

Abstract
Gram-negative bacteria such as Escherichia coli build a peptidoglycan (PG) cell wall in their periplasm using the precursor known as lipid II. Lipid II is a large amphipathic molecule composed of undecaprenyl diphosphate and a disaccharide-pentapeptide that PG-synthesizing enzymes use to build the PG sacculus. During PG biosynthesis, lipid II is synthesized at the cytoplasmic face of the inner membrane and then flipped across the membrane. This translocation of lipid II must be assisted by flippases thought to shield the disaccharide-pentapeptide as it crosses the hydrophobic core of the membrane. The inner membrane protein MurJ is essential for PG biogenesis and homologous to known and putative flippases of the MOP (multidrug/oligo-saccharidyl-lipid/polysaccharide) exporter superfamily, which includes flippases that translocate undecaprenyl diphosphate-linked oligosaccharides across the cytoplasmic membranes of bacteria. Consequently, MurJ has been proposed to function as the lipid II flippase in E. coli. Here, we present a three-dimensional structural model of MurJ generated by the I-TASSER server that suggests that MurJ contains a solvent-exposed cavity within the plane of the membrane. Using in vivo topological studies, we demonstrate that MurJ has 14 transmembrane domains and validate features of the MurJ structural model, including the presence of a solvent-exposed cavity within its transmembrane region. Furthermore, we present functional studies demonstrating that specific charged residues localized in the central cavity are essential for function. Together, our studies support the structural homology of MurJ to MOP exporter proteins, suggesting that MurJ might function as an essential transporter in PG biosynthesis.
INTRODUCTION
The cell envelope of most bacteria contains a cell wall exoskeleton composed of peptidoglycan (PG) that surrounds the cytoplasmic membrane (1, 2). The rigid PG structure protects the bacterium from osmotic rupture, serves as a scaffold onto which other envelope components are attached, and defines cell shape. Underscoring the essentiality of the PG cell wall is the fact that many antibiotics target PG biosynthesis (3).
Bacteria build their PG sacculus by polymerizing an N-acetylglucosamine–N-acetylmuramic acid (GlcNAc-MurNAc) disaccharide-pentapeptide into long glycan chains that are cross-linked by peptide bonds between stem peptides (2). This GlcNAc-MurNAc disaccharide-pentapeptide is synthesized at the cytoplasmic side of the membrane as a polyisoprenyl lipid-linked precursor known as lipid II (Fig. 1A) (4). Because lipid II polymerization occurs at the extracytoplasmic side of the membrane, an obligatory step in PG biosynthesis is the translocation of the lipid-linked disaccharide-pentapeptide across the cytoplasmic membrane.
Fig 1.

Membrane translocation of undecaprenol pyrophosphate-linked precursors during PG and O-antigen biosynthesis. (A) PG biogenesis requires synthesis of lipid II from UDP-MurNAc-pentapeptide (M), UPD-GlcNAc (G), and undecaprenol pyrophosphate (UND-PP). Lipid II is subsequently flipped by a flippase to the outer leaflet of the inner membrane, where it can be used by transglycosylases (TGs) to build glycan chains that are cross-linked by transpeptidases (TPs). (B) After the precursor for O antigen is synthesized as an UND-PP–oligosaccharide at the inner leaflet, it is translocated by the Wzx flippase to the outer leaflet of the inner membrane. Oligosaccharide subunits are then polymerized by Wzy to a length determined by Wzz and linked to the rough lipopolysaccharide (LPS) by WaaL. The resulting smooth lipopolysaccharide is transported to the outer membrane (OM).
The use of polyisoprenyl lipid-linked precursors in the biogenesis of envelope glycopolymers is widespread in bacteria. Examples include the biogenesis of PG, certain capsules and exopolysaccharides, and O antigens (5, 6). In these systems, bacteria build each precursor by transferring, in orderly fashion, sugar moieties from nucleotide-sugar precursors to the C55 polyisoprenoid undecaprenyl phosphate (undecaprenyl-P); this synthesis occurs at the cytoplasmic side of the membrane. Therefore, the resulting precursor must be translocated across the cytoplasmic membrane for the biogenesis of the glycopolymer to proceed. Membrane transport of these large amphipathic precursors requires flippases hypothesized to provide a cavity through which the hydrophilic portion of the lipid-linked molecule translocates while being shielded from the hydrophobic core of the membrane (Fig. 1) (7).
We lack mechanistic details of how undecaprenyl diphosphate (undecaprenyl-PP)–oligosaccharides are translocated across membranes. Nevertheless, from what is perhaps the best-characterized system, the biosynthesis of lipopolysaccharide O antigen, we know that there are at least three different types of membrane translocation of undecaprenyl-PP–saccharides: the synthase-dependent, the ABC (ATP-binding cassette) transporter-dependent, and the Wzy-dependent systems. The synthase-dependent system is not widely conserved, and it relies on translocation that is coupled to the synthesis of the undecaprenyl-PP–polysaccharide by a polymerizing glycosyltransferase (8, 9). In the ABC transporter-dependent pathway, synthesis of the full-length polysaccharide as an undecaprenyl-PP–polysaccharide intermediate occurs in the cytoplasmic side of the membrane, and ATP-dependent translocation is mediated by an ABC transporter (10, 11). In the Wzy-dependent system, an undecaprenyl-PP–oligosaccharide is synthesized at the cytoplasmic side of the membrane, translocated by the Wzx flippase, and polymerized by Wzy into undecaprenyl-PP–polysaccharide at the extracytoplasmic side of the membrane (Fig. 1B) (5).
In Escherichia coli, lipid II is composed of GlcNAc-MurNAc (l-Ala–γ-d-Glu–meso-diaminopimelic acid–d-Ala–d-Ala)–undecaprenyl-PP (see Fig. S1 in the supplemental material) and synthesized from nucleotide-sugar precursors at the cytoplasmic side of the membrane (Fig. 1A) (4, 12). How this bacterium flips lipid II across its cytoplasmic membrane during PG biosynthesis is controversial. In recent years, three inner membrane proteins, RodA, FtsW, and MurJ, have been proposed to be lipid II flippases in E. coli (13–16). RodA and FtsW are paralogs thought to perform the same function in PG biogenesis but at different stages of development. While RodA is required for the synthesis of lateral PG, FtsW is required for the synthesis of septal PG (17–23). They are members of PG-synthetic multiprotein complexes and have been proposed to be required for the recruitment of the essential PG transpeptidases PBP 2 (by RodA) and PBP 3 (by FtsW) (17, 24, 25). Both RodA and FtsW have 10 transmembrane (TM) domains (TMDs) (17, 26, 27) and were suggested to be the lipid II flippases decades ago (13). Data supporting a flippase role for RodA are lacking, but a recent in vitro study showed that FtsW promotes lipid II translocation across membranes (15). However, this study did not rule out nonspecific flipping. Furthermore, analysis of PG precursors in FtsW-depleted cells suggests that precursors are still used in reactions downstream of lipid II translocation and that their profile resembles that of cells treated with transpeptidase inhibitors (28). Therefore, additional studies are needed to determine whether FtsW and RodA translocate lipid II in vivo.
MurJ is an essential inner membrane protein whose depletion inhibits PG sacculus biosynthesis and leads to the accumulation of PG precursors, including lipid-linked intermediates (14, 16). Orthologs in some Gram-positive bacteria are also required for PG biosynthesis (29, 30). A key finding strengthening the proposal that MurJ is the lipid II flippase in E. coli is the fact that MurJ is a member of the MOP (multidrug/oligo-saccharidyl-lipid/polysaccharide) exporter superfamily (14, 16, 31). The best-studied members of this superfamily are drug exporters of the MATE (multidrug and toxic compound extrusion) family, which export lipophilic and cationic drugs across cytoplasmic membranes by using an electrochemical gradient of either Na+ or H+ ions to drive substrate transport (32). Notably, the MOP exporter superfamily includes several proteins implicated in the flipping of polyisoprenyl-linked oligosaccharides. Among them are the aforementioned bacterial Wzx flippases, which participate in the biosynthesis of envelope polysaccharides, such as the O antigen, the enterobacterial common antigen, and certain capsules. Wzx flippases translocate undecaprenyl-PP–oligosaccharide intermediates across the cytoplasmic membrane (5). Although these findings suggest a role for MurJ in lipid II transport, we lack direct evidence showing that MurJ flips lipid II.
The structure and function of proteins are intimately related. To gain insight into the function of MurJ, we conducted structural-functional analyses of the E. coli MurJ protein. Our studies reveal the topological map of MurJ and define a solvent-exposed cavity within its transmembrane core. Furthermore, changes in residues within this cavity affect function, suggesting that MurJ functions as a transporter.
MATERIALS AND METHODS
Bacterial strains and growth conditions.
