

Abstract
Translation of influenza A virus PB1-F2 occurs in a second open reading frame (ORF) of the PB1 gene segment. PB1-F2 has been implicated in regulation of polymerase activity, immunopathology, susceptibility to secondary bacterial infection, and induction of apoptosis. Experimental evidence of PB1-F2 molecular function during infection has been collected primarily from human and avian viral isolates. As the 2009 H1N1 (H1N1pdm09) strain highlighted, some swine-derived influenza viruses have the capacity to infect human hosts and emerge as a pandemic. Understanding the impact that virulence factors from swine isolates have on both human and swine health could aid in early identification of viruses with pandemic potential. Studies examining PB1-F2 from swine isolates have focused primarily on H1N1pdm09, which does not encode PB1-F2 but was engineered to carry a full-length PB1-F2 ORF to assess the impact on viral replication and pathogenicity. However, experimental evidence of PB1-F2 protein expression from swine lineage viruses has not been demonstrated. Here, we reveal that during infection, PB1-F2 expression levels are substantially different in swine and human influenza viruses. We provide evidence that PB1-F2 expression is regulated at the translational level, with very low levels of PB1-F2 expression from swine lineage viruses relative to a human isolate PB1-F2. Translational regulation of PB1-F2 expression was partially mapped to two independent regions within the PB1 mRNA, located downstream of the PB1-F2 start site. Our data suggest that carrying a full-length PB1-F2 ORF may not be predictive of PB1-F2 expression in infected cells for all influenza A viruses.
INTRODUCTION
Translation of influenza A virus (IAV) PB1-F2 initiates in a +1 open reading frame (ORF) relative to the PB1 gene via a leaky ribosomal scanning mechanism as a result of the 43S ribosomal complex bypassing the PB1 start codon and two additional intervening AUG codons that translate short ORFs (sORFs) (1, 2). Previously, Wise et al. showed that PB1-F2 translation initiation is regulated as a result of differences in Kozak sequence strength, with the PB1 and two intervening sORF start codons having weak to moderate-strength Kozak sequences, while the PB1-F2 ORF start codon has a strong Kozak sequence (2). Leaky ribosomal scanning, in combination with reinitiation of PB1 mRNA, is also responsible for translation of a third protein known as N40, which is encoded from a fifth AUG located in the same ORF as the PB1 gene but missing 39 amino (N)-terminal amino acids from the PB1 protein (3). Modification of the upstream start codons, Kozak sequences, and ORF nucleotide length has been shown to affect the level of PB1-F2 expression (2, 3).
In early studies, PB1-F2 was found to have proapoptotic activity when expressed either independently or during influenza virus infection. Chen et al. first determined that PB1-F2 was proapoptotic either when exposed to cells in synthetic form or in the context of cells infected with either wild-type or PB1-F2-deficient A/Puerto Rico/8/34 (PR8) IAV (1). In addition, the carboxyl (C)-terminal portion of PB1-F2 has also been shown to interact with mitochondrial antiviral stimulating protein (MAVS), resulting in decreased mitochondrial membrane potential and prevention of transcriptional upregulation of type I interferon (IFN), as well as other interferon-stimulated genes (4, 5). Conversely, other studies have shown that PB1-F2 promotes the production of type I IFN, particularly IFN-β, in human lung epithelial cells and is detrimental to successful virus infection (6). PB1-F2 has also been shown to increase susceptibility to secondary bacterial infection through promotion of proinflammatory cytokine expression, which leads to increases in viral and bacterial replication rates, infiltration of monocytes into the lungs, severity of disease, and mortality (7–10). These divergent findings may suggest that the IFN modulation properties of PB1-F2 are cell type or virus isolate specific.
PB1-F2 proteins from different IAV isolates do not possess each function described for the protein. In fact, much of the molecular work done on PB1-F2 has been performed using a limited number of viral isolates, such as PR8, A/WSN/1933 (WSN), A/Brevig Mission/1918 (1918 Spanish flu), and avian H5N1 isolates (1, 7, 11, 12). Because full-length PB1-F2 ORFs are present at various levels of prevalence in IAV, it is necessary to have a complete understanding of how PB1-F2 proteins from IAV isolates from different host species contribute to IAV replication or virulence. The 2009 swine origin H1N1 pandemic virus (H1N1pdm09) was a stark reminder that swine-derived IAV is capable of infecting and adapting to humans and emerging as a pandemic (13). Although not highly pathogenic, H1N1pdm09 illustrates the potential threat that swine IAV poses to human health (14). While H1N1pdm09 did not express full-length PB1-F2, recent IAV reassortants between H1N1pdm09 and H3N2 strains circulating in swine herds have infected humans (H3N2v strains), and although human-to-human transmission of these viruses is considered rare, they do appear to encode the capacity to express PB1-F2 (15). Thus, it is necessary to have a proper understanding of the molecular function and virulence potential of swine isolate-derived PB1-F2 proteins.
The majority of published studies examining the function of swine PB1-F2 have focused on measuring phenotypic differences in vitro and in vivo using wild-type isolates or recombinant H1N1pdm09 viruses engineered to carry a full-length PB1-F2 ORF. Conclusions from these studies about PB1-F2 effects on swine IAV infection conflict. For example, Hai et al. and Pena et al. found that restoring the PB1-F2 coding sequence to H1N1pdm09 resulted in increased growth kinetics and higher viral titers in cell culture, while Ozawa et al. showed that the presence of PB1-F2 had no affect on viral replication in vitro (16–19). Restoration of the PB1-F2 coding sequence had little to no effect on viral pathogenicity in infected mice or ferrets in these studies, although a slight increase in pathogenicity was noted at early times in infection of pigs (16–18). Other work using recombinant A/swine/Wisconsin/14094/99 viruses showed an increase of virus replication in primary alveolar macrophages but no differences in viral replication or pathogenicity in swine tissue explants or infected pigs (20). In all of the studies examining PB1-F2 function in swine IAV infection, PB1-F2 protein expression was not evaluated either in vitro or in vivo.
In this study, we determined that PB1-F2 protein expression from representative swine isolates is substantially decreased relative to that from a previously highly studied human isolate. Additionally, we determined that PB1-F2 expression is primarily regulated at the level of translation and identified two elements downstream from the PB1-F2 AUG that are involved in this regulation.
MATERIALS AND METHODS
Phylogenetic comparison of PB1-F2 nucleotide sequences.
IAV PB1-F2 nucleotide coding sequences (nucleotides [nt] 119 to 391) from A/swine/Ohio/511445/2007(H1N1) (OH07), A/Mexico/4108/2009(H1N1) (Mx09), A/swine/Nebraska/02013/2008(H1N1), A/swine/Indiana/A00968373/2012(H3N2), A/Puerto Rico/8/1934(H1N1) (PR8), A/Brevig Mission/1/1918(H1N1) (1918 Spanish Flu), A/Vietnam/1203/2004(H5N1), and A/Indonesia/CDC1047/2007(H5N1) were collected from NCBI. Using the Influenza Virus Resource database tool (NCBI), a nucleotide consensus sequence of swine PB1-F2 was established by comparing 310 unique PB1-F2 coding sequences from H1N1 and H3N2 viruses isolated from pigs in North America from 2000 to the present. A phylogram was then created using Phylogeny.fr (21, 22) and annotated using Adobe Illustrator.
Cells and reagents.
MDCK (Madin-Darby canine kidney) and PK-15 (porcine kidney epithelial) cells were maintained in Eagle's modified essential medium (EMEM) (Mediatech). A549 (human alveolar basal epithelial) cells were maintained in F-12K medium (Kaighn's Modification of Ham's F-12 Medium; Mediatech). 293T (human embryonic kidney) cells were maintained in Opti-MEM (Life Technologies). CEF (chicken embryo fibroblasts) were maintained in Dulbecco's modified essential medium (DMEM) (Life Technologies). 3D4/2 porcine alveolar macrophages were maintained in RPMI 1640 medium (ATCC). BHK-T7 cells (baby hamster kidney cells expressing T7 polymerase) (23) were maintained in DMEM supplemented with l-glutamine (Gibco), nonessential amino acids (Gibco), and amphotericin B (Fungizone; Gibco). All cell culture media were also supplemented with 10% fetal bovine serum (FBS) (Atlanta Biologicals) and penicillin/streptomycin (100 IU/ml; Mediatech). The primary antibodies used in this study include mouse anti-NP (α-NP) (ATCC) (24), rabbit α-PR8 PB1-F2 (Peter Palese, Mount Sinai School of Medicine), rabbit α-OH07 PB1-F2 (GenScript; peptide antibody against OH07 amino acids 13 to 26), goat α-PB1 (Santa Cruz Biotechnology, Inc.), mouse α-3×FLAG (Sigma-Aldrich), and rabbit α-β-actin (Cell Signaling Technologies). Prior to performing experiments, antibodies were titrated to identify antibody concentrations in which high specificity and low background signal were optimized. The secondary antibodies used in immunofluorescence and immunoblotting experiments in this study were as follows: Alexa 350-, Alexa 488-, or Alexa 594-conjugated donkey or goat α-mouse, α-rabbit, α-goat, or α-human IgG antibodies (Life Technologies) and alkaline phosphatase (AP)-conjugated goat α-mouse and α-rabbit or rabbit α-goat (Bio-Rad Laboratories). MG132 at a final concentration of 0.5 μM was added to cells where indicated.
Viruses and recombinant viruses.
A/swine/OH/511445/2007 (OH07), A/Puerto Rico/8/1934 (PR8), and A/Mexico/4108/2009 (Mx09; Alexander Klimov, CDC) were used to represent H1N1 swine, human, and pandemic 2009 isolates, respectively (Table 1). Eight reverse-genetics plasmids for PR8 (Richard Webby, St. Jude Children's Research Hospital) (25) and Mx09 (Daniel Perez, University of Maryland) were used for creation of recombinant viruses, as previously described (25). The 11-amino-acid-truncated Mx09 PB1-F2 sequence was replaced by the full-length OH07 PB1-F2 ORF sequence in the Mx09 PB1 gene to create PB1/MOM (details of the construction are provided below). Utilizing parental or mutant reverse-genetics plasmids, recombinant viruses rMx09, Mx09+MOM PB1 (rMx/MOM), Mx09+PR8 PB1 (rMx/PR8), rPR8, and PR8+MOM PB1 (rPR8/MOM) were created (Table 1.).
