

Abstract
Sulfate-reducing bacteria are characterized by a high number of hydrogenases, which have been proposed to contribute to the overall energy metabolism of the cell, but exactly in what role is not clear. Desulfovibrio spp. can produce or consume H2 when growing on organic or inorganic substrates in the presence or absence of sulfate. Because of the presence of only two hydrogenases encoded in its genome, the periplasmic HynAB and cytoplasmic Ech hydrogenases, Desulfovibrio gigas is an excellent model organism for investigation of the specific function of each of these enzymes during growth. In this study, we analyzed the physiological response to the deletion of the genes that encode the two hydrogenases in D. gigas, through the generation of ΔechBC and ΔhynAB single mutant strains. These strains were analyzed for the ability to grow on different substrates, such as lactate, pyruvate, and hydrogen, under respiratory and fermentative conditions. Furthermore, the expression of both hydrogenase genes in the three strains studied was assessed through quantitative reverse transcription-PCR. The results demonstrate that neither hydrogenase is essential for growth on lactate-sulfate, indicating that hydrogen cycling is not indispensable. In addition, the periplasmic HynAB enzyme has a bifunctional activity and is required for growth on H2 or by fermentation of pyruvate. Therefore, this enzyme seems to play a dominant role in D. gigas hydrogen metabolism.
INTRODUCTION
Hydrogenases are key enzymes in the hydrogen metabolism of Desulfovibrio spp. that catalyze the reversible oxidation of molecular hydrogen into protons and electrons (1). However, their role during sulfate respiration has not been clearly established. Odom and Peck proposed a hydrogen cycling model to explain energy conservation during growth on lactate and sulfate by Desulfovibrio spp., which belong to the deltaproteobacteria subgroup of the sulfate-reducing bacteria (SRB) (2). The model predicts that protons and electrons produced in the oxidation of lactate are used for the production of molecular hydrogen by a cytoplasmic hydrogenase. This hydrogen then diffuses across the membrane to the periplasm, where it is reoxidized by a periplasmic hydrogenase. Electrons are transferred back to the cytoplasm for sulfate reduction, thus creating a proton gradient across the membrane that leads to ATP formation. In this model, the presence of at least two hydrogenases on opposite sides of the membrane is a requirement for growth. In contrast, other studies suggested that the physiological role of these enzymes was to regulate the redox potential of the cell, controlling the flow of protons and electrons and generating a proton motive force (3). More recent models, proposed for Desulfovibrio vulgaris, suggested dual pathways for electron transfer from lactate to sulfate, one involving the cycling of H2 and the other a route involving a membrane-associated electron transfer chain (4, 5). Several membrane complexes have been identified in SRB that could be involved in this process (reviewed in reference 6). It has been estimated that about 48% of the electrons transported from lactate to sulfate involve H2 production (4).
Several studies have tried to elucidate the function of hydrogenases in Desulfovibrio spp., but because most of these organisms present a multiplicity of hydrogenases, in the periplasm and/or in the cytoplasm, identification of the role of each enzyme is complex. In addition, the expression patterns of different hydrogenases were shown to be different and to depend on the substrate, fermentative or respiratory growth, or metal availability (5, 7–13). Furthermore, the function of each hydrogenase in terms of hydrogen production or oxidation may vary depending on the conditions presented to the cell. Numerous studies have reported hydrogenase mutant strains of Desulfovibrio fructosovorans and in D. vulgaris Hildenborough (9, 14–17). However, in most cases, because of the multiplicity of enzymes present, these studies were not conclusive. This indicates that each hydrogenase may contribute to the overall energy metabolism of the cell and that the loss of one enzyme might be compensated for by the presence of the remaining ones. In spite of the extensive work performed with hydrogenase deletion strains and also transcriptomic analyses, the results obtained so far have not permitted a complete elucidation of the function and importance of each hydrogenase under different growth conditions.
Desulfovibrio gigas is an excellent biological model for investigation of the function and importance of hydrogenases in energy metabolism, since its genome, recently sequenced in our laboratory, encodes only two hydrogenases, the HynAB and Ech enzymes. Furthermore, because each hydrogenase is located in a different cell compartment, D. gigas is also an excellent model for the study of the importance of hydrogen cycling in energy conservation. The D. gigas periplasmic HynAB enzyme is one of the most extensively studied enzymes of the [NiFe] type and was the first [NiFe] hydrogenase to have its crystal structure solved (18). In Desulfovibrio spp., as in other bacteria, the periplasmic hydrogenases are believed to be involved in the consumption of hydrogen, coming either from the environment or from intracellular H2 cycling, generating protons and electrons. The electrons are then shuttled to the cytoplasm through the type I cytochrome c3 and the Qrc and Qmo complexes (19–21) to be used for sulfate reduction.
The D. gigas cytoplasmic Ech hydrogenase (22) belongs to the subgroup of multisubunit membrane-bound energy-conserving [NiFe] hydrogenases (23, 24), similar to the Ech hydrogenase from Methanosarcina barkeri (25, 26). These enzymes have subunits that show high similarity to energy-conserving complex I (NADH:quinone oxidoreductase). The Ech hydrogenase in methanogenic archaea can catalyze H2 formation from reduced ferredoxin, generating a proton motive force, or the reduction of ferredoxin by H2, driven by reverse electron transport (26–29). However, the function of the Ech hydrogenase in Desulfovibrio spp. is still not clear.
To elucidate the function of each of these two hydrogenases in D. gigas metabolism, we have generated single deletion (ΔechBC and ΔhynAB) mutants. These strains were tested under respiratory versus fermentative conditions with different energy sources (lactate, pyruvate, and H2), and their hydrogenase expression profiles were studied at the mRNA level.
MATERIALS AND METHODS
Generation of mutants.
To construct the mutant strains, recombinant cassettes containing the DNA fragments of the regions flanking the genes of interest were amplified from D. gigas wild-type (WT) ATCC 19364. A mutant D. gigas strain lacking a functional Ech hydrogenase (Δech) was produced by replacement of the echB (integral membrane subunit) and echC (electron transfer subunit) genes with a kanamycin resistance (kan) gene amplified from plasmid pJRD215 (27) by homologous recombination (30). Oligonucleotides (ech; see Table S1 in the supplemental material) were used to amplify ∼1-kb adjacent DNA regions upstream of subunit echB (flank I) and downstream of subunit echC (flank II), respectively, from a DNA fragment containing the ech operon. Pfx DNA polymerase (Invitrogen) was used for amplification. The PCR products were ligated with T4 DNA ligase (Fermentas) into plasmid pZErO-1 (Invitrogen), and this construct was transformed in Escherichia coli XL-I Blue. Plasmid DNA was extracted with the plasmid purification kit from Eppendorf.