Strains are listed in Table S1 in the supplemental material. Except for strains DY378 (33) and DH5α (Invitrogen Life Technologies), all strains were derived from strain NR754, an araD+ revertant of MC4100 (34, 35). Lysogeny broth (LB) and glucose M63 minimal broth and agar were prepared as described previously (36). Yeast tryptone (YT) agar contains 10 g/liter of tryptone, 5 g/liter of yeast extract, and 15 g/liter of agar. Unless indicated and for recombineering, all liquid cultures were grown under aeration at 37°C, and their growth was monitored by determination of the optical density at 600 nm (OD600). When appropriate, kanamycin (25 μg/ml), ampicillin (25 μg/ml for plasmid pRC7MurJ and 125 μg/ml for plasmid pMurJTOP), chloramphenicol (20 μg/ml), and 5-bromo-4-chloro-indolyl-β-d-galactopyranoside (X-Gal; 20 μg/ml) were added.
Construction of plasmids.
All plasmids and primers referenced below are listed in Tables S1 and S2 in the supplemental material, respectively. To construct pRC7MurJ, primers 5EcoR1PgfA and 3Hind3PgfA were used to amplify a PCR product that included the murJ open reading frame (ORF) flanked by 15 and 11 bp at the 5′ and 3′ ends, respectively, from the chromosome of NR754. This PCR product and pRC7 (37) were digested with EcoRI and HindIII and ligated to generate pRC7MurJ. Transformants containing pRC7MurJ were selected on LB agar containing 25 μg/ml ampicillin.
To construct pMurJTOP plasmids, primers 5MurJ10UPXba and 3MurJ11DOWNBamH were used to amplify a PCR product that includes the murJ and the 10- and 11-bp upstream and downstream sequences, respectively, of the murJ ORF. The resulting PCR product and pPLE01 (38) were digested with XbaI and BamHI and ligated to generate pMurJTOP1533. DH5α transformants harboring pMurJTOP1533 were selected on LB agar containing 125 μg/ml ampicillin. The pMurJTOP1533 plasmid expresses a tribrid fusion protein consisting of full-length MurJ fused at its C terminus to the alkaline phosphatase (AP)-LacZα (α fragment of β-galactosidase) dual-reporter fusion (39). A collection of plasmids carrying 3′-end deletions in murJ were generated so that the dual AP-LacZα reporter could be fused to MurJ derivatives (MurJTRUNC) that had been C-terminally truncated at different residues. To construct this collection of plasmids, pMurJTOP1533 was digested with EcoRV and used as the template in PCR mixtures containing primer 5PhoAEcoRV and each of the 3MurJEcoRV primers (see Table S2 in the supplemental material). After digestion with EcoRV, PCR products were ligated to generate the pMurJTOP collection (see Table S1 in the supplemental material). DH5α transformants harboring these pMurJTOP-derived plasmids were selected on LB agar containing 125 μg/ml ampicillin.
Plasmid pFLAGMurJ, which encodes full-length MurJ with an N-terminal FLAG epitope, was derived as follows from the ASKA collection plasmid carrying murJ (40). The ASKA collection plasmid, which complements depletion of MurJ (16, 30), encodes a MurJ variant with an N-terminal 6× His tag and linker and 5 additional residues at the C terminus. First, we restored the wild-type 3′ end of this plasmid-borne his-murJ allele by deleting the 15 bp encoding the aforementioned 5 C-terminal residues using PCR with primers pEcMurJwtCterm and 3pEcMurJwtCterm to generate pEcMurJwtCterm. Codons encoding the N-terminal 6× His tag and linker were subsequently deleted by PCR using the primers 5MurJNtHisDEL and 3pEcMurJntHISdel to construct pMurJ. Finally, pFLAGMurJ was generated by inserting codons encoding an N-terminal FLAG tag using PCR with primers 5NtermFlgMmurJ and 3NtermFlgMmurJ. All of the above in-frame deletions and insertions were made using site-directed mutagenesis (SDM) PCR (98°C for 2 min, followed by 30 cycles of 98°C for 30 s, 60°C for 30 s, and 72°C for 3 min and a final extension of 5 min at 72°C) with the Phusion polymerase (New England BioLabs), as per the manufacturer's instructions, with the exception that PCR products were treated with T4 polynucleotide kinase (New England BioLabs) prior to ligation in lieu of using phosphorylated primers. DH5α transformants harboring these plasmids were selected for on LB agar containing chloramphenicol.
Plasmids carrying alleles of flag-murJ encoding amino acid substitutions were constructed by SDM PCR (95°C for 2 min, followed by 18 cycles of 96°C for 1 min, 56°C for 1 min, and 72°C for 12 min and a final extension of 12 min at 72°C) using the Pfu Turbo polymerase (Agilent Technologies), as per the manufacturer's instructions. To generate pFLAGMurJΔCys, the two native Cys codons in flag-murJ (C314, C419) were changed to Ser codons in two consecutive steps. The mixture for the first PCR contained pFLAGMurJ as the template and mutagenic primers 5pEcMurJC314S and 3pEcMurJC314S, and the PCR yielded pFLAGMurJC314S. Subsequently, primers 5pEcMurJC419S and 3pEcMurJC419S were used to construct pFLAGMurJΔCys. NR2117-derived transformants harboring these plasmids were selected on medium containing chloramphenicol. Plasmids derived from pFLAGMurJΔCys that carry single Cys-codon substitutions in flag-murJ were generated by SDM using the Pfu Turbo polymerase in PCR mixtures with complementary mutagenic primers (see Table S2 in the supplemental material), as described above.
Construction of ΔmurJ strains.
A ΔmurJ::kan allele was constructed by deleting codons 2 to 511 from the 511-codon murJ chromosomal gene using recombineering, as follows: primers 5murJP1 and 3murJP2 were used to amplify a PCR product that contains a kanamycin resistance cassette flanked by FLP recombination target (FRT) sites and 73 bp of homology to the target sequences in the murJ locus from pKD4 (41). This PCR product was introduced into DY378(pRC7MurJ) via electroporation for recombineering, and recombinants were selected at 30°C on LB agar containing kanamycin (33). The resulting ΔmurJ::kan allele was introduced into NR754(pRC7MurJ) using P1 transduction (36), and transductants were selected on medium containing kanamycin to generate NR1648. The kan cassette was excised using pCP20 (42) to construct the ΔmurJ::FRT strain NR2066. To allow direct transformation with SDM-generated plasmids, ΔhsdR::kan from the Keio collection (43) was introduced into NR2066 by P1 transduction to generate NR2117.
Functionality test of flag-murJ alleles.
Plasmid pRC7MurJ is defective in partitioning functions, so it is easily lost from a population of cells under nonselective conditions (37). Consequently, in the absence of ampicillin, pRC7MurJ is readily lost in murJ+ cells. However, because murJ is essential, pRC7MurJ is stably maintained, even in the absence of ampicillin, in cells where pRC7MurJ provides the only functional allele of murJ. We took advantage of the partitioning defect of pRC7MurJ to screen for the functionality of the murJ alleles. Plasmids encoding functional flag-murJ alleles were identified because when they were introduced into NR2117 (a ΔmurJ strain carrying pRC7MurJ), they allowed survival of transformants that lost pRC7MurJ. This was evident by the loss of blue color when colonies were obtained on solid medium containing X-Gal. The resulting haploid flag-murJ single-Cys mutant strains were used as described below. NR2117 cells transformed with mutagenized plasmids encoding nonfunctional (i.e., total-loss-of-function) murJ alleles yielded blue colonies in the presence of X-Gal because they could survive only if they retained pRC7MurJ. We observed that the presence of pRC7MurJ yielded a lower level of expression of flag-murJ from pFLAGMurJ-derived plasmids, for unknown reasons. Therefore, to conduct the substituted cysteine accessibility method (SCAM) and assess the levels of nonfunctional FLAG-MurJ mutant proteins, we constructed merodiploid strains by introducing the noncomplementing plasmids into wild-type strain NR754.
Sensitivity to low-osmolarity conditions.
Haploid NR754 ΔmurJ ΔhsdR::kan strains carrying pFLAGMurJΔCys-derived plasmids with single Cys substitutions were tested for growth defects in low-osmolarity medium by examining the colony morphology and comparing it to that of the NR2131 parent strain after overnight growth on YT agar at 37°C. Strains that exhibited any growth defects were further tested by an efficiency-of-plating (EOP) assay. For this assay, overnight cultures of NR2131 and NR754 ΔmurJ ΔhsdR::kan carrying pFLAGmurJΔCys-derived plasmids with single Cys substitutions were grown in LB. Ten-fold serial dilutions of cultures were prepared, and LB and YT agar plates were inoculated with approximately 2 μl of each dilution using a 48-pin manifold. After overnight growth at 37°C, EOP values were calculated by dividing the number of colonies obtained for each strain under each condition by the number of colonies that NR2131 yielded on LB agar. Data represent the average ± standard deviation of three independent experiments.
