapin expression mediated by siRNA had no effect on cell viability (22, 24). The level of Snapin protein was reduced by more than 90% in different cells transfected with H2-siRNA compared to that in cells that were either not transfected with any siRNAs or transfected with control C-siRNA (Fig. 6; compare lanes 2, 5, and 6). In the presence of HCMV infection, UL105 was predominantly localized in the nuclei of the H2-siRNA-treated cells (92% nuclear versus 8% cytoplasmic) but was distributed in both the nuclei and cytoplasm of the untreated and the control siRNA-treated cells (60% nuclear versus 40% cytoplasmic) (Fig. 7m to o; Table 2).
The effect of H2-siRNA, which targets the mRNA region coding for the H2 domain, appeared to be specific. The treatment of U251-SΔH1 cells with H2-siRNA inhibited the expression of endogenous full-length Snapin and mutant SΔH1(37-65aa), which contained a deletion in the H1 domain and exhibited UL105 binding activity (Fig. 6, lane 3). In U251-SΔH2 cells, H2-siRNA treatment inhibited the expression of endogenous full-length Snapin but had no significant effect on the expression of mutant SΔH2(81-126aa), which contained a deletion of the H2 domain and exhibited no UL105 binding activity (lane 4). Upon treatment with H2-siRNA, the accumulation of nuclear UL105 was observed in both U251-SΔH1 and U251-SΔH2 cells (Table 2). These results suggest that the treatment of H2-siRNA downregulated Snapin expression by binding to its target mRNA, leading to a reduction of the cytoplasmic accumulation of UL105 and an increase of its nuclear localization.
To confirm the effects of Snapin expression on the localization of UL105, we determined the distribution of UL105 by cell fractionation and Western blot analysis (Fig. 8). Western blot analysis confirmed the altered expression levels of Snapin in these cells (Fig. 6; data not shown). Infected cells were subjected to differential centrifugation into nuclear and cytoplasmic fractions. The purity of the fractions was confirmed by immunoblotting for histone H1 (nuclear marker) and actin (cytoplasmic marker) (Fig. 8). In parental U251 cells that were infected with HCMV, 60% of UL105 was found in the nuclei, while 40% was in the cytoplasm (Fig. 8, bottom set of gels, lanes 4 and 11). Similar results were also found in control cells that either contained the empty expression vector (U251-C cells) or were treated with siRNA molecules designed not to recognize any viral or cellular transcripts (C-siRNA) (Fig. 8, bottom set of gels). Downregulation of the expression of Snapin resulted in an increase (more than 95%) of the level of UL105 in the nuclear fractions from the H2-siRNA-treated cells (Fig. 8, lanes 8 to 10). In contrast, less than 5% of UL105 was found in the nuclear fractions in cells overexpressing full-length Snapin protein and Snapin mutant SΔH1(37-65aa), which are capable of binding to UL105 (i.e., U251-S and U251-SΔH1 cells, respectively; lanes 14 and 13, respectively), but not in cells expressing Snapin mutant SΔH2(81-126aa), which exhibits no UL105 binding activity (i.e., U251-SΔH2 cells; lane 12). These observations confirm our results from the microscopy experiments (Table 2) and suggest that Snapin plays an important role in the cellular localization of UL105.
Fig 8.

Effect of altered expression of Snapin and its mutants on the nuclear and cytoplasmic distribution of UL105. Different cells (e.g., parental U251 cells, cells overexpressing Snapin and its mutants [U251-S, U251-ΔH1, and U251-ΔH2]) were either transfected with anti-Snapin H2-siRNA molecules (lanes 1 to 3 and 8 to 10) or not transfected with any siRNAs (lanes 4 to 7 and 11 to 14). Cells were infected with HCMV (MOI = 1) at 48 h transfection, harvested at 48 to 72 h postinfection, and separated into nuclear and cytoplasmic fractions. (Top three panels) Equivalent amounts of each fraction were analyzed by immunoblotting with anti-UL105 and anti-UL70, while the purity of the nuclear and cytoplasmic fractions was assayed by immunoblotting with anti-histone H1 and antiactin, respectively. (Bottom) The analyses were repeated three times, and the standard deviations are indicated by the error bars.
Modulation of UL105 cellular localization by Snapin is independent of its interaction with HCMV UL70.