For the hynAB gene, a D. gigas mutant strain lacking the entire dicistronic operon was produced by replacement with kan as in the Δech mutant. Oligonucleotides (hynAB; see Table S1 in the supplemental material) were used to amplify ∼1-kb adjacent DNA regions upstream and downstream of the operon directly from WT D. gigas genomic DNA (gDNA). Phusion high-fidelity DNA polymerase (Thermo Scientific) was used for amplifications. The PCR products were ligated and transformed into the vector YipLac211 according to the In-Fusion HD cloning kit (Clontech) protocol.
Kanamycin-resistant colonies of both constructs were selected, and their plasmids were analyzed by restriction enzyme pattern. These plasmids were used for D. gigas transformation.
D. gigas cells to be transformed were prepared as previously described (30) from 500 ml of an early stationary-phase culture. Immediately before transformation, 6 μg of the plasmid construct was mixed with the cells. Transformation was done aerobically in 0.1-cm cuvettes by electroporation in a Bio-Rad Gene Pulser Apparatus, setting the resistance to ∞ and using a 0.7-kV voltage and a 3-μF capacitance.
Immediately after electroporation, cells were inoculated into lactate-sulfate medium at 37°C. After a 5-h recovery period, kanamycin was added to the medium (50 μg/ml) and the cultures were allowed to grow overnight. Cells were then subcultured in lactate-sulfate medium with kanamycin (50 μg/ml) three consecutive times.
Colonies were grown for approximately 15 days to 1 month in medium supplemented with agar (15 g/liter) and kanamycin (50 μg/ml) in Hungate culture tubes by the roll tube technique and/or in plates inside an AnaeroPack Rectangular Jar 7L (Mitsubishi Gas Chemical Company, Inc.) with AnaeroPack System sachets (bioMérieux).
The deletions of the echBC and hynAB genes from the respective mutant strains were confirmed by PCR and Southern blot analyses (see the supplemental material).
Culture media and growth conditions.
D. gigas cells were grown anaerobically at 37°C in 100-ml flasks with 50 ml of medium. All media were inoculated with 10% (vol/vol) fresh preculture cells grown in lactate-sulfate medium.
For most phenotypic analyses, WT and mutant strains were grown in basal medium modified from reference 14 and containing (per liter) 1 g NH4Cl, 0.15 g CaCl2 · 2H2O, 1 g NaCl, 0.5 g KCl, 0.4 g MgCl2 · 7H2O, 4.9 g KH2PO4, 0.1 g yeast extract, and 1.5 ml of trace elements (31). This medium was then supplemented with either lactate or pyruvate as an electron donor at a concentration of 40 mM. Sulfate was added as an electron acceptor at either 40 or 5 mM or not added. The pH of all solutions was brought to 7.0 with NaOH. The growth conditions tested were lactate and sulfate at 40 and 40 mM, respectively; pyruvate and sulfate at 40 and 40 mM; pyruvate and limiting sulfate at 40 and 5 mM; and pyruvate at 40 mM.
For growth with H2 as the sole energy source, cells were grown on modified Postgate medium C containing (per liter) 0.82 g sodium acetate, 0.5 g KH2PO4, 1 g NH4Cl, 1.14 g Na2SO4, 0.05 g CaCl2 · 2H2O, 0.5 g MgSO4 · 7H2O, 0.2 g yeast extract, 0.0071 g FeSO4 · 7H2O, 0.3 g sodium citrate, 0.1 g ascorbic acid, 0.1 g sodium thioglycolate, 1 μM Ni, 1 μM Se, and 0.1 μM Mo. The cultures were inoculated in 100-ml flasks containing 50 ml of medium and then gassed with H2-CO2 (80:20, vol/vol) at a pressure of 1 atm. The cultures were grown with the flasks at 37°C in a horizontal position to enhance the gas-liquid surface area.
Growth of the cultures was monitored by determining the optical density at 600 nm (OD600). Biomass was determined by measuring the dry cell weight (dcw) and correlating it with the OD600. One unit of OD600 corresponded to 0.365 g (dcw)/liter.
Analytical procedures.
H2 quantification in the headspace of culture-containing serum bottles was performed by gas chromatography with a Thermo Electron Corporation TRACE GC Ultra gas chromatograph fitted with a Alltech Molecular Sieve 5A 80/100 column. The carrier gas was N2, and measurements were done at 130°C. Headspace volumes of 30 μl were withdrawn with a gas-tight syringe and injected into the gas chromatograph. The detection limit was 5 nmol of molecular hydrogen. Cultures of WT D. gigas and both mutant strains were compared.
Identification and quantification of the organic substrates and products generated during growth were performed by high-performance liquid chromatography (HPLC) analyses with a Waters chromatograph (Waters Chromatography, Milford, MA) consisting of a Waters 510 pump, a Waters 715 Autosampler, and a Waters temperature control module connected to an LKB 2142 Differential Refractometer detector (LKB, Bromma, Sweden). Chromatographic separation was undertaken with an Aminex HPX-87H column (300 by 7.8 mm) with a 9-μm particle size (Bio-Rad, Hercules, CA) at 45°C. Elution was carried out isocratically at a flow rate of 0.6 ml·min−1 with 0.005 N H2SO4, and the injection volume was 20 μl. The retention times of the compounds were compared with standards for identification, and the peak area was used for quantification.
Sulfate concentration was measured by HPLC analysis (32) and/or by the SulfaVer4 method (Hach-Lange). HPLC analyses were performed with a Hitachi LaChrom Elite HPLC apparatus with a photodiode array detector. Injections of 20 μl were made into a 10-μl loop operated in full-loop mode, and separation was achieved on a PRP-X100 (4.1 by 150 mm) with a 10-μm-particle-size column (Hamilton Company, Reno, NV) and a thermostat set at 25°C. Isocratic conditions of the mobile phase consisted of 3% (vol/vol) methanol and 97% (vol/vol) 4 mM 4-hydroxybenzoic acid (pH adjusted to 10). The flow rate of the eluent was 2 ml min−1. Indirect UV detection was done at 310 nm. The retention time of the compound was compared with a standard for identification, and the peak area was used for quantification.