Table 1.
Wild-type and recombinant viruses
| Name | Description |
|---|---|
| wtOH07 | Wild-type A/swine/Ohio/511445/2007(H1N1) |
| rMx09 | Recombinant A/Mexico/4108/2009(H1N1) |
| rPR8 | Recombinant A/Puerto Rico/8/1934(H1N1) |
| rMx/MOM | Mx09 expressing PB1/MOM |
| rMx/PR8 | Mx09 expressing PB1/PR8 |
| rPR8/MOM | PR8 expressing PB1/MOM |
Plasmid construction. (i) Reverse-genetics plasmids.
The OH07 PB1 gene sequence was amplified from virus stock by reverse transcription (RT)-PCR using universal primers that have been previously described (26). The resulting PCR product and pHW2000 were digested with BsmBI and ligated to create pHW2000/OH07/PB1. To create pDP2002/MOM/PB1, the OH07 PB1-F2 ORF was PCR amplified using gene-specific primers flanking the PB1-F2 sequence and pHW2000/OH07 PB1 as the template. The resulting PCR product and pDP2002/Mx09/PB1 plasmid were digested with BciVI and BstAPI and ligated.
(ii) Expression plasmids.
To create pOH07/PB1-F2/EGFP and pPR8/PB1-F2/EGFP, the PB1-F2 sequence from pHW2000/OH07/PB1 and pHW2000/PR8/PB1 was amplified using gene-specific primers containing XhoI and HindIII restriction sites. pEGFP-N1 and amplification products were digested with XhoI and HindIII and ligated. pOH07/PB1-F2/pET21a was created by PCR using gene-specific primers containing NdeI and XhoI digestion sites and pHW2000/OH07/PB1 as the template. The resulting PCR product and pET21a were digested with NdeI and XhoI and ligated. To create pOH07/PB1-F2/3×FLAG, a gene-specific 3×FLAG tag containing forward and reverse primers and a pHW2000/OH07/PB1-F2 template were used to amplify PB1-F2. The resulting PCR product and pCi-Neo were digested with XhoI and XbaI and ligated. pPR8/PB1-F2/3×FLAG was created by inserting annealed oligonucleotides containing the 3×FLAG tag flanked by KpnI and XbaI sites into KpnI- and XbaI-digested pPR8/PB1-F2/EGFP.
(iii) Translational-mapping plasmids.
pOOO/3×FLAG and pMOM/3×FLAG were created by inserting annealed oligonucleotides containing the 3×FLAG tag flanked by PasI and BstAPI sites into PasI- and BstAPI-digested pHW2000/OH07/PB1 and pDP2002/MOM/PB1. pPPP/3×FLAG was created by PCR amplification using gene-specific primers and pPR8/PB1-F2/3×FLAG as a template. The amplified product was ligated into pHW2000/PR8/PB1 using BstAPI and BfuAI digestion sites. pMMM/3×FLAG was created using gene-specific primers and pDP2002/Mx09/PB1 and pMOM/3×FLAG as the templates in an overlap PCR. The resulting product was inserted into pDP2002/Mx09/PB1 using BsaAI and BstAPI sites. pMxKI/3×FLAG was created using pMMM/3×FLAG as a template in overlap PCR mutagenesis to remove stop codons within PB1-F2 (27). Nucleotides that introduced stop codons were replaced as follows: 152A to C, 291A to G, and 381A to G. The PCR product was then ligated into pDP2002/Mx09/PB1 using BsaAI and BstAPI restriction sites. Additional translational plasmids constructed for this study to map PB1-F2 translational regulatory regions are listed in Table 2. The vector, insert PB1 nucleotide sequence, and methods used to create each plasmid are shown. These methods are PCR amplification with gene-specific primers, overlap PCR using gene-specific primers for amplification and fusion of sequences (27), and subcloning between plasmids containing identical restriction sites. All plasmids were selected by restriction digest and confirmed by sequencing. Primer sequences are available upon request.
Table 2.
3×FLAG translational-mapping plasmids
| Plasmid | Vector | Inserta | Restriction sites | Methodb |
|---|---|---|---|---|
| pPOP/3×FLAG | pHW2000/PR8/PB1 | pOOO/3×FLAG (nt 71–444) | BstAPI, BfuAI | P |
| pMPM/3×FLAG | pDP2002/Mx09/PB1 | pDP2002/Mx09/PB1 (nt 1–391); pPPP/3×FLAG (nt 392–445) | BsaAI, BstAPI | O |
| pOPO/3×FLAG | pHW2000/OH07/PB1 | pHW2000/OH07/PB1 (nt 1–391); pPPP/3×FLAG (nt 392–445) | BsaAI, BstAPI | O |
| pPPM/3×FLAG | pDP2002/Mx09/PB1 | pPPP/3×FLAG (nt 1–391); pDP2002/Mx09/PB1 (nt 392–445) | BsaAI, BstAPI | O |
| pPOM/3×FLAG | pDP2002/Mx09/PB1 | pPOP/3×FLAG (nt 1–391); pDP2002/Mx09/PB1 (nt 392–445) | BsaAI, BstAPI | O |
| pPPO/3×FLAG | pHW2000/OH07/PB1 | pPPO/3×FLAG (nt 1–391); pH 2002/OH07/PB1 (nt 392–445) | BsaAI, BstAPI | O |
| pPOO/3×FLAG | pHW2000/OH07/PB1 | pPOO/3×FLAG (nt 1–391); pHW2000/OH07/PB1 (nt 392–445) | BsaAI, BstAPI | O |
| pMOO/3×FLAG | pHW2000/OH07/PB1 | pMOM/3×FLAG (nt 1–445) | BsaAI, BstAPI | P |
| pOOM/3×FLAG | pDP2002/Mx09/PB1 | pOOO/3×FLAG (nt 1–445) | BsaAI, BstAPI | P |
| pMPO/3×FLAG | pHW2000/OH07/PB1 | pMPM/3×FLAG (nt 1–445) | BsaAI, BstAPI | P |
| pOPM/3×FLAG | pDP2002/Mx09/PB1 | pOPO/3×FLAG (nt 1–445) | BsaAI, BstAPI | P |
| pMOP/3×FLAG | pHW2000/PR8/PB1 | pMOM/3×FLAG (nt 1–444) | NheI, BfuAI | P |
| pMPP/3×FLAG | pHW2000/PR8/PB1 | pMPM/3×FLAG (nt 1–444) | NheI, BfuAI | P |
| pOOP/3×FLAG | pHW2000/PR8/PB1 | pOOO/3×FLAG (nt 1–444) | NheI, BfuAI | P |
| pOPP/3×FLAG | pHW2000/PR8/PB1 | pOPO/3×FLAG (nt 1–444) | NheI, BfuAI | P |
| pMPM(P1121-2298)/3×FLAG | pMPP/3×FLAG | pMPM/3×FLAG (nt 1–1140) | NheI | S |
| pMPM(P392-1140 M)3×FLAG | pMPM/3×FLAG | pMPP/3×FLAG (nt 1–1140) | NheI | S |
| pMPM(P392-816)/3×FLAG | pDP2002/Mx09/PB1 | pMPM(P392–1140 M)3×FLAG (nt 1–817) | BsaAI, HindIII | P |
| pMPM(P392-586)/3×FLAG | pMPM(P392-816)/3×FLAG | pDP2002/Mx09/PB1 (nt 583–817) | BsmBI, HindIII | P |
| pMPM(P582-816)/3×FLAG | pMPM(P392-816)/3×FLAG | pMPM/3×FLAG (nt 1–582) | BsaAI, BsmBI | P |
| pMO(P267-391)M/3×FLAG | pDP2002/Mx09/PB1 | pMOM/3×FLAG (nt 1–266); pMPM/3×FLAG (nt 267–445) | BsaAI, BstAPI | O |
| pM(P119-252)OM/3×FLAG | pDP2002/Mx09/PB1 | pMPM/3×FLAG (nt 1–252); pMOM/3×FLAG (nt 253–445) | BsaAI, BstAPI | O |
| pMO(P344-816)M/3×FLAG | pDP2002/Mx09/PB1 | pMOM/3 × FLAG (nt 1–336); pMPM(P582-816)/3×FLAG (nt 337–817) | BsaAI, EcoRI, HindIII | P |
| pMO(P267-335)O(P392-816)M/3×FLAG | pDP2002/Mx09/PB1 | pMO(P267-391)M/3×FLAG (nt 1–336); pMOP/3×FLAG (nt 337–817) | BsaAI, EcoRI, HindIII | P |
| pMOMF2UP/3×FLAG | pMPM(P392-816)/3×FLAG | pMO(P267-335)O(P392-816)M/3×FLAG (nt 1–336); pMOM/3×FLAG (nt 337–582) | BsaAI, EcoRI, BsmBI | P |
| pPP(O267-343)P/3×FLAG | pHW2000/PR8/PB1 | pPPP/3×FLAG (nt 71–266); pMO(P344-816)M/3×FLAG (nt 267–444) | BstAPI, BfuAI | O |
| pPR8F2dwn/3×FLAG | pPP(O267-343)P/3×FLAG | pDP2002/Mx09/PB1 (nt 583–817); pHW2000/PR8/PB1 (nt 818–1134) | BsmBI, HindIII, PstI | P |
The numbers correspond to the PB1 nucleotides inserted from the parent plasmid(s) into the vector and are in reference to the beginning of the influenza virus 5′ untranslated region prior to the PB1 mRNA sequence; they do not include the 3×FLAG sequence.
Method used to generate the insert. P, PCR; O, overlap PCR; S, subcloning.
Production of recombinant PB1-F2.