We have recently shown that Snapin also specifically interacts with HCMV primase UL70 and that up- and downregulation of Snapin expression leads to an accumulation of UL70 protein in the cytoplasm and nuclei, respectively (21). Because UL70 forms a primase-helicase complex with UL105 during HCMV infection, it is possible that the observed modulation of UL105 cellular localization by Snapin is mediated by its interaction with UL70. However, three lines of evidence suggest that this is unlikely and that the modulation of UL105 localization by Snapin is independent of its interaction with UL70.
First, to determine if Snapin may bind to UL105 independently of UL70, yeast two-hybrid assays and co-IP experiments were carried out to study if UL70 can interact with the Snapin mutants generated in this study. The results, summarized in Fig. 3, indicate that (i) the conserved H1 sequence of Snapin is essential for its interaction with UL70 and (ii) mutants SΔH1(37-65aa) and SΔH2(81-126aa), with deletion of the different conserved H1 and H2 domains, respectively, failed to interact with UL70 and UL105, respectively, suggesting that Snapin can bind to UL105 independently of UL70. These results are consistent with our observations of the interactions of Snapin and UL105 in the absence of UL70 and HCMV infection (Fig. 1, 3, and 5).
Second, the effect of the modulation of Snapin expression on UL105 localization appeared to be independent of the interaction of Snapin with UL70. In HCMV-infected U251-S, U251-SΔH1, and U251-SΔH2 cells expressing Snapin and its mutants SΔH1(37-65aa) and SΔH2(81-126aa), respectively, the total cellular levels of UL70 and UL105 were similar to those shown in Western analysis (Fig. 6). However, compared to the levels in parental U251 cells, the levels of UL70 but not those of UL105 were increased in the cytoplasm and decreased in the nuclei of U251-SΔH2 cells that expressed SΔH2(81-126aa) and that were capable of binding to UL70 but not UL105 (Fig. 8, lanes 5 and 12). In contrast, the levels of UL105 but not UL70 were increased in the cytoplasm and decreased in the nuclei of U251-SΔH1 cells that expressed SΔH1(37-65aa) and that were capable of binding to UL105 but not UL70 (Fig. 8, lanes 6 and 13). As expected, an accumulation of UL70 and UL105 was found in the cytoplasm of the U251-S cells expressing full-length Snapin due to its interaction with these two HCMV proteins (Fig. 8, bottom set of gels, lanes 7 and 14). These results suggest that Snapin may modulate the cellular localization of UL105 without interacting with UL70 and vice versa.
Third, the notion that Snapin modulates the cellular localization of UL105 independently of its interaction with UL70 is further supported by our siRNA experiments downregulating the expression of Snapin and its deletion mutants. The total cellular levels of UL70 and UL105 in cells treated with H2-siRNA were similar to those of UL70 and UL105 in cells that were not treated or treated with the control siRNA, respectively (Fig. 6; data not shown). The reduction of Snapin expression in H2-siRNA-treated U251-S and U251-SΔH1 cells appeared to lead to an increase of nuclear import of UL105 and UL70 (Fig. 8, lanes 2 and 3 and lanes 9 and 10). In the H2-siRNA-treated U251-SΔH2 cells, overexpression of mutant SΔH2(81-126aa), the expression of which was not affected by H2-siRNA due to the lack of the target H2 domain sequence, led to an increase of the UL70 level in the cytoplasm (lanes 1 and 8). The reduced expression of endogenous full-length Snapin protein led to an increase of UL105 in the nuclei, even in the presence of overexpressed SΔH2(81-126aa), as this mutant does not bind to UL105 (Fig. 8, lanes 1 and 8). Thus, UL70 and UL105 appeared to be accumulated in the cytoplasm and nuclei, respectively, in the H2-siRNA-treated U251-SΔH2 cells, consistent with our notion that Snapin interacts with UL105 and modulates its cellular localization independently from its interaction with UL70.
Effect of the interaction of UL105 by Snapin on HCMV DNA synthesis and lytic infection.
It is expected that a change of the UL105 level in the nucleus as a result of altered expression of Snapin may affect the level of HCMV DNA synthesis and lytic infection. To determine the effect of altered expression of Snapin on HCMV infection, we examined the profiles of viral gene expression by assaying the production of viral proteins of different kinetic classes during replication in cells where the expression of Snapin had been altered.
