

Abstract
Autographa californica multiple nucleopolyhedrovirus (AcMNPV) ac83 is a baculovirus core gene whose function in the AcMNPV life cycle is unknown. In the present study, an ac83-knockout AcMNPV (vAc83KO) was constructed to investigate the function of ac83 through homologous recombination in Escherichia coli. No budded virions were produced in vAc83KO-transfected Sf9 cells, although viral DNA replication was unaffected. Electron microscopy revealed that nucleocapsid assembly was aborted due to the ac83 deletion. Domain-mapping studies revealed that the expression of Ac83 amino acid residues 451 to 600 partially rescued the ability of AcMNPV to produce infectious budded virions. Bioassays indicated that deletion of the chitin-binding domain of Ac83 resulted in the failure of oral infection of Trichoplusia ni larvae by AcMNPV, but AcMNPV remained infectious following intrahemocoelic injection, suggesting that the domain is involved in the binding of occlusion-derived virions to the peritrophic membrane and/or to other chitin-containing insect tissues. It has been demonstrated that Ac83 is the only component with a chitin-binding domain in the per os infectivity factor complex on the occlusion-derived virion envelope. Interestingly, a functional inner nuclear membrane sorting motif, which may facilitate the localization of Ac83 to the envelopes of occlusion-derived virions, was identified by immunofluorescence analysis. Taken together, these results demonstrate that Ac83 plays an important role in nucleocapsid assembly and the establishment of oral infection.
INTRODUCTION
Baculoviruses are a family of enveloped double-stranded DNA viruses that are specifically pathogenic to arthropods, mainly affecting insects of the orders Lepidoptera, Hymenoptera, and Diptera (1). Based on their whole-genome sequences, Baculoviridae can be divided into four genera: Alphabaculovirus, Betabaculovirus, Gammabaculovirus, and Deltabaculovirus (2). Alphabaculovirus encompasses lepidopteran nucleopolyhedroviruses (NPVs) and can be further subdivided into two groups, group I and group II NPVs, based on phylogenetic analysis of the polyhedrin (polh) gene (3). This grouping is coincident with the utilization of different envelope fusion proteins for spreading infection (4). Autographa californica multiple NPV (AcMNPV) is the best-characterized baculovirus and the first to be completely sequenced. AcMNPV has a genome of approximately 134 kbp that encodes 156 putative open reading frames (ORFs) (5). To date, 59 baculovirus genomes have been completely sequenced, and these genomes have 37 core genes in common (1, 6, 7).
A unique feature during the life cycle of a baculovirus is the production of two morphologically distinct but genetically identical progeny virion phenotypes, budded virions (BVs) and occlusion-derived virions (ODVs). BVs are produced early in infection. Nucleocapsids assemble in the nucleus, migrate through the cytoplasm, and bud out of the plasma membrane to form BVs. BVs are able to spread infections within infected insect tissues and are responsible for establishing systemic infection. During the late phase of infection, nucleocapsids are retained in the nucleus and are enveloped by the virus-induced intranuclear microvesicles to form ODVs. ODVs are subsequently occluded within a proteinaceous crystal matrix to form occlusion bodies (OBs).
Although the nucleocapsids of BVs and ODVs are similar, their major differences are reflected in the composition of their envelopes, which accommodate their distinct roles (8, 9). Baculoviruses initiate infection when their insect hosts orally consume OB-contaminated food. A number of ODV envelope proteins, designated per os infectivity factors (PIFs), play roles in oral infectivity. Currently, seven PIFs have been identified: P74 (PIF0), PIF1, PIF2, PIF3, PIF4, PIF5, and PIF6 (10–16). All the PIF genes are baculovirus core genes (17), indicating that the per os infection processes have been conserved during evolution. Recently, the Spodoptera frugiperda NPV ORF58 (Sf58), which is conserved only in lepidopteran baculoviruses and is homologous to Ac108 in AcMNPV, was shown to be a PIF (18). Proteomic mass spectrometric analysis revealed that at least six proteins, P74, PIF1, PIF2, PIF3, PIF4, and Ac83, together with three other potential components, Ac5, PIF6, and Ac108, are present as a complex on the ODV envelope (19). PIF1, PIF2, and PIF3 form a stable core. PIF4 interacts strongly with the core, whereas P74 and Ac83 interact loosely with it (19).
Ac83 is encoded by the baculovirus core gene ac83. Sequence analysis revealed that homologs of Ac83 from Alphabaculovirus and Gammabaculovirus contain a type II chitin-binding domain (CBD) at the N terminus (20). The type II CBD is characterized by the presence of a 6-cysteine motif with the consensus sequence C-X13–20-C-X5–6-C-X9–19-C-X10–14-C-X4–14-C (where X is any amino acid other than cysteine) (21). The type II CBD is also referred to as the peritrophin-A domain because it is found in peritrophin-A chitin-binding proteins, particularly in the peritrophins of insect peritrophic membranes (PM) (21). In addition, type II CBDs have also been identified in proteins from the ecdysozoan clade that are frequently involved in the interaction with chitin (21, 22).
As the only component with a CBD in the PIF complex, it is thought that Ac83 plays a role in the oral infectivity of ODVs, but such a role has never been confirmed (19). ac83 encodes a putative protein of 847 amino acids (aa) with a predicted molecular mass of approximately 96 kDa (5). Proteomic analyses of the protein compositions of the BVs and ODVs of AcMNPV suggest that Ac83 is associated only with the ODV envelope (23, 24). However, the Ac83 homolog in Orgyia pseudotsugata NPV, OpP91, has been observed to localize predominantly to the capsid structure in BVs and ODVs (25). The closely related ac83 homolog in Bombyx mori NPV, BmP95, has been shown to be essential for nucleocapsid assembly, and deletion of BmP95 led to a defect in the spreading of the infection in cultured cells (20, 26).
In this study, an ac83-knockout virus was constructed in order to investigate the role of ac83 in the AcMNPV life cycle. We demonstrated that deletion of ac83 resulted in the failure of nucleocapsid assembly and of the subsequent morphogenesis of progeny BVs and ODVs. The rescue of Ac83 amino acid residues 451 to 600, which make up only approximately one-fifth of the full-length Ac83 protein (847 aa), was sufficient to produce infectious BVs. Most importantly, we observed that the CBD of Ac83 was essential for the establishment of efficient per os infection by AcMNPV. Considering the ubiquitous presence of chitin in the PM and the microvilli of epithelial cells of the insect midgut, and given that Ac83 is the only known component with a CBD in the PIF complex (19), we suggest that the CBD of Ac83 may assist in initiating primary viral infection by enhancing the binding of ODVs to the chitin components associated with the PM or microvilli.
MATERIALS AND METHODS
Bioinformatic analyses.
The homologs of Ac83 were searched against the nonredundant protein sequences at the NCBI database (http://blast.ncbi.nlm.nih.gov/Blast.cgi?PROGRAM=blastp&BLAST_PROGRAMS=blastp&PAGE_TYPE=BlastSearch&SHOW_DEFAULTS=on&LINK_LOC=blasthome) with the position-specific iterated (PSI) BLAST algorithm. The conserved domains of Ac83 were predicted from the UniProt database (http://www.uniprot.org/uniprot/Q06670), the NCBI Conserved Domain Search database (http://www.ncbi.nlm.nih.gov/Structure/cdd/wrpsb.cgi) (27), SMART (28), and HHpred (29). Multiple sequence alignments were constructed using Clustal X (30) with default settings and were edited with GeneDoc (31).
Viruses and cell lines.
The bacmid bMON14272 (Invitrogen), which contains an AcMNPV genome, was maintained in DH10B cells as described previously (32). Sf9 (Spodoptera frugiperda IPLB-Sf21-AE clonal isolate 9) insect cells were cultured at 27°C in TNM-FH medium (Invitrogen) supplemented with 10% fetal bovine serum (Invitrogen), penicillin (100 μg/ml), and streptomycin (30 μg/ml). The titers of BVs were determined by a 50% tissue culture infective dose (TCID50) endpoint dilution assay in Sf9 cells as described previously (33). The viral inoculum was allowed to adsorb to cells for 1 h of infection or 5 h of transfection at 27°C and was then replaced with fresh medium. Time zero was defined as the time when the viral inoculum was replaced.
Transcriptional analysis of ac83 and rapid amplification of 5′ cDNA ends (5′ RACE).
Sf9 cells (1 × 106) either were mock infected or were infected with AcMNPV at a multiplicity of infection (MOI) of 5 TCID50s/cell. The cells were collected at different hours postinfection (p.i.). Total RNA was extracted using an RNeasy Mini Kit (Qiagen) and was quantified by optical density measurements at 260 nm. After the RNA samples were treated with RNase-Free DNase (Promega), the first strand of cDNA was synthesized using an iScript cDNA synthesis kit (Bio-Rad). Reverse transcription-PCR (RT-PCR) with the ac83-specific primer pair ac83504/ac83304 was used to detect the ac83 transcripts. All PCR primers are listed in Table 1.
Table 1.
