

Abstract
The anoxygenic phototroph Rhodobacter sphaeroides uses 3-hydroxypropionate as a sole carbon source for growth. Previously, we showed that the gene (RSP_1434) known as acuI, which encodes a protein of the medium-chain dehydrogenase/reductase (MDR) superfamily, was involved in 3-hydroxypropionate assimilation via the reductive conversion to propionyl-coenzyme A (CoA). Based on these results, we speculated that acuI encoded acrylyl-CoA reductase. In this work, we characterize the in vitro enzyme activity of purified, recombinant AcuI using a coupled spectrophotometric assay. AcuI from R. sphaeroides catalyzes the NADPH-dependent acrylyl-CoA reduction to produce propionyl-CoA. Two other members of the MDR012 family within the MDR superfamily, the products of SPO_1914 from Ruegeria pomeroyi and yhdH from Escherichia coli, were shown to also be part of this new class of NADPH-dependent acrylyl-CoA reductases. The activities of the three enzymes were characterized by an extremely low Km for acrylyl-CoA (<3 μM) and turnover numbers of 45 to 80 s−1. These homodimeric enzymes were highly specific for NADPH (Km = 18 to 33 μM), with catalytic efficiencies of more than 10-fold higher for NADPH than for NADH. The introduction of codon-optimized SPO_1914 or yhdH into a ΔacuI::kan mutant of R. sphaeroides on a plasmid complemented 3-hydroxypropionate-dependent growth. However, in their native hosts, SPO_1914 and yhdH are believed to function in the metabolism of substrates other than 3-hydroxypropionate, where acrylyl-CoA is an intermediate. Complementation of the ΔacuI::kan mutant phenotype by crotonyl-CoA carboxylase/reductase from R. sphaeroides was attributed to the fact that the enzyme also uses acrylyl-CoA as a substrate.
INTRODUCTION
Acrylyl-coenzyme A (CoA) reductase is an enzyme that catalyzes the reduction of acrylyl-CoA to propionyl-CoA. To date, three different classes of acrylyl-CoA reductases have been recognized. Members of these different classes share little or no significant similarity in their primary structures. The characterized members of each class are (i) NADPH-dependent acrylyl-CoA reductases from autotrophic crenarchaea (Sulfolobus tokodaii and Metallosphaera sedula) (1), (ii) the C-terminal domain of propionyl-CoA synthase from the anoxygenic photosynthetic green nonsulfur bacterium Chloroflexus aurantiacus (2), and (iii) an NADH-dependent acrylyl-CoA reductase from the low-G/C Gram-positive bacterium Clostridium propionicum (3).
These acrylyl-CoA reductases are involved in central metabolic pathways of the organisms (see Fig. S1 in the supplemental material). The enzymes from Sulfolobus and Chloroflexus are involved in autotrophic CO2 fixation cycles known as the 3-hydroxypropionate/4-hydroxypropionate cycle (4, 5) and the 3-hydroxypropionate bi-cycle (6, 7, 8). While the two autotrophic CO2 fixation cycles share the same intermediates in the conversion of acetyl-CoA and CO2 to propionyl-CoA, the specific reaction steps are catalyzed by different enzymes. Sulfolobus acrylyl-CoA reductase is a bona fide reductase that acts on acrylyl-CoA as its substrate (1). In contrast, the Chloroflexus propionyl-CoA synthase is a large fusion protein in which the 3-hydroxypropionyl–CoA synthetase, 3-hydroxypropionyl–CoA dehydratase, and acrylyl-CoA reductase activities each occur in an independent domain (2). Because this fusion enzyme catalyzes an Mg2+ ATP-, K+-, CoA-, and NADPH-dependent conversion of 3-hydroxypropionate to propionyl-CoA, acrylyl-CoA is actually an intermediate during catalysis. Evidence that acrylyl-CoA is indeed the substrate of the C-terminal acrylyl-CoA reductase domain comes from the observation that the truncated propionyl-CoA synthase, whose C terminus is removed by partial trypsin digestion, releases acrylyl-CoA as a product (2).