Different cells were infected with HCMV and then harvested at several time points postinfection. Western blot analysis confirmed the altered levels of Snapin expression in these cells (Fig. 6; data not shown). For example, at 48 to 72 h postinfection, a reduction of more than 90% in the expression of Snapin and its deletion mutant was found in the U251-S and U251-SΔH1 cells treated with H2-siRNA molecules, while the levels of Snapin and its deletion mutants in U251-S, U251-SΔH1, and U251-SΔH2 cells were about 5-fold higher than those in U251 cells (Fig. 6; data not shown). The up- and downregulation of the expression of Snapin or its deletion mutants did not appear to affect the protein levels of either the immediately early viral gene product IE1 or the early gene product UL44 (Fig. 9A). However, a decrease in the protein level of glycoprotein H (gH) was detected in cells that exhibited overexpressed levels of Snapin and its mutants (i.e., the U251-S, U251-SΔH1, U251-SΔH2, and U251-SΔH2 cells treated with H2-siRNA), while an increase in the level of gH was found in cells that exhibited lower levels of Snapin and its mutants (i.e., U251-S and U251-SΔH1 cells treated with H2-siRNA) (Fig. 9A). Glycoprotein H is the product of a viral late gene whose expression is dependent upon viral DNA synthesis (1). We speculated whether the change of gH levels in these cells might be an effect of modulation of viral DNA synthesis, as a result of a change of the nuclear UL105 as well as UL70 level due to altered expression of Snapin.
Fig 9.

Effect of altered expression of Snapin on HCMV gene expression and viral genomic synthesis and production. (A) Western blot analysis was used to assay the levels of HCMV IE1, UL44, and gH in the parental U251 cells (U251; lanes 4, 11, 18, and 25), cells overexpressing full-length Snapin (U251-S; lanes 3, 7, 10, 14, 17, 21, 24, and 28), and Snapin mutants SΔH1(37-65aa) (U251-ΔH1; lanes 2, 6, 9, 13, 16, 20, 23, and 27) and SΔH2(81-126aa) (U251-SΔH2; lanes 1, 5, 8, 12, 15, 19, 22, and 26). Cells were either transfected with anti-Snapin H2-siRNA (H2-siRNA; lanes 1 to 3, 8 to 10, 15 to 17, and 22 to 24) or not transfected with any siRNAs (lanes 4 to 7, 11 to 14, 18 to 21, and 25 to 28), infected with HCMV at an MOI of 1 at 48 h posttransfection, and harvested at 48 to 72 h postinfection. The expression of cellular actin was used as the internal loading control. (B) To assay the level of intracellular viral DNA, cells were harvested at 72 h postinfection. (C) Total infection cultures were collected at 5 days postinfection, and viral titers were determined. The values of the relative HCMV titers and levels of DNA, which are the means from three independent experiments, represent the ratios of the viral titers or levels of DNA in different cells to those in the parental U251 cells (U251), respectively. The viral titers in parental U251 cells were 5 × 104 PFU/ml. The analyses were repeated three times, and the standard deviations are indicated by the error bars.
Two sets of experiments were carried out in these cells to determine whether this is the case. In the first set of experiments, total intracellular DNAs were isolated and the levels of viral DNA synthesis were assayed. The number of viral genomes present in each sample was determined by quantitative real-time PCR (qPCR), with the copy number of the viral UL83 sequence normalized to the copy number of the cellular β-actin gene sequence. At 72 h postinfection, a reduction of more than 90% in the number of copies of UL83/β-actin was observed in cells that exhibited overexpressed levels of Snapin and its mutants (i.e., U251-S, U251-SΔH1, U251-SΔH2, and H2-siRNA-treated U251-SΔH2 cells), while an increase of about 3.5- to 4-fold was found in cells that exhibited lower levels of Snapin and its mutants (i.e., the H2-siRNA-treated U251-S and U251-SΔH1 cells) (Fig. 9B). In contrast, no significant difference in the number of copies of UL83/β-actin was found between the parental U251 cells and the empty expression vector-containing U251-C cells or cells treated with C-siRNA molecules (Fig. 9B). These results suggest that altered expression of Snapin mutants that specifically interact with UL70 [e.g., SΔH2(81-126aa)] and UL105 [e.g., SΔH1(37-65aa)] independently modulates HCMV DNA synthesis.