Primer sequences used in this study
| Application | Primer | Sequence |
|---|---|---|
| Amplification of ac83 flanking sequences | ac83502 | 5′-GAGCTCATGATGTCTGGCGTAATGTTGCTC-3′ |
| ac83302 | 5′-GGATCCGTTGGGATTTTCATCATTTGCTCTA-3′ | |
| ac83503 | 5′-CTGCAGGCACGTCAAAAACGGCCAATAC-3′ | |
| ac83303 | 5′-AAGCTTCTTTACGAGTAGAATTCTACTTGTAAAACATAATC-3′ | |
| Amplification of ac83 promoter | ac83501 | 5′-GAATTCGCTCATCGATAGCGGCGGCGGA-3′ |
| Amplification of the 3′ end of ac83 tagged with HA | ac83301 | 5′-GGATCCTTAGGCGTAATCTGGGACGTCGTATGGGTATACAATGGAATCTTCTTGTAAATTATCCAA-3′ |
| Reverse transcription analysis of ac83 | ac83504 | 5′-CAAGCTAACCAGTTGTCATGCGGAA-3′ |
| ac83304 | 5′-TTATACAATGGAATCTTCTTGTAAATTATCC-3′ | |
| 5′ RACE analysis of ac83 | ac83305 | 5′-CACGGGAACACATTTTAGTTGTGCGTA-3′ |
| ac83306 | 5′-GTCGCCGCTCAAAATAAACTCG-3′ | |
| Deletion of Ac83 INM-SM | ac83307 | 5′-TATTCAGTCATCACTTGCATCGTGAACGCCCAAATCA-3′ |
| ac83505 | 5′-GGCGTTCACGATGCAAGTGATGACTGAATATGTGAAG-3′ | |
| Deletion of Ac83 CBD | ac83308 | 5′-GTGTCCGGCGCCGTTAACTTTACACTGGCCGGTTTC-3′ |
| ac83506 | 5′-GGCCAGTGTAAAGTTAACGGCGCCGGACACACGTAC-3′ | |
| Deletion of Ac83 PR region | ac83309 | 5′-ATAAAATAAATCTTTGTTGTCTAATATTAGCGCCACTACCCAATGGTCG-3′ |
| ac83507 | 5′-CTAATATTAGACAACAAAGATTTATTTTATTCATG-3′ | |
| Deletion of Ac83 aa 2–290 | ac83310 | 5′-CGATATCGGCCGTCATCGTGAACGCCCAAATCA-3′ |
| ac83508 | 5′-GCGTTCACGATGACGGCCGATATCGGCGAC-3′ | |
| Deletion of Ac83 aa 2–450 | ac83311 | 5′-GGCGTCGTTGTTTAACATCGTGAACGCCCAAATCA-3′ |
| ac83509 | 5′-TGGGCGTTCACGATGTTAAACAACGACGCCATCTTTG-3′ | |
| Deletion of Ac83 aa 742–847 | ac83312 | 5′-GGATCCTTAGGCGTAATCTGGGACGTCGTATGGGTATTTAACGTTGTTGCGTAACTCGT-3′ |
| Deletion of Ac83 aa 601–847 | ac83313 | 5′-GGATCCTTAGGCGTAATCTGGGACGTCGTATGGGTATTTGGGCCGTTTTACGAGAG-3′ |
| Deletion of Ac83 aa 551–847 | ac83314 | 5′-GGATCCTTAGGCGTAATCTGGGACGTCGTATGGGTATTCGTTTTCTACGATTTTTTGG-3′ |
| Amplification of Ac83 INM-SM | ac83510 | 5′-GGATCCATGATGTCTGGCGTAATGTTGCTC-3′ |
| ac83316 | 5′-GAATTCAGTCATCACTTGGAGCCGCTTG-3′ | |
| Amplification of gfp | GFP-F | 5′-GAATTCATGGGCAAAGGAGAAGAACTTTTCACTG-3′ |
| GFP-R | 5′-TCTAGATTATTTGTATAGTTCATCCATGCCATG-3′ | |
| Real-time PCR specific for AcMNPV DNA | gp41-F | 5′-CGTAGTGGTAGTAATCGCCGC-3′ |
| gp41-R | 5′-AGTCGAGTCGCGTCGCTTT-3′ |
5′ RACE analysis was performed to map the transcription start site of ac83. The ac83-specific reverse primers ac83305 and ac83306, together with the adaptor primers provided in the 5′/3′ RACE Kit, 2nd Generation (Roche), were used for PCR amplification, and 1 μg of purified total RNA isolated from the AcMNPV-infected Sf9 cells at 24 h p.i. was used as the template. The PCR products were purified with a gel extraction kit (Omega) and were cloned into the pMD18-T vector (TaKaRa) for sequencing.
Time course analysis of Ac83 synthesis.
Sf9 cells (1 × 106) were infected with vAc83:HA (a recombinant AcMNPV in which Ac83 was tagged with a hemagglutinin [HA] epitope; see details below) at an MOI of 10 TCID50s/cell. At the designated time points p.i., cells were scraped, collected, and centrifuged at 3,000 × g for 5 min at room temperature. Western blotting was performed using a mouse monoclonal anti-HA antibody (1:3,000; Abcam) as the primary antibody and a horseradish peroxidase (HRP)-conjugated goat anti-mouse antibody (1:5,000; Amersham Biosciences) as the secondary antibody to detect the expression profile of Ac83.
Generation of an ac83-knockout AcMNPV bacmid.
An ac83-knockout AcMNPV bacmid, designated bAc83KO, was generated as described previously, using primer pairs ac83502/ac83302 and ac83503/ac83303 (34). To avoid affecting adjacent genes, 360 nucleotides (nt) from the 5′ region of the ac83 ORF (AcMNPV nt 67884 to 68243) and 127 nt from the 3′ region of the ac83 ORF (AcMNPV nt 70301 to 70427) were preserved.
Construction of an ac83-knockout virus.
To facilitate the examination of viral infection, the AcMNPV polh and enhanced green fluorescence protein (egfp; referred to as gfp in this study) genes were inserted into the polh locus of bAc83KO to generate the ac83-knockout virus vAcac83KO-PH-GFP (vAc83KO) as described previously (35). A wild-type control virus, vAcWT-PH-GFP (vAcWT), was generated by the insertion of polh and gfp into bMON14272.
To generate an ac83-repaired virus, the donor plasmid pFB1-Ac83:HA-PG was constructed as follows. A 2,984-bp fragment containing an ac83 native promoter and the ac83 ORF tagged with an HA epitope prior to the ac83 stop codon (Ac83POHA) was PCR amplified using bMON14272 as the template and ac83501 and ac83301 as the primers. The PCR product was ligated into pUC18-SV40 (36) to generate pUC18-Ac83:HA. pUC18-Ac83:HA was subsequently digested with EcoRI and XbaI, and the resulting Ac83:HA fragment was ligated into pFB1-PH-GFP (34) to generate the donor plasmid pFB1-Ac83:HA-PG. Electrocompetent DH10B cells containing the helper plasmid pMON7124 and bAc83KO were transformed with pFB1-Ac83:HA-PG to generate the ac83-repaired virus vAcac83REP-HA-PH-GFP (vAc83:HA).
Construction of different truncated forms of ac83 rescue viruses.
A set of recombinant AcMNPVs with different ac83 gene truncations was generated to investigate the portion of Ac83 that is important for viral replication. The overlap PCR technique was used to construct the truncated Ac83 protein variants as described previously (37). Briefly, to delete the predicted inner nuclear membrane sorting motif (INM-SM) of Ac83, primers ac83501/ac83307 and ac83505/ac83301 were used to amplify the upper and lower overlap fragments of ac83, respectively, from pFB1-Ac83:HA-PG. Overlap PCR was conducted by mixing the resulting PCR products with the outmost primers ac83501 and ac83301. In the same manner, with primers ac83501/ac83308 and ac83506/ac83301, the predicted CBD of Ac83 was deleted, and the predicted proline-rich (PR) region of Ac83 was deleted using primers ac83501/ac83309 and ac83507/ac83301. To delete aa 2 to 290 (numbering is from the N terminus of Ac83 in this paper) from Ac83, primers ac83501/ac83310 and ac83508/ac83301 were used, while primers ac83501/ac83311 and ac83509/ac83301 were used for the deletion of Ac83 aa 2 to 450. The ac83 gene-specific primer ac83501 was used in combination with the ac83 gene truncation-specific primers ac83312, ac83313, and ac83314 to generate C-terminally truncated Ac83 protein variants. Finally, Ac83 aa 2 to 450 and aa 601 to 847 were deleted from Ac83 using primers ac83501 and ac83313, and the Ac83:del2-450 fragment was used as the template. The PCR products were cloned between EcoRI and BamHI sites in pUC18-SV40, and the resulting constructs were digested with EcoRI and XbaI and were ligated into the donor plasmid pFB1-PH-GFP. Electrocompetent DH10B cells containing the pMON7124 helper plasmid and the bAc83KO bacmid were transformed with the donor plasmids mentioned above to generate different truncated forms of the ac83 rescue viruses: vAcac83REP-delINM-SM-PH-GFP (vAc83:delSM), vAcac83REP-delCBD-PH-GFP (vAc83:delCBD), vAcac83REP-delPR-PH-GFP (vAc83:delPR), vAcac83REP-del2-290-PH-GFP (vAc83:del2-290), vAcac83REP-del2-450-PH-GFP (vAc83:del2-450), vAcac83REP-del742-847-PH-GFP (vAc83:del742-847), vAcac83REP-del601-847-PH-GFP (vAc83:del601-847), vAcac83REP-del551-847-PH-GFP (vAc83:del551-847), and vAcac83REP-del2-450/601-847-PH-GFP (vAc83:del2-450/601-847).
Time course analysis of BV production.
Sf9 cells (1 × 106) either were transfected in triplicate with 1.0 μg of bacmid DNA using Cellfectin liposome reagent (Invitrogen) or were infected with the indicated viruses at an MOI of 0.1 TCID50/cell. At the designated time points posttransfection (p.t.) or p.i., the supernatants containing BVs were harvested, and the titers were determined in duplicate using a TCID50 endpoint dilution assay (33). To determine whether any noninfectious BVs were produced in vAc83KO-transfected cells, the supernatants and cell pellets were harvested at 72 h p.t. for Western blotting as described previously (35). A polyclonal primary antibody against AcMNPV VP39 (dilution, 1:3,000) (38) was used to detect the presence of the major capsid protein VP39.
Analysis of viral DNA synthesis by qPCR.
To evaluate any effect of ac83 deletion on viral DNA replication, quantitative real-time PCR (qPCR) analysis was performed as described previously (6).
Electron microscopy analysis.