RNA preparation and real-time quantitative reverse transcription-PCR (qRT-PCR) analyses.
WT D. gigas and both mutant strains were grown at 37°C in the same medium composition as described for the growth experiments. Cells were harvested after 16 h (mid-exponential phase) or 32 h (stationary phase) of growth. Total RNA was extracted as described in references 33 and 34. However, DNase treatment with Turbo DNase (Ambion) had to be performed three times to avoid gDNA contamination in the RNA extracts. For each sample, 1 μg of total RNA was reverse transcribed with Transcriptor reverse transcriptase (Roche Diagnostics). Specific primers amplifying an ∼100-bp region of echE and hynB were designed (see Table S2 in the supplemental material). The 16S rRNA gene was used as an internal reference gene for each sample analyzed. qRT-PCRs were performed in a LightCycler 480 real-time PCR system (Roche) with LightCycler Fast Start DNA Master SYBR green I (Roche). Relative standard curves and gene expression were calculated as described in reference 31. For the final results, three biological replicates and two technical replicates were used for each condition.
Nucleotide sequence accession numbers.
The NCBI accession number of the D. gigas genome sequence described in this work is CP006585. In addition, the accession numbers of the hydrogenases and specific membrane complexes are as follows: aprAB qmoABCD, KF113859; dsrMKJOP, KF113860; echABCDEF, AY282786; hdrABC floxABCD, KF113861; hynAB, M18083; qrcABCD, KF113862.
RESULTS
A search of the D. gigas genome sequenced in our laboratory revealed the presence of genes encoding only the two known hydrogenases, the periplasmic HynAB and cytoplasmic Ech hydrogenases. Two mutant strains with these enzymes deleted, the ΔechBC and ΔhynAB mutant strains, were generated and then tested under different growth conditions. The deletions of echBC and hynAB were confirmed by both Southern blot and PCR analyses (see Fig. S1 and S2 in the supplemental material).
Mutant phenotype in sulfate respiration.
The growth of WT D. gigas was compared to the growth of both the Δech and ΔhynAB single mutant strains. In the presence of an excess of sulfate (40 mM) with either lactate (Fig. 1A) or pyruvate (Fig. 1B), both mutant strains reached a final cell density similar to that of the WT. However, the doubling times of the ΔechBC mutant strain growing on lactate and the ΔhynAB mutant strain growing on pyruvate were higher than those of the WT (Table 1). Also, the cell yield coefficient observed for the ΔechBC mutant was higher than that of the WT, especially during growth on pyruvate.
Fig 1.
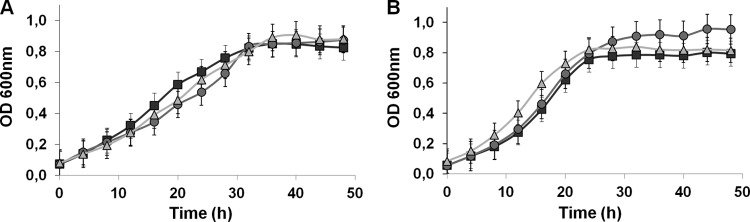
Growth curves of WT D. gigas and the ΔechBC and ΔhynAB mutant strains in medium containing 40 mM lactate and 40 mM sulfate (A) or 40 mM pyruvate and 40 mM sulfate (B). WT, squares; ΔechBC mutant, circles; ΔhynAB mutant, triangles. Each datum point is the average of three independent growth experiments.
Table 1.
Doubling times and cell yield coefficients of WT D. gigas and ΔechBC and ΔhynAB hydrogenase mutant strains during growth under different conditions
| Growth condition | Doubling time (h) |
YS (g [dry wt] cells/mol) |
||||
|---|---|---|---|---|---|---|
| WT | ΔechBC mutant | ΔhynAB mutant | WT | ΔechBC mutant | ΔhynAB mutant | |
| Lactate-sulfate | 8.7 | 11.1 | 9.0 | 6.8 | 7.2 | 6.8 |
| Pyruvate-sulfatea | 6.7 | 6.7 | 7.9 | 6.0 | 7.3 | 6.1 |
| Pyruvate-sulfateb | 6.9 | 7.1 | 8.0 | 5.2 | 6.3 | 4.7 |
| Pyruvate | 14.2 | 16.7 | 4.8 | 6.7 | ||
| H2-sulfate | 33.3 | 24.1 | ||||
Pyruvate-sulfate (40 mM–40 mM).
Pyruvate-limiting sulfate (40 mM–5 mM).
Metabolite quantification (see Table S3 in the supplemental material) revealed that both mutations did not affect substrate consumption or product formation, compared to the WT. The electron acceptor, sulfate, was reduced in agreement with the expected stoichiometries of 2:1 and 4:1, relative to lactate and pyruvate, respectively, and at the same rate by the mutant strains and the WT. No significant amounts of H2 accumulated during growth whenever sulfate was present. These results indicate that neither of the two hydrogenases is essential for growth under sulfate respiratory conditions with either lactate or pyruvate as the carbon source.
However, with H2 as the energy source in the presence of sulfate, a different phenotype was observed (Fig. 2). The doubling time of the WT under this respiratory condition was much higher than that seen when an organic acid (lactate or pyruvate) was the energy source (Table 1). Also, the behavior of the mutant strains was quite different. The ΔechBC mutant strain reached a final cell density similar to that of the WT and had an even higher doubling time. In contrast, the ΔhynAB mutant strain did not grow at all, revealing that, as expected, the periplasmic hydrogenase is essential for growth under these conditions.
Fig 2.