The Clostridium acrylyl-CoA reductase enzyme complex is involved in fermentation of alanine (9, 10, 11) (see Fig. S1 in the supplemental material). The enzyme is composed of propionyl-CoA dehydrogenase and electron-transferring flavoprotein subunits (3). The latter is required for shuttling electrons from NADH to propionyl-CoA dehydrogenase in acrylyl-CoA reduction. A similar enzyme complex appears to function during propionate fermentation of (R)-lactate by the anaerobic ruminal bacterium Megasphaera elsdenii (12, 13).
Previously, we showed that the anoxygenic photosynthetic bacterium Rhodobacter sphaeroides could use 3-hydroxypropionate as a sole carbon source for growth (14). Reductive conversion of 3-hydroxypropionate to propionyl-CoA was a necessary route for assimilation of this C3 compound and ultimately supplied succinyl-CoA, a precursor metabolite required for cell carbon biosynthesis (Fig. 1; see also Fig. S1 in the supplemental material). This was supported by the observation that an NADPH-, CoA-, and Mg2+ ATP-dependent conversion of 3-hydroxypropionate to propionyl-CoA (Fig. 1) was upregulated in cell extracts of R. sphaeroides grown with 3-hydroxypropionate compared to those grown with succinate as a sole carbon source. Furthermore, the gene (RSP_1434) encoding a member of the medium-chain dehydrogenase/reductase (MDR) superfamily was involved in this reductive conversion as shown by mutant analysis (14). Accordingly, we speculated that RSP_1434 might encode an NADPH-dependent acrylyl-CoA reductase. RSP_1434 is also known as acuI and is cotranscribed with the gene dddL (RSP_1433) that is believed to encode a dimethylsulfoniopropionate (DMSP) lyase which catalyzes the cleavage of DMSP into acrylate and dimethylsulfide (15, 16). DddL is proposed to be involved in the so-called cleavage pathway of DMSP metabolism, and AcuI is supposedly involved in acrylate utilization (15); however, its precise biochemical function was unknown.
Fig 1.
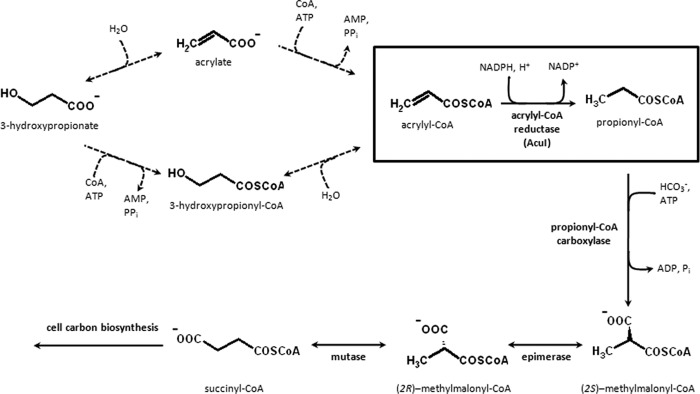
Reactions of the proposed reductive pathway for 3-hydroxypropionate assimilation by Rhodobacter sphaeroides 2.4.1. Acrylyl-CoA reductase (AcuI) is encoded by RSP_1434 (shown in the box). The reactions indicated by the dotted arrows are hypothetical; however, at least part of these reactions should exist, based on the fact that the overall conversion of 3-hydroxypropionate or acrylate to propionyl-CoA can be measured in cell extracts as described previously (14). In the case of R. sphaeroides, propionyl-CoA is assimilated via the methylmalonyl-CoA pathway and enters the citric acid cycle at succinyl-CoA. The methylmalonyl-CoA pathway consists of the following enzymes: propionyl-CoA carboxylase (alpha and beta subunits encoded by RSP_2191 and RSP_2189, respectively), ethylmalonyl-CoA/methylmalonyl-CoA epimerase (encoded by RSP_0812), and (2R)-methylmalonyl–CoA mutase (encoded by RSP_2192).