Escherichia coli Rosetta was transformed with pOH07/PB1-F2/pET21a using an Eppendorf Multiporator (Eppendorf) set to 1,700 V with a time constant of 5 ms. The cells were resuspended in 1 ml of Luria broth (LB), incubated in a 37°C shaking incubator for 30 min, and plated on Luria agar containing 50 μg/ml ampicillin (AMP) for overnight incubation at 37°C. Colonies were picked and grown in LB containing 50 μg/ml AMP. The following day, the cultures were diluted 1:100 in the same medium formulation with the addition of 1 mM isopropyl-β-d-l-thiogalactopyranoside (IPTG) for 4 h. Pellets were collected and purified using a BioLogic protein purification system (28). Expression of PB1-F2 in each 5-ml fraction of the purified protein was confirmed using immunoblotting.
Infections and transfections.
MDCK, A549, PK-15, 3D4/2, or BHK-T7 cells or CEF were plated to a final density of 2.5 × 105/well (6-well plate) or 5 × 105/60-mm dish and 293T cells to a final density of 2 × 106/60-mm dish for transfections and infections. Infections were carried out using EMEM or F-12K medium containing 0.4% bovine serum albumin (BSA), penicillin-streptomycin (100 IU/ml), and 1 μg/μl tosylsulfonyl phenylalanyl chloromethyl ketone (TPCK)-treated trypsin. Viral titers were determined for each cell line using the 50% tissue culture infective dose (TCID50) and a working estimate of the number of PFU/ml was calculated (PFU/ml = TCID50 × 0.7; ATCC [http://www.atcc.org/support/faqs/48802/Converting%2bTCID50%2bto%2bplaque%2bforming%2bunits%2bPFU-124.aspx]). Infections were carried out at a multiplicity of infection (MOI) of either 2 for immunofluorescence (IF) assay and RT-quantitative PCR (qPCR) or 5 for immunoblots. For PB1-F2 degradation assays, infected MDCK cells were incubated with MG132 for 4 h prior to harvest. MDCK, A549, and PK-15 cells were transfected using Lipofectamine 2000 (Invitrogen), and 293T, BHK-T7, and MDCK (pulse-chase experiments only) cells were transfected using TransIT-LT1 (Mirus) according to the manufacturer's instructions.
Immunofluorescence microscopy.
Cells plated on coverslips were transfected for 24 h or infected for the indicated times and then fixed with 4% paraformaldehyde in phosphate-buffered saline (PBS) (137 mM NaCl, 3 mM KCl, 8 mM Na2HPO4 [pH 7.5]), permeabilized with PBS containing 0.2% Triton-X, and blocked by incubation in 2% BSA in PBS. Samples were incubated with primary antibodies in 2% BSA in PBS for 45 min and washed three times with PBS-Tween (PBS-T), followed by incubation with secondary antibodies in 2% BSA in PBS for 45 min. The cells were washed an additional three times with PBS and mounted on slides using Prolong Antifade with or without DAPI (4′,6-diamidino-2-phenylindole) (Life Technologies). Stained cells were visualized using a Zeiss Axiovert 200 inverted microscope equipped with fluorescence optics and Axiovision software (Zeiss). Images were prepared using Adobe Photoshop and Illustrator software (Adobe). PB1-F2 expression was quantified by counting NP (infected)- or PB1 (transfected)-expressing cells and then counting PB1-F2 or 3×FLAG-expressing cells within this subset to determine the percentage of infected or transfected cells expressing PB1-F2. All quantifications were done in triplicate on independent experimental samples to calculate averages and standard deviations and for statistical comparison.
RT-qPCR.
MDCK cells were infected with either OH07 or PR8 virus for 12 h. Total RNA was extracted from each sample using TRIzol reagent according to the manufacturer's directions (Invitrogen). cDNA was created using Superscript III First-Strand Synthesis SuperMix (Invitrogen) with oligo(dT) primer. RNA was removed by treating samples with RNase A for 3 h. Tenfold serial dilutions of sample cDNA were quantified using an iScript One-Step RT-PCR with SYBR green kit (Bio-Rad). Primers against either PB1 (Forward, 5′-GGCCCTTCAGTTGTTCATC-3′; Reverse, 5′-GCAGACTTCAGGAATGTG-3′) or NP (Forward, 5′-GCGTCTCAAGGCACCAAAC-3′; Reverse, 5′-TCAAAAGCAGAGAGCACCATT-3′) were used for amplification of cDNA samples. The qPCR mixture for each sample consisted of 7.5 μl 2× SYBR green RT-PCR mixture, 5 μl of nuclease-free water, 0.6 μl of each primer, 0.3 μl iScript RNase H+ reverse transcriptase, hot-start iTaq DNA polymerase mixture, and 1 μl of diluted cDNA template. The qPCR samples were detected with a MyiQ iCycler real-time PCR detection system (Bio-Rad), and each experiment contained three replicates of each sample, with two experimental replicates performed. Samples were then compared using the Pfaffl method in reference to PR8 readings (29).
Immunoprecipitation.
Twenty microliters of MagnaBind protein A beads (Thermo Scientific) per transfected sample was added to 400 μl RAF buffer (20 mM Tris [pH 8.0], 137 mM NaCl, 10% glycerol, 1% NP-40) containing Halt Protease Inhibitor cocktail (Thermo Scientific). The beads were washed three times with RAF buffer. Immunoprecipitating antibodies were added to aliquots of washed beads and incubated at 4°C with constant inversion mixing for 2 h. Antibody-bead complexes were washed six times and resuspended in 100 μl of RAF buffer per sample; 48 h posttransfection, the cells were washed using PBS, harvested, and pelleted into a 1.5-ml tube. RAF buffer with protease inhibitors (800 μl) was added to each sample and vortexed four times for 30 s at 10-min intervals with incubation on ice between vortexing periods. Cellular debris from lysed transfected samples was removed by centrifugation, and 400 μl of supernatant was added to new 1.5-ml tubes with antibody-bead complexes. Samples were incubated at 4°C with constant inversion mixing overnight and then washed six times with RAF buffer with protease inhibitors and resuspended in 15 μl of 2× sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) loading buffer (100 mM Tris [pH 6.8], 200 mM dithiothreitol, 4% SDS, 0.2% bromophenol blue, and 10% glycerol).
Immunoblotting.
Immunoprecipitated (IP) samples, infected or transfected cell lysates, or recombinant OH07 PB1-F2 protein was separated by 10% (PB1 and actin) or 15% (PB1-F2 and FLAG) Tris-glycine SDS-PAGE. The gels were transferred via electroblotting in transfer buffer (25 mM Tris, 192 mM glycine, 20% methanol [pH 8.3]) onto 0.45-μm nitrocellulose (Bio-Rad) at 70 V for 70 min. The membranes were blocked using 5% milk in Tris-buffered saline with 0.5% Tween 20 (TBS-T) for 30 min and incubated with 5% milk in TBS-T containing primary antibodies at room temperature overnight. The blots were washed three times with TBS-T and incubated for 4 h with the appropriate AP-conjugated secondary antibody in 1% milk in TBS-T. The blots were washed three times with TBS-T and exposed to chemiluminescent reagent (Lumiphos; Thermo Scientific). Images were captured using a ChemiDocXRS imager and Quantity One imaging software (Bio-Rad). The images were processed using Adobe Photoshop and Illustrator software (Adobe).
Pulse-chase analysis.
MDCK cells were mock transfected or transfected with either pOH07/PB1-F2/3×FLAG or pPR8/PB1-F2/3×FLAG. Twenty-four hours posttransfection, 100 μg/ml cycloheximide was added to samples, and at 0, 30, 60, 120, and 240 min post-drug treatment, lysates were collected using 1% SDS in 50 mM Tris-HCl, pH 8.0, and immunoblotted with 3×FLAG antibody as described above. As a control to confirm cycloheximide efficacy, additional mock-transfected samples were either left untreated or treated with cycloheximide for 30 min, at which point l-azidohomoalanine (l-AHA) (Invitrogen) was added at a final concentration of 50 μM for an additional 4 h. Samples were lysed with 1% SDS in 50 mM Tris-HCl, pH 8.0, and proteins that had incorporated l-AHA in the absence and presence of cycloheximide were labeled with biotin-alkyne as previously described (30), The biotin-labeled proteins were separated on SDS-PAGE, transferred to nitrocellulose membranes, and detected via incubation with alkaline phosphatase-conjugated streptavidin (Invitrogen), followed by exposure to chemiluminescence reagent.
Statistical analysis.
Samples from infection studies were compared using paired t test analysis, and statistically different groups (P < 0.05) are indicated. Translation clone and mapping studies were compared using single-factor analysis of variance (ANOVA) and were grouped based upon sequence. Statistically significant groups (P < 0.01) are indicated. All statistical analyses were computed using Excel 2007 with the Analysis ToolPak add-on (Microsoft).
RESULTS
Detection of swine PB1-F2 in transfected and infected cells.
A phylogram was created comparing PB1-F2 nucleotide coding sequences from OH07, Mx09, PR8, a swine consensus sequence (derived from unique PB1-F2 sequences from 2000 to the present), 1918 Spanish flu, two highly pathogenic avian H5N1 isolates, and two other swine IAVs. Comparison of the OH07 PB1-F2 nucleotide sequence with a consensus sequence and other swine PB1-F2 sequences using the phylogram indicated that the OH07 PB1-F2 sequence used in our studies is highly representative of PB1-F2 sequences from swine influenza virus isolates, including a swine-origin H3N2 variant strain (H3N2v; A/Swine/Indiana/A00968373/2012) recently isolated from an infected pig.