Sf9 cells (1 × 106) were either transfected with 1 μg of bacmid DNA or infected with virus at an MOI of 5 TCID50s/cell. At 72 h p.t. or 60 h p.i., the cells were collected and were prepared for electron microscopy as described previously (6). The samples were examined with a JEOL JEM-1400 transmission electron microscope at an accelerating voltage of 120 kV.
In vivo infectivity assays.
The infectivity of BVs in vivo was examined by hemocoelically injecting different doses of BVs of vAcWT, vAc83:HA, or vAc83:delCBD into 4th-instar Trichoplusia ni larvae. H2O was used as a blank control. For the oral infectivity bioassays, OBs of vAcWT, vAc83:HA, or vAc83:delCBD purified from infected Sf9 cells were administered to newly molted 4th-instar T. ni larvae via oral inoculation as described previously (13). To destroy the PM in the midgut, newly molted 4th-instar T. ni larvae were treated with calcofluor white as described previously (39). A cohort of 24 larvae was used for each treatment, and the treatment was repeated in triplicate. At 24 h postinoculation, fresh diet was provided to the larvae except when the diet had not been entirely consumed. Infected larvae were reared individually in 24-well plates and were monitored daily until all larvae had either pupated or died. The data were analyzed by one-way analysis of variance (ANOVA) followed by Tukey's test.
BV and ODV purification.
BVs and ODVs were purified, and ODVs were fractionated into envelopes and nucleocapsids, as described previously (6). Immunoblotting was performed as described above with a monoclonal anti-HA antibody or one of the following primary antibodies: (i) a polyclonal antibody against AcMNPV ODV-E25 (dilution, 1:2,000) (24) or (ii) a polyclonal antibody against AcMNPV VP39. An HRP-conjugated goat anti-rabbit antibody (1:8,000; Amersham Biosciences) or an HRP-conjugated goat anti-mouse antibody was used as the secondary antibody.
Generation of a transient expression plasmid expressing INM-SM-GFP and a recombinant virus hyperexpressing INM-SM-GFP.
To investigate whether the INM-SM of Ac83 is functional, a transient expression assay was performed. A gfp gene was PCR amplified from pFB1-PH-GFP using primers GFP-F and GFP-R. The PCR product was digested with EcoRI and XbaI and was inserted into pIB/V5-His (Invitrogen) to generate pIB-GFP. The INM-SM of Ac83 was PCR amplified from bMON14272 using primers ac83510 and ac83316. The PCR product was digested with BamHI and EcoRI and was ligated into pIB-GFP to construct pIB-SM-GFP.
To generate a recombinant virus expressing green fluorescent protein (GFP) in frame with the Ac83 INM-SM under the control of the polh promoter, pIB-SM-GFP was digested with BamHI and XbaI, and the resulting fragment was inserted into pFastBac1 (Invitrogen) to generate donor plasmid pFB1-SM-GFP. The site-specific transposition of INM-SM-GFP into bMON14272 was performed as described above. The recombination of the resulting virus, vAcSM-GFP, in which INM-SM-GFP was placed under the control of the polh promoter for hyperexpression, was confirmed by PCR analysis.
Immunofluorescence.
Immunofluorescence analysis was performed as described previously (6). Cells were directly stained with 4′,6-diamidino-2-phenylindole (DAPI; Roche) to detect the distribution of DNA. GFP fluorescence and the immunofluorescence of HA were examined using a Leica TCS SP5 laser scanning confocal microscope with the same parameter settings as those used for the mock-infected cells in each experiment.
RESULTS
Evolutionary conservation of Ac83 domains.
The full-length Ac83 amino acid sequence was used as a query to scan the UniProt catalog, the NCBI Conserved Domain Search database, and the SMART server. In addition, the amino acid sequences of Ac83 homologs were aligned in order to analyze their evolutionary conservation. In summary, all Ac83 homologs contained the Baculo_VP91_N domain (aa 22 to 192) and a transmembrane domain (TM) at the N terminus (aa 5 to 27). A typical type II CBD (aa 224 to 282) was highly conserved in all Ac83 homologs from NPVs but was weakly conserved in granulosis viruses (GVs) (Fig. 1). The CBD domain could be predicted in two Ac83 homologs of GVs (Cydia pomonella GV and Cryptophlebia leucotreta GV) by UniProt but not by NCBI, although sequence alignment showed that the CBD domains predicted in the Ac83 homologs of these two GVs were profoundly diversified (Fig. 1). In addition, a PR region (aa 671 to 698) was present in the Ac83 homologs from group I NPVs and some group II NPVs but was absent from Ac83 homologs of the lepidopteran GVs and Hymenoptera and Diptera NPVs (Fig. 1).
Fig 1.

Overview of the conservation of Ac83 domains. The predicted domains of Ac83 are indicated in the scheme. Alignments of the CBD, TM, PR, and putative C2HC zinc finger regions of Ac83 homologs are shown. The Ac83 homologs of GVs are marked with asterisks. The cysteine and histidine residues that define the C2HC zinc finger are shown below the alignment. Black shading denotes 100% conservation. Dark gray and light gray shading represent 80 and 60% conservation, respectively. The alignment was performed using ClustalX and was edited with GeneDoc software. PlxyNPV, Plutella xylostella NPV; BmNPV, Bombyx mori NPV; OpNPV, Orgyia pseudotsugata NPV; SfNPV, Spodoptera frugiperda NPV; SpltNPV, Spodoptera litura NPV; TnSNPV, Trichoplusia ni single NPV; HearNPV, Helicoverpa armigera NPV; NeabNPV, Neodiprion abietis NPV; NeleNPV, Neodiprion lecontei NPV; NeseNPV, Neodiprion sertifer NPV; CuniNPV, Culex nigripalpus NPV; CpGV, Cydia pomonella GV; CrleGV, Cryptophlebia leucotreta GV.
After careful inspection, we hypothesized that a putative C2HC-type zinc finger (aa 148 to 197) with the consensus sequence C-X5-C-Xn-H-X6-C (where X is any amino acid) was present in the N-terminal regions of all the Ac83 homologs (Fig. 1). In agreement with our prediction, it has been suggested previously that BmP95 possesses two putative zinc fingers in its N terminus (40). Since the zinc finger motif has been observed to be widely involved in protein-DNA and protein-protein interactions (41, 42), its presence in Ac83 homologs may suggest a role for the Ac83 homologs in the interaction with viral DNA or other proteins.
ac83 is a late gene.
The temporal transcriptional pattern of ac83 was determined by RT-PCR using the total RNA extracted from AcMNPV-infected Sf9 cells at various time points p.i. As shown in Fig. 2A, ac83 transcripts were first detected at 24 h p.i. and remained detectable up to 48 h p.i. pe38 was used to represent baculovirus early genes, and vp39 was used as a late gene. As expected, pe38 transcripts were first detected at 3 h p.i., and vp39 transcripts were first detected at 24 h p.i. (data not shown). No signals could be detected in mock-infected cells or by using the templates without reverse transcription prior to PCR, indicating that there was no contamination of genomic DNA in the RT-PCR analysis (data not shown).
Fig 2.
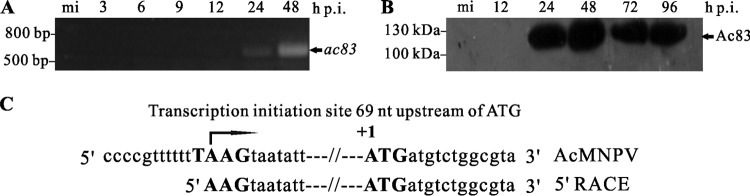
Transcription and expression kinetics of ac83. (A) RT-PCR analysis of ac83 transcription. Total RNA was extracted from AcMNPV-infected cells at the designated time points and was used for RT-PCR analysis. A DNA marker is shown on the left. (B) Time expression profile of Ac83. Cells were either mock infected (mi) or infected with vAc83:HA at an MOI of 5 TCID50s/cell. At different hours p.i., cells were collected and were prepared for Western blotting. Transferred proteins were detected with an anti-HA antibody. A protein marker is shown on the left. (C) Transcriptional mapping of the initiation site of ac83 using 5′ RACE analysis. The AcMNPV genome sequence and 5′ RACE sequencing results are shown. The translation start codon of Ac83 is shown in boldface and is designated +1, and the typical baculovirus late gene promoter consensus motif TAAG is also shown in boldface. The arrow indicates the location of the transcription initiation site of ac83.
Sequence analysis of the 300 nt upstream of the predicted start codon of the ac83 ORF (ATG) indicated that there were two canonical baculovirus late gene promoter motif TAAG elements located at nucleotide positions −215 and −70 and four early baculovirus promoter motifs, CAGT or CATT, located at nucleotide positions −265, −234, −226, and −218. To determine the transcription initiation site of ac83, 5′ RACE analysis was performed using the total RNA isolated from AcMNPV-infected cells at 24 h p.i. Three clones derived from the 5′ RACE products were sequenced, and the results indicated that the transcription of ac83 was initiated at the first A of the proximal canonical baculovirus late gene promoter motif TAAG (nt −69) (Fig. 2C). This agrees with the data on ac83 from another study (43).
To determine the time course of Ac83 expression in virus-infected cells, vAc83:HA-infected Sf9 cells were collected at the time points p.i. indicated in Fig. 2B and were analyzed by immunoblotting with a monoclonal anti-HA antibody. An immunoreactive band of approximately 115 kDa, larger than the predicted molecular mass of Ac83 (96 kDa), was first observed at 24 h p.i. and persisted at 96 h p.i. (Fig. 2B). The expression profile of Ac83 was similar to that of the baculovirus late gene vp39 (data not shown). Thus, these results demonstrated that ac83 is a baculovirus late gene.
Construction of an ac83-knockout AcMNPV.
To investigate the role of Ac83 in the baculovirus life cycle, the AcMNPV bacmid bMON14272 was used to replace a 2,057-bp region of ac83 with a 1,038-bp Cm cassette via the λ Red recombination system as described previously (6). In the resulting ac83-knockout bacmid (named bAc83KO), the expression of ac83 was thought to be completely disrupted. In addition, 360 nt from the 5′ end and 127 nt from the 3′ end of the ac83 ORF were retained to preserve the putative promoter of ac82 and homologous region 3 (Fig. 3A).