An acuI-like gene is also found in a marine chemoheterotrophic alphaproteobacterium, Ruegeria pomeroyi DSS-3, that can grow with DMSP as a sole carbon and energy source (17). For Rg. pomeroyi, the acuI-like gene (SPO_1914) is located immediately downstream of dmdA (SPO_1913), encoding DMSP demethylase, which is involved in the demethylation pathway of DMSP metabolism (18, 19). Rg. pomeroyi is thought to metabolize DMSP using both the cleavage and demethylation pathways (20). Therefore, even though SPO_1914 is clustered with a gene involved in the demethylation pathway, the product of SPO_1914 is required for the metabolism of acrylate that results as a product of the cleavage pathway (21) (see Fig. S1 in the supplemental material). A recent study by Johnston and colleagues (22) has found that Escherichia coli also possesses an acuI homolog known as yhdH (EcoGene accession number EG11315), which was postulated to function in resistance to acrylate.
The products of acuI, SPO_1914, and yhdH belong to the MDR012 family of the MDR superfamily (23). As of the year 2010, over 400 of mostly eubacterial protein sequences comprised the MDR012 family. However, despite their prevalence, the nature of the catalytic activities of the MDR012 enzymes remains elusive.
In this paper, we present biochemical evidence demonstrating that AcuI is an NADPH-dependent acrylyl-CoA reductase. For this, AcuI from R. sphaeroides was heterologously produced, and the kinetic properties were studied using a coupled spectrophotometric assay. The product of the reaction was identified as propionyl-CoA. Furthermore, two additional members of the MDR012 family, the product of SPO_1914 from Rg. pomeroyi as well as YhdH from E. coli, were shown to be active as NADPH-dependent acrylyl-CoA reductases and exhibit properties similar to those of AcuI. While AcuI has a role in anaplerosis (i.e., replenishing tricarboxylic acid cycle intermediates) during growth with 3-hydroxypropionate by R. sphaeroides, the same role cannot be simply extrapolated to the other members of the MDR012 family. In Rg. pomeroyi, SPO_1914 is involved in DMSP metabolism (21), while in E. coli, the physiological role of YhdH remains unknown.
MATERIALS AND METHODS
Materials.
3-Hydroxypropionate was purchased from Tokyo Chemical Industry (TCI) America (Portland, OR). Acetyl-CoA, propionyl-CoA, and crotonyl-CoA were synthesized from their anhydrides (24). (2S)-Methylmalonyl–CoA was synthesized enzymatically using crotonyl-CoA carboxylase/reductase (25).
Bacterial strains and growth conditions.
R. sphaeroides 2.4.1 (DSMZ 158) was used. The ΔacuI::kan LB2 mutant strain and the LB2 strain carrying the acuI-complementing plasmid pMA5-1 or the empty vector pBBRsm2MCS5 were isolated as described previously (14). For growth studies, the cells of R. sphaeroides wild type or mutants were pregrown for 12 to 24 h phototrophically (anoxic/light) in 6-ml minimal medium containing 10 mM sodium succinate as a sole carbon source (26). Pregrowth cultures were inoculated into anoxic stoppered screw-cap (Hungate) tubes containing 6-ml mineral medium supplemented with the appropriate carbon source and incubated under a light (3,000 lx) at 30°C. Growth was followed as the optical density at 578 nm (OD578). The mutant strains were grown in the presence of 20 μg ml−1 kanamycin and/or 25 μg ml−1 spectinomycin, where appropriate. E. coli strains DH5α, SM10, and Rosetta2 (DE3) were grown aerobically in Luria-Bertani (LB) broth containing the appropriate antibiotics (100 μg ml−1 ampicillin, 34 μg ml−1 chloramphenicol, 50 μg ml−1 kanamycin, and/or 50 μg ml−1 spectinomycin). For conjugation, the cells of R. sphaeroides were grown aerobically in LB medium at 30°C in darkness.
Complementation plasmids.
