

Abstract
The newly emerged Middle East respiratory syndrome coronavirus (MERS-CoV) has infected at least 77 people, with a fatality rate of more than 50%. Alarmingly, the virus demonstrates the capability of human-to-human transmission, raising the possibility of global spread and endangering world health and economy. Here we have identified the receptor-binding domain (RBD) from the MERS-CoV spike protein and determined its crystal structure. This study also presents a structural comparison of MERS-CoV RBD with other coronavirus RBDs, successfully positioning MERS-CoV on the landscape of coronavirus evolution and providing insights into receptor binding by MERS-CoV. Furthermore, we found that MERS-CoV RBD functions as an effective entry inhibitor of MERS-CoV. The identified MERS-CoV RBD may also serve as a potential candidate for MERS-CoV subunit vaccines. Overall, this study enhances our understanding of the evolution of coronavirus RBDs, provides insights into receptor recognition by MERS-CoV, and may help control the transmission of MERS-CoV in humans.
INTRODUCTION
Since the summer of 2012, a novel coronavirus, Middle East respiratory syndrome coronavirus (MERS-CoV), has emerged from the Middle East and spread to parts of Europe. MERS-CoV infection often leads to acute pneumonia and renal failure, and the human fatality rate is more than 50% (1, 2). To date, MERS-CoV has infected at least 77 people and was able to be transmitted from human to human. The genomic sequence of MERS-CoV is closely related to the sequences of certain bat coronaviruses (3–5), raising concerns over persistent bat-to-human cross-species transmission of the virus. The clinical signs and epidemic patterns of MERS-CoV are reminiscent of the severe acute respiratory syndrome coronavirus (SARS-CoV), the etiological agent of the worldwide SARS epidemic in 2002-2003 that infected more than 8,000 people with a ∼10% fatality rate (6, 7). MERS-CoV poses a significant threat to global health and economy.
Coronaviruses are enveloped and positive-stranded RNA viruses and can be divided into three major genera, α, β, and γ (8). They mainly cause respiratory, gastrointestinal, and central nervous system diseases in mammals and birds. Coronaviruses recognize a variety of host receptors. Human NL63 respiratory coronavirus (HCoV-NL63) from α-genus and SARS-CoV from β-genus both recognize angiotensin-converting enzyme 2 (ACE2) as their host receptor (9, 10). Porcine respiratory coronavirus (PRCV) and some other coronaviruses from α-genus recognize aminopeptidase N (APN) (11, 12). Mouse hepatitis coronavirus (MHV) from β-genus recognizes carcinoembryonic antigen-related cell adhesion molecule 1 (CEACAM1) (13, 14), although certain MHV strains also recognize heparan sulfate (15, 16). Some coronaviruses from each of the three genera recognize sugars (17–20). MERS-CoV belongs to the β-genus and uses human dipeptidyl peptidase 4 (DPP4) as its host receptor (21). Receptor recognition is a major determinant of coronavirus host range and tropism.
An envelope-anchored trimeric spike protein is responsible for coronavirus entry into host cells via binding to the host receptor and subsequently fusing viral and host membranes (22). The spike protein consists of a receptor-binding S1 subunit and a membrane fusion S2 subunit. The S1 subunit contains two independent domains, an N-terminal domain (NTD) and a C-domain, both of which can potentially function as receptor-binding domains (RBD) (Fig. 1A) (23). Specifically, coronavirus S1 C-domains can function as ACE2-, APN-, or heparan sulfate-binding RBDs, whereas S1 NTDs can function as CEACAM1- or sugar-binding RBDs. To date, crystal structures have been determined for a number of coronavirus RBDs by themselves or in complex with their host receptors, revealing how coronaviruses have evolved to recognize host receptors and thereby traffic between different species (24–28). It is not known which one of MERS-CoV S1 domains is the DPP4-binding RBD or how the tertiary structure of MERS-CoV RBD fits into the landscape of coronavirus evolution.
Fig 1.
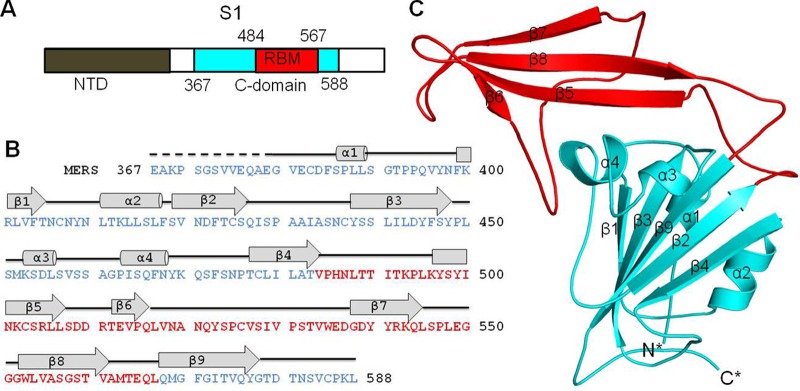
Crystal structure of the MERS-CoV S1 C-domain. (A) Domain structure of MERS-CoV S1 subunit that contains an N-terminal domain (NTD) and a C-domain. The boundaries of the C-domain and proposed receptor-binding motif (RBM) of MERS-CoV were determined by sequence and structural comparisons with the SARS-CoV S1 subunit (see Fig. 2). (B) Sequence and secondary structure of the MERS-CoV C-domain. Helices are drawn as cylinders, and strands are drawn as arrows. The disordered region is shown as a dashed line. (C) Crystal structure of the MERS-CoV C-domain. The core structure is in cyan, and the proposed RBM region is in red.