Topology reporter assays.
Cells carrying pMurJTOP plasmids were grown overnight in LB with ampicillin. One milliliter of these cultures was pelleted and resuspended in 250 μl 1 M Tris-HCl (pH 8.0) to assay for AP and β-galactosidase (BG) activities (44, 45). Detection of AP activity was performed as follows: 100 μl of cell suspension was added to 900 μl of 1 M Tris-HCl (pH 8.0); cells were permeabilized after the addition of 1 drop of 0.1% SDS and 2 drops of CHCl3 and vortexed for 10 s. In a 96-well plate, 150 μl of p-nitrophenyl phosphate substrate (11.1 mg/ml in 1 M Tris-HCl, pH 8.0) was added to 100 μl of permeabilized cells, and the absorbance at 410 nm was measured every 15 s for 10 min. For detection of BG activity, 100 μl of cell suspension was added to 900 μl of Z buffer (60 mM Na2HPO4·7H2O, 40 mM NaH2PO4·H2O, 10 mM KCl, 1 mM MgSO4·7H2O, 50 mM β-mercaptoethanol); cells were permeabilized as described above for the AP activity assay. In a 96-well plate, 100 μl of o-nitrophenyl-β-d-galactopyranoside (5 mg/ml in Z buffer) was added to 100 μl of permeabilized cells and 50 μl of Z buffer. The absorbance at 420 nm was measured every 15 s for 10 min. Relative activities (changes in absorbance over time) were calculated from three independent assays, and an average activity was obtained. For each reporter set, the highest average activity value was identified as the maximal activity. The average value for each fusion was used to calculate the percentage of the maximal AP (AP max) and maximal BG (BG max) activities. A normalized AP/BG activity ratio was calculated for each fusion by dividing its percent AP max by its percent BG max (39).
SCAM.
Logarithmically growing cells were collected, washed with phosphate-buffered saline (PBS; pH 7.4), and resuspended in a volume of PBS normalized to the OD600 (100 μl for an OD600 of 1.0). For the blocking step, four tubes containing 50 μl of cell suspension were either untreated (positive- and negative-label control tubes) or treated with 5 mM the blocking agent N-ethylmaleimide (NEM; Santa Cruz Biotechnology) or sodium (2-sulfonatoethyl) methanethiosulfonate (MTSES; Santa Cruz Biotechnology). After rotating at room temperature for 1 h, cells were washed with PBS and resuspended in 50 μl of lysis buffer (15 mM Tris-HCl, pH 7.4, 1% SDS, 6 M urea). Both of the blocked samples and the positive-label control sample were labeled with 5 mM maleimide-polyethylene glycol (Mal-PEG; molecular weight [MW], 2,000; Laysan Bio); nothing was added to the negative-label control tube. After rotating the samples for 1 h with protection from light, 50 μl of 2× AB buffer (6.84 mM Na2HPO4, 3.16 mM NaH2PO4, 50 mM Tris-HCl, pH 6.8, 6 M urea, 1% β-mercaptoethanol, 3% SDS, 10% glycerol, 0.1% bromophenol blue) was added. Samples were incubated at 45°C for 30 min prior to immunoblotting. Eighty-two of the single-Cys-substitution mutants were additionally tested with a 5-min blocking incubation; in all but 11 of these mutants, results from both blocking times were comparable. Representative results are shown in Fig. S6A in the supplemental material. All results are presented in Fig. 5 and in Table S3 and Fig. S6B and C in the supplemental material. The SCAM topology map (see Fig. 4) was generated using the TeXtopo package in the LaTeX program (46).
Fig 5.

MurJ has 14 TMDs and a solvent-exposed cavity within the transmembrane region. SCAM results for 110 MurJ single-Cys-substitution mutants are consistent with the model structure generated by I-TASSER for the first 12 TMDs and further define TMDs 13 and 14. TMD numbers are shown on top of the topology model. Amino acids in TMDs 1, 2, 7, and 8 are accessible to aqueous Cys-blocking agents, supporting the existence of a solvent-exposed cavity within the membrane plane. Substituted Cys are defined as not blocked (0% block with NEM, 0% block with MTSES), partially NEM blocked (<100% block with NEM, 0% block with MTSES), NEM blocked (100% block with NEM, 0% block with MTSES), partially blocked (<100% block with NEM, <100% block with MTSES), partially MTSES blocked (100% block with NEM, <100% block with MTSES), and MTSES blocked (100% block with NEM, 100% block with MTSES).
Fig 4.

Native Cys residues are dispensable for MurJ function. (A) Anti-FLAG immunoblot analysis shows equivalent levels of FLAG-MurJ (wild type [WT]) and FLAG-MurJΔCys C314S C419S (ΔCys) in their respective haploid strains (ΔmurJ carrying pFLAGMurJ plasmids). The slower mobility of the ΔCys derivative is caused by the C314S substitution. To test sample loading, blots were reprobed with anti-LptB antiserum. (B) Haploid ΔmurJ strains complemented with pFLAGMurJ or pFLAGMurJΔCys exhibit no growth defects in LB compared to the growth of a wild-type strain (murJ+).
Detection of FLAG-MurJ by immunoblotting.
Samples were either prepared as described above for the SCAM experiments or as follows for determining the levels of loss-of-function mutants. After overnight growth, cells were normalized by the OD600, collected by centrifugation, resuspended in 100 μl of 1× AB buffer containing 1 μl of Benzonase (Novagen), and incubated for 30 min at 45°C. Samples were loaded onto a 10% SDS-polyacrylamide gel and subjected to electrophoresis. Proteins were electrophoretically transferred from the gel to nitrocellulose membranes in a semidry transfer apparatus (Bio-Rad). Membranes were incubated with anti-FLAG M2 (1:20,000; Sigma-Aldrich) and antimouse-horseradish peroxidase (HRP; 1:10,000; GE Amersham) antibodies. As controls for loading, these membranes were blotted with anti-LptB (1:50,000; our laboratory collection) and antirabbit-HRP (1:10,000, GE Amersham) antibodies. The signal was developed with a VisiGlo Plus HRP chemiluminescent substrate kit (Amresco) and detected using a ChemiDoc XRS+ system (Bio-Rad).
In silico modeling.
The E. coli MurJ amino acid sequence (GenBank accession no. NP_415587) was submitted to the I-TASSER server (47, 48). The model presented here was obtained from a 14 April 2013 submission. Figures were prepared from Protein Data Bank (PDB) files downloaded from the I-TASSER server using the PyMOL molecular graphics system, version 1.5.0.4 (Schrödinger, LLC, Portland, OR). Solvent-accessible electrostatic potential was calculated using the adaptive Poisson-Boltzmann solver (APBS) program (49).
The E. coli MurJ amino acid sequence was also submitted to the TOPPRED (50), HMMTOP (51), TMHMM (52), and SOSUI (53) servers for topological predictions and MINNOU (54) for TMD predictions.
RESULTS
In silico structural model of MurJ.
We want to understand the essential function that MurJ has in PG biogenesis (14, 16). Because of the strong relationship between protein structure and function, we first focused on obtaining a structural analysis of MurJ that could guide and complement functional studies. We also reasoned that if MurJ is a lipid II flippase, as it has been proposed (14, 16), its structure might contain features supportive of this function.
Recent advances in computational biology are facilitating structural modeling efforts (55). Therefore, we investigated whether obtaining an in silico structural model of MurJ was possible using the I-TASSER server, which was ranked the number one server for structural prediction in a recent Critical Assessment of Techniques for Protein Structure Prediction competitions (CASP7-10) (47, 48). The best structural model that I-TASSER generated for MurJ (Fig. 2) had a confidence score (C) of −1.08 (C scores range from −5 to 2; the higher that the score is, the better that the quality of the model is) and a template modeling (TM) score of 0.58 (a TM score of >0.5 indicates a model of correct topology) (48, 56). The top 10 templates used in modeling are 5 regions of PfMATE from Pyrococcus furiosus (PDB accession no. 3VVN), 3 regions of NorM-VC from Vibrio cholerae (PDB accession no. 3MKT), 1 region of the PR65/A subunit of protein phosphatase 2A (PDB accession no. 1B3U), and 1 region of NorM-NG from Neisseria gonorrhoeae (PDB accession no. 4HUK) (57–60). In these alignments, the normalized Z scores, which reflect the quality of the threading alignments, with a value of >1 being a confident alignment (47), were 1.06 to 3.29 for PfMATE, 1.25 to 9.40 for NorM-VC, 2.36 for PR65/A, and 1.08 for NorM-NG.
Fig 2.