In order to study swine PB1-F2 expression, peptide antibodies against OH07 PB1-F2 amino acid sequence residues 13 to 26 (TEHTNIQKKGNGRQ) were obtained from a commercial vendor (GenScript). The antibody was first tested for its ability to recognize swine PB1-F2 in transfected cells. Plasmids were constructed that express OH07 PB1-F2 fused in frame with a C-terminal enhanced green fluorescent protein (EGFP) (pOH07/PB1-F2/EGFP) or 3×FLAG (pOH07/PB1-F2/3×FLAG) tag. MDCK cells were transfected with these plasmids, and the fusion proteins were visualized using the OH07 PB1-F2 antibody, the 3×FLAG antibody, or the inherent fluorescence of EGFP. The PB1-F2 antibody staining revealed that the OH07-derived fusion proteins were localized throughout the nucleus and cytoplasm of cells in a pattern that was similar to that of the 3×FLAG antibody and EGFP fluorescence (Fig. 1B), strongly suggesting the antibody recognized the OH07 PB1-F2 fusion proteins. Additional plasmid constructs encoding N-terminal 3×FLAG and EGFP PB1-F2 fusion proteins showed similar colocalization of the PB1-F2 antibody staining with the 3×FLAG antibody or inherent EGFP fluorescence (data not shown).
Fig 1.

Swine PB1-F2 expression in transfected and infected cells. (A) Nucleotide phylogram comparing A/swine/Ohio/511445/2007(H1N1), A/Mexico/4108/2009(H1N1), A/swine/Nebraska/02013/2008(H1N1), A/swine/Indiana/A00968373/2012(H3N2), A/Puerto Rico/8/1934(H1N1), A/Brevig Mission/1/1918(H1N1), A/Indonesia/CDC1047/2007(H5N1), A/Vietnam/1203/2004(H5N1), and swine consensus PB1-F2 sequence. The swine consensus sequence was established using unique H1N1 and H3N2 PB1-F2 sequences collected from 2000 to the present. Bootstrap values are either shown above the branches or indicated by the dotted lines. (B) MDCK cells were transfected with pOH07/PB1-F2/3×FLAG (top row) or pOH07/PB1-F2/EGFP (bottom row). Twenty-four hours posttransfection, the cells were fixed and stained with swine PB1-F2 antibody (middle column) and anti-FLAG antibody (top left) or inherent EGFP fluorescence (bottom left). A merged image is also shown. (C) Diagram of reverse-genetics plasmid encoding the OH07 PB1-F2 in the context of the Mx09 PB1 gene. pro, promoter; term, terminator. (D) MDCK, A549, and PK-15 cells were infected with OH07 (top row), rMx/MOM (middle row), and rMx09 (bottom row). At 12 h p.i., the cells were immunostained with antibodies against NP (left columns) and OH07 PB1-F2 (right columns), followed by Alexa Fluor-conjugated secondary antibodies. Bars, 10 μm.
To examine swine PB1-F2 expression in infected cells, we wanted to utilize both wild-type and recombinant viruses. However, for reasons that are unclear, we were unable to create a recombinant OH07 virus from reverse-genetics plasmids despite multiple attempts using a variety of protocols. Because of this, we instead developed a recombinant virus system in which we replaced the truncated PB1-F2 nucleotide sequences in the PB1 reverse-genetics plasmid from an H1N1pdm09 (Mx09) viral isolate with the OH07 PB1-F2 nucleotide sequence (nt 128 to 445, denoted PB1/Mexico/Ohio/Mexico or pDP2002/MOM/PB1) (Fig. 1C). The seven other Mx09 plasmids plus pDP2002/MOM/PB1 were then transfected into cells to produce recombinant virus (rMx/MOM). It should be noted that the PB1 protein produced from pDp2002/MOM/PB1 contains a single conserved mutation of isoleucine to valine compared to pDp2002/Mx09/PB1 using Blastp (NCBI). MDCK, A549, and PK-15 cells were infected with wild-type OH07 (wtOH07), Mx09, or rMx/MOM viruses, and at 12 h postinfection (p.i.), the cells were stained with antibodies against the NP protein and OH07 PB1-F2 to visualize swine PB1-F2 expression in infected cells (Fig. 1D). Distinct nuclear PB1-F2 staining was visible in some wtOH07- and rMx/MOM-infected cells, whereas no PB1-F2 staining was detected in Mx09-infected cells, strongly suggesting our antibody specifically recognized swine PB1-F2 in cells infected with wild-type and recombinant viruses expressing PB1-F2 from OH07.
PB1-F2 expression levels differ between viral isolates.
Upon examination of PB1-F2 expression in infected cells, we noticed a striking difference in the number of OH07-infected cells expressing detectable amounts of PB1-F2 relative to cells infected with the human laboratory-adapted strain PR8. This led us to hypothesize that there may be a strain-specific difference in the number of infected cells expressing PB1-F2 between swine and human origin isolates. To examine this hypothesis, we infected cells with OH07, rMx/MOM, or rPR8 and determined the number of infected cells expressing detectable amounts of PB1-F2 in each sample over a 48-h time course (Fig. 2A). Surprisingly, these studies suggested that at all times p.i., the number of cells expressing PB1-F2 in the swine virus-infected samples (<1% of infected cells expressing PB1-F2 for every sample) was statistically significantly lower than in PR8 virus-infected cells (peaking at just over 14% of infected cells expressing PB1-F2 at 12 h p.i.) (Fig. 2A). These experiments were repeated in A549, PK-15, and 3D4/2 cells and CEF with similar results (Fig. 2B and data not shown), suggesting that this phenotype is not cell type specific.
Fig 2.

PB1-F2 expression is differentially regulated in a strain-specific manner. (A) MDCK cells were infected with OH07, rMx/MOM, or rPR8 virus at an MOI of 2. At the indicated times p.i., the cells were immunostained with NP and PB1-F2 antibodies, followed by Alexa Fluor-conjugated secondary antibodies. (B) MDCK, A549, and PK-15 cells were infected with OH07, rMx/MOM, or rPR8 virus at an MOI of 2, and at 12 h p.i., the cells were immunostained for NP and PB1-F2. The percentage of NP-expressing cells also expressing PB1-F2 was determined and plotted on a graph. Statistically significant groups (P < 0.05) compared to PR8-infected samples are denoted with asterisks. The error bars indicate standard deviations. (C) Sample lysates (400 μg) from mock- or pOH07/PB1-F2/3×FLAG-transfected BHK-T7 cells and OH07-infected (MOI = 5; 12 h p.i.) MDCK cells (left) or serial dilutions of pOH07/PB1-F2/pET21 bacterially expressed purified protein ranging from 25 to 400 ng (right) were separated on SDS-PAGE, transferred to nitrocellulose membranes, and immunoblotted with antibodies against OH07 PB1-F2 or β-actin, as indicated, followed by AP-conjugated secondary antibodies.
Levels of PB1-F2 protein expression in cells infected with OH07 were further examined using immunoblot analysis. In these experiments, we were unable to detect OH07 PB1-F2 from infected cells prepared under multiple experimental conditions using the OH07 PB1-F2 antibody, even though the samples were confirmed to be 95 to 100% infected (MOI = 5) by immunofluorescence using a monoclonal antibody against the influenza virus nucleoprotein (Fig. 2C and data not shown). We could easily detect a recombinant OH07 PB1-F2/3×FLAG protein at the appropriate molecular weight from transfected cell lysates with the OH07 PB1-F2 antibody (Fig. 2C), suggesting the antibody has the capacity to recognize OH07 PB1-F2 using immunoblot analysis. Moreover, serial dilutions of the OH07 PB1-F2 protein purified from E. coli were detected down to 25 ng (Fig. 2C), further illustrating that the antibody has the capacity to detect low levels of OH07 PB1-F2 by immunoblot analysis. These findings support our immunofluorescence data showing that there is little to no detectible PB1-F2 protein present in cells infected with the virus.
To confirm that the low expression levels of PB1-F2 observed in these experiments were representative of swine IAV isolates, MDCK cells were infected for 12 h with two additional swine viruses that are predicted at the sequence level to express a full-length PB1-F2 protein [A/Swine/Nebraska/02013/2008(H1N1) and A/Swine/Indiana/A00968373/2012(H3N2)], and the percentage of PB1-F2-expressing infected cells was determined. Similar to OH07, less than 0.5% of the cells infected with these swine viruses expressed PB1-F2 (data not shown), suggesting that a low expression level of PB1-F2 is a common swine IAV phenotype.
Strain-specific differences in PB1-F2 expression are dependent on the PB1 gene.
To determine if any of the other IAV gene segments impact the different PB1-F2 protein levels measured in Fig. 2, additional recombinant viruses were constructed that combined either the PR8 PB1 gene with the seven other Mx09 gene segments (rMx/PR8) or the MOM PB1 gene with the seven other PR8 gene segments (rPR8/MOM). MDCK cells were inoculated with rPR8, rMx/PR8, rMx/MOM, or rPR8/MOM, and at 12 h p.i., the cells were immunostained and the percentage of infected cells expressing detectable amounts of PB1-F2 was determined (Fig. 3). While there was a small but significant (P < 0.05) decrease in the number of cells expressing PB1-F2 in the rMx/PR8 virus-infected cells relative to rPR8-infected cells, there was no significant increase in the number of cells expressing PB1-F2 in the rPR8/MOM-infected cells relative to rMx/MOM-infected cells. Moreover, the rPR8- and rMx/PR8-infected cells had greatly increased numbers of PB1-F2-expressing cells compared to rMx/MOM- or rPR8/MOM-infected cells. These results strongly suggest that differences outside the PB1 gene do not contribute substantially to PB1-F2 protein expression levels. Taken together with the results in Fig. 2A, these findings suggest that PB1-F2 expression levels are differently regulated between influenza virus isolates in a manner dependent on the PB1 gene and independent of the cell line and virus genetic backbone. Importantly, these data also suggest that these isolates of swine influenza virus do not express substantial amounts of PB1-F2, even though they encode the capacity to do so at the nucleotide level.
Fig 3.