In the second set of experiments, viral progeny production in these cells was determined. Virus stocks were prepared from the infected cultures (cells and culture medium together), and their titers were determined. At 5 days after infection, a reduction in viral yield of at least 20-fold was observed in cells that exhibited overexpressed levels of Snapin and its mutants (i.e., U251-S, U251-SΔH1, U251-SΔH2, and H2-siRNA-treated U251-SΔH2 cells), while an increase of about 5-fold was found in cells that exhibited lower levels of Snapin and its mutants (i.e., the H2-siRNA treated U251-S and U251-SΔH1 cells) (Fig. 9C). In contrast, no significant difference in viral yield was found between the parental U251 cells and the empty expression vector-containing U251-C cells or cells treated with C-siRNA molecules (Fig. 9C). These results suggest that altered expression of Snapin mutants SΔH1(37-65aa) and SΔH2(81-126aa), which interact specifically with UL105 and UL70, respectively, independently modulates viral lytic replication and production.
DISCUSSION
In this study, we provide the first direct evidence that the HCMV UL105 protein specifically interacts with human Snapin. Snapin is a highly conserved and predominantly cytoplasmic protein that is ubiquitously expressed in many types of cells, such as adipocytes and neuronal cells (22–24). This protein functions as a component of the SNARE complex of proteins that is required for synaptic vesicle docking and fusion in neuronal cells (23). Snapin is also found in other types of cells to interact with components of the biogenesis of lysosome-related organelles complex 1 (BLOC-1), which is a ubiquitously expressed multisubunit protein complex required for normal biogenesis of specialized organelles of the endosomal-lysosomal system, such as melanosomes and platelet-dense granules (35, 36). Our results suggest that the specific interaction of Snapin sequesters UL105 in the cytoplasm and reduces its nuclear import, leading to the inhibition of HCMV DNA synthesis and particle production. It will be interesting to investigate if such inhibition mediated by the Snapin interaction with UL105 construes an antiviral defense for the benefit of the host or represents a means of viral temperance (28) for the benefit of HCMV, which is well-known for its ability to control its replication in a highly regulated manner during persistent and latent infections. Regulation of viral genomic DNA replication without affecting immediate early and early gene expression represents a means for viral persistent infection and plays an important role in viral pathogenesis. Our results from studies on the effect of Snapin on the cellular localization of UL105 suggest a novel mechanism for regulating the cellular distribution of helicase homologues in other herpesviruses, leading to a modulation of viral DNA replication/growth.
Our findings of the UL105-Snapin interaction may have general implications regarding the pathways for localization and assembly of the components of the DNA replication machinery of herpesviruses. Each component of the tripartite helicase-primase complex is highly conserved in all herpesviruses (1, 37, 38). For example, the helicase-primase complex in HSV-1 is encoded by UL52 (helicase), UL5 (primase), and UL8 (primase-associated factor), and that in KSHV is encoded by ORF44 (helicase), ORF56 (primase), and ORF40/41 (primase-associated factor), while that in EBV is encoded by BBLF (helicase), BSLF1 (primase), and BBLF2/3 (primase-associated factor) (11, 12, 39–41). It is generally believed that the process for proper localization of the helicase-primase complex in the nuclei, the site of viral DNA synthesis, is conserved among all herpesviruses (1). In EBV, when expressed individually in human cells, BBLF, BSLF1, and BBLF2/3 were localized in the cytoplasm (12). However, when all three were expressed in the same cells, these proteins were localized in the nuclei. Similar results were also observed in the localization of UL5, UL8, and UL52 of HSV-1 (11, 13, 42). In KSHV, when expressed individually in cells, ORF56 (primase), ORF44, and ORF40/41 were localized in the cytoplasm (41). However, these three proteins were localized in the nuclei only when they were coexpressed with other core components of the viral DNA replication machinery, including the DNA polymerase, the polymerase processivity factor, and single-strand DNA-binding protein (41). UL105 has been shown to interact with UL70 and UL102 (8). However, little is currently known about how UL105 (and its counterparts in other viruses) interacts with UL70 and UL102 (and their counterparts in other viruses) to achieve proper localization in the nuclei. No cellular factors that interact with UL105 have been reported. In this study, we provide the first direct evidence that UL105, when expressed alone in human cells, is exclusively localized in the cytoplasm (Fig. 5). This is consistent with the cytoplasmic localization pattern of its counterparts in HSV, EBV, and KSHV that was found when these proteins were expressed alone in the absence of other viral proteins (11, 12, 39–41). Furthermore, we showed for the first time that UL105 specifically interacts with Snapin, which is predominantly localized in the cytoplasm. Snapin may play a key role in the cellular localization of UL105 by binding to and sequestering this protein in the cytoplasmic compartment. Whether Snapin also interacts with the counterparts of UL105 in other viruses is currently unknown. Our results indicate that the conserved coiled-coil H2 domain of Snapin is essential for its binding to UL105. It will be interesting to determine whether this domain is also required for the interaction of Snapin with helicase proteins of other herpesviruses.