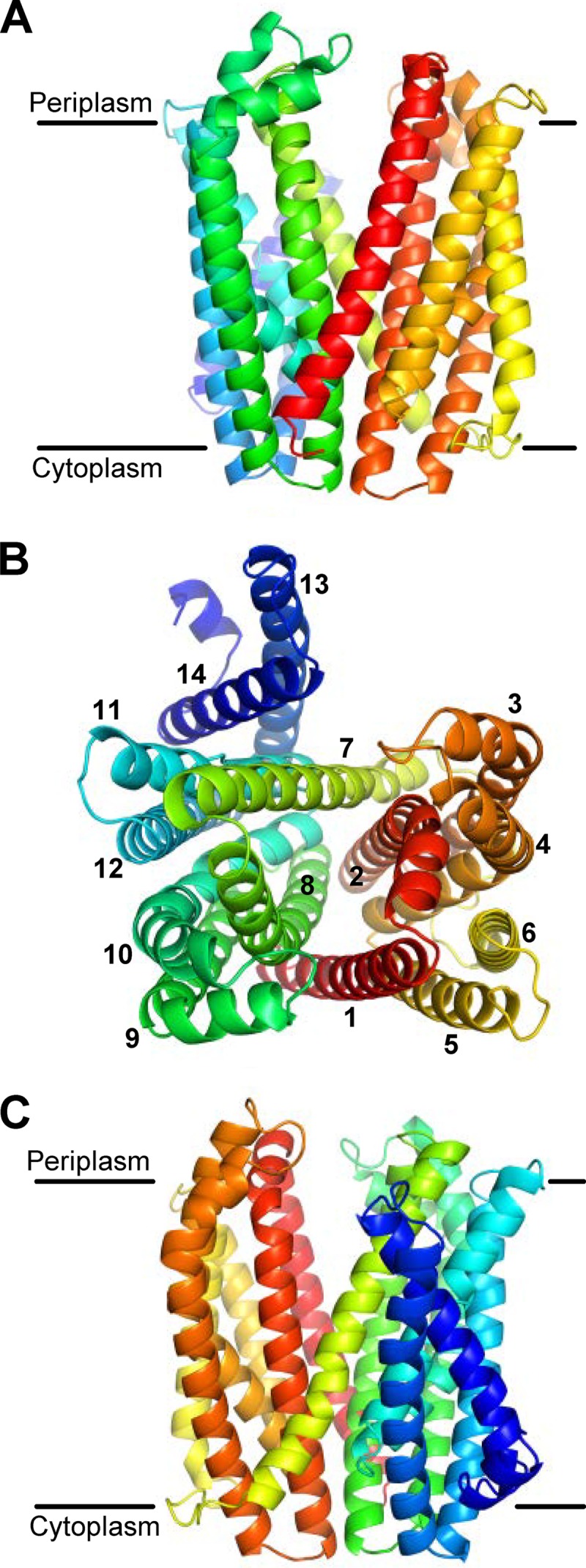
I-TASSER model structure of MurJ. (A) Front view from the membrane plane. (B) View from the periplasm with TMDs numbered 1 to 14. I-TASSER predicts a structure for TMDs 1 to 12 modeled after the crystal structures of MOP exporters; the last two C-terminal α helices are demonstrated herein to comprise TMDs 13 and 14. (C) Back view from the membrane plane. The molecule is colored from red (N terminus) to blue (C terminus) using a rainbow spectrum. Approximations of the membrane boundaries are marked with black lines.
The best structural analogs identified by I-TASSER with TM scores of >0.5 were PfMATE (TM = 0.83), NorM-VC (TM = 0.77), and NorM-NG (TM = 0.74). These structural analogs have 459 to 461 amino acids, while MurJ has 511 amino acids; in all three cases, the solved MATE protein structures align with the first 86% (i.e., up to amino acid 439) of the MurJ model structure. Even though this region of MurJ and these proteins share only ca. 21% amino acid sequence identity (see Fig. S2 in the supplemental material), alignments of the MurJ model and these crystal structures show high degrees of structural similarity (see Fig. S3 in the supplemental material). Noteworthy is the fact that although PfMATE and NorM-VC share only approximately 22% sequence identity, their structures are very similar (57).
PfMATE and NorM are members of the MATE family of proteins, which, as described above, belongs to the MOP exporter superfamily (31). Both transporters translocate hydrophobic and amphipathic toxic compounds across the cytoplasmic membrane (32), but while PfMATE is H+ driven, most characterized NorM proteins are Na+ driven (57, 58, 60, 61). Their respective X-ray crystal structures reveal that PfMATE and NorM have 12 TMDs organized into two 6-helix bundles (TMDs 1 to 6 and TMDs 7 to 12, respectively) forming a V-shaped structure containing a central cavity open to the periplasm and two lateral portals open into the membrane (57, 58, 60). In addition, several structures have revealed binding of these proteins to substrates and cations. Models for antiport transport through this cavity have been developed from structures and functional data (57, 58, 60).
In the MurJ structural model, amino acids 1 to 439 of MurJ align with the PfMATE structure representing the outward-open state (see Fig. S3 in the supplemental material) (57). Accordingly, this portion of the model includes 12 TMDs arranged into two 6-helix bundles that form a V-shaped structure with a central cavity and two lateral portals (Fig. 2; see Fig. S3 in the supplemental material). With respect to the last 71 amino acids of MurJ (amino acids 440 to 511), which do not align with PfMATE, the I-TASSER model shows them folded into two additional α helices that could potentially constitute two TMDs (Fig. 2). The central cavity in the MurJ model structure is within the predicted transmembrane region of the protein, and it is lined by four α helices that contain putative TMDs 1, 2, 7, and 8 (Fig. 2B). Therefore, I-TASSER predicts that MurJ has 12 to 14 TMDs arranged into a structure highly similar to that of MATE exporters, suggesting a role for MurJ in the transport of substrates across the inner membrane of E. coli.
MurJ has 14 TMDs.
To test the validity of the structural model generated by I-TASSER, we first determined the topology of MurJ in silico and in vivo. Various commonly used topology algorithms predict that MurJ has 12 to 14 TMDs (Fig. 3A). All programs tested agree that the C terminus of MurJ is cytoplasmic, and two also predict the existence of 13 TMDs between residues 26 to 503. However, there are some discrepancies among the in silico topological predictions, especially regarding the existence of a TMD within the first 25 amino acids of MurJ. This is likely because this putative TMD contains several charged and polar residues. This case illustrates the importance of using more than one topology prediction algorithm and, ultimately, determining topology experimentally.
Fig 3.

Topological models predict 12 to 14 TMDs in MurJ. (A) Alignment of in silico topology predictions generated from various servers (as noted). Numbered black line, MurJ amino acid positions; black boxes TMDs; blue line, cytoplasmic localization; red line, periplasmic localization. The number of TMDs obtained from each prediction is given at the right. (B) Topological model (top) derived from in vivo MurJ truncation-fusion data (bottom). Domains and the numbers of the TMDs are represented as described for panel A. The enzymatic activity for each tribrid MurJ truncation-reporter fusion was assessed to determine the location of the MurJ amino acid at the fusion joint. Data are presented as the normalized activity ratio (percent AP max/percent BG max; see Materials and Methods for details).
We used two complementary approaches to determine the topology of MurJ experimentally: reporter fusion technology and the substituted cysteine accessibility method (SCAM). Similar approaches have been used for the MOP exporter and O-antigen flippase Wzx (38, 62, 63). For the reporter strategy, we employed a dual-reporter fusion system (39). For this system, we generated plasmid-encoded (38) tribrid proteins by fusing a hybrid protein composed of alkaline phosphatase (AP) and the α fragment of β-galactosidase (LacZα) to different C-terminal truncations of MurJ (MurJTRUNC). Because AP is active only in the periplasm and LacZα is functional only in the cytoplasm, the AP and LacZ activities produced in every MurJTRUNC-AP-LacZα tribrid can be used to determine the topological location of the last amino acid in MurJTRUNC. Specifically, high AP and low LacZ activities reflect periplasmic localization, low AP and high LacZ activities reflect cytoplasmic localization, and low AP and LacZ activities reflect transmembrane localization (64). We constructed 52 reporter fusion alleles that maximized coverage throughout murJ (see Table S1 in the supplemental material). The average distance between adjacent joints was 10 residues, with the location of the fusion joint ultimately being dependent on optimal primer design for plasmid construction (see Table S2 in the supplemental material).
Using this method, we determined that MurJ contains 12 TMDs after residue 80 and that its C terminus is cytoplasmic; both findings are in agreement with three of the four topology server predictions (Fig. 3). Although the MurJ structural model and topology prediction servers predict 1 to 2 TMDs within the first 60 amino acids of MurJ, our reporter fusion data suggested that this region is cytoplasmic (Fig. 2 and 3). This disagreement is likely to result from improper targeting or insertion into the membrane of truncated proteins containing <80 amino acids of the N terminus of MurJ. This targeting/insertion defect might explain why a MurJ variant truncated after residue 80 is unstable (see Fig. S4 in the supplemental material).