Here we have identified the MERS-CoV S1 C-domain as the RBD, characterized its interaction with human DPP4, and determined its crystal structure. This study provides structural insights into the evolution and receptor recognition of MERS-CoV. The identified MERS-CoV RBD also has therapeutic implications.
MATERIALS AND METHODS
Protein preparation and crystallization.
The MERS-CoV S1 C-domain (residues 367 to 588) was expressed and purified as previously described for the SARS-CoV C-domain (24, 29, 30). Briefly, the MERS-CoV C-domain containing an N-terminal honeybee melittin signal peptide and a C-terminal His6 tag was expressed in insect cells using the Bac-to-Bac expression system (Invitrogen), secreted into cell culture medium, and subsequently purified on a nickel-nitrilotriacetic acid (Ni-NTA) affinity column and a Superdex 200 gel filtration column (GE Healthcare). The protein was concentrated to 10 mg/ml and stored in buffer containing 20 mM Tris (pH 7.4) and 200 mM NaCl. Crystallization of the MERS-CoV C-domain was set up using the sitting drop vapor diffusion method, with 2 μl protein solution added to 2 μl reservoir buffer containing 27% (vol/vol) polyethylene glycol (PEG) 3350, 0.2 M MgCl2, and 0.1 M bis-Tris (pH 7.0) at 20°C. After 2 weeks, crystals of the MERS-CoV C-domain were harvested in buffer containing 25% (vol/vol) ethylene glycol, 27% (vol/vol) PEG 3350, 0.2 M MgCl2, and 0.1 M bis-Tris (pH 7.0) and flash-frozen in liquid nitrogen. Heavy-atom derivatives of MERS-CoV C-domain crystals were prepared by soaking crystals for 15 min in buffer containing 1 M NaI, 25% (vol/vol) ethylene glycol, 27% (vol/vol) PEG 3350, 0.2 M MgCl2, and 0.1 M bis-Tris (pH 7.0).
Data collection and structure determination.
Data were collected at the Advanced Photon Source beamline 24-ID-C at 1.0716 Å for native crystals and 1.4586 Å for iodine-derivatized crystals. The crystal structure was determined using SIRAS (single isomorphous replacement with anomalous signal). X-ray diffraction data were processed using HKL2000 (31). Sixteen iodine sites were identified with the program HYSS (32) followed by phase refinement and solvent flattening with RESOLVE (33). The model was built and refined with Refmac (34) at 2.13 Å to a final Rwork and Rfree of 14.9% and 20.6%, respectively. In the final model of the MERS-CoV C-domain, 98% of residues are in the favored regions of the Ramachandran plot, and 0% of residues are in the disallowed regions.
MERS-CoV infection assay.
Vero cells were incubated with different concentrations of recombinant MERS-CoV C-domain in minimum essential medium (GIBCO) for 1 h at 37°C. Medium containing the protein was removed, and the cells were infected with MERS-CoV at a multiplicity of infection (MOI) of 0.01 for 1 h at 37°C. Unbound viruses were washed with 1× phosphate-buffered saline (PBS), and the medium with MERS-CoV C-domain was added back to the cells. Supernatant was sampled at various time points, and the virus replication was determined by plaque assays on Vero cells as described previously (35).
Pseudotyped-virus infection assay.
Pseudotyped-virus infection was carried out using murine leukemia viruses (MLVs) expressing β-galactosidase and pseudotyped with MERS-CoV spike protein as described previously (36). Briefly, to prepare pseudotyped viruses, HEK293T cells were cotransfected with spike protein-encoding plasmid pcDNA3.1(+), p3240, expressing murine leukemia virus gag and pol genes, and a murine leukemia virus β-galactosidase-transducing vector, pBAG. At 48 h posttransfection, virus supernatants were harvested and concentrated 20 times using Amicon Ultra-4 centrifugal filter units (Millipore) with 100-kDa molecular mass cutoff. HEK293T cells transiently expressing human DPP4 in pcDNA3.1(+) were inoculated in 96-well plates by adding 10 μl of concentrated viral supernatant to 100 μl Dulbecco's modified Eagle medium (DMEM) per well. To examine the inhibition of MERS-CoV C-domain on infection, HEK293T cells transiently expressing human DPP4 were infected by pseudotyped virus in the presence of 20 μg/ml MERS-CoV C-domain or bovine serum albumin (BSA). Infection efficiency was quantified 48 h postinfection by measuring β-galactosidase activity. As a negative control, HEK293T cells were transfected with empty pcDNA3.1(+) plasmid, instead of a DPP4 insertion-containing plasmid. As another control, MLVs pseudotyped with vesicular stomatitis virus glycoprotein (VSV-G) were used to infect HEK293T cells transiently expressing human DPP4, in both the absence and presence of 20 μg/ml MERS-CoV C-domain protein or BSA.
DPP4 pulldown assay.
HEK293T cells were transfected with either empty pcDNA3.1(+) vector or pcDNA3.1(+) vector containing the human DPP4 gene. At 48 h posttransfection, cells were harvested and lysed in PBS with 300 mM NaCl and 0.25% SDS. Cell lysate was mixed with MERS-CoV C-domain. The protein complex was then precipitated with Ni-NTA agarose (Thermo Scientific). DPP4 and MERS-CoV C-domain contain a C-terminal hemagglutinin (HA) tag and His6 tag, respectively, and were detected by anti-HA and anti-His6 antibodies (Santa Cruz Biotechnology), respectively.
Protein structure accession number.
Coordinates and structure factors have been submitted to the PDB, accession number 4L3N.