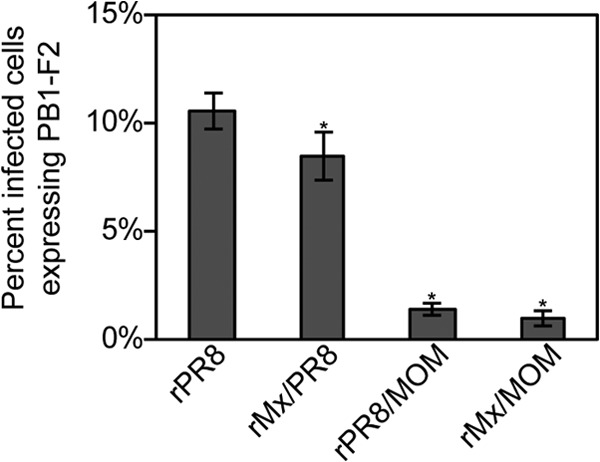
Differential PB1-F2 expression is dependent on the PB1 gene. MDCK cells were infected with rPR8, rMx/PR8, rPR8/MOM, or rMx/MOM at an MOI of 2, and at 12 h p.i., the cells were fixed and immunostained with antibodies against NP and PB1-F2, followed by Alexa Fluor-conjugated secondary antibodies. The percentage of NP-expressing cells also expressing detectable amounts of PB1-F2 was determined and plotted on a graph. Statistically significant groups (P < 0.05) compared to PR8-infected samples are denoted with asterisks. The error bars indicate standard deviations.
PB1-F2 expression is primarily regulated at the level of translation.
To identify the step in which PB1-F2 expression is differently regulated in PR8 versus OH07, we examined PB1 mRNA levels and PB1-F2 protein stability in infected cells. In order to determine if OH07 PB1 mRNA levels were significantly decreased relative to PR8, MDCK cells were infected with rPR8 or wtOH07 at an MOI of 2. At 12 h p.i., total RNA was isolated from infected cells and subjected to reverse transcription and RT-qPCR using PB1 and NP gene-specific primers to determine the OH07 PB1/NP RNA ratios relative to PR8 in infected cells (Fig. 4A). These experiments indicated that the wtOH07-infected cells contained slightly higher PB1/NP mRNA ratios than rPR8-infected samples, suggesting that decreased OH07 PB1-F2 protein levels do not result from a decrease in PB1 mRNA levels.
Fig 4.

Differential PB1-F2 expression is regulated primarily at the level of translation. (A) MDCK cells were infected with OH07 or PR8 at an MOI of 2. RNA was extracted at 12 h p.i. and used in two-step RT-qPCRs. A 10-fold dilution series of rPR8 samples was utilized to develop a standard and used for comparison to OH07. Triplicate samples from two experimental duplicates were tested and compared using the Pfaffl method. (B) MDCK cells were infected with OH07 or PR8 at an MOI of 2. At 8 h p.i., MG132 (0.5 μM) was added to the cells, and at 12 h p.i., the cells were fixed and immunostained for NP and PB1-F2, followed by Alexa Fluor-conjugated secondary antibodies. The percentage of NP-expressing cells also expressing PB1-F2 was determined and plotted on a graph. Statistically significant groups (P < 0.05) are indicated by asterisks. (C) (Left) MDCK cells were mock, pOH07/PB1-F2/3×FLAG, or pPR8/PB1-F2/3×FLAG transfected, and at 24 h posttransfection were incubated in the presence of 100 μg/ml of cycloheximide for pulse-chase analysis of PB1-F2 degradation. At the indicated times post-drug addition, samples were harvested and proteins were separated on SDS-PAGE and immunoblotted with antibodies against 3×FLAG or β-actin. (Right) Mock-transfected samples were left untreated (U) or treated with cycloheximide for 30 min (T), and then l-AHA was added for 4 h to label the proteins. Cells were harvested, and the labeled proteins were biotinylated, precipitated, and separated on SDS-PAGE and detected using AP-conjugated streptavidin. (D) OH07 and PR8 PB1-F2/3×FLAG protein levels were normalized to β-actin levels, and the percent PB1-F2 protein at each time point relative to that at time zero was quantified. Means and standard deviations were determined from five independent experiments and graphed, and statistically significant differences (P < 0.05) compared to PR8 PB1/3×FLAG samples at each time point are denoted with an asterisk. The error bars indicate standard deviations.
It has previously been reported that the PB1-F2 protein from some IAV isolates is unstable and in some cases can be stabilized by pharmaceutical inhibition of the proteasome (1). To determine the inherent stability of the OH07 PB1-F2 protein, we investigated its accumulation in the absence and presence of proteasome inhibition. While we measured a significant increase in the number of rPR8-infected cells expressing PB1-F2 following proteasome inhibition (P < 0.05), MG132 had no significant effect on the number of cells expressing PB1-F2 in wtOH07-infected cells (Fig. 4B). We were unable to detect OH07 PB1-F2 from infected cells to examine protein stability via immunoblot analysis (see above), and therefore, we instead examined OH07/PB1-F2/3×FLAG stability relative to PR8/PB1-F2/3×FLAG. A pulse-chase analysis was performed on MDCK cells transfected with either pOH07/PB1-F2/3×FLAG or pPR8/PB1-F2/3×FLAG and incubated with cycloheximide for 0, 30, 60, 120, and 240 min (Fig. 4C). Control samples treated or not with cycloheximide and labeled with the methionine analog l-azidohomoalanine confirmed that cycloheximide inhibited protein synthesis. These experiments showed that both OH07 and PR8 PB1-F2/3×FLAG proteins undergo degradation over time, with OH07 PB1-F2 appearing, qualitatively, to degrade more rapidly than PR8 PB1-F2. However, aside from a slight but statistically significant difference at the 60-min time point, quantitative analysis of immunoblots from multiple experiments show that the degradation rate of OH07 PB1-F2 is not statistically significantly different from that of PR8 PB1-F2 (Fig. 4D). These results also show that the OH07 PB1-F2 half-life is between 30 and 60 min. While this instability may marginally impact our ability to detect the protein in infected cells, it should not result in the near absence of expression that we see by both immunofluorescence and immunoblot analyses in highly infected cells. Taken together with the results from Fig. 4A, this suggests OH07 PB1-F2 protein levels are likely regulated in a strain-dependent manner at the level of protein translation.
Validation of a plasmid-based system to identify PB1 sequences involved in regulation of PB1-F2 expression.
In order to determine if specific sequences within the PB1 gene played a role in differentially regulating PB1-F2 protein expression between influenza virus isolates, we developed a method to evaluate PB1-F2 expression independent of infection using plasmid expression vectors. Parental plasmids were constructed from the full-length PB1 gene in the reverse-genetics plasmid backbones of OH07 (pOOO/3×FLAG), PB1/MOM (pMOM/3×FLAG), and PR8 (pPPP/3×FLAG) in which a 3×FLAG tag was inserted in frame at the C terminus of the PB1-F2 ORF (Fig. 5A). The insertion of the 3×FLAG tag resulted in the addition of 25 non-PB1 amino acids between residues 121 and 122 of the PB1 protein but did not disrupt the PB1 protein ORF. In order to validate our plasmid-based translational system, we examined the expression levels of PB1-F2/3×FLAG relative to PB1 using pPPP/3×FLAG, pOOO/3×FLAG, and pMOM/3×FLAG plasmids. MDCK cells were transfected with each plasmid, and at 24 h posttransfection, the cells were fixed and immunostained with FLAG- and PB1-specific antibodies. Cells expressing PB1 were examined for PB1-F2/3×FLAG protein expression, and the number of cells expressing PB1-F2/3×FLAG relative to the total number of PB1-expressing cells was determined. In these experiments, similar to what was measured in infection, cells transfected with pPPP/3×FLAG expressed PB1-F2 in a much higher percentage of cells (66%) than either pOOO/3×FLAG (13%) or pMOM/3×FLAG (11%) (Fig. 5B). To confirm these data, immunoprecipitation of PB1-F2/3×FLAG and PB1 proteins expressed in transfected 293T cells was performed using 3×FLAG- and PB1-specific antibodies, and protein expression levels were analyzed using immunoblot analysis. Again, while cells transfected with pPPP/3×FLAG expressed detectable levels of PB1-F2, there was no detectable PB1-F2 expression in pOOO/3×FLAG- or pMOM/3×FLAG-transfected cells. The levels of PB1 protein immunoprecipitated were similar between plasmids, suggesting (i) that there were no substantial differences in PB1 mRNA levels between plasmids and (ii) that the differences in PB1-F2 protein expression from these plasmids were specific to the PB1-F2 ORF (Fig. 5C). Interestingly, a second faint band was detected below the PB1 protein in these experiments that may be the N40 protein, a protein product of the PB1 mRNA that initiates downstream of the PB1-F2 start codon (2).
Fig 5.

Differential PB1-F2 expression is replicated in a plasmid model system. (A) Diagrams of plasmids created to examine PB1-F2 expression in transfected cells. A C-terminal 3×FLAG tag was inserted in frame with the PB1-F2 ORF in the pHW2000/PB1 or pDP2002/PB1 plasmid from PR8 (pPPP/3×FLAG), OH07 (pOOO/3×FLAG), MOM (pMOM/3×FLAG), and an Mx09 PB1 gene mutated to restore the PB1-F2 ORF (pMxKI/3×FLAG). (B) The indicated plasmids were transfected into MDCK cells, and at 24 h posttransfection, the cells were immunostained with antibodies against PB1 and 3×FLAG, followed by Alexa Fluor-conjugated secondary antibodies. The percentage of PB1-expressing cells also expressing PB1-F2/3×FLAG was determined and plotted on a graph. Statistically significant groups (P < 0.01) compared to PPP/3×FLAG are indicated with asterisks. The error bars indicate standard deviations. (C) The indicated plasmids were transfected into 293T cells, and at 48 h posttransfection, the cells were lysed and immunoprecipitated with antibodies against 3×FLAG or PB1. The immunoprecipitated proteins were separated on SDS-PAGE, transferred to nitrocellulose membranes, and immunoblotted with 3×FLAG or PB1 antibodies. Lysates from the transfections were immunoblotted with antibodies against β-actin as a control for protein input levels in immunoprecipitation.