The second and complementary method that we used to experimentally determine the topology of MurJ is SCAM, which provides topological information on a specific Cys residue within a protein (64–66). In this approach, cells expressing a collection of mutant alleles each encoding a specific single Cys residue are treated (or not) with one of two Cys-reactive chemicals, NEM and MTSES (66–68). Because NEM crosses the inner membrane but MTSES does not, a cytoplasmic Cys reacts only with NEM, while a periplasmic Cys reacts with both NEM and MTSES. In contrast, a membrane-buried Cys that is inaccessible to the aqueous environment will not react with either chemical. Cells are lysed after being treated (or not) with either NEM or MTSES, and their proteins are denatured and exposed to Mal-PEG, which reacts with free thiols, causing a mass increase that can be detected by immunoblotting. Therefore, a mass increase can be observed only in proteins containing a thiol that was not previously blocked by NEM or MTSES.
Two requirements for using SCAM are that the protein be detectable and initially Cys-less. Therefore, we developed a system that relies on FLAG-MurJΔCys, an N-terminally FLAG-tagged MurJ derivative where the two native Cys residues (C314 and C419) were changed to Ser. The resulting FLAG-MurJΔCys protein is detectable by probing for the FLAG tag in immunoblots and is functional, since it complements the deletion of chromosomal murJ (Fig. 4). A total of 110 different single Cys substitutions in FLAG-MurJΔCys were constructed and analyzed by SCAM using NEM and MTSES (see Table S3 in the supplemental material). We maximized coverage by using in silico topological and structural predictions as a guide. We probed TMDs 1, 2, 7, and 8 at a higher density because of the results that we describe below.
Using SCAM, we determined that MurJ has 14 TMDs and that its N and C termini are in the cytoplasm (Fig. 5). Thus, our data support cytoplasmic localization (100% block with NEM and 0% block with MTSES) and periplasmic localization (100% block with each of NEM and MTSES) of segments flanking all TMDs that had been predicted by topology prediction servers in toto and implied by the MurJ structural model (Fig. 2, 3, and 5; see Table S3 in the supplemental material). The topological model derived from SCAM corresponding to the MurJ sequence encompassing TMDs 3 to 14 also correlated with the topological model derived from the reporter fusion approach (see Fig. S5A in the supplemental material).
Membranes are composed of an ∼30-Å hydrophobic domain flanked by two 15-Å interface domains; a challenge in topological studies is to determine the boundaries of these domains. To obtain our topology model (Fig. 5), we combined our in vivo data, the known frequency of occurrence at a particular depth in the lipid bilayer for each particular amino acid (69), and the MurJ structural model. Figure S5B in the supplemental material highlights the transmembrane core of our topology model on the three-dimensional (3-D) model.
A comparison of our topology model to in silico topology predictors revealed that SOSUI was most similar to our model (see Table S4 in the supplemental material) (53). Most of the disagreements between our model and the topology predictors occurred in TMDs 1 to 4 and 7 to 9. This is likely because, in the model structure, these TMDs contain 27 to 35 amino acids owing to the predicted tilting and bending of their helices (see Table S4 in the supplemental material); therefore, their length is longer than that of a canonical ∼20-residue TMD (70), a parameter that constrains some of these algorithms, such as 20 amino acids for TOPPRED and 25 amino acids for HMMTOP and TMHMM (50, 52, 71, 72). It is worth noting that although the MINNOU server does not provide information regarding the orientation of TMDs with respect to the membrane, the model predicted by this algorithm for the location of TMDs correlated best with our model (see Table S4 in the supplemental material) (54).
MurJ contains a solvent-exposed cavity within the membrane region.
A key feature of the MurJ structural model is that it contains a central cavity mostly lined by TMDs 1, 2, 7, and 8 (Fig. 2). We obtained SCAM results that support this model, since many residues located within the plane of the membrane in these TMDs react with MTSES and/or NEM (Fig. 5; see Fig. S6 and Table S3 in the supplemental material). Specifically, membrane-permeant NEM reacted with residues in TMDs 1, 2, 4, 5, 7, 8, and 11.
When mapped onto the MurJ structural model, residues that fully reacted with NEM were located as follows: (i) facing the cavity along TMDs 1, 2, 7, and 8; (ii) at the cytoplasmic end of TMDs 4 and 5; (iii) in TMDs 7 and 11, where both helices face each other; and (iv) in TMD 1 in a region facing TM5 (see Fig. S6B and C and Table S3 in the supplemental material). Of these, some residues also fully reacted with membrane-impermeant MTSES (see Fig. S6B and C and Table S3 in the supplemental material). These residues are located in the MurJ structural model in TMDs 1, 2, 7, and 8 and at a single location (S276) in TMD 8 facing TMD 10. Together, these SCAM results validate the I-TASSER structural model in three ways. First, residues that react with both NEM and MTSES concentrate with respect to the membrane on the top half of the 3-D model, while those that react only with NEM concentrate in the bottom half (see Fig. S6C in the supplemental material); this would be expected of a correct model, since NEM reaches the cytoplasm but MTSES does not. Second, these SCAM results confirm the existence of the central cavity predicted by the MurJ structural model. Third, they also confirm that this cavity is mainly lined by TMDs 1, 2, 7, and 8 (Fig. 2).
We found additional labeling patterns that probably reflect the differential accessibility of certain residues to these Cys-reacting reagents. Sixteen residues were fully labeled with NEM but only partially with MTSES (see Fig. S6B and C and Table S3 in the supplemental material). Because MTSES (MW, 242.27) is larger than NEM (MW, 125.13) and charged, we interpret these results to mean that these residues are not as accessible to the aqueous periplasm as those that are fully labeled with MTSES. In the MurJ structural model, these residues are located as follows: (i) within TMDs 1, 2, and 8, facing the cavity; (ii) in TMD 2 facing TMDs 3 and 4; (iii) at the membrane interface in the periplasmic end of TMD 3; and (iv) in the cytoplasmic end of TM7, facing the cavity (residue 228) and possibly delineating the limit of MTSES accessibility within the cavity (<50% MTSES reactivity). It is noteworthy that 9 of these 16 residues that fully reacted with NEM but that only partially reacted with MTSES are located in TMD 2, even in locations that face away from the cavity. This set of data suggests that TMD 2 might move within the core of MurJ, as this has been proposed for various TMDs lining the central cavity of MATE transporters (57, 60).
In addition, two residues in TMD 8 (A268C and R270C) reacted partially with both MTSES and NEM and showed decreased reactivity when blocked with either reagent for 5 min versus 1 h, suggesting minimal accessibility to the aqueous environment. The MurJ structural model shows that A268 faces away from the cavity and toward TMD 10, which could explain the partial reactivity; however, it is not clear what could prevent reactivity with R270C, since this residue faces the cavity. We also found 15 residues that only partially reacted with NEM (see Fig. S6B and C and Table S3 in the supplemental material). Interestingly, in the MurJ structural model, none of these residues face the central cavity. Instead, most are located in the top half of the protein (with respect to the membrane), and with the exception of 3 residues in TMD 1 (facing the membrane) and one in TMD 3 (at the periplasmic end at the membrane interface), they are dispersed along TMDs 7 to 12.
In agreement with the idea that partial reactivity or blocking with NEM and/or MTSES reflects limited accessibility to the aqueous environment, we observed an increase in blocking when incubation with these agents was extended from 5 min to 45 min for residues 109, 249, 268, 270, 272, 350, 387, and 389. With the exception of residue 109 (in TMD 3), these residues are in TMDs 7, 8, 10, and 11. Most of these residues face away from the cavity but are located near the predicted periplasm-membrane interface in the model, which could explain the observed reduced reactivity. Alternatively, it is possible that reduced reactivity reflects partial movement of helices within MurJ. In contrast, we cannot explain why NEM reaches residue 389 over time, since it is predicted to be facing the membrane.
Interestingly, we found that MTSES blocking of Cys residues at positions 14, 57, and 281 decreases with time, indicating loss of the MTSES label. Strikingly, these residues (T14, E57, and V281 in TMDs 1, 2, and 8, respectively) are positioned near the bottom of the V-shaped cavity. Since MTSES forms a disulfide bond by reacting with the thiol of a Cys residue, MTSES blocking is sensitive to reducing agents. It is therefore possible that the loss of the MTSES label with time reflects exposure of these residues to the reducing environment of the cytoplasm, as suggested by their location in the MurJ structural model.
In summary, data generated by SCAM demonstrate that the MurJ structure contains a central cavity. The pattern of reactivity and the degree of accessibility further validate the in silico 3-D model presented here.
The central cavity in MurJ contains charged residues essential for function.