RESULTS AND DISCUSSION
Identification and functional characterization of MERS-CoV RBD.
We designed a construct of the MERS-CoV S1 C-domain (residues 367 to 588), based on the sequence alignment of MERS-CoV and SARS-CoV S1 subunits (Fig. 2A). Although the two S1 subunits have low sequence similarity, sequence alignment was guided by conserved cysteines. Based on its crystal structure, SARS-CoV C-domain contains eight essential cysteines that form four disulfide bonds (24). Our designed MERS-CoV C-domain also contains eight cysteines, seven of which are conserved between SARS-CoV and MERS-CoV (Fig. 2A). The MERS-CoV C-domain was expressed and purified using the protocol that we used for the SARS-CoV C-domain (24, 29, 30). The protein was a monomer in solution, based on the gel filtration chromatography profile.
Fig 2.

Sequence and structural comparisons of MERS-CoV and SARS-CoV S1 C-domains. (A) Sequence alignment of MERS-CoV and SARS-CoV C-domains. Residues corresponding to C-domain core structures are in cyan, and residues corresponding to the proposed RBM region are in red. Cysteine residues are highlighted. Asterisks indicate positions that have fully conserved residues; colons indicate positions that have strongly conserved residues; periods indicate positions that have weakly conserved residues. (B) Crystal structure of MERS-CoV C-domain. The secondary structures are colored in the same way as the corresponding sequence in panel A. Disulfide bond-linked cysteine residues are shown as sticks in yellow, and N-linked glycans are shown as sticks in green. (C) Crystal structures of SARS-CoV C-domain (PDB 2AJF). Two disordered loops are drawn as dashed lines. Although cysteines 377 and 511 are disordered in this structure, they were shown to form a disulfide bond in another study (46).
We characterized the interactions between the recombinant MERS-CoV C-domain and human DPP4 on human cell surfaces, using three alternative approaches. First, the recombinant MERS-CoV C-domain inhibits MERS-CoV infection of permissive monkey cells (Fig. 3A). The inhibition efficiency increased with the concentration of MERS-CoV C-domain. At a 13 μM concentration, the MERS-CoV C-domain completely inhibits MERS-CoV infection. Second, the recombinant MERS-CoV C-domain inhibits the infection of MERS-CoV spike-pseudotyped retroviruses, but not VSV-G-pseudotyped retroviruses, in cells that express human DPP4 (Fig. 3B). Finally, the MERS-CoV C-domain pulled down human DPP4 from cells that express human DPP4 (Fig. 3C). These results all suggest that the recombinant MERS-CoV C-domain interacts directly with cell surface human DPP4 and competes with virus surface MERS-CoV spike protein for the binding site on human DPP4. Therefore, we conclude that the MERS-CoV S1 C-domain is the RBD that binds human DPP4.
Fig 3.
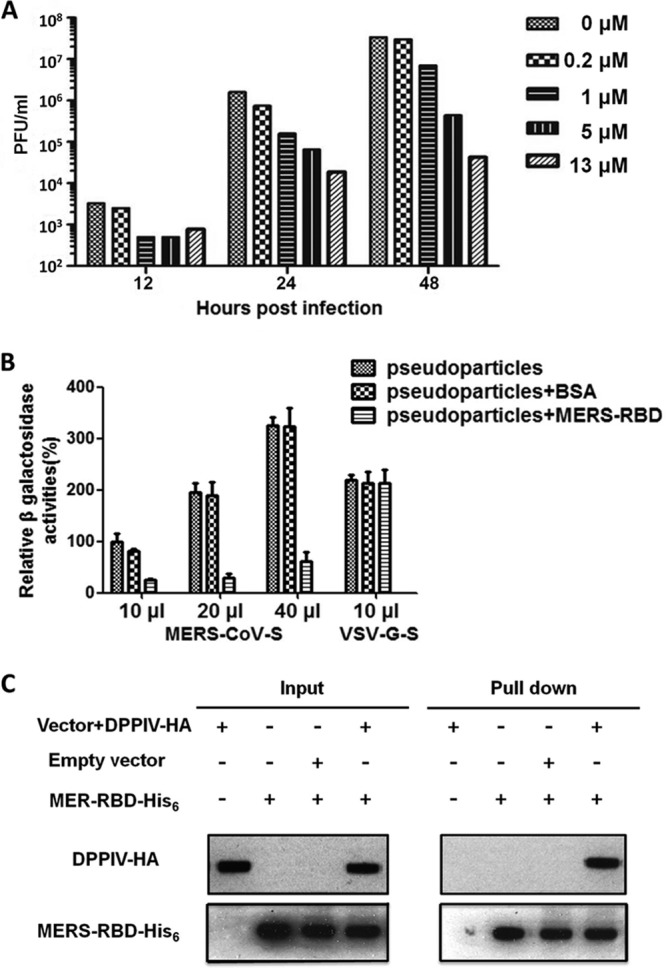
Interactions between the MERS-CoV S1 C-domain and human DPP4. (A) Dose-dependent inhibition of MERS-CoV infection by the MERS-CoV C-domain. Vero cells were treated with increasing concentrations of the MERS-CoV C-domain prior to infection. Virus titers were determined by plaque assay and are shown as PFU/ml. (B) Inhibition of MERS-CoV spike-pseudotyped retrovirus infection by MERS-CoV C-domain. MERS-CoV spike-pseudotyped MLV and VSV-G-pseudotyped MLV were mixed with the MERS-CoV C-domain and BSA, respectively. HEK293T cells transiently expressing human DPP4 were infected with different amount of pseudotyped virus alone or its mixture with proteins. At 48 h postinfection, cells were lysed and β-galactosidase activities were measured. The β-galactosidase activity of cell lysate infected with 10 μl pseudotyped virus alone was taken as 100%. Values are means and standard errors of the means (SEM). (C) Pulldown of cell surface DPP4 by MERS-CoV C-domain. 293T cells were transfected with either empty vector or vector containing DPP4-HA gene. At 48 h posttransfection, cells were harvested and lysed. Cell lysate was mixed with the MERS-CoV C-domain containing a C-terminal His6 tag. The C-domain/DPP4 complex was then precipitated with Ni-NTA beads.