Based on the similarity between OH07 and Mx09 sequences (Fig. 1A), we hypothesized that knock-in of PB1-F2 in Mx09 may not result in substantial increases in PB1-F2 expression. To examine this, we created a plasmid in which single nucleotide changes in the third position of the premature stop codons within the Mx09 PB1-F2 ORF were replaced with coding sequence (152A to C, 291A to G, and 381A to G) and cloned into the PB1/3×FLAG plasmid backbone (pMxKI/3×FLAG) (Fig. 5A). Similar to OOO/3×FLAG and MOM/3×FLAG, levels of PB1-F2 expression from the MxKI/3×FLAG plasmid were very low in transfected cells compared to PPP/3×FLAG (Fig. 5B and C), suggesting that reintroduction of the full-length PB1-F2 ORF into the Mx09 PB1 gene does not result in substantial expression of PB1-F2.
The PB1-F2 ORF and downstream PB1 sequences regulate PB1-F2 expression levels.
To determine if sequences in the PB1 gene play a role in regulation of PB1-F2 expression, PB1-F2/3×FLAG plasmids were constructed in which the PB1 gene was divided into three regions: the 5′ region prior to the PB1-F2 AUG (5′ PB1), the PB1-F2 ORF (F2 ORF), and the 3′ region downstream of the PB1-F2 stop codon (3′ PB1). It should be noted that though large portions of sequence are being interchanged between plasmids, relatively few changes are made to the amino acid sequence of the PB1 proteins, because all three PB1 proteins (OH07, PR8, and Mx09) are relatively conserved, with 95 to 98% of the amino acids identical and with 98 to 99% either identical or conserved (data not shown). Utilizing this approach, all possible combinations of these PB1 chimeric genes between Mx09, PR8, and OH07 were created (Fig. 6A). Each construct was named based on the first letter of the virus from which each PB1 sequence section is derived. For example, the parental PR8 plasmid described above is denoted pPPP/3×FLAG because the 5′ region before the PB1-F2 ORF is from PR8, the PB1-F2 ORF is from PR8, and the 3′ region following the PB1-F2 stop codon is from PR8.
Fig 6.
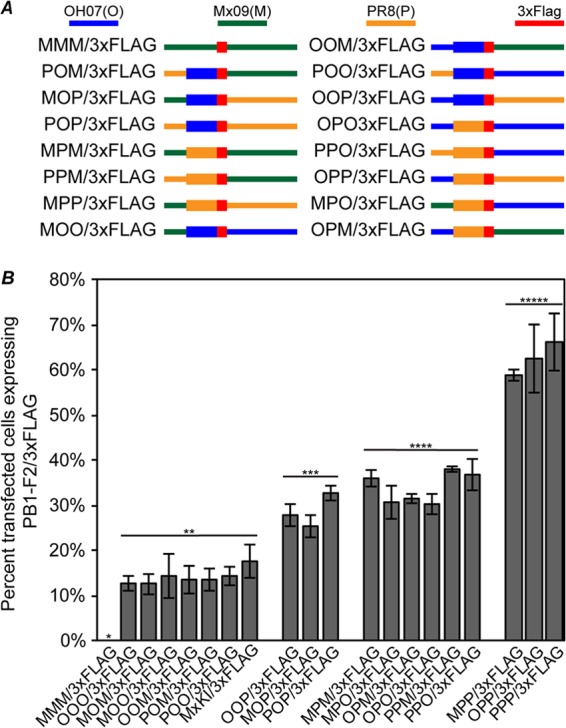
PB1-F2 expression is differentially regulated by the PB1-F2 ORF and 3′ PB1 regions. (A) Diagrams of chimeric PB1 gene plasmids constructed to identify sequence requirements for differential PB1-F2 expression. The PB1 gene was divided into three sections consisting of the 5′ region upstream of PB1-F2, the PB1-F2 ORF, and the 3′ region downstream of PB1-F2 in the 3×FLAG-tagged PB1-F2 plasmids created for Fig. 5. All possible combinations of the PB1 gene using these regions from OH07, Mx09, or PR8 were created. (B) MDCK cells were transfected with the indicated plasmids, and at 24 h posttransfection, the cells were immunostained with antibodies against PB1 and 3×FLAG, followed by Alexa Fluor-conjugated secondary antibodies. The percentage of PB1-expressing cells also expressing detectable amounts of PB1-F2/3×FLAG was determined and plotted on a graph. Statistically significant groups (P < 0.01) as determined by ANOVA are indicated with a line and two, three, four, or five asterisks. The error bars indicate standard deviations.
Each of the chimeric plasmids was transfected into MDCK cells, and at 24 h posttransfection, the cells were fixed and stained with PB1- and FLAG-specific antibodies and the percentage of cells expressing PB1 that also expressed PB1-F2/3×FLAG was determined. PB1-F2 expression from these plasmids fits into three distinct groups, i.e., low expressers (13 to 18% positive cells), medium expressers (26 to 38% positive cells), and high expressers (59 to 66% positive cells) (Fig. 6B). Grouping of the low-, medium-, and high-PB1-F2-expressing plasmids resulted in the identification of a clear pattern of PB1-F2 expression. The 5′ region upstream of PB1-F2, which harbors the PB1 AUG and Kozak sequence, 2 additional AUGs that include short ORFs, and the PB1-F2 Kozak sequence, had no significant impact on the numbers of cells expressing detectable amounts of PB1-F2 in this system, as low, medium, and high expressers were found to have this region from both swine and PR8 origins. However, when both the F2 ORF and 3′ PB1 regions originated from swine lineage gene segment 2 (OH07 and Mx09), PB1-F2 expression was low. Similarly, when both the F2 ORF and 3′ PB1 regions originated from PR8, PB1-F2 expression was high. Finally, when the origins of the F2 ORF and the 3′ PB1 regions were mixed (one swine origin and one PR8 origin), PB1-F2 was expressed at a medium level. Thus, sequences within the F2 ORF and the 3′ PB1 regions play roles in the regulation of PB1-F2 expression from the influenza A virus PB1 gene.
PB1 nucleotides 267 to 335 and 582 to 816 are necessary and sufficient to modulate PB1-F2 protein expression.
In order to identify specific sequences within the F2 ORF and 3′ PB1 region involved in regulation of PB1-F2 expression, a series of additional plasmids were constructed using the MPM/3×FLAG (medium-expresser) plasmid as the parent and making incremental swaps of swine and PR8 origin sequence within one or both of these regions. To examine the 3′ PB1 region, it was first divided in half, and Mx09 nt 392 to 1140 [pMPM(P392-1140)/3×FLAG] or 1131 to 2298 [pMPM(P1131-2298)/3×FLAG] from MPM/3×FLAG was replaced with the same sequence from PR8 (Fig. 7A). While pMPM(P1131-2298)/3×FLAG remained a medium expresser like the parent plasmid, pMPM(P392-1140)/3×FLAG expressed PB1-F2 at high levels, implicating PB1 nt 392 to 1140 in PB1-F2 translational regulation (Fig. 7A). PB1 nt 392 to 816 [pMPM(P392-816)/3×FLAG] and 392 to 586 [pMPM(P392-586)/3×FLAG] from Mx09 were then replaced with PR8 sequence in the pMPM/3×FLAG construct and tested for PB1-F2 expression. pMPM(P392-816)/3×FLAG was a high expresser; however, pMPM(P392-586)/3×FLAG was a medium expresser, suggesting that PB1 nt 587 to 816 were necessary for PB1-F2 translational regulation. To determine if PB1 nt 587 to 816 were sufficient for modulation of PB1-F2 expression, PR8 PB1 nt 582 to 816 were cloned into pMPM/3×FLAG [pMPM(P582-816)/3×FLAG]. This plasmid was also found to be a high PB1-F2 expresser. These data implicate PB1 nt 582 to 816 in the 3′ PB1 region as necessary and sufficient for differential regulation of PB1-F2 translation.
Fig 7.

PB1 nucleotides 267 to 343 and 582 to 816 are necessary and sufficient for translational regulation of PB1-F2. (A to C) Diagrams and results for plasmids used to map sequences necessary and sufficient for PB1-F2 translational regulation. MDCK cells were transfected with the indicated plasmids, and at 24 h posttransfection, the cells were immunostained with antibodies against PB1 and 3×FLAG, followed by Alexa Fluor-conjugated secondary antibodies. The percentage of PB1-expressing cells also expressing PB1-F2/3×FLAG was determined and plotted on a graph. Statistically similar groups based on ANOVA (P < 0.01) are indicated with one or two asterisks. The error bars indicate standard deviations. (D) The indicated plasmids were transfected into 293T cells, and at 48 h posttransfection, the cells were lysed and immunoprecipitated with antibodies against 3×FLAG or PB1. The immunoprecipitated proteins were separated on SDS-PAGE, transferred to nitrocellulose membranes, and immunoblotted with 3×FLAG or PB1 antibodies. Lysates from transfections were immunoblotted with antibodies against β-actin as a control for protein input levels in immunoprecipitation.
A similar strategy was used to map the F2 ORF translational regulatory element. In these experiments, the MOM/3×FLAG plasmid was initially used as the parental construct. Clones were made in which OH07 PB1 nt 119 to 252 [pM(P119-252)OM/3×FLAG] or 267 to 391 [pMO(P267-391)M/3×FLAG] from pMOM/3×FLAG were replaced with PR8 PB1 nucleotides, and PB1-F2 expression was measured as previously described (Fig. 7B). While pM(P119-252)OM/3×FLAG remained a low expresser, pMO(P267-391)M/3×FLAG expressed medium levels of PB1-F2, suggesting PB1 nt 267 to 391 modulate PB1-F2 translation (Fig. 7B).