The similarity of the MurJ structural model to the crystal structure of known MATE transporters suggests that MurJ might also function similarly to MATE transporters. Because the central cavity is a feature of MATE transporters that is key for their function (32), we predicted that if MurJ functions as a transporter, we might find residues within its cavity that are functionally important.
We tested the functionality of the 110 flag-murJ alleles generated for SCAM and identified both total- and partial-loss-of-function alleles. Total-loss-of-function alleles were identified by their inability to complement the deletion of chromosomal murJ. When plasmids encoding nonfunctional flag-murJ alleles were introduced into a ΔmurJ/pRC7MurJ strain, only those cells that retained pRC7MurJ survived (see Materials and Methods for details). These alleles encode Cys substitutions for charged residues located in TMD 1 (R18 and R24), periplasmic loop 1 (D39), TMD 2 (R52), and TMD 8 (R270). Detection by immunoblotting to the FLAG tag was significantly reduced only for the D39C variant (Fig. 6A), indicating that changes in residues R18, R24, R52, and R270 affect MurJ activity. Whether residue D39 is required only for structural stability or also for function remains to be determined. To test whether these effects on MurJ function were specific to replacements with Cys, we also constructed derivatives with replacements with Ala. Like those with Cys substitutions, MurJ variants with an Ala residue at positions R18, R24, D39, or R270 were nonfunctional. For R52, while a replacement with Cys rendered MurJ nonfunctional, a replacement with Ala rendered it partially functional, according to the high sensitivity to low osmolarity exhibited by a haploid strain expressing this R52A derivative (determined as described below; data not shown). Clearly, R52 plays a key role in MurJ function, but additional studies are required to determine the specific requirements at this position.
Fig 6.

Detection of FLAG-MurJ variants bearing Cys substitutions that affect MurJ function. (A) Anti-FLAG immunoblot analysis of a wild-type (murJ+) strain with pFLAGMurJΔCys variants expressing total-loss-of-function single-Cys murJ alleles revealed that only the D39C change results in lowers levels of detection of FLAG-MurJ derivatives than of the parent allele flag-murJΔCys (wild type). (B) Anti-FLAG immunoblot showing that haploid (ΔmurJ/pFLAGMurJΔCys) strains with pFLAGMurJΔCys variants expressing partial loss-of-function single-Cys murJ alleles contain lower levels of FLAG-MurJ derivatives than of the parent allele flag-murJΔCys (wild type). To test sample loading, blots were reprobed with anti-LptB antiserum.
To identify mutants with altered MurJ function among those carrying a complementing flag-murJ allele, we screened haploid strains carrying single Cys substitutions in pFLAGMurJΔCys for growth defects in low-osmolarity medium since the PG sacculus protects cells from osmotic lysis. Among the 105 mutants tested, we found 3 with increased sensitivity to low osmolarity (Table 1). These alleles encode changes in TMD 2 (K46) and TMD 8 (D269 and E273), and they are partial-loss-of-function alleles because they are recessive to wild-type murJ. Similar phenotypes were observed when these residues were changed to Ala instead of Cys. We also found that the K46C substitution resulted in a phenotype milder than the phenotypes caused by the D269C and E273C substitutions (Table 1) and that each of these changes decreased detection of their respective FLAG-MurJ variants, with the D269C and E273C substitutions causing a stronger reduction in detection than the K46C substitution (Fig. 6B).
Table 1.
Sensitivity of selected mutants to low-osmolarity medium
| Amino acid change | TMD | EOPa |
|
|---|---|---|---|
| LB | YT | ||
| None | 1.00 ± 0.00 | 0.70 ± 0.52 | |
| K46C | 2 | 0.40 ± 0.52 | 0.01 ± 0.00 |
| D269C | 8 | 0.70 ± 0.52 | <10−5 |
| E273C | 8 | 4.00 ± 5.20 | <10−5 |
EOP values for haploid strains with flag-murJ alleles expressing single Cys substitutions were calculated as described in Materials and Methods.
Together, these functional data show that after changing 110 residues in MurJ to Cys, we detected significant phenotypes only in specific charged residues located in TMDs 1, 2, and 8 and the periplasmic loop that connects TMD 1 and TMD 2. These regions are integral components of the solvent-exposed cavity that we have identified through SCAM (Fig. 5). Therefore, our in vivo data demonstrate an essential role of this central cavity in MurJ structure and function.
DISCUSSION
The inner membrane protein MurJ is essential for PG biogenesis and, therefore, the viability of E. coli (14, 16). Here we have combined in silico and in vivo approaches to generate the first structural and topological model of MurJ. Our results demonstrate that MurJ has 14 TMDs and suggest that the first 12 TMDs form a structure highly similar to the crystal structure of transporters of the MOP exporter superfamily (31, 57, 58, 60). A key feature of this model structure is a central, solvent-exposed cavity that extends into the hydrophobic core of the membrane. Importantly, we present further in vivo evidence supporting the suggestion that this cavity contains several charged residues essential for MurJ function. Together, our data suggest a model where MurJ is a transporter essential for PG biogenesis.
Obtaining high-resolution structural information for cytoplasmic membrane proteins composed of multiple TMDs is difficult. Nevertheless, in recent years the number of membrane proteins that have been crystallized has increased, and this structural information has aided the development and improvement of algorithms that generate de novo topological and structural models (55). These models are crucial for understanding the biogenesis, function, and structure of these proteins. A dilemma that investigators face is which of these in silico modeling algorithms to use. Our studies with MurJ illustrate how crucial it is that investigators use more than one topology prediction algorithm and that they also conduct topological studies in vivo to ultimately develop topological models. In cases where structural homologs have been crystallized, a structural prediction algorithm, like I-TASSER, can be extremely powerful in developing such models (47).
In these studies, we obtained a high-confidence structural model of MurJ with I-TASSER because three proteins that also belong to the MOP exporter superfamily were crystallized (31, 47, 57, 58, 60). We confirmed in vivo important features of this proposed structure. We showed that MurJ has 14 TMDs, which was previously suggested to be a feature that makes MurJ differ from most 12-TMD MOP exporters (31). The relevance of the additional TMDs remains to be elucidated (see below). Importantly, we demonstrated using SCAM the existence of a central, solvent-exposed cavity lined by TMDs 1, 2, 7, and 8. This type of architecture is conserved among crystallized MATE proteins (57, 58, 60) and proposed in a structural model for the O-antigen flippase Wzx (62), despite the low level of primary sequence homology among these proteins. The Wzx proteins belong to the polysaccharide transporter (PST) family of proteins; notably, among the MOP exporter superfamily, MurJ is most related to the PST and MATE families, with the greatest sequence similarity being to PST proteins (31). Based on this homology and the fact that the central cavity of MurJ contains several polar and charged residues, some of which are essential for function, we propose a model where MurJ is a transporter that uses its central cavity as a conduit to translocate, across the cytoplasmic membrane, a substrate containing a hydrophilic moiety.
The crystal structures of MATE proteins have revealed that the outward-opened cavity in each of these exporters contains two side portals that open into the outer leaflet of the cytoplasmic membrane. This architecture suggests a model where amphipathic substrates might exit the cavity and enter the membrane after transport (57, 58, 60). Analogous side portals at the cytoplasmic face might allow substrate engagement into the transporter cavity for export; however, a high-resolution structure in the inward-opened state is not available. The structural model of MurJ, which is in the outward-opened conformation, also predicts two side portals that open into the periplasmic leaflet of the membrane. However, it is possible that TMDs 13 and 14, which are absent in MATE proteins, are not properly oriented in this structural model with respect to the remaining MATE-like structural component and they might occlude one of these portals.
There are two pressing questions regarding the function of MurJ as a transporter. What is the energy source driving transport, and what is the substrate(s) that MurJ transports? Regarding the energy source, it is likely that MurJ is a secondary active antiporter that utilizes the electrochemical potential of either Na+ or H+, since this is the mechanism that powers other members of the MOP exporter superfamily (5, 31, 32). Regarding the substrate(s), the structural model predicts that the central cavity in MurJ has an overall positive charge (see Fig. S7 in the supplemental material). Interestingly, Wzx has a cationic lumen that mediates the translocation of its anionic substrate, the O antigen (62). Our functional data show the essentiality of some positively charged residues located in the MurJ cavity, but it remains to be determined whether the cationic nature of its lumen reflects the ability of MurJ to transport a negatively charged substrate, including lipid II (see Fig. S1 in the supplemental material). Nevertheless, the work presented here suggests that MurJ is a transporter; therefore, we continue to favor a model for PG biogenesis where MurJ functions as a lipid II flippase (14, 16). Future work is needed to test this model and elucidate the essential role of MurJ in PG biogenesis.