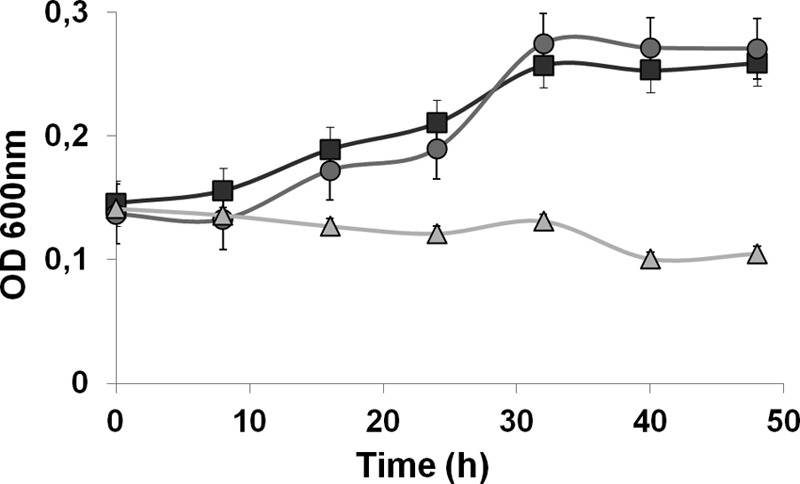
Growth curves of WT D. gigas and the ΔechBC and ΔhynAB mutant strains in H2-sulfate. Cells were grown on modified medium C containing 10 mM acetate and 20 mM sulfate with a gas phase composed of H2-CO2 (80:20, vol/vol) at a pressure of 1 atm. WT, squares; ΔechBC mutant, circles; ΔhynAB mutant, triangles. Each datum point is the average of three independent growth experiments.
Mutant phenotype in sulfate limitation and fermentation.
Desulfovibrio spp. are unable to grow fermentatively in the presence of lactate, unless H2 partial pressures are kept low by hydrogenotrophic organisms such as methanogenic archaea. This is due to the fact that oxidation of lactate to pyruvate is an endergonic reaction. However, these bacteria can grow fermentatively with pyruvate. Thus, we evaluated the effect of deleting each hydrogenase under fermentative conditions in pyruvate. The WT and both mutant strains were grown in pyruvate with sulfate limitation (5 mM) or in its absence (Fig. 3A and B, respectively). In the first case, although sulfate was limiting, the growth of the D. gigas WT and mutant strains reached cell densities similar to those seen when sulfate was present in excess (Fig. 1), revealing that enough sulfate was present to allow respiratory growth. In agreement with this, the doubling time was similar to pyruvate respiratory conditions and once again the deletion of the hynAB genes slowed growth (Table 1). When yield coefficients were compared, a decrease in cell yield was observed for all of the strains under this condition, especially in the case of the ΔhynAB mutant, which possibly correlates with the lower sulfate concentration available. Once again, it is noticeable that the absence of the echBC genes appears to increase the cell yield of the mutant strain over that of the WT.
Fig 3.

WT D. gigas and the ΔechBC and ΔhynAB mutant strains grown in pyruvate with sulfate limitation (A, C, and E) or with no sulfate added (B, D, and F). A and B, growth curves (filled symbols) and sulfate quantification (open symbols); C and D, quantification of pyruvate (filled symbols) and acetate (open symbols); E and F, hydrogen accumulation in the headspace. WT D. gigas, squares; ΔechBC mutant, circles; and ΔhynAB mutant, triangles.
Interestingly, after sulfate was completely reduced at 20 to 24 h (Fig. 3A), the cells stopped growing and started to accumulate H2 (Fig. 3E), and the ΔechBC mutant strain, containing only the periplasmic hydrogenase, accumulated much more H2 than did the WT strain. On the other hand, the ΔhynAB mutant strain, having the cytoplasmic hydrogenase, did not accumulate any H2. No difference in pyruvate consumption or acetate formation was observed, as shown in Fig. 3C, indicating that the difference in H2 accumulation was due to HynAB activity.
Under fermentative conditions when no sulfate was added, a small amount of sulfate (∼1 to 3 mM) was nevertheless present in the beginning of growth, coming from the inoculum. After this sulfate was reduced (∼16 h), we observed a phenotype different from that seen during respiratory growth on pyruvate. The ΔhynAB mutant strain did not show fermentative growth once the small amount of residual sulfate was consumed, whereas the ΔechBC mutant and WT strains were able to grow, albeit to lower cell densities (Fig. 3B) and with reduced cell yields (Table 1) than in pyruvate-sulfate. Furthermore, the doubling times of these strains in pyruvate fermentation were at least double those seen when sulfate was present. From the metabolite analysis, it is clear that after the residual sulfate was reduced, the absence of the HynAB hydrogenase prevented the cells from fermenting pyruvate, resulting in the absence of fermentative growth of this strain (Fig. 3B and D). Furthermore, the expression of the ech genes is also lower under this condition in the ΔhynAB mutant strain than in the WT strain (see below), which may also have contributed to the lack of growth.
Similarly to what happened under sulfate-limiting conditions, H2 accumulation in the ΔechBC strain was much higher than that in the WT (Fig. 3F). Interestingly, this accumulation started only after all of the pyruvate was consumed, as previously reported for D. vulgaris (39). A slight accumulation of H2 could be observed in the ΔhynAB mutant strain as well, showing that the Ech hydrogenase can contribute to some H2 production under these conditions.
These results indicate that under fermentative conditions, HynAB is an essential enzyme for cell growth and that cells are unable to ferment pyruvate in its absence. Furthermore, periplasmic HynAB is the main hydrogenase responsible for the H2 accumulation observed under fermentative conditions.
Expression profiles of the ech and hyn genes.
The higher H2 accumulation by the ΔechBC mutant strain, relative to that by the WT, during pyruvate fermentation is very surprising, since it could be expected that the cytoplasmic Ech hydrogenase would be responsible for H2 production from reduced ferredoxin produced by the pyruvate:ferredoxin oxidoreductase. To evaluate if this was due to an increase in HynAB transcripts in the deletion strain, we analyzed the expression levels of genes coding for both the HynAB and Ech hydrogenases in WT D. gigas and the deletion strains. Real-time qRT-PCR was performed to analyze the mRNA expression levels of the HynAB hydrogenase in both WT D. gigas (Fig. 4A) and the ΔechBC mutant strain (Fig. 4B) in the mid-exponential (16 h) and stationary (32 h) phases in pyruvate-limiting sulfate and pyruvate fermentation, as well as under lactate-sulfate and H2-sulfate conditions, for which microarray expression data have been reported for other Desulfovibrio spp. (5, 7). Expression of the Ech hydrogenase was similarly evaluated in WT D. gigas (Fig. 5A) and the ΔhynAB mutant strain (Fig. 5B). Quantification of all samples was performed relative to the 16S rRNA gene.
Fig 4.