Our co-IP experiments showed that UL105 potentially interacts with Snapin in several different types of cells, such as U251, U373MG, and 293T cells, foreskin fibroblasts, and HeLa cells. Furthermore, we investigated the roles of Snapin in UL105 localization and HCMV infection in U251 cells. U251 cells were derived from neuronal tissues that can naturally be infected with HCMV in vivo and have been used to study HCMV infection (1). Further experiments in fibroblasts and other cells permissive to HCMV infection should elucidate the role of Snapin in these cells.
We have recently shown that Snapin also specifically interacts with HCMV primase UL70 and modulates its cellular localization (21). Given the fact that UL70 is known to form a primase-helicase complex with UL105 during HCMV infection, it is possible that the observed modulation of UL105 cellular localization by Snapin is mediated by its interaction with UL70. However, several lines of evidence suggest that this is unlikely and that the modulation of UL105 localization by Snapin is via specific interaction of Snapin with UL105 and is independent of its interaction with UL70. First, the Snapin-UL105 interaction was identified by both yeast two-hybrid assays and co-IP experiments in human cells in the absence of UL70 expression (Fig. 1, 3, and 5). Second, different domains of Snapin are essential for interactions with UL70 and UL105. Snapin mutant SΔH2(81-126aa), with the deletion of the conserved H2 domain, bound to UL70 but not UL105, while mutant SΔH1(37-65aa), with the deletion of the different H1 domain, bound to UL105 but not UL70. Third, Snapin may modulate the cellular localization of UL105 in the absence of its interaction with UL70 and vice versa. Compared to the levels in parental U251 cells, the levels of UL105 but not UL70 were increased in the cytoplasm and decreased in the nuclei of U251-SΔH1 cells, which expressed SΔH1(37-65aa), which is capable of binding to UL105 but not UL70 (Fig. 8). Fourth, altered expression of Snapin mutants that specifically interact with UL105 [e.g., SΔH1(37-65aa)] and UL70 [e.g., SΔH2(81-126aa)] independently modulates HCMV DNA synthesis and virus progeny production, respectively (Fig. 9).
In addition to the coding sequence of Snapin, the coding sequences of several human proteins were also identified to be present in the yeast colonies that were positive in the UL105 yeast two-hybrid screen experiments. Additional and independent experiments (e.g., coimmunoprecipitation) will be performed to confirm the interactions of these proteins with UL105. Further studies on these potential interactions will provide insight into how host proteins modulate UL105 function in HCMV infection.
It is possible that Snapin interacts with HCMV proteins other than UL105 and UL70, leading to a modulation of viral DNA replication, as observed in our experiments. However, our yeast two-hybrid experiments revealed no positive interaction between Snapin and more than 100 HCMV ORFs other than UL105 and UL70 (data not shown). Co-IP experiments using antibodies against several HCMV proteins that are currently commercially available (e.g., UL44, IE1, and UL83) did not reveal any positive interactions (Fig. 2; data not shown). Furthermore, the Snapin mutant with the deletion of the H1 domain [i.e., SΔH1(37-65aa)] interacted only with UL105 and not other HCMV proteins tested. While we cannot completely exclude the possibility that Snapin may affect HCMV DNA replication by interacting with HCMV proteins other than UL105 and UL70, our results for mock-infected and HCMV-infected cells (e.g., Fig. 5 and 7 and Table 2) suggest that Snapin may interact with UL105 and alters its cellular localization in the absence of other HCMV proteins, possibly leading to a modulation of viral DNA replication during HCMV infection.
Our observations suggest that the proper cellular localization of UL105 in either the cytoplasm or nucleus is at a delicate balance and may be dictated by the cellular expression level of Snapin. How UL105 is retained in the cytoplasm by Snapin is currently unknown. It is conceivable that Snapin sequesters UL105 in specific cytoplasmic compartments so that UL105 is not available for interaction with other viral or cellular protein carriers for nuclear transport. When Snapin expression is reduced, UL105 is available to interact with the cellular nuclear transport protein(s), leading to increased nuclear import of UL105. In contrast, Snapin, when overexpressed, interacts with and sequesters UL105, leading to its accumulation in the cytoplasm. This hypothesis is supported by our observations that up- and downregulation of Snapin promotes cytoplasmic and nuclear accumulation of UL105, respectively. It will be interesting to determine whether the cellular localization of the homologues of UL105 in other viruses also employs similar cellular pathways. These and future studies on the interactions of the herpesvirus helicase proteins with other viral and cellular proteins will provide insight into the mechanism of proper localization and assembly of the helicase-primase complex during lytic infection of herpesviruses.