Crystal structure of MERS-CoV RBD.
Having identified the MERS-CoV RBD, we further determined its crystal structure (Fig. 1B and C). The MERS-CoV RBD was crystallized in space group P212121, with 2 molecules per asymmetric unit. The structure was determined using an iodine derivative obtained by a short cryosoak (37) and refined at a 2.13-Å resolution (Table 1). The two copies of MERS-CoV RBD occupying the same asymmetric unit have a small buried interface (∼500 Å2), consistent with the observation during protein purification that the MERS-CoV RBD is a monomer in solution.
Table 1.
Data collection and refinement statistics
| Parameter | MERS-RBD nativea | NaI derivativea |
|---|---|---|
| Data collection | ||
| Space group | P212121 | P212121 |
| Cell dimensions | ||
| a, b, c (Å) | 45.361, 108.065, 124.287 | 45.822, 108.817, 124.328 |
| α, β, γ (°) | 90, 90, 90 | 90, 90, 90 |
| Resolution (Å) | 50–2.13 (2.17–2.13) | 50–2.32 (2.36–2.32) |
| Rsym or Rmerge | 0.065 (0.459) | 0.119 (0.574) |
| I/σI | 31.8 (2.0) | 18.3 (2.0) |
| Completeness (%) | 97.8 (62.3) | 98.5 (90.5) |
| Redundancy | 6.9 (4.3) | 4.3 (2.7) |
| Refinement | ||
| Resolution (Å) | 49.6–2.13 | |
| No. of reflections | 34770 | |
| Rwork/Rfree | 0.149/0.206 | |
| No. of atoms | 3,701 | |
| Protein | 3,261 | |
| Ligand | 120 | |
| Water | 320 | |
| B factors (Å2) | 43.3 | |
| Protein | 40.1 | |
| Ligand | 104.3 | |
| Water | 53.1 | |
| RMSD | ||
| Bond lengths (Å) | 0.008 | |
| Bond angles (°) | 1.33 |
Values in parentheses are for the highest-resolution shell.
The crystal structure of the MERS-CoV RBD contains a core structure and an accessory subdomain (Fig. 1C). The core structure is a five-stranded antiparallel β-sheet with several short α-helices. The accessory subdomain lies on one edge of the core structure and consists of a four-stranded antiparallel β-sheet. The eight cysteines in the RBD form four disulfide bonds (Fig. 2B). Three of the disulfide bonds stabilize the core structure by connecting cysteine 383 to 407, 425 to 478, and 437 to 585; the remaining disulfide bond strengthens the accessory subdomain by connecting cysteine 503 to 526. The RBD also contains 2 glycans that are N-linked to Asn410 in the core structure and Asn487 in the accessory subdomain, respectively (Fig. 2B). We were able to trace all of the RBD residues (except for the N-terminal 12 residues) with an average B factor of 40.1 Å2. Overall, the MERS-CoV RBD has a well-folded tertiary structure that is stabilized by disulfide bonds and glycans.
Comparisons of MERS-CoV and SARS-CoV S1 C-domains.
The C-domains of β-genus MERS-CoV and SARS-CoV (both function as RBDs) have similar tertiary structures, despite their low amino acid sequence similarity. The Z score between the two RBDs is 11.9, as calculated by the protein folding Dali server (38). Like the MERS-CoV RBD, the SARS-CoV RBD contains a core structure and an accessory subdomain (Fig. 2C). The two core structures are highly similar, both consisting of a five-stranded antiparallel β-sheet. The three disulfide bonds that strengthen the core structure are all conserved between the two RBDs. On the other hand, the accessory subdomains of the two RBDs are markedly different. The accessory subdomain of the SARS-CoV RBD mainly consists of loops, with a short two-stranded antiparallel β-sheet in the middle of the accessory subdomain. In contrast, the same region in the MERS-CoV RBD mainly consists of a four-stranded antiparallel β-sheet. The disulfide bond that stabilizes the accessory subdomain of MERS-CoV RBD is arranged differently in SARS-CoV RBD. Overall, the similar core structures of the two RBDs suggest that the two RBDs share an evolutionary origin and their different accessory subdomains resulted from divergent evolution.
The accessory subdomain of β-coronavirus S1 C-domains has been shown to be a hypervariable region that β-coronaviruses often use for receptor recognition. This region in SARS-CoV C-domain binds its receptor, ACE2, and thus has been named the receptor-binding motif (RBM) (24). The same region in MHV often contains long insertions or deletions, depending on the MHV strain. In certain MHV strains, a six-amino-acid insertion in this region allows the C-domain to recognize heparan sulfate (15, 16). We propose that the accessory subdomain of MERS-CoV C-domain also functions as RBM by binding to DPP4 and that MERS-CoV and SARS-CoV have diverged in their RBMs to recognize different receptors (Fig. 4).
Fig 4.