Fig 3.

Construction of recombinant viruses and analysis of viral replication. (A) Schematic diagram of the recombinant viruses used in this study. The bacmid bAc83KO was constructed by replacing a 2,057-bp fragment of ac83 in the bMON14272 genome with a 1,038-bp Cm cassette via ET homologous recombination in Escherichia coli. The ac83 deletion virus (vAc83KO) was constructed by inserting the polh and gfp genes into the polh locus of bAc83KO by Tn7-mediated transposition. The ac83 rescue virus (vAc83:HA) was constructed by transposing the ac83 ORF tagged with an HA epitope (triangle) under the control of its native promoter together with the polh and gfp genes into the polh locus of bAc83KO. The wild-type control virus (vAcWT) was constructed by transposing the polh and gfp genes into the polh locus of bMON14272. (B) Sf9 cells were transfected with vAcWT, vAc83KO, or vAc83:HA. At the indicated time points p.t., cells were monitored for infection by a fluorescence microscope. (C) The production of infectious BVs in the supernatants of transfected cells was determined by TCID50 endpoint dilution assays. Each data point was determined from the average for three independent transfections. Error bars represent standard deviations. (D) The supernatants (Sup) and cell extracts (Cell) of transfected cells at 96 h p.t. were separated by SDS-PAGE and were analyzed by Western blotting with anti-VP39 to detect the major capsid protein VP39.
To examine whether the deletion of ac83 had any effects on OB morphogenesis and to facilitate the observation of the progress of viral infection, an ac83-knockout AcMNPV, named vAc83KO, was constructed by inserting the polh gene of AcMNPV and the gfp gene into the polh locus of bAc83KO via Tn7-mediated transposition as described previously (34). Similarly, the two genes were inserted into bMON14272 to generate a wild-type AcMNPV control, named vAcWT. To confirm the phenotype resulting from the deletion of ac83, an HA tag prior to the ac83 stop codon was inserted, together with the two genes, into bAc83KO to generate a rescue virus, named vAc83:HA (Fig. 3A).
ac83 is essential for BV production.
To determine the effect of ac83 deletion on viral replication, Sf9 cells were transfected with vAc83KO, vAc83:HA, or vAcWT, and the infection was monitored using fluorescence microscopy. No obvious differences in the number of fluorescent cells were observed between these viruses at 24 h p.t., indicating comparable transfection efficiencies of approximately 20% (Fig. 3B). By 72 h p.t., nearly all cells transfected with vAcWT or vAc83:HA displayed fluorescence. However, the number of vAc83KO-transfected cells displaying fluorescence did not increase from 24 to 72 h p.t. (Fig. 3B). Light microscopy revealed no differences in OB formation in any of the three viruses up to 48 h p.t. However, by 96 h p.t., most of the cells transfected with vAcWT or vAc83:HA contained OBs, whereas the number of vAc83KO-transfected cells containing OBs did not increase (Fig. 3B). These results suggested that ac83 is essential for the production of infectious BVs. The deletion of ac83 did not affect the progression of infection into the very late phase, as evidenced by the formation of OBs in vAc83KO-transfected cells.
To further assess the effect of ac83 deletion on virus replication, virus growth curve analysis was performed. Sf9 cells were transfected with vAc83KO, vAc83:HA, or vAcWT individually; the supernatants were collected at the time points indicated in Fig. 3C; and the BV titers were determined using a TCID50 endpoint dilution assay. No BV titers could be detected in the vAc83KO-transfected cells at any time p.t., indicating that no infectious BVs were produced (Fig. 3C). In contrast, Sf9 cells transfected with vAc83:HA or vAcWT displayed normal and comparable increases in BV titers (Fig. 3C). These results demonstrated that deletion of ac83 led to a defect in infectious BV production.
In order to determine whether noninfectious BVs were produced in vAc83KO-transfected Sf9 cells, Western blotting was performed to compare the levels of the major capsid protein VP39 in the supernatants of the cells transfected with each of the bacmids. As shown in Fig. 3D, VP39 was detected in the extracts of cells transfected with vAc83KO, vAc83:HA, or vAcWT, indicating that ac83 deletion did not affect vp39 expression. However, VP39 was detected in the supernatants of vAc83:HA- and vAcWT-transfected cells only. No band was detected in the supernatants of Sf9 cells transfected with vAc83KO (Fig. 3D), indicating that ac83 deletion resulted in a defect in BV production.
ac83 is not required for viral DNA replication.
To determine whether ac83 deletion affects viral DNA replication, qPCR was performed. A 38K-knockout AcMNPV (vAc38kKO), in which the 38K gene was replaced with a Cm gene cassette, was used as a noninfectious control, because vAc38kKO is unable to spread infection from cell to cell but does not affect viral DNA replication (34). Total cellular DNA was isolated from cells transfected with vAc83KO or vAc38kKO at 12, 24, 48, and 72 h p.t. and was treated with DpnI to eliminate input bacmid DNA. The amount of viral DNA replication in the vAc83KO-transfected Sf9 cells was comparable to that in vAc38kKO-transfected cells (Fig. 4), indicating that ac83 deletion did not affect viral DNA synthesis.
Fig 4.
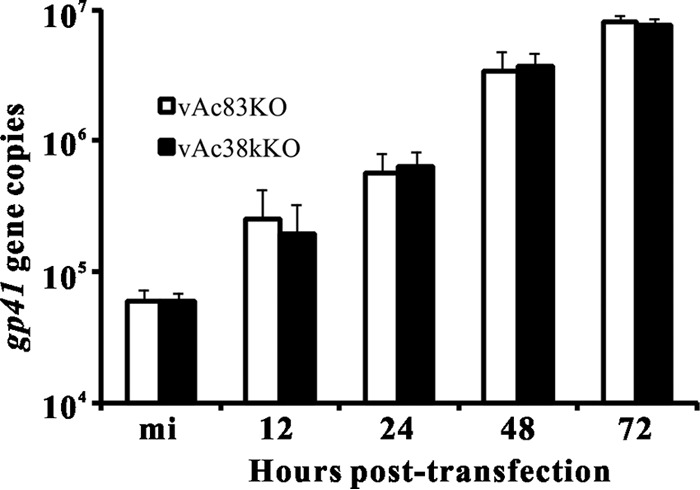
Real-time PCR analysis of viral DNA synthesis. Sf9 cells were transfected in triplicate with vAc83KO or vAc38kKO. At the designated time points p.t., total intracellular DNA was isolated, digested with the restriction enzyme DpnI to eliminate input bacmid DNA, and analyzed by real-time PCR. The results are averages for three independent replication assays. Error bars indicate standard deviations.
ac83 is essential for nucleocapsid assembly.
To determine the effect of ac83 deletion on virion morphogenesis, thin sections of vAcWT-, vAc83KO-, and vAc83:HA-transfected Sf9 cells at 60 h p.t. were observed with an electron microscope. As expected, cells transfected with vAc83:HA displayed characteristics similar to those of vAcWT-transfected cells, as demonstrated by the formation of a net-shaped virogenic stroma interspersed with rod-shaped electron-dense nucleocapsids (Fig. 5A and B) and by multiply enveloped mature nucleocapsids within the ring zone or embedded in the developing polyhedra (Fig. 5C). However, in vAc83KO-transfected cells, although a typical VS and virus-induced microvesicles could be formed (Fig. 5D and E), masses of elongated, rod-shaped electron-lucent nucleocapsid-like structures were present in the ring zone and adjacent to the inner nuclear membrane (Fig. 5F). These structures seemed to lack an electron-dense core indicative of nucleoprotein that contains viral DNA, suggesting that nucleocapsid assembly might be interrupted. A large number of circular electron-dense bodies appeared near the VS stromal matte (Fig. 5G); these are often observed in cells with abnormal nucleocapsids (44). In addition, although most vAc83KO-transfected cells contained elongated electron-lucent structures that were apart from the virogenic stroma and localized to the inner nuclear membrane, these aberrant structures could occasionally be observed at the electron-dense edges of the virogenic stroma in a few vAc83KO-transfected cells (Fig. 5H). No virions were embedded in the polyhedra in the vAc83KO-transfected cells (Fig. 5I). Taken together, these observations indicated that deletion of ac83 affected nucleocapsid assembly.
Fig 5.

Electron microscopy of Sf9 cells transfected with vAc83KO or vAc83:HA. (A to C) Cells transfected with vAc83:HA. (A) Portion of a cell showing the virogenic stroma (VS), polyhedra (P), and nuclear membrane (nm) (arrow). (B) Rod-shaped nucleocapsids (arrow) within the electron-dense edges of the VS. (C) Multiply enveloped mature ODVs (arrow) and polyhedra with ODVs embedded. (D to I) Cells transfected with vAc83KO. (D) VS. (E) Portion of the cell region showing microvesicles (arrows) in the nucleus. (F) Cluster of elongated, electron-lucent tubular structures that localized near the inner nuclear membrane (arrows). (G) VS devoid of nucleocapsids and exhibiting electron-dense bodies (arrows). (H) Nucleus with aberrant tubular structures (arrows) within the VS. (I) Polyhedra devoid of embedded virions.
Expression of Ac83 aa 451 to 600 is sufficient to rescue infectious BV production.