Supplementary Material
ACKNOWLEDGMENTS
We thank Joseph Lam and Thomas Bernhardt for providing pPLE01 and pRC7, respectively, and Lea Murphy for helping with the construction of pPLE01 derivatives.
This research was supported by funds from The Ohio State University (to N.R.) and the National Institute of General Medical Sciences of the National Institutes of Health under award number R01GM100951 (to N.R.).
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Published ahead of print 9 August 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JB.00731-13.
REFERENCES
- 1.Silhavy TJ, Kahne D, Walker S. 2010. The bacterial cell envelope. Cold Spring Harb. Perspect. Biol. 2:a000414. 10.1101/cshperspect.a000414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Vollmer W, Bertsche U. 2008. Murein (peptidoglycan) structure, architecture and biosynthesis in Escherichia coli. Biochim. Biophys. Acta 1778:1714–1734 [DOI] [PubMed] [Google Scholar]
- 3.Silver LL. 2013. Viable screening targets related to the bacterial cell wall. Ann. N. Y. Acad. Sci. 1277:29–53 [DOI] [PubMed] [Google Scholar]
- 4.van Heijenoort J. 2007. Lipid intermediates in the biosynthesis of bacterial peptidoglycan. Microbiol. Mol. Biol. Rev. 71:620–635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Islam ST, Lam JS. 2013. Wzx flippase-mediated membrane translocation of sugar polymer precursors in bacteria. Environ. Microbiol. 15:1001–1015 [DOI] [PubMed] [Google Scholar]
- 6.Whitfield C. 2006. Biosynthesis and assembly of capsular polysaccharides in Escherichia coli. Annu. Rev. Biochem. 75:39–68 [DOI] [PubMed] [Google Scholar]
- 7.Sanyal S, Menon AK. 2009. Flipping lipids: why an' what's the reason for? ACS Chem. Biol. 4:895–909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cuthbertson L, Mainprize IL, Naismith JH, Whitfield C. 2009. Pivotal roles of the outer membrane polysaccharide export and polysaccharide copolymerase protein families in export of extracellular polysaccharides in gram-negative bacteria. Microbiol. Mol. Biol. Rev. 73:155–177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Whitney JC, Howell PL. 2013. Synthase-dependent exopolysaccharide secretion in Gram-negative bacteria. Trends Microbiol. 21:63–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cuthbertson L, Kos V, Whitfield C. 2010. ABC transporters involved in export of cell surface glycoconjugates. Microbiol. Mol. Biol. Rev. 74:341–362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Greenfield LK, Whitfield C. 2012. Synthesis of lipopolysaccharide O-antigens by ABC transporter-dependent pathways. Carbohydr. Res. 356:12–24 [DOI] [PubMed] [Google Scholar]
- 12.Umbreit JN, Strominger JL. 1972. Isolation of the lipid intermediate in peptidoglycan biosynthesis from Escherichia coli. J. Bacteriol. 112:1306–1309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ehlert K, Holtje JV. 1996. Role of precursor translocation in coordination of murein and phospholipid synthesis in Escherichia coli. J. Bacteriol. 178:6766–6771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Inoue A, Murata Y, Takahashi H, Tsuji N, Fujisaki S, Kato J. 2008. Involvement of an essential gene, mviN, in murein synthesis in Escherichia coli. J. Bacteriol. 190:7298–7301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mohammadi T, van Dam V, Sijbrandi R, Vernet T, Zapun A, Bouhss A, Diepeveen-de Bruin M, Nguyen-Disteche M, de Kruijff B, Breukink E. 2011. Identification of FtsW as a transporter of lipid-linked cell wall precursors across the membrane. EMBO J. 30:1425–1432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ruiz N. 2008. Bioinformatics identification of MurJ (MviN) as the peptidoglycan lipid II flippase in Escherichia coli. Proc. Natl. Acad. Sci. U. S. A. 105:15553–15557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ikeda M, Sato T, Wachi M, Jung HK, Ishino F, Kobayashi Y, Matsuhashi M. 1989. Structural similarity among Escherichia coli FtsW and RodA proteins and Bacillus subtilis SpoVE protein, which function in cell division, cell elongation, and spore formation, respectively. J. Bacteriol. 171:6375–6378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Henriques AO, Glaser P, Piggot PJ, Moran CP., Jr 1998. Control of cell shape and elongation by the rodA gene in Bacillus subtilis. Mol. Microbiol. 28:235–247 [DOI] [PubMed] [Google Scholar]
- 19.Ishino F, Jung HK, Ikeda M, Doi M, Wachi M, Matsuhashi M. 1989. New mutations fts-36, lts-33, and ftsW clustered in the mra region of the Escherichia coli chromosome induce thermosensitive cell growth and division. J. Bacteriol. 171:5523–5530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Iwaya M, Jones CW, Khorana J, Strominger JL. 1978. Mapping of the mecillinam-resistant, round morphological mutants of Escherichia coli. J. Bacteriol. 133:196–202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Khattar MM, Begg KJ, Donachie WD. 1994. Identification of FtsW and characterization of a new ftsW division mutant of Escherichia coli. J. Bacteriol. 176:7140–7147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Matsuzawa H, Hayakawa K, Sato T, Imahori K. 1973. Characterization and genetic analysis of a mutant of Escherichia coli K-12 with rounded morphology. J. Bacteriol. 115:436–442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tamaki S, Matsuzawa H, Matsuhashi M. 1980. Cluster of mrdA and mrdB genes responsible for the rod shape and mecillinam sensitivity of Escherichia coli. J. Bacteriol. 141:52–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mercer KL, Weiss DS. 2002. The Escherichia coli cell division protein FtsW is required to recruit its cognate transpeptidase, FtsI (PBP3), to the division site. J. Bacteriol. 184:904–912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ishino F, Park W, Tomioka S, Tamaki S, Takase I, Kunugita K, Matsuzawa H, Asoh S, Ohta T, Spratt BG, Matsuhashi M. 1986. Peptidoglycan synthetic activities in membranes of Escherichia coli caused by overproduction of penicillin-binding protein 2 and RodA protein. J. Biol. Chem. 261:7024–7031 [PubMed] [Google Scholar]
- 26.Lara B, Ayala JA. 2002. Topological characterization of the essential Escherichia coli cell division protein FtsW. FEMS Microbiol. Lett. 216:23–32 [DOI] [PubMed] [Google Scholar]
- 27.Pastoret S, Fraipont C, den Blaauwen T, Wolf B, Aarsman ME, Piette A, Thomas A, Brasseur R, Nguyen-Disteche M. 2004. Functional analysis of the cell division protein FtsW of Escherichia coli. J. Bacteriol. 186:8370–8379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lara B, Mengin-Lecreulx D, Ayala JA, van Heijenoort J. 2005. Peptidoglycan precursor pools associated with MraY and FtsW deficiencies or antibiotic treatments. FEMS Microbiol. Lett. 250:195–200 [DOI] [PubMed] [Google Scholar]
- 29.Huber J, Donald RG, Lee SH, Jarantow LW, Salvatore MJ, Meng X, Painter R, Onishi RH, Occi J, Dorso K, Young K, Park YW, Skwish S, Szymonifka MJ, Waddell TS, Miesel L, Phillips JW, Roemer T. 2009. Chemical genetic identification of peptidoglycan inhibitors potentiating carbapenem activity against methicillin-resistant Staphylococcus aureus. Chem. Biol. 16:837–848 [DOI] [PubMed] [Google Scholar]
- 30.Ruiz N. 2009. Streptococcus pyogenes YtgP (Spy_0390) complements Escherichia coli strains depleted of the putative peptidoglycan flippase MurJ. Antimicrob. Agents Chemother. 53:3604–3605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hvorup RN, Winnen B, Chang AB, Jiang Y, Zhou XF, Saier MH., Jr 2003. The multidrug/oligosaccharidyl-lipid/polysaccharide (MOP) exporter superfamily. Eur. J. Biochem. 270:799–813 [DOI] [PubMed] [Google Scholar]
- 32.Kuroda T, Tsuchiya T. 2009. Multidrug efflux transporters in the MATE family. Biochim. Biophys. Acta 1794:763–768 [DOI] [PubMed] [Google Scholar]
- 33.Yu D, Ellis HM, Lee EC, Jenkins NA, Copeland NG, Court DL. 2000. An efficient recombination system for chromosome engineering in Escherichia coli. Proc. Natl. Acad. Sci. U. S. A. 97:5978–5983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ruiz N, Gronenberg LS, Kahne D, Silhavy TJ. 2008. Identification of two inner-membrane proteins required for the transport of lipopolysaccharide to the outer membrane of Escherichia coli. Proc. Natl. Acad. Sci. U. S. A. 105:5537–5542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Casadaban MJ. 1976. Transposition and fusion of the lac genes to selected promoters in Escherichia coli using bacteriophage lambda and Mu. J. Mol. Biol. 104:541–555 [DOI] [PubMed] [Google Scholar]