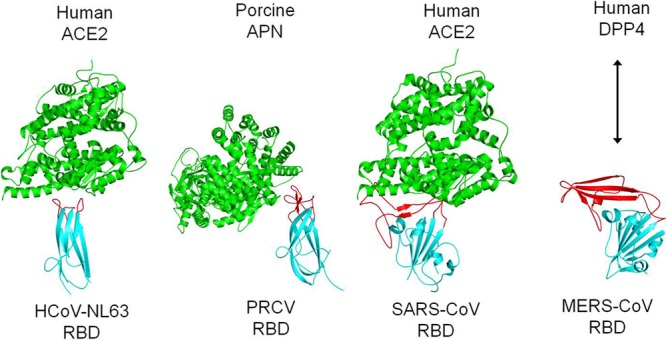
Comparison of receptor-binding mechanisms of coronavirus S1 C-domains. These C-domains include HCoV-NL63 RBD complexed with human ACE2 (PDB 3KBH), PRCV RBD complexed with porcine APN (4F5C), and SARS-CoV RBD complexed with human ACE2 (2AJF). The arrow represents interaction between the MERS-CoV RBD and human DPP4. RBMs and core structures of coronavirus C-domains are in red and cyan, respectively, and receptors are in green.
Why is the accessory subdomain in β-coronavirus C-domain hypervariable and often involved in receptor binding? The reason is probably related to its location on the trimeric spike protein. As we have shown previously using electron microscopy, the S1 C-domain is located on the membrane-distal tip of the trimeric spike protein, with the accessory subdomain sitting on the very tip (39). Hence, the accessory subdomain is the most exposed and protruding region on the whole spike protein, which has two results. First, this region is exposed to the host immune system and thus evolves at an increased pace to evade the host immune pressure. Second, this region is easily accessible to potential host receptors. Consequently, β-coronaviruses can take advantage of the increased evolution rate in this region to screen for and, if circumstances allow, switch to new receptors.
Structural comparisons of MERS-CoV and α-coronavirus S1 C-domains.
The C-domains of β-genus MERS-CoV and α-genus HCoV-NL63 and PCRV (all function as RBDs) have different primary, secondary, and tertiary structures. Both HCoV-NL63 and PCRV RBDs contain a β-sandwich core structure consisting of two β-sheet layers that stack together through hydrophobic interactions, which is significantly different from the core structure of MERS-CoV RBD that consists of a single β-sheet layer (Fig. 5). The Z scores between MERS-CoV and HCoV-NL63 RBDs and between MERS-CoV and PRCV RBDs are both less than zero, suggesting no obvious similarity in their tertiary structures. Furthermore, the proposed DPP4-binding RBM in MERS-CoV RBD is also different from the ACE2-binding RBM in HCoV-NL63 and the APN-binding RBM in PRCV (Fig. 4 and 5). The proposed RBM in MERS-CoV RBD and the RBM in SARS-CoV RBD are both single long and continuous subdomains, while the RBMs in HCoV-NL63 and PRCV RBDs both consist of three short and discontinuous loops. Despite their different tertiary structures, these C-domains of α- and β-coronaviruses are located in the same region of their S1 subunits, suggesting that they are evolutionarily related.
Fig 5.

Comparison of tertiary structures of coronavirus S1 C-domains. (Top) Tertiary structures of coronavirus C-domains (PDB IDs are the same as in Fig. 4). (Bottom) Schematic illustration of the structural topologies of coronavirus C-domains. β-strands are depicted as arrows and α-helices as cylinders. The secondary structures of all of the coronavirus C-domains are colored and numbered in the same way as for the HCoV-NL63 C-domain.
A closer inspection of these coronavirus C-domains reveals that the MERS-CoV RBD has a structural topology (connectivity of secondary structural elements) related to those of α-coronaviruses, consistent with what we showed previously with the SARS-CoV RBD (23). Although one of the β-sheet layers in the RBD core structure of α-coronaviruses has become α-helices in β-coronaviruses, all of the secondary structural elements in these C-domains are connected in the same order (Fig. 5). These results suggest that MERS-CoV and α-coronavirus C-domains share an evolutionary origin but have diverged dramatically to assume tertiary structures with a similarity that is barely recognizable. The evolutionary relationship between MERS-CoV and α-coronavirus C-domains may also be explained by the location of the C-domain on the tip of the trimeric spike protein and hence an increased evolution rate of this domain.
Position of MERS-CoV S1 C-domains on landscape of coronavirus evolution.
Here we summarize possible evolutionary relationships between MERS-CoV and other coronavirus C-domains (Fig. 6). All coronavirus C-domains likely share an evolutionary origin because of their related structural topology. They have diverged into the β-sandwich core structure in α-coronaviruses and the β-sheet core structure in β-coronaviruses. The three RBM loops in α-coronaviruses further diverged into ACE2-binding RBMs in HCoV-NL63 and APN-binding RBMs in PRCV. The RBM subdomain in β-coronaviruses diverged into ACE2-binding RBM in SARS-CoV, heparan sulfate-binding RBM in some strains of MHV, and DPP4-binding RBM in MERS-CoV. Therefore, this study has successfully positioned MERS-CoV RBD on the landscape of coronavirus evolution and, during this process, has also advanced our understanding of the evolution and receptor recognition of coronaviruses.
Fig 6.