AcMNPV ac83 encodes a polypeptide of 847 aa residues with a predicted molecular mass of approximately 96 kDa. To define the functional domains of Ac83 required for infectious BV production, a gene truncation strategy generating N- and C-terminal deletion variants of Ac83 was implemented. A set of fragments encoding both N- and C-terminally truncated variants of Ac83 was produced by using the overlap PCR technique, cloned into the donor plasmid pFB1-PH-GFP, and then inserted into bAc83KO. The abilities of the ac83-truncated variants to rescue infectious BV production were monitored by fluorescence microscopy and were determined by a TCID50 endpoint dilution assay. As shown in Fig. 6A and B, although protein database mining demonstrated that Ac83 contains a putative INM-SM, a CBD, and a PR region (Fig. 1), none of these play a role in infectious BV production, as evidenced by the fact that similar growth kinetics were found for vAc83:delSM, vAc83:delCBD, vAc83:delPR, and vAc83:HA. In addition, the deletion of the N-terminal 290 aa residues of Ac83 (vAc83:del2-290) had no effect on infectious BV production, as evidenced by similar growth kinetics for vAc83:del2-290 and vAc83:HA. Further truncation of Ac83 up to the N-terminal 450 aa residues (vAc83:del2-450) still resulted in almost complete rescue of infectious BV production. At 96 h p.i., the BV titer of vAc83:del2-450 was only 4-fold lower than that of vAc83:HA. C-terminal truncation of Ac83 revealed that the deletion of Ac83 aa 742 to 847 (vAc83:del742-847) had no significant effect on infectious BV production. After the removal of Ac83 aa 601 to 847 (vAc83:del601-847), the virus propagated, although the ability to produce progeny virus was attenuated to some extent: the titer at 96 h p.i. was approximately 100-fold lower than that of vAc83:HA. An Ac83 construct with a further truncation, of Ac83 aa 551 to 847 (vAc83:del551-847), completely failed to rescue infectious BV production. Finally, unexpectedly, a construct from which both the N-terminal amino acid residues 2 to 450 and the C-terminal amino acid residues 601 to 847 of Ac83 were removed (vAc83:del2-450/601-847) still showed virus propagation, although the ability of this truncation form of Ac83 to rescue virus production was severely compromised. The expression of truncated Ac83 was examined by Western blotting and exhibited a level comparable to that of full-length Ac83 (data not shown). In the viruses with C-terminal truncations (vAc83:del742-847, vAc83:del601-847, vAc83:del551-847, and vAc83:del2-450/601-847), there are no overlaps between the repaired fragments and the remaining 3′-terminal sequences of ac83 in the deletion locus, so intragenomic recombination would be impossible. Furthermore, no intragenomic recombination was detected in other Ac83 truncation variants (vAc83:delSM, vAc83:delCBD, vAc83:delPR, vAc83:del2-290, and vAc83:del2-450) by PCR analysis (data not shown). Taken together, these results demonstrate that the most important region of Ac83 for infectious BV production is located between Ac83 aa 451 and 600.
Fig 6.

Expression of Ac83 aa 451 to 600 is sufficient to rescue infectious BV production. (A) Abilities of different Ac83 truncation viruses to rescue infectious BV production. Sf9 cells were transfected with the indicated bacmid, and virus propagation was monitored by fluorescence microscopy at the indicated time points p.t. (B) One-step growth curves of the bAc83KO genome repaired with a virus from which the INM-SM, the CBD, or the PR region of Ac83 was deleted (a) or with a virus in which Ac83 was N-terminally truncated (b), C-terminally truncated (c), or both N- and C-terminally truncated (d). Sf9 cells were infected with the indicated viruses at an MOI of 0.1 TCID50/cell, and at the designated time points p.i., the supernatants were harvested and were analyzed for the release of infectious BVs by a TCID50 endpoint dilution assay. Data points are averages of titers derived from three independent infections. Error bars represent standard deviations.
The CBD of Ac83 is essential for the efficient establishment of per os infection.
Ac83 contains a typical type II CBD that possibly interacts with the chitin component of insects and has been postulated to play a role in baculovirus primary infection (17). A recent study found that Ac83 was a component of the PIF complex on the ODV envelope, which further suggested that Ac83 might play a role in baculovirus per os infection (19). To determine the in vivo effect of deletion of the Ac83 CBD, BVs and OBs of vAc83:delCBD, vAc83:HA, and vAcWT from transfected Sf9 cells were bioassayed in 4th-instar T. ni larvae. Intrahemocoelic injection of the vAc83:delCBD BV supernatant into larvae at 5 TCID50s/larva resulted in a mortality rate similar to that with vAc83:HA or vAcWT (Table 2). Similar results were obtained at doses of 10 or 50 TCID50s/larva (data not shown), suggesting that deletion of the Ac83 CBD did not affect BV infectivity. However, bioassays with OBs revealed significant differences in oral infectivity between the three viruses. OBs of vAc83:HA and vAcWT produced similar mortality rates, and no significant difference in the median lethal dose (LD50) was observed (Table 2). In contrast, feeding the larvae with as many as 1 × 104 OBs of vAc83:delCBD caused almost no mortality, and only 58.3% mortality was achieved when the larvae were administered a high dose of 5 × 106 OBs/larva (Table 2), suggesting that deletion of the Ac83 CBD severely attenuated the oral infectivity of AcMNPV.
Table 2.
Infectivities of vAcWT, vAc83:HA, and vAc83:delCBD in 4th-instar T. ni larvaea and the effect of calcofluor white on the infectivity of vAc83:delCBD
| Inoculation method and virus or control | Dosage | Mortality (%) by 6 days p.i. | LD50b (95% confidence interval) |
|---|---|---|---|
| Injection | |||
| vAcWT | 5 TCID50s | 100 | |
| vAc83:HA | 5 TCID50s | 100 | |
| vAc83:delCBD | 5 TCID50s | 100 | |
| H2O | 4.1c | ||
| Per os | |||
| vAc83:delCBD | 1 × 104 OBs | 8.3 | |
| 5 × 106 OBs | 58.6 | ||
| H2O | 0 | ||
| vAcWT | 123.2 (92.5–164.1) A | ||
| vAc83:HA | 144.9 (108.3–194.1) A | ||
| vAc83:delCBD + calcofluor white | 1.5 × 105 (7.9 × 104-2.7 × 105) B |
A total of 24 larvae were used for each treatment.
Expressed as OBs/larva. Values followed by different capital letters are significantly different from each other.
Larval death not due to virus infection (microscopy and PCR analysis).
Since deletion of the Ac83 CBD severely compromised oral infectivity, it is possible that any changes in OB morphogenesis upon the deletion of the Ac83 CBD might account for the defect. Thus, OB formation was observed using electron microscopy. Mature OBs containing ODVs were observed in both vAc83:delCBD- and vAc83:HA-infected Sf9 cells and were morphologically indistinguishable (Fig. 7).
Fig 7.
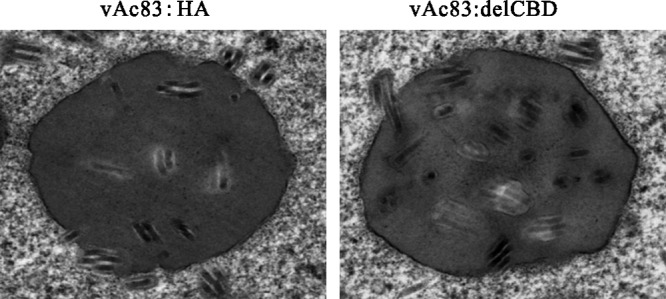
Electron microscopy of Sf9 cells infected with vAc83:HA or vAc83:delCBD. Sf9 cells were infected with vAc83:HA or vAc83:delCBD at an MOI of 5 TCID50s/cell. At 60 h p.t., cells were prepared for electron microscopy analysis.
Previous studies have shown that disruption of the PM enhances larval susceptibility to baculovirus infection (39), and some baculoviruses encode a kind of protease called enhancin, which disrupts the PM (45). Moreover, the chemical reagent calcofluor white is able to bind chitin so as to disrupt the PM, thus enhancing baculovirus infectivity (39). Considering the presence of the CBD, Ac83 may play a role in the disruption of the PM to facilitate the passage of ODVs. If so, disruption of the PM should enhance the oral infectivity of vAc83:delCBD. Therefore, bioassays of vAc83:delCBD in the presence of calcofluor white were performed. Larvae treated with calcofluor white displayed higher susceptibility than untreated larvae (data not shown). The LD50 of vAc83:delCBD in calcofluor white-treated 4th-instar T. ni larvae was 1.5 × 105 OBs/larva. However, the calcofluor white treatment did not completely rescue the oral infectivity of vAc83:delCBD; the LD50 of vAc83:delCBD in insects treated with calcofluor white was still approximately 3 log units higher than that of vAcWT (123.2 OBs/larva) or vAc83:HA (144.9 OBs/larva) (Table 2). Therefore, the bioassay results suggested that the Ac83 CBD might have other targets in addition to the PM, such as microvilli.
Ac83 localizes specifically to the ODV envelope.
Previous results concerning the localization profile of Ac83 and its homologs in virions were ambiguous. Although OpP91 has been demonstrated to be a component of both the BVs and the ODVs of Orgyia pseudotsugata NPV and localized predominantly to capsids, proteomic analyses of the BVs and ODVs of AcMNPV and Helicoverpa armigera NPV suggested that Ac83 and VP91 (the Helicoverpa armigera NPV homolog) belonged to the ODV envelope-specific component (23, 25, 46). In the present study, BVs and ODVs purified from vAc83:HA-infected Sf9 cells were analyzed by Western blotting to determine the localization of Ac83. As shown in Fig. 8A, Ac83 is specifically localized to the purified AcMNPV ODVs but not to BVs. The purified ODVs were further fractionated into envelope and nucleocapsid fractions, and Ac83 was detected in the ODV envelope fraction. The major capsid protein VP39 and the BV/ODV envelope-associated protein ODV-E25, used as controls, could be detected only in the expected fractions (Fig. 8B). Thus, these results demonstrated that Ac83 is an ODV envelope-specific protein, a finding consistent with previous proteomic results (23, 24).
Fig 8.