ACKNOWLEDGMENTS
We are indebted to Paul Rider, Gia-Phong Vu, Luyao Shao, and Jun Huang for technical assistance and invaluable suggestions. We also thank John Wu of Promab Inc. for excellent assistance with antibody production.
A.S. was the recipient of a China Graduate Student Scholarship from the Chinese Ministry of Education (NSFC; no. 30528022). E.Y. was partially supported by a block grant predoctoral fellowship (University of California, Berkeley). This research has been supported by grants from the National Basic Research Program of China (973 Program, no. 2011CB504800 and 2012CB518900), the National Natural Science Foundation of China (no. 31100128 and 81030031), and NIH (RO1-AI050468 and RO1-DE014145).
Footnotes
Published ahead of print 24 July 2013
REFERENCES
- 1.Mocarski ES, Shenk T, Pass RF. 2007. Cytomegalovirus, p 2701–2772 In Knipe DM, Howley PM, Griffin DE, Martin MA, Lamb RA, Roizman B, Straus SE. (ed), Fields virology, 5th ed. Lippincott William & Wilkins, Philadelphia, PA [Google Scholar]
- 2.Britt WJ. 1999. Congenital cytomegalovirus infection, p 269–281 In Hitchcock H, MacKay T, Wasserheit JN. (ed), Sexually transmitted diseases and adverse outcomes of pregnancy. ASM Press, Washington, DC [Google Scholar]
- 3.Britt W. 2008. Manifestations of human cytomegalovirus infection: proposed mechanisms of acute and chronic disease. Curr. Top. Microbiol. Immunol. 325:417–470 [DOI] [PubMed] [Google Scholar]
- 4.Wills MR, Ashiru O, Reeves MB, Okecha G, Trowsdale J, Tomasec P, Wilkinson GW, Sinclair J, Sissons JG. 2005. Human cytomegalovirus encodes an MHC class I-like molecule (UL142) that functions to inhibit NK cell lysis. J. Immunol. 175:7457–7465 [DOI] [PubMed] [Google Scholar]
- 5.Pari GS. 2008. Nuts and bolts of human cytomegalovirus lytic DNA replication. Curr. Top. Microbiol. Immunol. 325:153–166 [DOI] [PubMed] [Google Scholar]
- 6.Biron KK. 2006. Antiviral drugs for cytomegalovirus diseases. Antiviral Res. 71:154–163 [DOI] [PubMed] [Google Scholar]
- 7.Woon HG, Scott GM, Yiu KL, Miles DH, Rawlinson WD. 2008. Identification of putative functional motifs in viral proteins essential for human cytomegalovirus DNA replication. Virus Genes 37:193–202 [DOI] [PubMed] [Google Scholar]
- 8.McMahon TP, Anders DG. 2002. Interactions between human cytomegalovirus helicase-primase proteins. Virus Res. 86:39–52 [DOI] [PubMed] [Google Scholar]
- 9.Smith JA, Jairath S, Crute JJ, Pari GS. 1996. Characterization of the human cytomegalovirus UL105 gene and identification of the putative helicase protein. Virology 220:251–255 [DOI] [PubMed] [Google Scholar]
- 10.Smith JA, Pari GS. 1995. Expression of human cytomegalovirus UL36 and UL37 genes is required for viral DNA replication. J. Virol. 69:1925–1931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Barnard EC, Brown G, Stow ND. 1997. Deletion mutants of the herpes simplex virus type 1 UL8 protein: effect on DNA synthesis and ability to interact with and influence the intracellular localization of the UL5 and UL52 proteins. Virology 237:97–106 [DOI] [PubMed] [Google Scholar]
- 12.Gao Z, Krithivas A, Finan JE, Semmes OJ, Zhou S, Wang Y, Hayward SD. 1998. The Epstein-Barr virus lytic transactivator Zta interacts with the helicase-primase replication proteins. J. Virol. 72:8559–8567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Marsden HS, Cross AM, Francis GJ, Patel AH, MacEachran K, Murphy M, McVey G, Haydon D, Abbotts A, Stow ND. 1996. The herpes simplex virus type 1 UL8 protein influences the intracellular localization of the UL52 but not the ICP8 or POL replication proteins in virus-infected cells. J. Gen. Virol. 77(Pt 9):2241–2249 [DOI] [PubMed] [Google Scholar]