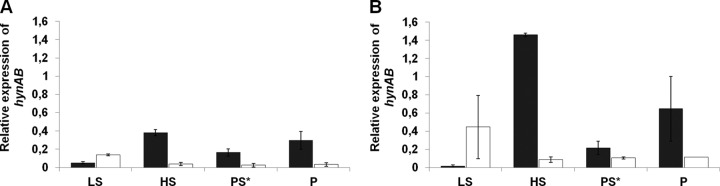
Quantification of the mRNA levels of the hynB hydrogenase gene by qRT-PCR in the mid-exponential (16 h) and stationary (32 h) phases of both the WT (A) and ΔechBC mutant (B) strains grown under the following conditions: LS, 40 mM lactate–40 mM sulfate; PS*, 40 mM pyruvate–5 mM sulfate; P, 40 mM pyruvate; HS, 1 atm H2–20 mM sulfate. Black bars, 16 h; white bars, 32 h. The expression of the hynAB gene was normalized to that of the 16S rRNA gene. The standard errors of all values are shown.
Fig 5.
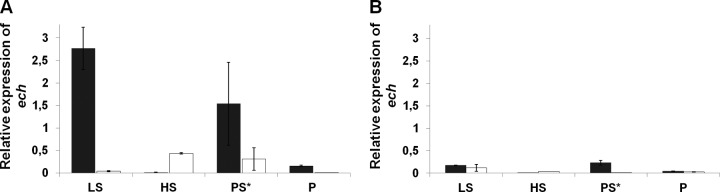
Quantification of the mRNA levels of the echE hydrogenase gene by qRT-PCR in the mid-exponential (16 h) and stationary (32 h) phases of both the WT (A) and ΔhynAB mutant (B) strains grown under the following conditions: LS, 40 mM lactate–40 mM sulfate; PS*, 40 mM pyruvate–5 mM sulfate; P, 40 mM pyruvate; HS, 1 atm H2–20 mM sulfate. Black bars, 16 h; white bars, 32 h. The expression of the echE gene was normalized to that of the 16S rRNA gene. The standard errors of all values are shown.
The expression of the hynB gene was highest in the WT strain during exponential growth in hydrogen respiration and in pyruvate fermentation (Fig. 4A), whereas it was lower in pyruvate-sulfate and even lower in lactate-sulfate. After cells entered stationary phase, hynB gene expression dropped to very low levels in hydrogen-sulfate, pyruvate-sulfate, and pyruvate fermentation, whereas it increased under lactate-sulfate conditions.
An increase in hynB gene expression, compared to that of the WT, was observed in the ΔechBC mutant strain under conditions in which the hynAB deletion prevented growth, i.e., hydrogen respiration and pyruvate fermentation (compare Fig. 4B and A). This increase was more prominent during the exponential phase but was also observed during the stationary phase, where we saw a more accentuated drop in the expression of hynB in the WT. Under respiratory conditions with organic substrates (lactate-sulfate and pyruvate-limiting sulfate), hynB expression in the exponential phase was similar to that of the WT, and once again an increase was observed in the stationary phase relative to the WT level. These increased levels of hynB in the stationary phase observed in the ΔechBC mutant may account for the greater accumulation of H2 observed in pyruvate limitation fermentation during physiological analyses. Therefore, in the ΔechBC mutant strain, the absence of the Ech hydrogenase appears to be compensated for by an increased level of HynAB (except during exponential-phase growth in lactate-sulfate).
We also measured the levels of echE expression in the WT and ΔhynAB mutant strains. The echE mRNA levels were highest in the D. gigas WT strain during exponential growth on an organic substrate (lactate or pyruvate) in the presence of sulfate (Fig. 4A, 16 h). After cells entered the stationary phase (32 h), the levels of echE gene expression decreased significantly, especially during growth on lactate. During respiratory growth with H2 as the energy source, the opposite behavior was observed, where the echE mRNA level was almost undetectable during the exponential phase (16 h) but increased after cells entered the stationary phase. During fermentative growth on pyruvate, the echE transcript levels were low in the exponential phase and decreased further to undetectable levels after cells entered the stationary phase. This suggests that Ech activity is related mainly to the initial phase of respiratory growth (i.e., when sulfate is still present) with organic substrates, whereas Ech appears to play a much less important role during fermentative growth.
When echE gene expression in the ΔhynAB mutant strain was analyzed (Fig. 5B), a major overall decrease in expression was observed under all conditions, compared to the WT level (Fig. 5A), suggesting that the HynAB hydrogenase is involved in the regulation of Ech expression. These results indicate that during hydrogen-sulfate respiration and pyruvate fermentation, the Ech hydrogenase is poorly expressed and thus cannot compensate for the absence of the HynAB hydrogenase, preventing growth under these conditions.
DISCUSSION
Previous studies have addressed the role of hydrogenases in Desulfovibrio spp. In a D. vulgaris strain with the periplasmic Hyn1 [NiFe] hydrogenase deleted, cells were able to grow to almost the same level as the WT in lactate-sulfate medium (17). Similar results were obtained with a D. fructosovorans periplasmic hynABC and cytoplasmic hnd hydrogenase double mutant that was able to grow under all of the conditions tested (14). A further triple mutant lacking all described periplasmic hydrogenases was also shown to grow in fructose-sulfate medium (16). However, in these studies, there were additional hydrogenases present that could compensate for the absence of the missing genes.
D. gigas is uniquely positioned among Desulfovibrio spp. for use in testing the function of hydrogenases and the importance of hydrogen cycling in energy conservation, since it contains only two enzymes, one periplasmic and one cytoplasmic. Furthermore, the genomic analyses revealed no sequences related to the pyruvate:formate lyase gene in D. gigas, and therefore, formate cycling should not be participating in the energy conservation processes as an alternative to hydrogen cycling.
The results obtained in this work demonstrate that neither of the two hydrogenases is essential for the growth of D. gigas under respiratory conditions on an organic substrate (lactate or pyruvate) in the presence of sulfate. In addition, the absence of the hynAB or echBC genes also did not lead to any significant accumulation of molecular hydrogen. Since no other hydrogenase, either periplasmic or cytoplasmic, is present in the genome of D. gigas and formate is not participating in the energy conservation process, this suggests that the hydrogen cycling pathway does not play a major part in the bioenergetics of D. gigas. The cell yield coefficients calculated for sulfate respiration with lactate and pyruvate are almost identical for the WT and hydrogenase mutant strains. Nevertheless, a slightly lower growth rate is observed for the ΔechBC mutant strain growing on lactate-sulfate and for the ΔhynAB mutant strain in both lactate-sulfate and pyruvate-sulfate, suggesting that a small fraction of the electron flow to sulfate may involve the production of H2. In D. vulgaris, a mathematical model of metabolism, supported by experimental results, indicated that two simultaneous pathways for electron flow during growth in lactate-sulfate coexist, one requiring the obligate cycling of H2 and the other not requiring it (4). The model estimated that 48% of the electron flow from lactate to sulfate involved H2 production. Our results suggest that this value may be significantly lower for D. gigas. This would be in line with the reduced number of hydrogenases in this organism compared to D. vulgaris, which has seven hydrogenases. Nevertheless, we have been unable to date to generate a double hydrogenase mutant of D. gigas. However, given the difficulty in transforming this organism, we cannot clearly conclude that this is due to the required presence of at least one hydrogenase.