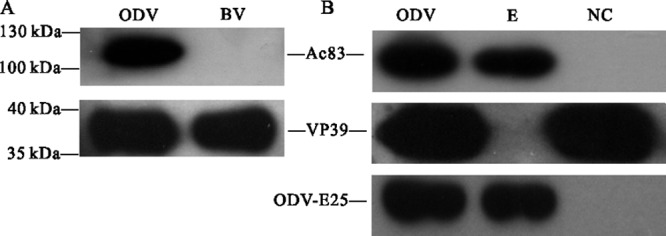
Localization of Ac83 in purified and fractionated virions. BVs and ODVs were purified from vAc83:HA-infected Sf9 cells, and the purified ODVs were further fractionated into envelope (E) and nucleocapsid (NC) fractions. Protein samples were subjected to Western blotting in order to determine the localization profile of Ac83 in purified BVs and ODVs (A) or fractionated ODVs (B). Proteins were detected by immunoblotting with an anti-HA antibody to detect HA-tagged Ac83, with anti-ODV-E25 to detect the BV/ODV envelope-associated protein ODV-E25, and with an anti-VP39 antibody to detect the major capsid protein VP39.
Ac83 contains a functional INM-SM.
It has been shown previously that the N-terminal 23 aa of ODV-E66, one of the ODV envelope proteins of AcMNPV, are sufficient to traffic fusion proteins to intranuclear membranes and the ODV envelope, and this sequence has been termed an INM-SM (47, 48). A typical INM-SM contains two features: (i) a hydrophobic domain of approximately 18 aa and (ii) a positively charged amino acid within aa 4 to 8 from the end of the hydrophobic domain that is positioned on the cytoplasmic or nucleoplasmic side of the membrane (48). The INM-SM is found not only in several ODV proteins but also in many inner nuclear envelope proteins of eukaryotes (47, 49). Ac83 has been predicted to have an INM-SM similar to that of ODV-E66 (Fig. 9A) (50). To investigate whether the INM-SM of Ac83 is functional, Ac83 INM-SM-GFP fusion proteins were monitored to determine their localization in relation to the absence or presence of viral infection. Sf9 cells either were transfected with pIB-SM-GFP or were infected with vAcSM-GFP at an MOI of 5 TCID50s/cell. When the GFP fusion protein was transiently expressed in Sf9 cells, it localized almost exclusively in the cytoplasm and formed a rim around the nucleus (Fig. 9B). In the context of infection, GFP fluorescence accumulated at the periphery of the nucleus and formed large foci near the INM (Fig. 9B). The localization pattern of the chimeric INM-SM-GFP was very similar to that of the well-characterized INM-SM of ODV-E66 (47, 49).
Fig 9.

Ac83 contains a functional inner nuclear membrane sorting motif and is localized to the nuclear periphery during infection. (A) Schematic representation of two characteristics of the INM-SM of Ac83. The highly hydrophobic TM is boxed and shown in red. The conserved positively charged amino acids are also shown in red and are marked with stars. (B and C) Confocal microscopy. (B) Sf9 cells were transfected with pIB-SM-GFP (top) or were infected with vAcSM-GFP (bottom) at an MOI of 5 TCID50s/cell. At 24 h p.t. or 48 h p.i., the cells were prepared for monitoring of the localization profile of GFP (green). (C) Sf9 cells were infected with vAc83:HA at an MOI of 5 TCID50s/cell. At 48 h p.i., the cells were fixed, probed with a mouse monoclonal anti-HA antibody to detect HA-tagged Ac83, and visualized using Alexa Fluor 647-conjugated goat anti-mouse IgG (red). The cells were stained with DAPI to directly visualize nuclear DNA (blue). BF, bright-field microscopy.
The subcellular localization pattern of Ac83 was also examined in vAc83:HA-infected Sf9 cells. The HA-tagged Ac83 was probed with a mouse monoclonal antibody against HA, visualized by Alexa Fluor 647-conjugated goat anti-mouse IgG, and examined using laser scanning confocal microscopy. Discrete foci with red fluorescence appeared near the INM at 24 h p.i. and became concentrated at 48 h p.i. (Fig. 9C). By 72 h p.i., the concentration of fluorescence in the nucleus was more pronounced, as larger foci formed around the periphery of the nucleus (data not shown). Taken together, the results described above suggested that Ac83 contains a functional INM-SM that guides it to anchor the INM and the ODV envelope.
DISCUSSION
ac83 is one of the 37 baculovirus core genes, but its role in the AcMNPV life cycle is unknown. Here we demonstrate that ac83 plays multiple roles in the baculovirus life cycle. ac83 is essential for AcMNPV nucleocapsid assembly and for efficient establishment of per os infection, thus providing a rationale for its essential role as a core gene.
To investigate the functional role of ac83 in the baculovirus life cycle, we generated an ac83-knockout virus and observed that ac83 was essential for nucleocapsid assembly (Fig. 5). In vAc83KO-transfected Sf9 cells, a mass of abnormal electron-lucent tubular structures was observed in the ring zone or adjacent to the inner nuclear membrane; however, deletion of ac83 did not affect viral DNA replication (Fig. 5). The results were entirely consistent with the most recent study on BmP95 (20).
Truncation analysis revealed that expression of Ac83 aa 451 to 600, which constitute approximately one-fifth of full-length (847-aa) Ac83, resulted in the formation of infectious BVs, although production was severely compromised (Fig. 6). It has been observed that repair of the N-terminal 500 aa of BmP95 could not rescue the function of BmP95 (20). Similarly, in our study, repair of the N-terminal 550 aa of Ac83, corresponding to the truncation of Ac83 aa 551 to 847 (vAc83:del551-847), completely failed to rescue infectious BV production (Fig. 6). These results indicated that the functional domains required for infectious BV production reside in Ac83 aa 451 to 600; while the other parts of Ac83 are not strictly essential, their presence contributes significantly to the fitness of the virus.
Examination of Ac83 aa 451 to 600 revealed a potential nuclear localization signal (NLS), 597KRPK600 (data not shown), which may function in the nuclear transport of Ac83. However, repair of Ac83 aa 451 to 596 (disruption of both the INM-SM and the predicted NLS) still resulted in infectious BV production (data not shown), implying that there are unidentified signals targeting this fragment of Ac83 to the nucleus.
Baculoviruses initiate infection through an oral infection route when the insects consume food contaminated with OBs. ODVs are released from OBs under the alkaline conditions of the midguts of larvae and carry with them a battery of proteins, named PIFs, to infect the midgut epithelia of host larvae. Before the infection of epithelia, ODVs encounter a barrier: the PM. A few lepidopteran NPVs and GVs encode a class of metalloproteinases called enhancins, which degrade the mucin component of the PM (45). However, it is not clear how baculoviruses without enhancins pass through the PM. For example, though highly infectious, AcMNPV does not encode an enhancin (17). The PM is not the functional target of identified PIFs (51). However, the PMs of T. ni larvae were degraded upon inoculation of either wild-type AcMNPV or AcMNPV with p74 deleted (10). Thus, there must be unidentified proteins in AcMNPV that play pivotal roles in the disruption of the PM or the passage of ODVs through the PM. In this study, we observed that deletion of the Ac83 CBD resulted in the failure of per os infection of T. ni with AcMNPV.
Chitin is generally essential for the function of chitin/protein structures (52). Interference with the chitin component of the PM often leads to a defect in the integrity and normal function of the PM (53, 54). Calcofluor white, a chemical reagent with chitin-binding properties, blocks PM formation, disassociates the PM by acting as a chitin-binding competitor, and concomitantly causes the loss of protection of the insect from baculovirus infection (39). Thus, targeting of the chitin component by using a chitin-binding reagent promises to be an ideal strategy for blocking PM protein formation (55). AcMNPV encodes four proteins with the potential to bind chitin: Ac83, Ac145, Ac150, and Ac64 (GP37) (17). Although Ac145 and Ac150 contain CBDs, these proteins have not been detected in the PIF complex (19). Ac64 (GP37), which resembles the entomopoxvirus protein spheroidin, has been shown to be associated with polyhedra but not with ODVs and is not essential for baculovirus replication in vivo or in vitro (56, 57). Ac83 contains a type II CBD and may possess the ability to bind chitin. Unfortunately, we did not observe any chitin-binding ability of Ac83 in vitro (data not shown). Nevertheless, because deletion of the Ac83 CBD resulted in the failure of per os AcMNPV infection and because Ac83 is a component of the PIF complex on the ODV envelope, we logically speculate that the affinity of Ac83 for chitin could possibly disrupt the spatial structure and, concomitantly, the integrity of the PM, thus facilitating the passage of ODVs. In accordance with this hypothesis, bioassays indicated that disruption of the PM with calcofluor white could partially rescue the oral infectivity of vAc83:delCBD, suggesting that the PM is a functional target of Ac83 (Table 2).
GVs have a set of 23 specific genes that are not present in NPVs (58), and one remarkable characteristic of GVs is the universal presence of metalloproteinase homologs in their genomes (17). One of these metalloproteinases is a stromelysin-1-like metalloproteinase encoded by a GV-specific gene (17). Stromelysin-1 is an enzyme that is involved in the breakdown of the extracellular matrix and in tissue remodeling (59, 60). Thus, the metalloproteinase homologs in GVs may facilitate the passage of GVs through the PM and may explain why the Ac83 CBD is conserved in NPVs but is poorly conserved in GVs (Fig. 1).