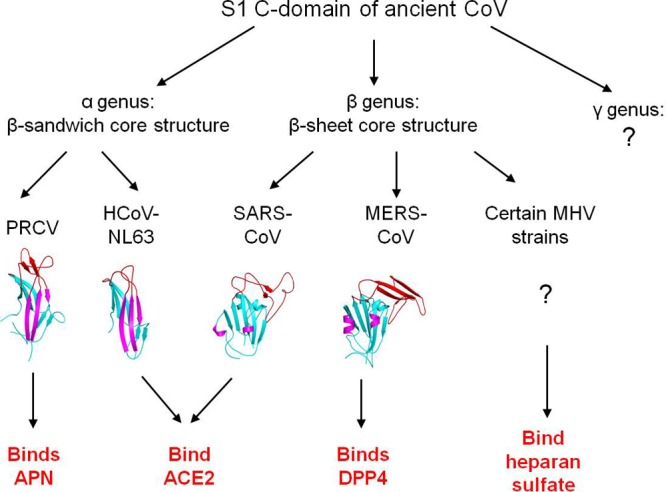
Proposed evolution of the structures and functions of coronavirus S1 C-domains.
Therapeutic implications.
MERS-CoV RBD has therapeutic implications. As we have shown in this study, MERS-CoV RBD functions as an effective inhibitor of MERS-CoV entry (Fig. 3A). In addition, MERS-CoV RBD is also a potential candidate for MERS-CoV subunit vaccines. Previous studies have shown that SARS-CoV RBD can elicit robust host immune responses against SARS-CoV infections (40, 41). Likewise, the MERS-CoV RBD identified in this study has a well-folded structure and interacts strongly with human DPP4, making it a potential vaccine candidate. The determined structure of MERS-CoV RBD provides a platform for structure-based design of effective MERS-CoV vaccines. For example, to help focus the antigenicity of the proposed RBM region, new glycosylation sites may be introduced to the surface of the core structure. In addition, to further stabilize the RBD structure, new disulfide bonds can be introduced into the RBD and some flexible loops can be modified. In sum, by identifying the MERS-CoV RBD and determining its crystal structure, this study may be used to control the transmission of MERS-CoV in humans.
ADDENDUM
During the submission and review of the present study, two other studies independently mapped the MERS-CoV RBD fragments (residues 358 to 588 and 377 to 662), both of which are similar to the one identified in the present study (residues 367 to 588) (42, 43). These studies also showed that the RBD efficiently elicits neutralizing antibodies, confirming that the crystal structure of MERS-CoV can be useful in structure-based vaccine design.
In addition, two other studies independently determined the crystal structure of MERS-CoV RBD (residues 367 to 606) complexed with human DPP4 (44, 45). These studies delineated the molecular interactions between the MERS-CoV RBD and its receptor and confirmed that the accessory subdomain in the MERS-CoV RBD is the DPP4-binding RBM. The structures of the MERS-CoV RBD as determined in these studies are a good match with the structure reported in the present study (e.g., MERS-CoV RBD in PDB 4KR0 can be superimposed onto the current structure with a root mean square deviation [RMSD] of 0.82 Å). Although these recently published studies are in general agreement with the present study, this study is unique in that it focuses on (i) the evolution of MERS-CoV and other coronavirus RBDs and (ii) the therapeutic implications of MERS-CoV RBD. Both of these issues are critical for understanding and controlling MERS-CoV.
ACKNOWLEDGMENTS
We thank Lang Chen for assistance and discussion.
This work was supported by NIH grant R01AI089728 (to F. Li), by NIH grants R01AI085524 and 1U19AI100625 (to R. S. Baric), and by NIH grant 8P41GM103403-10 (to the Northeastern Collaborative Access Team beamlines at the Advanced Photon Source). Use of the Advanced Photon Source was supported by the U.S. DOE under contract DE-AC02-06CH11357. Computer resources were provided by the Basic Sciences Computing Laboratory of the University of Minnesota Supercomputing Institute.
Footnotes
Published ahead of print 31 July 2013
REFERENCES
- 1.de Groot RJ, Baker SC, Baric RS, Brown CS, Drosten C, Enjuanes L, Fouchier RA, Galiano M, Gorbalenya AE, Memish Z, Perlman S, Poon LL, Snijder EJ, Stephens GM, Woo PC, Zaki AM, Zambon M, Ziebuhr ZJ. 2013. Middle East respiratory syndrome coronavirus (MERS-CoV); announcement of the Coronavirus Study Group. J. Virol. 87:7790–7792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zaki AM, van Boheemen S, Bestebroer TM, Osterhaus A, Fouchier RAM. 2012. Isolation of a novel coronavirus from a man with pneumonia in Saudi Arabia. N. Engl. J. Med. 367:1814–1820 [DOI] [PubMed] [Google Scholar]
- 3.Annan A, Baldwin HJ, Corman VM, Klose SM, Owusu M, Nkrumah EE, Badu EK, Anti P, Agbenyega O, Meyer B, Oppong S, Sarkodie YA, Kalko EK, Lina PH, Godlevska EV, Reusken C, Seebens A, Gloza-Rausch F, Vallo P, Tschapka M, Drosten C, Drexler JF. 2013. Human betacoronavirus 2c EMC/2012-related viruses in bats, Ghana and Europe. Emerg. Infect. Dis. 19:456–459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Holmes KV, Dominguez SR. 2013. The new age of virus discovery: genomic analysis of a novel human betacoronavirus isolated from a fatal case of pneumonia. mBio 4:e00548–12. 10.1128/mBio.00548-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lau SK, Li KS, Tsang AK, Lam CS, Ahmed S, Chen H, Chan KH, Woo PC, Yuen KY. 2013. Genetic characterization of betacoronavirus lineage C viruses in bats reveals marked sequence divergence in the spike protein of Pipistrellus bat coronavirus HKU5 in Japanese pipistrelle: implications for the origin of the novel Middle East respiratory syndrome coronavirus. J. Virol. 87:8638–8650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ksiazek TG, Erdman D, Goldsmith CS, Zaki SR, Peret T, Emery S, Tong SX, Urbani C, Comer JA, Lim W, Rollin PE, Dowell SF, Ling AE, Humphrey CD, Shieh WJ, Guarner J, Paddock CD, Rota P, Fields B, DeRisi J, Yang JY, Cox N, Hughes JM, LeDuc JW, Bellini WJ, Anderson LJ. 2003. A novel coronavirus associated with severe acute respiratory syndrome. N. Engl. J. Med. 348:1953–1966 [DOI] [PubMed] [Google Scholar]