Thus, as suggested before, the mechanism of hydrogen cycling does not seem to be strictly essential for the Desulfovibrio genus (9) and may make different contributions to the overall electron flow in different organisms. This is not entirely surprising since it was previously shown that some SRB do not have any hydrogenases at all (e.g., Desulfococcus oleovorans) or have no cytoplasmic hydrogenases (like Desulfomicrobium baculatum, which is closely related to Desulfovibrionaceae) (6).
Thus, H2 is not an obligatory intermediate in the oxidation of organic compounds by D. gigas since deletion of the hydrogenases does not affect the ability of cells to grow. This agrees with the observation by Lupton et al. that H2 added to lactate-sulfate medium did not competitively inhibit the oxidation of lactate or increase growth or substrate utilization (3). However, we cannot rule out the possibility that other electron transfer pathways substitute for hydrogen cycling when one of the hydrogenases is missing. It is plausible that the cells can reroute electron flow if one of several pathways is impaired. This would be the advantage of having dual or multiple pathways for electron flow, and sulfate reducers are recognized for their metabolic flexibility.
Chemiosmotic models of energy conservation have also been proposed in which electrons generated from substrate oxidation are transported through membrane-bound electron carriers to sulfate reduction (vectorial electron transport) and in the process translocate protons to the periplasm (vectorial proton transport) (6, 35). Two membrane complexes that were identified in the D. gigas genome, the Qmo and Dsr complexes, were proposed to perform this function (21, 36). An energy conservation process may result from electron transfer from the quinone pool to AprAB through the Qmo complex (19, 37), and also the Dsr complex may be involved in menaquinol oxidation with reduction of DsrC, associated with proton translocation (36, 38). Furthermore, other electron transfer pathways to sulfate may involve Hdr-related proteins that could provide a soluble pathway from different donors (such as lactate, pyruvate, ferredoxin, or H2) to the reduction of DsrC in a flavin-based electron bifurcation mechanism for energy conservation (6). Such a system may involve the HdrABC/FloxABCD proteins that are also encoded in the D. gigas genome.
Regarding the physiological role of hydrogenases, the periplasmic enzymes are generally presumed to be involved in H2 oxidation. Previous deletions of periplasmic hydrogenases in D. vulgaris demonstrated that in lactate-sulfate or H2-sulfate, the absence of one periplasmic hydrogenase could be compensated for by the others (9). In D. gigas, which has only the HynAB hydrogenase, the physiological and mRNA expression data indicate that this enzyme has a bidirectional role in terms of hydrogen metabolism. When hydrogen is the energy source for sulfate respiration, HynAB acts as the H2 uptake hydrogenase, as expected, and the electrons from the oxidation of H2 can then be shuttled to the cytoplasmic reduction of sulfate through cytochrome c3, Qrc, and Qmo (19). Thus, the absence of the hynAB genes impaired growth in hydrogen-sulfate. When sulfate is absent, i.e., under fermentative conditions, our results indicate that this enzyme acts in the opposite direction, as a hydrogen-producing enzyme, as the HynAB-lacking strain was not able to ferment pyruvate. Interestingly, Ech is not able to compensate for the absence of HynAB, as could be expected, but this may also be due to the low level of expression of this enzyme under fermentative conditions.
In agreement with the above, higher levels of hynAB expression were observed during growth with H2-sulfate and in pyruvate fermentation than during lactate-sulfate respiration. These levels were further increased under these conditions in the ΔechBC mutant strain, which can account for the greater accumulation of H2 when this strain is fermenting pyruvate. Our results are similar to what was observed in a D. vulgaris Hildenborough periplasmic [Fe] hydrogenase (Δhyd) mutant strain (39), where this hydrogenase was required for pyruvate fermentation, in addition to its function in hydrogen uptake in hydrogen-sulfate medium. Also, during syntrophic growth, a similar role of the periplasmic hydrogenase, acting as an H2-producing enzyme, was observed in D. vulgaris Hildenborough cultures grown in lactate with a hydrogenotrophic methanogen (8). Furthermore, also in Desulfovibrio alaskensis G20, the HynAB hydrogenase seems to be the main enzyme responsible for H2 production during syntrophic growth (12).
However, in these previous studies, the presence of multiple hydrogenases prevented the direct determination of whether a periplasmic hydrogenase is responsible for H2 production. In this paper, the inability of the ΔhynAB mutant strain to grow with pyruvate demonstrated that this periplasmic hydrogenase is undoubtedly essential for fermentative growth and H2 production. Furthermore, the absence of the hynAB gene caused a lower growth rate in pyruvate-sulfate, suggesting that this is the main hydrogenase in pyruvate metabolism.
Interestingly, accumulation of H2 started only after pyruvate was consumed, as previously observed in D. vulgaris (39), suggesting that some intracellular cycling of H2 may be occurring and/or that other fermentation products are accumulated inside the cell and later converted to H2. The fact that the ΔhynAB mutant strain cannot grow by pyruvate fermentation supports the idea that intracellular H2 cycling is required, but this inability could also be due to the reduced expression of the Ech hydrogenase.
Regarding the cytoplasmic Ech hydrogenase, our physiological analysis and mRNA expression results indicates that this hydrogenase is not essential for the growth of the organism under any of the conditions tested. Actually, the yield coefficients of the ΔechBC mutant strain indicate that the deletion of this gene even slightly increased the final cell yield, particularly whenever pyruvate was the energy source. Nevertheless, the expression of this enzyme is quite high during exponential growth in lactate-sulfate, suggesting that it is probably playing a role under this condition. Also, the growth rate of this mutant strain in lactate-sulfate is lower. In the stationary phase, Ech is virtually not expressed in lactate-sulfate and pyruvate fermentation. Interestingly, transcriptomic analysis in D. vulgaris indicated an upregulation of this enzyme under H2-sulfate compared to lactate-sulfate (5, 7). However, the Δech mutant strain of D. gigas was able to reach a final cell density similar to that of the WT with a shorter doubling time when growing in H2-sulfate.