- 14.Penfold ME, Mocarski ES. 1997. Formation of cytomegalovirus DNA replication compartments defined by localization of viral proteins and DNA synthesis. Virology 239:46–61 [DOI] [PubMed] [Google Scholar]
- 15.Alvisi G, Jans DA, Guo J, Pinna LA, Ripalti A. 2005. A protein kinase CK2 site flanking the nuclear targeting signal enhances nuclear transport of human cytomegalovirus ppUL44. Traffic 6:1002–1013 [DOI] [PubMed] [Google Scholar]
- 16.Alvisi G, Ripalti A, Ngankeu A, Giannandrea M, Caraffi SG, Dias MM, Jans DA. 2006. Human cytomegalovirus DNA polymerase catalytic subunit pUL54 possesses independently acting nuclear localization and ppUL44 binding motifs. Traffic 7:1322–1332 [DOI] [PubMed] [Google Scholar]
- 17.Giesen K, Radsak K, Bogner E. 2000. The potential terminase subunit of human cytomegalovirus, pUL56, is translocated into the nucleus by its own nuclear localization signal and interacts with importin alpha. J. Gen. Virol. 81:2231–2244 [DOI] [PubMed] [Google Scholar]
- 18.Pizzorno MC, Mullen MA, Chang YN, Hayward GS. 1991. The functionally active IE2 immediate-early regulatory protein of human cytomegalovirus is an 80-kilodalton polypeptide that contains two distinct activator domains and a duplicated nuclear localization signal. J. Virol. 65:3839–3852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wood LJ, Baxter MK, Plafker SM, Gibson W. 1997. Human cytomegalovirus capsid assembly protein precursor (pUL80.5) interacts with itself and with the major capsid protein (pUL86) through two different domains. J. Virol. 71:179–190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schmolke S, Kern HF, Drescher P, Jahn G, Plachter B. 1995. The dominant phosphoprotein pp65 (UL83) of human cytomegalovirus is dispensable for growth in cell culture. J. Virol. 69:5959–5968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shen A, Lei J, Yang E, Pei Y, Chen YC, Gong H, Xiao G, Liu F. 2011. Human cytomegalovirus primase UL70 specifically interacts with cellular factor Snapin. J. Virol. 85:11732–11741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bao Y, Lopez JA, James DE, Hunziker W. 2008. Snapin interacts with the Exo70 subunit of the exocyst and modulates GLUT4 trafficking. J. Biol. Chem. 283:324–331 [DOI] [PubMed] [Google Scholar]
- 23.Buxton P, Zhang XM, Walsh B, Sriratana A, Schenberg I, Manickam E, Rowe T. 2003. Identification and characterization of Snapin as a ubiquitously expressed SNARE-binding protein that interacts with SNAP23 in non-neuronal cells. Biochem. J. 375:433–440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Suzuki F, Morishima S, Tanaka T, Muramatsu I. 2007. Snapin, a new regulator of receptor signaling, augments alpha1A-adrenoceptor-operated calcium influx through TRPC6. J. Biol. Chem. 282:29563–29573 [DOI] [PubMed] [Google Scholar]
- 25.Salsman J, Zimmerman N, Chen T, Domagala M, Frappier L. 2008. Genome-wide screen of three herpesviruses for protein subcellular localization and alteration of PML nuclear bodies. PLoS Pathog. 4:e1000100. 10.1371/journal.ppat.1000100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kilani AF, Trang P, Jo S, Hsu A, Kim J, Nepomuceno E, Liou K, Liu F. 2000. RNase P ribozymes selected in vitro to cleave a viral mRNA effectively inhibit its expression in cell culture. J. Biol. Chem. 275:10611–10622 [DOI] [PubMed] [Google Scholar]