- 7.Peiris JSM, Lai ST, Poon LLM, Guan Y, Yam LYC, Lim W, Nicholls J, Yee WKS, Yan WW, Cheung MT, Cheng VCC, Chan KH, Tsang DNC, Yung RWH, Ng TK, Yuen KY. 2003. Coronavirus as a possible cause of severe acute respiratory syndrome. Lancet 361:1319–1325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Perlman S, Netland J. 2009. Coronaviruses post-SARS: update on replication and pathogenesis. Nat. Rev. Microbiol. 7:439–450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hofmann H, Pyrc K, van der Hoek L, Geier M, Berkhout B, Pohlmann S. 2005. Human coronavirus NL63 employs the severe acute respiratory syndrome coronavirus receptor for cellular entry. Proc. Natl. Acad. Sci. U. S. A. 102:7988–7993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li WH, Moore MJ, Vasilieva N, Sui JH, Wong SK, Berne MA, Somasundaran M, Sullivan JL, Luzuriaga K, Greenough TC, Choe H, Farzan M. 2003. Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature 426:450–454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Delmas B, Gelfi J, Lharidon R, Vogel LK, Sjostrom H, Noren O, Laude H. 1992. Aminopeptidase-N is a major receptor for the enteropathogenic coronavirus TGEV. Nature 357:417–420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yeager CL, Ashmun RA, Williams RK, Cardellichio CB, Shapiro LH, Look AT, Holmes KV. 1992. Human aminopeptidase-N is a receptor for human coronavirus-229E. Nature 357:420–422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dveksler GS, Pensiero MN, Cardellichio CB, Williams RK, Jiang GS, Holmes KV, Dieffenbach CW. 1991. Cloning of the mouse hepatitis virus (MHV) receptor: expression in human and hamster cell lines confers susceptibility to MHV. J. Virol. 65:6881–6891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Williams RK, Jiang GS, Holmes KV. 1991. Receptor for mouse hepatitis virus is a member of the carcinoembryonic antigen family of glycoproteins. Proc. Natl. Acad. Sci. U. S. A. 88:5533–5536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Watanabe R, Matsuyama S, Taguchi F. 2006. Receptor-independent infection of murine coronavirus: Analysis by spinoculation. J. Virol. 80:4901–4908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Watanabe R, Sawicki SG, Taguchi F. 2007. Heparan sulfate is a binding molecule but not a receptor for CEACAM1-independent infection of murine coronavirus. Virology 366:16–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cavanagh D, Davis PJ. 1986. Coronavirus IBV—removal of spike glycopolypeptide-S1 by urea abolishes infectivity and hemagglutination but not attachment to cells. J. Gen. Virol. 67:1443–1448 [DOI] [PubMed] [Google Scholar]
- 18.Schultze B, Cavanagh D, Herrler G. 1992. Neuraminidase treatment of avian infectious-bronchitis coronavirus reveals a hemagglutinating activity that is dependent on sialic acid-containing receptors on erythrocytes. Virology 189:792–794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schultze B, Gross HJ, Brossmer R, Herrler G. 1991. The S-protein of bovine coronavirus is a hemagglutinin recognizing 9-O-acetylated sialic acid as a receptor determinant. J. Virol. 65:6232–6237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schwegmann-Wessels C, Herrler G. 2006. Sialic acids as receptor determinants for coronaviruses. Glycoconj. J. 23:51–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Raj VS, Mou HH, Smits SL, Dekkers DHW, Muller MA, Dijkman R, Muth D, Demmers JAA, Zaki A, Fouchier RAM, Thiel V, Drosten C, Rottier PJM, Osterhaus A, Bosch BJ, Haagmans BL. 2013. Dipeptidyl peptidase 4 is a functional receptor for the emerging human coronavirus-EMC. Nature 495:251–254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bosch BJ, van der Zee R, de Haan CAM, Rottier PJM. 2003. The coronavirus spike protein is a class I virus fusion protein: structural and functional characterization of the fusion core complex. J. Virol. 77:8801–8811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li F. 2012. Evidence for a common evolutionary origin of coronavirus spike protein receptor-binding subunits. J. Virol. 86:2856–2858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li F, Li WH, Farzan M, Harrison SC. 2005. Structure of SARS coronavirus spike receptor-binding domain complexed with receptor. Science 309:1864–1868 [DOI] [PubMed] [Google Scholar]