Overall, it seems that the Ech hydrogenase does not play a central role in energy metabolism under the conditions tested, which agrees with the fact that no Ech hydrogenase has been identified in the genomes of many SRB, including Desulfovibrio spp. such as D. alaskensis G20 and Desulfovibrio piger (6).
Our results also provide compelling evidence of the importance of obtaining expression data to complement studies of gene deletions. Indeed, the expression of each hydrogenase was significantly altered in the mutants. In particular, the expression of the Ech hydrogenase was almost completely abolished upon deletion of the HynAB enzyme, regardless of the energy source or type of growth. This suggests that HynAB may be somehow involved in the regulation of the ech genes. One interesting observation was that the two hydrogenases have somewhat complementary expression, as the HynAB hydrogenase was expressed more during growth in H2-sulfate and pyruvate fermentation, whereas the Ech hydrogenase was expressed more during growth in lactate-sulfate and pyruvate-sulfate.
In conclusion, the HynAB hydrogenase appears to have a more predominant role in the metabolism of D. gigas and is essential for growth with H2-sulfate and pyruvate fermentation.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by Fundação para Ciência e Tecnologia (FCT) grants (PTDC/BIA-MIC/104030/2008 to IACP/CRP and Pest-OE/EQB/LA0004/2011 to the Instituto de Tecnologia Química e Biológica—António Xavier [ITQB], 049/BI-BI/2011 to C.I.S and SFRH/BD/45211/2008 to F.O.M.-S.).
We thank Maria Cristina Leitão for technical support with the HPLC quantifications, Ana Raquel Ramos for technical support in the Southern blot analysis, and Mónica Martins for methodological support and helpful discussions.
Footnotes
Published ahead of print 23 August 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JB.00411-13.
REFERENCES
- 1.Matias PM, Pereira IA, Soares CM, Carrondo MA. 2005. Sulphate respiration from hydrogen in Desulfovibrio bacteria: a structural biology overview. Prog. Biophys. Mol. Biol. 89:292–32915950057 [Google Scholar]
- 2.Odom JM, Peck HD., Jr 1981. Localization of dehydrogenases, reductases, and electron transfer components in the sulfate-reducing bacterium Desulfovibrio gigas. J. Bacteriol. 147:161–169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lupton FS, Conrad R, Zeikus JG. 1984. Physiological function of hydrogen metabolism during growth of sulfidogenic bacteria on organic substrates. J. Bacteriol. 159:843–849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Noguera DR, Brusseau GA, Rittmann BE, Stahl DA. 1998. A unified model describing the role of hydrogen in the growth of Desulfovibrio vulgaris under different environmental conditions. Biotechnol. Bioeng. 59:732–746 [DOI] [PubMed] [Google Scholar]
- 5.Keller KL, Wall JD. 2011. Genetics and molecular biology of the electron flow for sulfate respiration in Desulfovibrio. Front. Microbiol. 2:135. 10.3389/fmicb.2011.00135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pereira IA, Ramos AR, Grein F, Marques MC, da Silva SM, Venceslau SS. 2011. A comparative genomic analysis of energy metabolism in sulfate reducing bacteria and archaea. Front. Microbiol. 2:69. 10.3389/fmicb.2011.00069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pereira PM, He Q, Valente FM, Xavier AV, Zhou J, Pereira IA, Louro RO. 2008. Energy metabolism in Desulfovibrio vulgaris Hildenborough: insights from transcriptome analysis. Antonie Van Leeuwenhoek 93:347–362 [DOI] [PubMed] [Google Scholar]
- 8.Walker CB, He ZL, Yang ZK, Ringbauer JA, He Q, Zhou JH, Voordouw G, Wall JD, Arkin AP, Hazen TC, Stolyar S, Stahl DA. 2009. The electron transfer system of syntrophically grown Desulfovibrio vulgaris. J. Bacteriol. 191:5793–5801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Caffrey SM, Park HS, Voordouw JK, He Z, Zhou J, Voordouw G. 2007. Function of periplasmic hydrogenases in the sulfate-reducing bacterium Desulfovibrio vulgaris Hildenborough. J. Bacteriol. 189:6159–6167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Plugge CM, Scholten JC, Culley DE, Nie L, Brockman FJ, Zhang W. 2010. Global transcriptomics analysis of the Desulfovibrio vulgaris change from syntrophic growth with Methanosarcina barkeri to sulfidogenic metabolism. Microbiology 156(Pt 9):2746–2756 [DOI] [PubMed] [Google Scholar]
- 11.Li X, McInerney MJ, Stahl DA, Krumholz LR. 2011. Metabolism of H2 by Desulfovibrio alaskensis G20 during syntrophic growth on lactate. Microbiology 157(Pt 10):2912–2921 [DOI] [PubMed] [Google Scholar]
- 12.Meyer B, Kuehl J, Deutschbauer AM, Price MN, Arkin AP, Stahl DA. 2013. Variation among Desulfovibrio species in electron transfer systems used for syntrophic growth. J. Bacteriol. 195:990–1004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Valente FM, Almeida CC, Pacheco I, Carita J, Saraiva LM, Pereira IA. 2006. Selenium is involved in regulation of periplasmic hydrogenase gene expression in Desulfovibrio vulgaris Hildenborough. J. Bacteriol. 188:3228–3235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Malki S, De Luca G, Fardeau ML, Rousset M, Belaich JP, Dermoun Z. 1997. Physiological characteristics and growth behavior of single and double hydrogenase mutants of Desulfovibrio fructosovorans. Arch. Microbiol. 167:38–45 [DOI] [PubMed] [Google Scholar]
- 15.Casalot L, Valette O, De Luca G, Dermoun Z, Rousset M, de Philip P. 2002. Construction and physiological studies of hydrogenase depleted mutants of Desulfovibrio fructosovorans. FEMS Microbiol. Lett. 214:107–112 [DOI] [PubMed] [Google Scholar]