Combining findings from 5′ PB1 mapping with F2 ORF mapping, PR8 PB1 nt 344 to 816 in pMOM/3×FLAG were replaced with the OH07 sequence [pMO(P344-816)M/3×FLAG], which resulted in a decrease of PB1-F2 expression to medium levels. In contrast, replacing OH07 PB1 nt 267 to 335 with the PR8 sequence [pMO(P267-335)O(P392-816)M/3×FLAG] resulted in high-level expression of PB1-F2. Likewise, replacing PR8 PB1 nt 267 to 343 with the OH07 sequence in the pPPP/3×FLAG parent [pPP(O267-343)P/3×FLAG] resulted in medium levels of PB1-F2 expression. Altogether, these findings suggest PB1 nt 267 to 335 within the PB1-F2 ORF are necessary and sufficient for PB1-F2 translational modulation.
Using the above data, two final plasmids carrying the minimal sequences identified above for increasing PB1-F2 expression to high levels in the pMOM/3×FLAG background (pMOMF2up/3×FLAG; nt 267 to 335 and 582 to 816 from PR8 PB1 in OH07/Mx09 PB1), or, alternatively, for decreasing PB1-F2 expression to low levels in the pPPP/3×FLAG background (PR8F2down/3×FLAG; nt 267 to 343 and 587 to 819 from OH07/Mx09 PB1 in PR8 PB1) (nucleotide sequence discrepancies between these clones are the result of the availability of restriction digestion sites) were created and tested for PB1-F2 expression (Fig. 7C). MDCK cells transfected with pMOMF2up/3×FLAG expressed the swine-like PB1-F2 protein in similar numbers of cells as pPPP/3×FLAG-transfected cells. Moreover, MDCK cells transfected with pPR8F2down/3×FLAG expressed the human-like PB1-F2 protein in a similar number of cells as pMOM/3×FLAG, thus illustrating that these regions are necessary and sufficient to either increase or decrease the number of cells expressing detectable amounts of PB1-F2 to either PR8 or OH07 PB1-F2 levels, respectively.
These data were confirmed by immunoprecipitation of PB1-F2/3×FLAG and PB1 from 293T cells transfected with pMOM/3×FLAG, pMOMF2UP/3×FLAG, pPPP/3×FLAG, or pPR8F2dwn/3×FLAG, followed by immunoblot assay using 3×FLAG antibodies (Fig. 7D). In these experiments, it was evident that pPR8F2dwn/3×FLAG expresses substantially lower levels of PB1-F2 than pPPP/3×FLAG. Likewise, pMOMF2up/3×FLAG expresses substantially higher levels of PB1-F2 than pMOM/3×FLAG, supporting our findings that these sequences in the F2 ORF and 3′ region are sufficient to differentially regulate PB1-F2 expression. However, these experiments also show that the total levels of PB1-F2/3×FLAG protein expressed in pMOMF2up/3×FLAG do not reach the levels expressed by pPPP/3×FLAG, suggesting that while these regions regulate the number of cells expressing PB1-F2, further regulation of the total amounts of protein per cell may also be present. These data show that PB1-F2 expression levels are regulated at the translational level by two regions within the F2 ORF and 3′ PB1 region and can be modified to modulate PB1-F2 expression in the influenza A virus PB1 gene.
DISCUSSION
Our data demonstrate that carrying a full-length PB1-F2 ORF is not predictive of expression of the protein during infection and that IAV expresses dramatically different levels of the PB1-F2 protein in a strain-specific manner. In this study, we were only able to detect PB1-F2 derived from wild-type swine and recombinant MOM/PB1 strains of IAV in rare infected cells (less than 1%) (Fig. 2A and B and 3 and data not shown). Because of such low expression levels, the swine-derived protein may not have a measurable effect during infection in cells or animals. Our findings may help explain the lack of impact following introduction of the full-length PB1-F2 sequence into H1N1pdm09, as PB1-F2 expression was not demonstrated in the PB1-F2 knock-in mutants used in prior studies (16–19). It should be noted that in our infection studies the OH07 and PR8 PB1-F2 proteins were detected using two different antibodies with different reactivities to their respective proteins, which could feasibly contribute to the differences in protein expression measured in the study. To minimize this possibility, the concentration of each PB1-F2 antibody used was optimized to detect the maximum level of signal while minimizing background for experiments in which we compared OH07 and PR8 protein levels. Additionally, immunoblot detection of both purified OH07 PB1-F2 and OH07 PB1-F2 from transfected cells confirmed that the OH07 antibody had the capacity to detect PB1-F2 when the protein was present, even at relatively low levels. Moreover, the strain-specific differences in PB1-F2 levels we measured in infection were corroborated in our transfection studies, utilizing a single (3×FLAG) antibody, suggesting these results are not likely a result of antibody affinity differences. Finally, these results are also supported by previous studies that have demonstrated strain-specific differences in PB1-F2 expression levels using other methods (31, 32).
Our data show that the presence of the full-length PB1-F2 ORF in the IAV genome is not a predictor for PB1-F2 protein expression and suggests that restoring the PB1-F2 protein-coding sequence alone in viruses lacking a full-length ORF may not be sufficient to restore PB1-F2 expression. Additionally, inserting a PB1-F2 ORF sequence from one viral isolate into the genome sequence of another isolate, such as PR8, is likely also misleading with regard to the expression of the protein in its wild-type virus context because of the contribution of the 3′ PB1 translational regulatory region to PB1-F2 expression. As shown here, when the low-expressing OH07 PB1-F2 ORF was cloned into the PR8 PB1 gene (POP/3×FLAG), PB1-F2 was expressed at much higher levels than when it was present in a fully swine origin-derived PB1 gene (OOO/3×FLAG and MOM/3×FLAG). When such a chimeric gene is introduced into a recombinant virus, PB1-F2 may be expressed at higher levels than when located in its native PB1 gene context, and any phenotypic contribution to IAV infection attributed to PB1-F2 expression would be skewed as a result. Thus, it is necessary to consider all of the complexities of PB1-F2 translation in order to determine its level of expression and contribution, if any, to IAV infection.
There is strong evidence that PB1-F2 protein translation is regulated at the level of ribosomal leaky scanning and that altering the 5′ sequence upstream of the PB1-F2 ORF can modulate PB1-F2 expression. In particular, mutation of the third PB1 AUG so that translation initiation is prevented just prior to the PB1-F2 ORF results in increased expression of PB1-F2 (2). Indeed, we were able to recapitulate this finding in our studies (data not shown). However, because the Kozak sequences regulating this PB1-F2 modulation are conserved between OH07/Mx09 and PR8, interchanging the 5′ PB1 region using our chimeric PB1 translational clones had no effect on expression levels in our system. Instead, we have identified nucleotides 267 to 343 in the F2 ORF region and nucleotides 582 to 816 in the 3′ PB1 region as additional contributors to the regulation of PB1-F2 translation. Our studies suggest that each region can be utilized to increase or decrease PB1-F2 expression, but when both are present, the effect on PB1-F2 is more dramatic.
The mechanism by which the F2 ORF and 3′ PB1 regions affect PB1-F2 translation is currently not clear. The fact that these two regions can impact translation individually and additively suggests that the F2 ORF and 3′ PB1 regions act independently on translation regulation of PB1-F2 and, given the unknown nature of their regulation, that one of the regions could promote PB1-F2 translation while the other inhibits it. One possibility is that one or both of these regions could affect a previously predicted pseudoknot that incorporates the PB1-F2 start codon by either stabilizing the structure to aid in translation at the PB1-F2 start codon or inhibiting the formation of the pseudoknot to allow the translational machinery to scan past the PB1-F2 AUG (33). Alternatively, one or both of these regions could be involved in forming a novel RNA structure in the PB1 mRNA. Also, it is possible that one or both of the regions interact with PB1, N40, or a host RNA binding protein in order to facilitate PB1-F2 translational regulation. Thus, a significant amount of work remains in order to extend these initial mapping studies and fully understand these novel translational regulatory regions.
Though we do not understand the mechanism through which these translational regulatory regions contribute to PB1-F2 expression, they still may be useful as a tool for predicting PB1-F2 expression. As we have illustrated, the F2 ORF and 3′ PB1 regions described here can be interchanged to modify the level of PB1-F2 expression (Fig. 6 and 7). Therefore, using RNA sequences from the F2 ORF and 3′ PB1 regions of known PB1-F2-expressing viruses for comparison with viruses with unknown PB1-F2 expression may allow better prediction of biologically relevant PB1-F2 expression. Studies are under way to narrow the minimal RNA sequences necessary and sufficient for regulation of PB1-F2 in multiple virus strain backgrounds so that accurate predictions of PB1-F2 expression may be possible.
It is unclear why the full PB1-F2 ORF would be maintained in some swine origin IAVs and yet these viruses contain regulatory sequences that result in such low expression levels of the protein in infected cells. It is possible that PB1-F2 expression is either unnecessary or deleterious for productive infection in the swine host and has therefore been lost either through truncation or through this novel translational regulatory mechanism. These data corroborate a publication by Trifonov et al. which suggested that IAVs are evolving in a manner to make PB1-F2 unnecessary (34). Here, we have illustrated that expression of PB1-F2 from modern swine PB1 genes was minimal, and in contrast, expression of PB1-F2 from the PR8 PB1 gene, a laboratory-adapted virus isolated in 1934, expressed PB1-F2 at a higher level. While Trifonov et al. illustrated that this was occurring through truncation of the PB1-F2 ORF, our data suggest that downstream regulatory sequences may also play a role in suppression of PB1-F2 protein expression (34). Nonetheless, spillovers between swine- and human-adapted viruses are not infrequent events, and it will be important to further determine both the sequence determinants involved in expression of swine origin PB1-F2 and the consequences of that expression. Utilizing data from this work, future experiments will include creating recombinant viruses that express swine origin PB1-F2 at biologically significant levels so that the molecular function and pathogenic impact of the protein encoded by these viruses can be fully investigated.
ACKNOWLEDGMENTS
We thank Chelsie Sievers and Dana McCullough (Iowa State University) for their intellectual and technical assistance with this project. In addition, we thank Zhangqi Shen and Jing Yu (Iowa State University) for their assistance with qPCR and protein purification assays, respectively. Special thanks are due to Richard Webby (St. Jude Children's Research Hospital), Peter Palese (Mount Sinai School of Medicine), Alexander Klimov (Centers for Disease Control and Prevention), Daniel Perez (University of Maryland), and Michelle Harland (National Animal Disease Center) for providing reagents.