- 36.Silhavy TJ, Berman ML, Enquist LW. 1984. Experiments with gene fusions. Cold Spring Harbor Laboratory, Cold Spring Harbor, NY [Google Scholar]
- 37.de Boer PAJ, Crossley RE, Rothfield LI. 1989. A division inhibitor and a topological specificity factor coded for by the minicell locus determine proper placement of the division septum in E. coli. Cell 56:641–649 [DOI] [PubMed] [Google Scholar]
- 38.Islam ST, Taylor VL, Qi M, Lam JS. 2010. Membrane topology mapping of the O-antigen flippase (Wzx), polymerase (Wzy), and ligase (WaaL) from Pseudomonas aeruginosa PAO1 reveals novel domain architectures. mBio 1(3):e00189–10. 10.1128/mBio.00189-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Alexeyev MF, Winkler HH. 1999. Membrane topology of the Rickettsia prowazekii ATP/ADP translocase revealed by novel dual pho-lac reporters. J. Mol. Biol. 285:1503–1513 [DOI] [PubMed] [Google Scholar]
- 40.Kitagawa M, Ara T, Arifuzzaman M, Ioka-Nakamichi T, Inamoto E, Toyonaga H, Mori H. 2005. Complete set of ORF clones of Escherichia coli ASKA library (a complete set of E. coli K-12 ORF archive): unique resources for biological research. DNA Res. 12:291–299 [DOI] [PubMed] [Google Scholar]
- 41.Datsenko KA, Wanner BL. 2000. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. U. S. A. 97:6640–6645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cherepanov PP, Wackernagel W. 1995. Gene disruption in Escherichia coli: TcR and KmR cassettes with the option of Flp-catalyzed excision of the antibiotic-resistance determinant. Gene 158:9–14 [DOI] [PubMed] [Google Scholar]
- 43.Baba T, Ara T, Hasegawa M, Takai Y, Okumura Y, Baba M, Datsenko KA, Tomita M, Wanner BL, Mori H. 2006. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol. Syst. Biol. 2:2006.0008. 10.1038/msb4100050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Manoil C. 1991. Analysis of membrane protein topology using alkaline phosphatase and beta-galactosidase gene fusions. Methods Cell Biol. 34:61–75 [DOI] [PubMed] [Google Scholar]
- 45.Miller JH. 1972. Experiments in molecular genetics. Cold Spring Harbor Laboratory, Cold Spring Harbor, NY [Google Scholar]
- 46.Beitz E. 2000. T(E)Xtopo: shaded membrane protein topology plots in LAT(E)X2epsilon. Bioinformatics 16:1050–1051 [DOI] [PubMed] [Google Scholar]
- 47.Roy A, Kucukural A, Zhang Y. 2010. I-TASSER: a unified platform for automated protein structure and function prediction. Nat. Protoc. 5:725–738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhang Y. 2008. I-TASSER server for protein 3D structure prediction. BMC Bioinformatics 9:40. 10.1186/1471-2105-9-40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Baker NA, Sept D, Joseph S, Holst MJ, McCammon JA. 2001. Electrostatics of nanosystems: application to microtubules and the ribosome. Proc. Natl. Acad. Sci. U. S. A. 98:10037–10041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Claros MG, von Heijne G. 1994. TopPred II: an improved software for membrane protein structure predictions. Comput. Appl. Biosci. 10:685–686 [DOI] [PubMed] [Google Scholar]
- 51.Tusnady GE, Simon I. 2001. The HMMTOP transmembrane topology prediction server. Bioinformatics 17:849–850 [DOI] [PubMed] [Google Scholar]
- 52.Krogh A, Larsson B, von Heijne G, Sonnhammer EL. 2001. Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J. Mol. Biol. 305:567–580 [DOI] [PubMed] [Google Scholar]
- 53.Hirokawa T, Boon-Chieng S, Mitaku S. 1998. SOSUI: classification and secondary structure prediction system for membrane proteins. Bioinformatics 14:378–379 [DOI] [PubMed] [Google Scholar]
- 54.Cao B, Porollo A, Adamczak R, Jarrell M, Meller J. 2006. Enhanced recognition of protein transmembrane domains with prediction-based structural profiles. Bioinformatics 22:303–309 [DOI] [PubMed] [Google Scholar]
- 55.Helles G. 2008. A comparative study of the reported performance of ab initio protein structure prediction algorithms. J. R. Soc. Interface 5:387–396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Xu J, Zhang Y. 2010. How significant is a protein structure similarity with TM-score = 0.5? Bioinformatics 26:889–895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tanaka Y, Hipolito CJ, Maturana AD, Ito K, Kuroda T, Higuchi T, Katoh T, Kato HE, Hattori M, Kumazaki K, Tsukazaki T, Ishitani R, Suga H, Nureki O. 2013. Structural basis for the drug extrusion mechanism by a MATE multidrug transporter. Nature 496:247–251 [DOI] [PubMed] [Google Scholar]
- 58.He X, Szewczyk P, Karyakin A, Evin M, Hong WX, Zhang Q, Chang G. 2010. Structure of a cation-bound multidrug and toxic compound extrusion transporter. Nature 467:991–994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Groves MR, Hanlon N, Turowski P, Hemmings BA, Barford D. 1999. The structure of the protein phosphatase 2A PR65/A subunit reveals the conformation of its 15 tandemly repeated HEAT motifs. Cell 96:99–110 [DOI] [PubMed] [Google Scholar]
- 60.Lu M, Symersky J, Radchenko M, Koide A, Guo Y, Nie R, Koide S. 2013. Structures of a Na+-coupled, substrate-bound MATE multidrug transporter. Proc. Natl. Acad. Sci. U. S. A. 110:2099–2104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Rouquette-Loughlin C, Dunham SA, Kuhn M, Balthazar JT, Shafer WM. 2003. The NorM efflux pump of Neisseria gonorrhoeae and Neisseria meningitidis recognizes antimicrobial cationic compounds. J. Bacteriol. 185:1101–1106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Islam ST, Fieldhouse RJ, Anderson EM, Taylor VL, Keates RA, Ford RC, Lam JS. 2012. A cationic lumen in the Wzx flippase mediates anionic O-antigen subunit translocation in Pseudomonas aeruginosa PAO1. Mol. Microbiol. 84:1165–1176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Marolda CL, Li B, Lung M, Yang M, Hanuszkiewicz A, Rosales AR, Valvano MA. 2010. Membrane topology and identification of critical amino acid residues in the Wzx O-antigen translocase from Escherichia coli O157:H4. J. Bacteriol. 192:6160–6171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Islam ST, Lam JS. 2013. Topological mapping methods for alpha-helical bacterial membrane proteins—an update and a guide. Microbiologyopen 2:350–364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bogdanov M, Zhang W, Xie J, Dowhan W. 2005. Transmembrane protein topology mapping by the substituted cysteine accessibility method (SCAM(TM)): application to lipid-specific membrane protein topogenesis. Methods 36:148–171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Karlin A, Akabas MH. 1998. Substituted-cysteine accessibility method. Methods Enzymol. 293:123–145 [DOI] [PubMed] [Google Scholar]
- 67.Kuwabara N, Inoue H, Tsuboi Y, Nakamura N, Kanazawa H. 2004. The fourth transmembrane domain of the Helicobacter pylori Na+/H+ antiporter NhaA faces a water-filled channel required for ion transport. J. Biol. Chem. 279:40567–40575 [DOI] [PubMed] [Google Scholar]
- 68.Pailler J, Aucher W, Pires M, Buddelmeijer N. 2012. Phosphatidylglycerol::prolipoprotein diacylglyceryl transferase (Lgt) of Escherichia coli has seven transmembrane segments, and its essential residues are embedded in the membrane. J. Bacteriol. 194:2142–2151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Senes A, Chadi DC, Law PB, Walters RF, Nanda V, Degrado WF. 2007. E(z), a depth-dependent potential for assessing the energies of insertion of amino acid side-chains into membranes: derivation and applications to determining the orientation of transmembrane and interfacial helices. J. Mol. Biol. 366:436–448 [DOI] [PubMed] [Google Scholar]
- 70.Engelman DM, Steitz TA, Goldman A. 1986. Identifying nonpolar transbilayer helices in amino acid sequences of membrane proteins. Annu. Rev. Biophys. Biophys. Chem. 15:321–353 [DOI] [PubMed] [Google Scholar]
- 71.Tusnady GE, Kalmar L, Simon I. 2008. TOPDB: topology data bank of transmembrane proteins. Nucleic Acids Res. 36:D234–D239. 10.1093/nar/gkm751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Tusnady GE, Simon I. 1998. Principles governing amino acid composition of integral membrane proteins: application to topology prediction. J. Mol. Biol. 283:489–506 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.