- 27.To A, Bai Y, Shen A, Gong H, Umamoto S, Lu S, Liu F. 2011. Yeast two hybrid analyses reveal novel binary interactions between human cytomegalovirus-encoded virion proteins. PLoS One 6:e17796. 10.1371/journal.pone.0017796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dunn W, Chou C, Li H, Hai R, Patterson D, Stolc V, Zhu H, Liu F. 2003. Functional profiling of a human cytomegalovirus genome. Proc. Natl. Acad. Sci. U. S. A. 100:14223–14228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Trang P, Lee M, Nepomuceno E, Kim J, Zhu H, Liu F. 2000. Effective inhibition of human cytomegalovirus gene expression and replication by a ribozyme derived from the catalytic RNA subunit of RNase P from Escherichia coli. Proc. Natl. Acad. Sci. U. S. A. 97:5812–5817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zou H, Lee J, Umamoto S, Kilani AF, Kim J, Trang P, Zhou T, Liu F. 2003. Engineered RNase P ribozymes are efficient in cleaving a human cytomegalovirus mRNA in vitro and are effective in inhibiting viral gene expression and growth in human cells. J. Biol. Chem. 278:37265–37274 [DOI] [PubMed] [Google Scholar]
- 31.Miller AD, Rosman GJ. 1989. Improved retroviral vectors for gene transfer and expression. Biotechniques 7:980–982, 984–986, 989–990 [PMC free article] [PubMed] [Google Scholar]
- 32.Liu F, Altman S. 1995. Inhibition of viral gene expression by the catalytic RNA subunit of RNase P from Escherichia coli. Genes Dev. 9:471–480 [DOI] [PubMed] [Google Scholar]
- 33.Hanfler J, Kreuzer KA, Laurisch K, Rayes N, Neuhaus P, Schmidt CA, Oettle H. 2003. Quantitation of cytomegalovirus (hCMV) DNA and beta-actin DNA by duplex real-time fluorescence PCR in solid organ (liver) transplant recipients. Med. Microbiol. Immunol. 192:197–204 [DOI] [PubMed] [Google Scholar]
- 34.Dunn W, Trang P, Khan U, Zhu J, Liu F. 2001. RNase P-mediated inhibition of cytomegalovirus protease expression and viral DNA encapsidation by oligonucleotide external guide sequences. Proc. Natl. Acad. Sci. U. S. A. 98:14831–14836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ruder C, Reimer T, Delgado-Martinez I, Hermosilla R, Engelsberg A, Nehring R, Dorken B, Rehm A. 2005. EBAG9 adds a new layer of control on large dense-core vesicle exocytosis via interaction with Snapin. Mol. Biol. Cell 16:1245–1257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Starcevic M, Dell'Angelica EC. 2004. Identification of Snapin and three novel proteins (BLOS1, BLOS2, and BLOS3/reduced pigmentation) as subunits of biogenesis of lysosome-related organelles complex-1 (BLOC-1). J. Biol. Chem. 279:28393–28401 [DOI] [PubMed] [Google Scholar]
- 37.Kieff E, Rickinson AB. 2007. Epstein-Barr virus and its replication, p 2604–2654 In Knipe DM, Howley PM, Griffin DE, Lamb RA, Martin MA, Roizman B, Straus SEPM. (ed), Fields virology, 5th ed. Lippincott William & Wilkins, Philadelphia, PA [Google Scholar]
- 38.Roizman B, Knipe DM, Whitley RJ. 2007. Herpes simplex viruses, p 2503–2601 In Knipe DM, Howley PM, Griffin DE, Lamb RA, Martin MA, Roizman B, Straus SEPM. (ed), Fields virology, 5th ed, vol 2 Lippincott William & Wilkins, Philadelphia, PA [Google Scholar]
- 39.Pari GS, Anders DG. 1993. Eleven loci encoding trans-acting factors are required for transient complementation of human cytomegalovirus oriLyt-dependent DNA replication. J. Virol. 67:6979–6988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wu CA, Nelson NJ, McGeoch DJ, Challberg MD. 1988. Identification of herpes simplex virus type 1 genes required for origin-dependent DNA synthesis. J. Gen. Virol. 62:435–443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tortorella D, Gewurz BE, Furman MH, Schust DJ, Ploegh HL. 2000. Viral subversion of the immune system. Annu. Rev. Immunol. 18:861–926 [DOI] [PubMed] [Google Scholar]
- 42.Calder JM, Stow EC, Stow ND. 1992. On the cellular localization of the components of the herpes simplex virus type 1 helicase-primase complex and the viral origin-binding protein. J. Gen. Virol. 73:531–538 [DOI] [PubMed] [Google Scholar]