This project was funded through a U.S. Department of Agriculture collaborative agreement with C.L.M and grants to C.L.M. from the Iowa Pork Producers and Iowa State University College of Veterinary Medicine.
Footnotes
Published ahead of print 24 July 2013
REFERENCES
- 1.Chen W, Calvo PA, Malide D, Gibbs J, Schubert U, Bacik I, Basta S, O'Neill R, Schickli J, Palese P, Henklein P, Bennink JR, Yewdell JW. 2001. A novel influenza A virus mitochondrial protein that induces cell death. Nat. Med. 7:1306–1312 [DOI] [PubMed] [Google Scholar]
- 2.Wise HM, Foeglein A, Sun J, Dalton RM, Patel S, Howard W, Anderson EC, Barclay WS, Digard P. 2009. A complicated message: identification of a novel PB1-related protein translated from influenza A segment 2 mRNA. J. Virol. 83:8021–8031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wise HM, Barbezange C, Jagger BW, Dalton RM, Gog JR, Curran MD, Taubenberger JK, Anderson EC, Digard P. 2011. Overlapping signals for translational regulation and packaging of influenza A virus segment 2. Nucleic Acids Res. 39:7775–7790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dudek Sabine E, Wixler L, Nordhoff C, Nordmann A, Anhlan D, Wixler V, Ludwig S. 2011. The influenza virus PB1-F2 protein has interferon antagonistic activity. Biol. Chem. 392:1135. [DOI] [PubMed] [Google Scholar]
- 5.Varga ZT, Ramos I, Hai R, Schmolke M, García-Sastre A, Fernandez-Sesma A, Palese P. 2011. The influenza virus protein PB1-F2 inhibits the induction of type I interferon at the level of the MAVS adaptor protein. PLoS Pathog. 7:e1002067. 10.1371/journal.ppat.1002067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Le Goffic R, Bouguyon E, Chevalier C, Vidic J, Da Costa B, Leymarie O, Bourdieu C, Decamps L, Dhorne-Pollet S, Delmas B. 2010. Influenza A virus protein PB1-F2 exacerbates IFN-β expression of human respiratory epithelial cells. J. Immunol. 185:4812–4823 [DOI] [PubMed] [Google Scholar]
- 7.Conenello GM, Zamarin D, Perrone LA, Tumpey T, Palese P. 2007. A single mutation in the PB1-F2 of H5N1 (HK/97) and 1918 influenza A viruses contributes to increased virulence. PLoS Pathog. 3:e141. 10.1371/journal.ppat.0030141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mitzner D, Dudek SE, Studtrucker N, Anhlan D, Mazur I, Wissing J, Jänsch L, Wixler L, Bruns K, Sharma A, Wray V, Henklein P, Ludwig S, Schubert U. 2009. Phosphorylation of the influenza A virus protein PB1-F2 by PKC is crucial for apoptosis promoting functions in monocytes. Cell. Microbiol. 11:1502–1516 [DOI] [PubMed] [Google Scholar]
- 9.McAuley JL, Hornung F, Boyd KL, Smith AM, McKeon R, Bennink J, Yewdell JW, McCullers JA. 2007. Expression of the 1918 influenza A virus PB1-F2 enhances the pathogenesis of viral and secondary bacterial pneumonia. Cell Host Microbe 2:240–249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Conenello GM, Tisoncik JR, Rosenzweig E, Varga ZT, Palese P, Katze MG. 2011. A single N66S mutation in the PB1-F2 protein of influenza A virus increases virulence by inhibiting the early interferon response in vivo. J. Virol. 85:652–662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schmolke M, Manicassamy B, Pena L, Sutton T, Hai R, Varga ZT, Hale BG, Steel J, Perez DR, Garcia-Sastre A. 2011. Differential contribution of PB1-F2 to the virulence of highly pathogenic H5N1 influenza A virus in mammalian and avian species. PLoS Pathog. 7:e1002186. 10.1371/journal.ppat.1002186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zamarin D, Ortigoza MB, Palese P. 2006. Influenza A virus PB1-F2 protein contributes to viral pathogenesis in mice. J. Virol. 80:7976–7983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Neumann G, Noda T, Kawaoka Y. 2009. Emergence and pandemic potential of swine-origin H1N1 influenza virus. Nature 459:931–939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Belser JA, Wadford DA, Pappas C, Gustin KM, Maines TR, Pearce MB, Zeng H, Swayne DE, Pantin-Jackwood M, Katz JM, Tumpey TM. 2010. Pathogenesis of pandemic influenza A (H1N1) and triple-reassortant swine influenza A (H1) viruses in mice. J. Virol. 84:4194–4203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pearce MB, Jayaraman A, Pappas C, Belser JA, Zeng H, Gustin KM, Maines TR, Sun X, Raman R, Cox NJ, Sasisekharan R, Katz JM, Tumpey TM. 2012. Pathogenesis and transmission of swine origin A(H3N2)v influenza viruses in ferrets. Proc. Natl. Acad. Sci. U. S. A. 109:3944–3949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hai R, Schmolke M, Varga ZT, Manicassamy B, Wang TT, Belser JA, Pearce MB, Garcia-Sastre A, Tumpey TM, Palese P. 2010. PB1-F2 expression by the 2009 pandemic H1N1 influenza virus has minimal impact on virulence in animal models. J. Virol. 84:4442–4450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ozawa M, Basnet S, Burley LM, Neumann G, Hatta M, Kawaoka Y. 2011. Impact of amino acid mutations in PB2, PB1-F2, and NS1 on the replication and pathogenicity of pandemic (H1N1) 2009 influenza viruses. J. Virol. 85:4596–4601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pena L, Vincent AL, Loving CL, Henningson JN, Lager KM, Lorusso A, Perez DR. 2012. Restoring PB1-F2 into the 2009 pandemic H1N1 influenza virus has minimal effects in swine. J. Virol. 86:5523–5532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wanitchang A, Kramyu J, Jongkaewwattana A. 2010. Enhancement of reverse genetics-derived swine-origin H1N1 influenza virus seed vaccine growth by inclusion of indigenous polymerase PB1 protein. Virus Res. 147:145–148 [DOI] [PubMed] [Google Scholar]
- 20.Pena L, Vincent AL, Loving CL, Henningson JN, Lager KM, Li W, Perez DR. 2012. Strain-dependent effects of PB1-F2 of triple-reassortant H3N2 influenza viruses in swine. J. Gen. Virol. 93:2204–2214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dereeper A, Audic S, Claverie JM, Blanc G. 2010. BLAST-EXPLORER helps you building datasets for phylogenetic analysis. BMC Evol. Biol. 10:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dereeper A, Guignon V, Blanc G, Audic S, Buffet S, Chevenet F, Dufayard JF, Guindon S, Lefort V, Lescot M, Claverie JM, Gascuel O. 2008. Phylogeny.fr: robust phylogenetic analysis for the non-specialist. Nucleic Acids Res. 36:W465–W469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Buchholz UJ, Finke S, Conzelmann K-K. 1999. Generation of bovine respiratory syncytial virus (BRSV) from cDNA: BRSV NS2 is not essential for virus replication in tissue culture, and the human RSV leader region acts as a functional BRSV genome promoter. J. Virol. 73:251–259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kitikoon P, Nilubol D, Erickson BJ, Janke BH, Hoover TC, Sornsen SA, Thacker EL. 2006. The immune response and maternal antibody interference to a heterologous H1N1 swine influenza virus infection following vaccination. Vet. Immunol. Immunopathol. 112:117–128 [DOI] [PubMed] [Google Scholar]
- 25.Hoffmann E, Neumann G, Kawaoka Y, Hobom G, Webster RG. 2000. A DNA transfection system for generation of influenza A virus from eight plasmids. Proc. Natl. Acad. Sci. U. S. A. 97:6108–6113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hoffmann E, Stech J, Guan Y, Webster RG, Perez DR. 2001. Universal primer set for the full-length amplification of all influenza A viruses. Arch. Virol. 146:2275–2289 [DOI] [PubMed] [Google Scholar]
- 27.Higuchi R, Krummel B, Saiki R. 1988. A general method of in vitro preparation and specific mutagenesis of DNA fragments: study of protein and DNA interactions. Nucleic Acids Res. 16:7351–7367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Carrillo-Conde B, Schiltz E, Yu J, Chris Minion F, Phillips GJ, Wannemuehler MJ, Narasimhan B. 2010. Encapsulation into amphiphilic polyanhydride microparticles stabilizes Yersinia pestis antigens. Acta Biomater. 6:3110–3119 [DOI] [PubMed] [Google Scholar]
- 29.Pfaffl MW. 2001. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 29:e45. 10.1093/nar/29.9.e45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Qin Q, Carroll K, Hastings C, Miller CL. 2011. Mammalian orthoreovirus escape from host translational shutoff correlates with stress granule disruption and is independent of eIF2α phosphorylation and PKR. J. Virol. 85:8798–8810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chen C-J, Chen G-W, Wang C-H, Huang C-H, Wang Y-C, Shih S-R. 2010. Differential localization and function of PB1-F2 derived from different strains of influenza A virus. J. Virol. 84:10051–10062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Reis AL, McCauley JW. 2013. The influenza virus protein PB1-F2 interacts with IKKβ and modulates NF-κB signalling. PLoS One 8:e63852. 10.1371/journal.pone.0063852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Moss WN, Priore SF, Turner DH. 2011. Identification of potential conserved RNA secondary structure throughout influenza A coding regions. RNA 17:991–1011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Trifonov V, Racaniello V, Rabadan R. 2009. The contribution of the PB1-F2 protein to the fitness of influenza A viruses and its recent evolution in the 2009 influenza A (H1N1) pandemic virus. PLoS Curr. 1:RRN1006. 10.1371/currents.RRN1006 [DOI] [PMC free article] [PubMed] [Google Scholar]