- 16.Casalot L, De Luca G, Dermoun Z, Rousset M, de Philip P. 2002. Evidence for a fourth hydrogenase in Desulfovibrio fructosovorans. J. Bacteriol. 184:853–856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Goenka A, Voordouw JK, Lubitz W, Gartner W, Voordouw G. 2005. Construction of a [NiFe]-hydrogenase deletion mutant of Desulfovibrio vulgaris Hildenborough. Biochem. Soc. Trans. 33(Pt 1):59–60 [DOI] [PubMed] [Google Scholar]
- 18.Volbeda A, Charon MH, Piras C, Hatchikian EC, Frey M, Fontecilla-Camps JC. 1995. Crystal structure of the nickel-iron hydrogenase from Desulfovibrio gigas. Nature 373:580–587 [DOI] [PubMed] [Google Scholar]
- 19.Venceslau SS, Lino RR, Pereira IA. 2010. The Qrc membrane complex, related to the alternative complex III, is a menaquinone reductase involved in sulfate respiration. J. Biol. Chem. 285:22774–22783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li X, Luo Q, Wofford NQ, Keller KL, McInerney MJ, Wall JD, Krumholz LR. 2009. A molybdopterin oxidoreductase is involved in H2 oxidation in Desulfovibrio desulfuricans G20. J. Bacteriol. 191:2675–2682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pires R, Lourenço A, Morais F, Teixeira M, Xavier A, Saraiva L, Pereira I. 2003. A novel membrane-bound respiratory complex from Desulfovibrio desulfuricans ATCC 27774. Biochim. Biophys. Acta 1605:67–82 [DOI] [PubMed] [Google Scholar]
- 22.Rodrigues R, Valente FM, Pereira IA, Oliveira S, Rodrigues-Pousada C. 2003. A novel membrane-bound Ech [NiFe] hydrogenase in Desulfovibrio gigas. Biochem. Biophys. Res. Commun. 306:366–375 [DOI] [PubMed] [Google Scholar]
- 23.Vignais PM, Billoud B, Meyer J. 2001. Classification and phylogeny of hydrogenases. FEMS Microbiol. Rev. 25:455–501 [DOI] [PubMed] [Google Scholar]
- 24.Hedderich R, Forzi L. 2005. Energy-converting [NiFe] hydrogenases: more than just H2 activation. J. Mol. Microbiol. Biotechnol. 10:92–104 [DOI] [PubMed] [Google Scholar]
- 25.Künkel A, Vorholt JA, Thauer RK, Hedderich R. 1998. An Escherichia coli hydrogenase-3-type hydrogenase in methanogenic archaea. Eur. J. Biochem. 252:467–476 [DOI] [PubMed] [Google Scholar]
- 26.Meuer J, Kuettner HC, Zhang JK, Hedderich R, Metcalf WW. 2002. Genetic analysis of the archaeon Methanosarcina barkeri Fusaro reveals a central role for Ech hydrogenase and ferredoxin in methanogenesis and carbon fixation. Proc. Natl. Acad. Sci. U. S. A. 99:5632–5637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Soboh B, Linder D, Hedderich R. 2004. A multisubunit membrane-bound [NiFe] hydrogenase and an NADH-dependent Fe-only hydrogenase in the fermenting bacterium Thermoanaerobacter tengcongensis. Microbiology 150(Pt 7):2451–2463 [DOI] [PubMed] [Google Scholar]
- 28.Welte C, Kratzer C, Deppenmeier U. 2010. Involvement of Ech hydrogenase in energy conservation of Methanosarcina mazei. FEBS J. 277:3396–3403 [DOI] [PubMed] [Google Scholar]
- 29.Welte C, Kallnik V, Grapp M, Bender G, Ragsdale S, Deppenmeier U. 2010. Function of Ech hydrogenase in ferredoxin-dependent, membrane-bound electron transport in Methanosarcina mazei. J. Bacteriol. 192:674–678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Broco M, Rousset M, Oliveira S, Rodrigues-Pousada C. 2005. Deletion of flavoredoxin gene in Desulfovibrio gigas reveals its participation in thiosulfate reduction. FEBS Lett. 579:4803–4807 [DOI] [PubMed] [Google Scholar]
- 31.Varela-Raposo A, Pimentel C, Morais-Silva F, Rezende A, Ruiz JC, Rodrigues-Pousada C. 2013. Role of NorR-like transcriptional regulators under nitrosative stress of the delta-proteobacterium, Desulfovibrio gigas. Biochem. Biophys. Res. Commun. 431:590–596 [DOI] [PubMed] [Google Scholar]
- 32.Rethmeier J, Rabenstein A, Langer M, Fischer U. 1997. Detection of traces of oxidized and reduced sulfur compounds in small samples by combination of different high-performance liquid chromatography methods. J. Chromatogr. 760:295–302 [Google Scholar]
- 33.Rodrigues R, Vicente JB, Felix R, Oliveira S, Teixeira M, Rodrigues-Pousada C. 2006. Desulfovibrio gigas flavodiiron protein affords protection against nitrosative stress in vivo. J. Bacteriol. 188:2745–2751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Silva G, Oliveira S, LeGall J, Xavier AV, Rodrigues-Pousada C. 2001. Analysis of the Desulfovibrio gigas transcriptional unit containing rubredoxin (rd) and rubredoxin-oxygen oxidoreductase (roo) genes and upstream ORFs. Biochem. Biophys. Res. Commun. 280:491–502 [DOI] [PubMed] [Google Scholar]
- 35.Wood P. 1978. A chemiosmotic model for sulphate respiration. FEBS Lett. 95:12–18 [DOI] [PubMed] [Google Scholar]
- 36.Pires RH, Venceslau SS, Morais F, Teixeira M, Xavier AV, Pereira IA. 2006. Characterization of the Desulfovibrio desulfuricans ATCC 27774 DsrMKJOP complex—a membrane-bound redox complex involved in the sulfate respiratory pathway. Biochemistry 45:249–262 [DOI] [PubMed] [Google Scholar]
- 37.Ramos AR, Keller KL, Wall JD, Pereira IA. 2012. The membrane QmoABC complex interacts directly with the dissimilatory adenosine 5′-phosphosulfate reductase in sulfate reducing bacteria. Front. Microbiol. 3:137. 10.3389/fmicb.2012.00137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Oliveira TF, Vonrhein C, Matias PM, Venceslau SS, Pereira IA, Archer M. 2008. The crystal structure of Desulfovibrio vulgaris dissimilatory sulfite reductase bound to DsrC provides novel insights into the mechanism of sulfate respiration. J. Biol. Chem. 283:34141–34149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Voordouw G. 2002. Carbon monoxide cycling by Desulfovibrio vulgaris Hildenborough. J. Bacteriol. 184:5903–5911 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.