It is currently unclear whether ODVs diffuse randomly to access the midgut epithelium. Previous studies have demonstrated that infected midgut cells are unstable and frequently are sloughed into the gut lumen, an effective strategy used by lepidopteran hosts to defend against virus infection (61). In addition, midgut cells are also prone to apoptosis during development (62). Therefore, ODVs must establish infection efficiently and effectively, because the infection of target cells is a race against time. Not only is chitin a component of the PM; it is also associated with midgut epithelial cells. The chitin component of the PM is produced by chitin synthase, a plasma membrane-embedded enzyme located at the apical tips of brush border microvilli (17). Therefore, an affinity of Ac83 for the chitin of the midgut epithelial cells may offer evolutionary advantages by facilitating the passage of ODVs through the PM or the interaction of ODVs with the insect epithelial cells, possibilities consistent with our bioassay results. Another ODV envelope protein, ODV-E66, may also have a function similar to that of Ac83. ODV-E66 has been shown to play an important role in oral infection with AcMNPV (63). ODV-E66 encodes a novel chondroitinase that efficiently digests chondroitin and has been postulated to play a role in viral infection of the epithelial cells or in the disintegration of the infected host (64). Therefore, from an evolutionary viewpoint, we do not suppose random diffusion is utilized by baculovirus ODVs in the process of infecting midgut epithelial cells, because it is not an efficient mechanism for the rapid establishment of a primary infection. In view of the roles of Ac83 and ODV-E66 in the per os infection process of AcMNPV, we hypothesize that Ac83 and ODV-E66 may represent a class of proteins that facilitate the efficient establishment of infection with ODVs in the insect midgut.
Perusal of the N-terminal sequences of Ac83 homologs revealed a C2HC-type zinc finger that was conserved in all Ac83 homologs (Fig. 1). The C2HC-type zinc finger was first identified in myelin transcription factor I, a transcription factor that binds to the cis-regulatory elements and thus regulates the expression of a gene specific to glia, the myelin proteolipid protein gene (65). Subsequently, it was found that the C2HC-type zinc finger could be directly involved in protein-protein interactions (41). Interestingly, protein mining using Uniprot or ExPASy identified two type II CBDs in Ac83, the first of which overlaps with the predicted zinc finger of Ac83 (data not shown). Analysis indicated that the first type II CBD identified is incomplete compared with the second one (data not shown). Considering that the zinc finger motif and type II CBDs are both rich in cysteine, we hypothesized that the first type II CBD is a putative zinc finger motif. Most importantly, sequence analysis of other components of the PIF complex revealed that nearly all of them potentially contain one or more types of zinc finger (data not shown). In addition, Ac83 also contains a PR region that, while it plays no role in infectious BV production (Fig. 6), may function in protein-protein interactions, such as the interaction of Ac83 with other PIF components. Considering that the PIF complex components are possibly arranged in a noncovalent manner (66), it is intriguing to hypothesize that the putative C2HC zinc finger motif, together with the PR region, may play a pivotal role in the association of Ac83 with other PIF complex components.
In summary, it is intriguing to speculate that Ac83 may represent another type of PIF that not only participates in per os infection but may also perform other pivotal roles in the baculovirus life cycle. To the best of our knowledge, Ac83 is the first baculovirus protein that functions both in nucleocapsid assembly and in per os infectivity.
ACKNOWLEDGMENTS
This research was supported by the National Basic Research Program of China (973 Program; grant 2009CB118903), the National Nature Science Foundation of China (grant 30900941), the Hi-Tech Research and Development Program of China (863 Program; grant 2011AA10A204), and the Fundamental Research Funds for the Central Universities (grant 09lgpy39).
Footnotes
Published ahead of print 17 July 2013
REFERENCES
- 1.Herniou EA, Olszewski JA, Cory JS, O'Reilly DR. 2003. The genome sequence and evolution of baculoviruses. Annu. Rev. Entomol. 48:211–234 [DOI] [PubMed] [Google Scholar]
- 2.Jehle JA, Blissard GW, Bonning BC, Cory JS, Herniou EA, Rohrmann GF, Theilmann DA, Thiem SM, Vlak JM. 2006. On the classification and nomenclature of baculoviruses: a proposal for revision. Arch. Virol. 151:1257–1266 [DOI] [PubMed] [Google Scholar]
- 3.Zanotto PM, Kessing BD, Maruniak JE. 1993. Phylogenetic interrelationships among baculoviruses: evolutionary rates and host associations. J. Invertebr. Pathol. 62:147–164 [DOI] [PubMed] [Google Scholar]
- 4.Lung O, Westenberg M, Vlak JM, Zuidema D, Blissard GW. 2002. Pseudotyping Autographa californica multicapsid nucleopolyhedrovirus (AcMNPV): F proteins from group II NPVs are functionally analogous to AcMNPV GP64. J. Virol. 76:5729–5736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ayres MD, Howard SC, Kuzio J, Lopez-Ferber M, Possee RD. 1994. The complete DNA sequence of Autographa californica nuclear polyhedrosis virus. Virology 202:586–605 [DOI] [PubMed] [Google Scholar]
- 6.Yuan M, Huang Z, Wei D, Hu Z, Yang K, Pang Y. 2011. Identification of Autographa californica nucleopolyhedrovirus ac93 as a core gene and its requirement for intranuclear microvesicle formation and nuclear egress of nucleocapsids. J. Virol. 85:11664–11674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Garavaglia MJ, Miele SA, Iserte JA, Belaich MN, Ghiringhelli PD. 2012. The ac53, ac78, ac101, and ac103 genes are newly discovered core genes in the family Baculoviridae. J. Virol. 86:12069–12079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Braunagel SC, Summers MD. 1994. Autographa californica nuclear polyhedrosis virus, PDV, and ECV viral envelopes and nucleocapsids: structural proteins, antigens, lipid and fatty acid profiles. Virology 202:315–328 [DOI] [PubMed] [Google Scholar]
- 9.Slack J, Arif BM. 2007. The baculoviruses occlusion-derived virus: virion structure and function. Adv. Virus Res. 69:99–165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Faulkner P, Kuzio J, Williams GV, Wilson JA. 1997. Analysis of p74, a PDV envelope protein of Autographa californica nucleopolyhedrovirus required for occlusion body infectivity in vivo. J. Gen. Virol. 78(Part 12):3091–3100 [DOI] [PubMed] [Google Scholar]
- 11.Kikhno I, Gutierrez S, Croizier L, Croizier G, Ferber ML. 2002. Characterization of pif, a gene required for the per os infectivity of Spodoptera littoralis nucleopolyhedrovirus. J. Gen. Virol. 83:3013–3022 [DOI] [PubMed] [Google Scholar]
- 12.Pijlman GP, Pruijssers AJ, Vlak JM. 2003. Identification of pif-2, a third conserved baculovirus gene required for per os infection of insects. J. Gen. Virol. 84:2041–2049 [DOI] [PubMed] [Google Scholar]
- 13.Fang M, Nie Y, Harris S, Erlandson MA, Theilmann DA. 2009. Autographa californica multiple nucleopolyhedrovirus core gene ac96 encodes a per os infectivity factor (pif-4). J. Virol. 83:12569–12578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ohkawa T, Washburn JO, Sitapara R, Sid E, Volkman LE. 2005. Specific binding of Autographa californica M nucleopolyhedrovirus occlusion-derived virus to midgut cells of Heliothis virescens larvae is mediated by products of pif genes Ac119 and Ac022 but not by Ac115. J. Virol. 79:15258–15264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Harrison RL, Sparks WO, Bonning BC. 2010. Autographa californica multiple nucleopolyhedrovirus ODV-E56 envelope protein is required for oral infectivity and can be substituted functionally by Rachiplusia ou multiple nucleopolyhedrovirus ODV-E56. J. Gen. Virol. 91:1173–1182 [DOI] [PubMed] [Google Scholar]
- 16.Nie Y, Fang M, Erlandson MA, Theilmann DA. 2012. Analysis of the Autographa californica multiple nucleopolyhedrovirus overlapping gene pair lef3 and ac68 reveals that AC68 is a per os infectivity factor and that LEF3 is critical, but not essential, for virus replication. J. Virol. 86:3985–3994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rohrmann GF. 2011. Baculovirus molecular biology. U.S. National Library of Medicine, National Center for Biotechnology Information, Bethesda, MD [Google Scholar]
- 18.Simon O, Palma L, Williams T, Lopez-Ferber M, Caballero P. 2012. Analysis of a naturally-occurring deletion mutant of Spodoptera frugiperda multiple nucleopolyhedrovirus reveals sf58 as a new per os infectivity factor of lepidopteran-infecting baculoviruses. J. Invertebr. Pathol. 109:117–126 [DOI] [PubMed] [Google Scholar]
- 19.Peng K, van Lent JW, Boeren S, Fang M, Theilmann DA, Erlandson MA, Vlak JM, van Oers MM. 2012. Characterization of novel components of the baculovirus per os infectivity factor complex. J. Virol. 86:4981–4988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Xiang X, Shen Y, Yang R, Chen L, Hu X, Wu X. 27 March 2013. Bombyx mori nucleopolyhedrovirus BmP95 plays an essential role in budded virus production and nucleocapsid assembly. J. Gen. Virol. 10.1099/vir.0.050583-0 [DOI] [PubMed] [Google Scholar]
- 21.Tellam RL, Wijffels G, Willadsen P. 1999. Peritrophic matrix proteins. Insect Biochem. Mol. Biol. 29:87–101 [DOI] [PubMed] [Google Scholar]
- 22.Aguinaldo AM, Turbeville JM, Linford LS, Rivera MC, Garey JR, Raff RA, Lake JA. 1997. Evidence for a clade of nematodes, arthropods and other moulting animals. Nature 387:489–493 [DOI] [PubMed] [Google Scholar]