- 25.Peng GQ, Sun DW, Rajashankar KR, Qian ZH, Holmes KV, Li F. 2011. Crystal structure of mouse coronavirus receptor-binding domain complexed with its murine receptor. Proc. Natl. Acad. Sci. U. S. A. 108:10696–10701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Peng GQ, Xu LQ, Lin YL, Chen L, Pasquarella JR, Holmes KV, Li F. 2012. Crystal structure of bovine coronavirus spike protein lectin domain. J. Biol. Chem. 287:41931–41938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Reguera J, Santiago C, Mudgal G, Ordoño D, Enjuanes L, Casasnovas JM. 2012. Structural bases of coronavirus attachment to host aminopeptidase N and its inhibition by neutralizing antibodies. PLoS Pathog. 8:e1002859. 10.1371/journal.ppat.1002859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wu KL, Li WK, Peng GQ, Li F. 2009. Crystal structure of NL63 respiratory coronavirus receptor-binding domain complexed with its human receptor. Proc. Natl. Acad. Sci. U. S. A. 106:19970–19974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li F. 2008. Structural analysis of major species barriers between humans and palm civets for severe acute respiratory syndrome coronavirus infections. J. Virol. 82:6984–6991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wu KL, Peng GQ, Wilken M, Geraghty RJ, Li F. 2012. Mechanisms of host receptor adaptation by severe acute respiratory syndrome coronavirus. J. Biol. Chem. 287:8904–8911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Otwinowski Z, Minor W. 1997. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 276:307–326 [DOI] [PubMed] [Google Scholar]
- 32.Grosse-Kunstleve RW, Adams PD. 2003. Substructure search procedures for macromolecular structures. Acta Crystallogr. D Biol. Crystallogr. 59:1966–1973 [DOI] [PubMed] [Google Scholar]
- 33.Terwilliger TC. 2000. Maximum-likelihood density modification. Acta Crystallogr. D Biol. Crystallogr. 56:965–972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Murshudov GN, Vagin AA, Lebedev A, Wilson KS, Dodson EJ. 1999. Efficient anisotropic refinement of macromolecular structures using FFT. Acta Crystallogr. D Biol. Crystallogr. 55:247–255 [DOI] [PubMed] [Google Scholar]
- 35.Josset L, Menachery VD, Gralinski LE, Agnihothram S, Sova P, Carter VS, Yount BL, Graham RL, Baric RS, Katze MG. 2013. Cell host response to infection with novel human coronavirus EMC predicts potential antivirals and important differences with SARS coronavirus. mBio 4:e00165–13. 10.1128/mBio.00165-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wu K, Chen L, Peng G, Zhou W, Pennell CA, Mansky LM, Geraghty RJ, Li F. 2011. A virus-binding hot spot on human angiotensin-converting enzyme 2 is critical for binding of two different coronaviruses. J. Virol. 85:5331–5337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dauter Z, Dauter M, Rajashankar KR. 2000. Novel approach to phasing proteins: derivatization by short cryo-soaking with halides. Acta Crystallogr. D Biol. Crystallogr. 56:232–237 [DOI] [PubMed] [Google Scholar]
- 38.Holm L, Sander C. 1998. Touring protein fold space with Dali/FSSP. Nucleic Acids Res. 26:316–319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li F, Berardi M, Li WH, Farzan M, Dormitzer PR, Harrison SC. 2006. Conformational states of the severe acute respiratory syndrome coronavirus spike protein ectodomain. J. Virol. 80:6794–6800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.He YX, Lu H, Siddiqui P, Zhou YS, Jiang SB. 2005. Receptor-binding domain of severe acute respiratory syndrome coronavirus spike protein contains multiple conformation-dependent epitopes that induce highly potent neutralizing antibodies. J. Immunol. 174:4908–4915 [DOI] [PubMed] [Google Scholar]
- 41.He YX, Zhou YS, Wu H, Luo BJ, Chen JM, Li WB, Jiang SB. 2004. Identification of immunodominant sites on the spike protein of severe acute respiratory syndrome (SARS) coronavirus: implication for developing SARS diagnostics and vaccines. J. Immunol. 173:4050–4057 [DOI] [PubMed] [Google Scholar]
- 42.Du L, Zhao G, Kou Z, Ma C, Sun S, Poon VK, Lu L, Wang L, Debnath AK, Zheng BJ, Zhou Y, Jiang S. 2013. Identification of receptor-binding domain in S protein of the novel human coronavirus MERS-CoV as an essential target for vaccine development. J. Virol. [Epub ahead of print.] 10.1128/JVI.01048-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mou H, Raj VS, van Kuppeveld FJ, Rottier PJ, Haagmans BL, Bosch BJ. 2013. The receptor binding domain of the new MERS coronavirus maps to a 231-residue region in the spike protein that efficiently elicits neutralizing antibodies. J. Virol. [Epub ahead of print.] 10.1128/JVI.01277-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lu G, Hu Y, Wang Q, Qi J, Gao F, Li Y, Zhang Y, Zhang W, Yuan Y, Bao J, Zhang B, Shi Y, Yan J, Gao GF. 2013. Molecular basis of binding between novel human coronavirus MERS-CoV and its receptor CD26. Nature [Epub ahead of print.] 10.1038/nature12328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang N, Shi X, Jiang L, Zhang S, Wang D, Tong P, Guo D, Fu L, Cui Y, Liu X, Arledge KC, Chen YH, Zhang L, Wang X. 2013. Structure of MERS-CoV spike receptor-binding domain complexed with human receptor DPP4. Cell Res. [Epub ahead of print.] 10.1038/cr.2013.92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Prabakaran P, Gan JH, Feng Y, Zhu ZY, Choudhry V, Xiao XD, Ji XH, Dimitrov DS. 2006. Structure of severe acute respiratory syndrome coronavirus receptor-binding domain complexed with neutralizing antibody. J. Biol. Chem. 281:15829–15836 [DOI] [PMC free article] [PubMed] [Google Scholar]