- 23.Braunagel SC, Russell WK, Rosas-Acosta G, Russell DH, Summers MD. 2003. Determination of the protein composition of the occlusion-derived virus of Autographa californica nucleopolyhedrovirus. Proc. Natl. Acad. Sci. U. S. A. 100:9797–9802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang R, Deng F, Hou D, Zhao Y, Guo L, Wang H, Hu Z. 2010. Proteomics of the Autographa californica nucleopolyhedrovirus budded virions. J. Virol. 84:7233–7242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Russell RL, Rohrmann GF. 1997. Characterization of P91, a protein associated with virions of an Orgyia pseudotsugata baculovirus. Virology 233:210–223 [DOI] [PubMed] [Google Scholar]
- 26.Ono C, Kamagata T, Taka H, Sahara K, Asano S, Bando H. 2012. Phenotypic grouping of 141 BmNPVs lacking viral gene sequences. Virus Res. 165:197–206 [DOI] [PubMed] [Google Scholar]
- 27.Marchler-Bauer A, Anderson JB, Cherukuri PF, DeWeese-Scott C, Geer LY, Gwadz M, He S, Hurwitz DI, Jackson JD, Ke Z, Lanczycki CJ, Liebert CA, Liu C, Lu F, Marchler GH, Mullokandov M, Shoemaker BA, Simonyan V, Song JS, Thiessen PA, Yamashita RA, Yin JJ, Zhang D, Bryant SH. 2005. CDD: a Conserved Domain Database for protein classification. Nucleic Acids Res. 33:D192–D196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Letunic I, Doerks T, Bork P. 2009. SMART 6: recent updates and new developments. Nucleic Acids Res. 37:D229–D232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Soding J. 2005. Protein homology detection by HMM-HMM comparison. Bioinformatics 21:951–960 [DOI] [PubMed] [Google Scholar]
- 30.Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, Thompson JD, Gibson TJ, Higgins DG. 2007. Clustal W and Clustal X version 2.0. Bioinformatics 23:2947–2948 [DOI] [PubMed] [Google Scholar]
- 31.Nicholas KB, Nicholas HB, Deerfield DW. 1997. GeneDoc: analysis and visualization of genetic variation. EMBNEW News 4:14 [Google Scholar]
- 32.Luckow VA, Lee SC, Barry GF, Olins PO. 1993. Efficient generation of infectious recombinant baculoviruses by site-specific transposon-mediated insertion of foreign genes into a baculovirus genome propagated in Escherichia coli. J. Virol. 67:4566–4579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.O'Reilly DR, Miller LK, Luckow VA. 1992. Baculovirus expression vectors: a laboratory manual. Oxford University Press, New York, NY [Google Scholar]
- 34.Wu W, Lin T, Pan L, Yu M, Li Z, Pang Y, Yang K. 2006. Autographa californica multiple nucleopolyhedrovirus nucleocapsid assembly is interrupted upon deletion of the 38K gene. J. Virol. 80:11475–11485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yuan M, Wu W, Liu C, Wang Y, Hu Z, Yang K, Pang Y. 2008. A highly conserved baculovirus gene p48 (ac103) is essential for BV production and ODV envelopment. Virology 379:87–96 [DOI] [PubMed] [Google Scholar]
- 36.Cai Y, Long Z, Qiu J, Yuan M, Li G, Yang K. 2012. An ac34 deletion mutant of Autographa californica nucleopolyhedrovirus exhibits delayed late gene expression and a lack of virulence in vivo. J. Virol. 86:10432–10443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Daimon T, Katsuma S, Iwanaga M, Kang W, Shimada T. 2005. The BmChi-h gene, a bacterial-type chitinase gene of Bombyx mori, encodes a functional exochitinase that plays a role in the chitin degradation during the molting process. Insect Biochem. Mol. Biol. 35:1112–1123 [DOI] [PubMed] [Google Scholar]
- 38.Li L, Li Z, Chen W, Pang Y. 2007. Cloning, expression of Autographa californica nucleopolyhedrovirus vp39 gene in Escherichia coli and preparation of its antibody. Biotechnology 17:5–7 [Google Scholar]
- 39.Wang P, Granados RR. 2000. Calcofluor disrupts the midgut defense system in insects. Insect Biochem. Mol. Biol. 30:135–143 [DOI] [PubMed] [Google Scholar]
- 40.Lu M, Swevers L, Iatrou K. 1998. The p95 gene of Bombyx mori nuclear polyhedrosis virus: temporal expression and functional properties. J. Virol. 72:4789–4797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Akhtar A, Becker PB. 2001. The histone H4 acetyltransferase MOF uses a C2HC zinc finger for substrate recognition. EMBO Rep. 2:113–118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Klug A, Rhodes D. 1987. Zinc fingers: a novel protein fold for nucleic acid recognition. Cold Spring Harbor Symp. Quant. Biol. 52:473–482 [DOI] [PubMed] [Google Scholar]
- 43.Chen YR, Zhong S, Fei Z, Hashimoto Y, Xiang JZ, Zhang S, Blissard GW. 2013. The transcriptome of the baculovirus Autographa californica multiple nucleopolyhedrovirus in Trichoplusia ni cells. J. Virol. 87:6391–6405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Olszewski J, Miller LK. 1997. Identification and characterization of a baculovirus structural protein, VP1054, required for nucleocapsid formation. J. Virol. 71:5040–5050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang P, Granados RR. 1997. An intestinal mucin is the target substrate for a baculovirus enhancin. Proc. Natl. Acad. Sci. U. S. A. 94:6977–6982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hou D, Zhang L, Deng F, Fang W, Wang R, Liu X, Guo L, Rayner S, Chen X, Wang H, Hu Z. 2013. Comparative proteomics reveal fundamental structural and functional differences between the two progeny phenotypes of a baculovirus. J. Virol. 87:829–839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hong T, Summers MD, Braunagel SC. 1997. N-terminal sequences from Autographa californica nuclear polyhedrosis virus envelope proteins ODV-E66 and ODV-E25 are sufficient to direct reporter proteins to the nuclear envelope, intranuclear microvesicles and the envelope of occlusion derived virus. Proc. Natl. Acad. Sci. U. S. A. 94:4050–4055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Saksena S, Shao Y, Braunagel SC, Summers MD, Johnson AE. 2004. Cotranslational integration and initial sorting at the endoplasmic reticulum translocon of proteins destined for the inner nuclear membrane. Proc. Natl. Acad. Sci. U. S. A. 101:12537–12542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Braunagel SC, Williamson ST, Saksena S, Zhong Z, Russell WK, Russell DH, Summers MD. 2004. Trafficking of ODV-E66 is mediated via a sorting motif and other viral proteins: facilitated trafficking to the inner nuclear membrane. Proc. Natl. Acad. Sci. U. S. A. 101:8372–8377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Braunagel SC, Summers MD. 2007. Molecular biology of the baculovirus occlusion-derived virus envelope. Curr. Drug Targets 8:1084–1095 [DOI] [PubMed] [Google Scholar]
- 51.Song J, Wang R, Deng F, Wang H, Hu Z. 2008. Functional studies of per os infectivity factors of Helicoverpa armigera single nucleocapsid nucleopolyhedrovirus. J. Gen. Virol. 89:2331–2338 [DOI] [PubMed] [Google Scholar]
- 52.Elorza MV, Rico H, Sentandreu R. 1983. Calcofluor white alters the assembly of chitin fibrils in Saccharomyces cerevisiae and Candida albicans cells. J. Gen. Microbiol. 129:1577–1582 [DOI] [PubMed] [Google Scholar]
- 53.Eisemann CH, Donaldson RA, Pearson RD, Cadogan LC, Vuocolo T, Tellam RL. 1994. Larvicidal activity of lectins on Lucilia cuprina: mechanism of action. Entomol. Exp. App. 72:1–10 [Google Scholar]
- 54.Harper MS, Hopkins TL, Czapla TH. 1998. Effect of wheat germ agglutinin on formation and structure of the peritrophic membrane in European corn borer (Ostrinia nubilalis) larvae. Tissue Cell 30:166–176 [DOI] [PubMed] [Google Scholar]
- 55.Wang P, Granados RR. 2001. Molecular structure of the peritrophic membrane (PM): identification of potential PM target sites for insect control. Arch. Insect Biochem. Physiol. 47:110–118 [DOI] [PubMed] [Google Scholar]
- 56.Vialard JE, Yuen L, Richardson CD. 1990. Identification and characterization of a baculovirus occlusion body glycoprotein which resembles spheroidin, an entomopoxvirus protein. J. Virol. 64:5804–5811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cheng X, Krell P, Arif B. 2001. P34.8 (GP37) is not essential for baculovirus replication. J. Gen. Virol. 82:299–305 [DOI] [PubMed] [Google Scholar]
- 58.Wormleaton S, Kuzio J, Winstanley D. 2003. The complete sequence of the Adoxophyes orana granulovirus genome. Virology 311:350–365 [DOI] [PubMed] [Google Scholar]
- 59.Sternlicht MD, Lochter A, Sympson CJ, Huey B, Rougier JP, Gray JW, Pinkel D, Bissell MJ, Werb Z. 1999. The stromal proteinase MMP3/stromelysin-1 promotes mammary carcinogenesis. Cell 98:137–146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ko R, Okano K, Maeda S. 2000. Structural and functional analysis of the Xestia c-nigrum granulovirus matrix metalloproteinase. J. Virol. 74:11240–11246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Volkman LE. 2007. Baculovirus infectivity and the actin cytoskeleton. Curr. Drug Targets 8:1075–1083 [DOI] [PubMed] [Google Scholar]
- 62.Uwo MF, Ui-Tei K, Park P, Takeda M. 2002. Replacement of midgut epithelium in the greater wax moth, Galleria mellonela, during larval-pupal moult. Cell Tissue Res. 308:319–331 [DOI] [PubMed] [Google Scholar]
- 63.Xiang X, Chen L, Hu X, Yu S, Yang R, Wu X. 2011. Autographa californica multiple nucleopolyhedrovirus odv-e66 is an essential gene required for oral infectivity. Virus Res. 158:72–78 [DOI] [PubMed] [Google Scholar]
- 64.Sugiura N, Setoyama Y, Chiba M, Kimata K, Watanabe H. 2011. Baculovirus envelope protein ODV-E66 is a novel chondroitinase with distinct substrate specificity. J. Biol. Chem. 286:29026–29034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kim JG, Hudson LD. 1992. Novel member of the zinc finger superfamily: a C2-HC finger that recognizes a glia-specific gene. Mol. Cell. Biol. 12:5632–5639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Peng K, van Oers MM, Hu Z, van Lent JW, Vlak JM. 2010. Baculovirus per os infectivity factors form a complex on the surface of occlusion-derived virus. J. Virol. 84:9497–9504 [DOI] [PMC free article] [PubMed] [Google Scholar]