For constitutive expression of the codon-optimized SPO_1914 from a tetA promoter derived from plasmid pRL27 (27), the complementation plasmid pMA31-1 was constructed by replacing the insert acuI in pMA5-1 (14) with SPO_1914 from pMA28 (described below) using NdeI-BamHI restriction and subsequent ligation. For constitutive expression of the codon-optimized yhdH from the tetA promoter, the complementation plasmid pMA44-4 was constructed by replacing the insert acuI in pMA5-1 with yhdH from pMA37-4 (described below) using NdeI-HindIII restriction and subsequent ligation. For constitutive expression of ccr, the complementation plasmid pMA30-1 was constructed by replacing the insert acuI in pMA5-1 with ccr from pHD1 using NdeI-EcoRI restriction and subsequent ligation. For plasmid pHD1, the coding region for ccr was amplified from chromosomal DNA of R. sphaeroides 2.4.1 using primers 5′-CTA TAC TCA TAT GGC CCT CGA CGT GCA G-3′ (NdeI site is underlined) and 5′-CGT CTA CTA AGC TTT GCG GAT CGC TCC GAT CAG G-3′ (HindIII site is underlined) (annealing temperature, 59°C) and cloned into pET16b using NdeI-HindIII restriction and subsequent ligation. The complementation plasmids were mobilized to the ΔacuI::kan LB2 strain by conjugation with E. coli SM10 carrying the plasmid.
Heterologous expression of acuI and production of the N-terminal decahistidine-tagged (His10-tagged) enzyme in E. coli.
The coding region of acuI was amplified from the chromosomal DNA of R. sphaeroides 2.4.1 using the primers 5′-GGA GAA GCA TAT GAG AGC CGT TCT GAT AG-3′ (NdeI site is underlined) and 5′-CTA CGC GTT CGG ATC CGG AGT GCA TAC-3′ (BamHI site is underlined) (annealing temperature, 59°C). The product was cloned into pET16b using NdeI-BamHI restriction, resulting in the expression plasmid pMA2-1. E. coli Rosetta2 (DE3) was transformed with pMA2-1 and grown aerobically at 37°C until an OD578 of 0.6 to 0.8 was reached. At this point, 0.4 mM isopropyl-β-d-thiogalactopyranoside (IPTG) was added to the culture and further incubated aerobically at 30°C for 17 h. The cells were harvested by centrifugation and stored at −80°C until use.
Heterologous expression of the synthetic SPO_1914 and production of the His10-tagged enzyme in E. coli.
SPO_1914 from Rg. pomeroyi DSS-3 was chemically synthesized with codons modified for optimal expression in R. sphaeroides 2.4.1 (see Fig. S2 in the supplemental material). For this, codons used less than 5% of the time were replaced with more frequently used codons deduced from a codon bias map of the bacterium. The synthesized SPO_1914 was cloned into pUC57 using XbaI-BamHI restriction sites (GenScript, Piscataway, NJ). A fragment containing SPO_1914 was cloned into pET16b following NdeI-BamHI restriction, resulting in the expression plasmid pMA29-1. Transformation and induction of pMA29-1 in E. coli Rosetta2 (DE3) were carried out as described for pMA2-1.
Expression of the synthetic yhdH and production of the His10-tagged enzyme in E. coli.
The gene yhdH (EcoGene accession no. EG11315) from E. coli K-12 substrain MG1655 was chemically synthesized with codons modified for optimal expression in R. sphaeroides 2.4.1 (see Fig. S3 in the supplemental material). The synthesized yhdH was cloned into pJET1.2 by blunt-end cloning (GenScript, Piscataway, NJ). A fragment containing yhdH was cloned into pET16b following NdeI-HindIII restriction, resulting in the expression plasmid pMA37-4. Transformation and induction of pMA37-4 in E. coli Rosetta2 (DE3) were performed as described for pMA2-1.
Purification of the recombinant enzymes.
Harvested cells of E. coli Rosetta2 (DE3) (2 to 3 g [wet weight]) were suspended in 10 ml 20 mM Tris-Cl (pH 8.0) buffer containing 0.1 mg ml−1 DNase I and passed twice through a chilled French pressure cell at 137 MPa. Cell lysate was centrifuged at 4°C and 10,000 × g for 15 min, followed by 100,000 × g for 1 h. Glycerol was added to the cell extract to a final concentration of 15%.
Step 1: nickel affinity chromatography.
Cell extract (9 to 12 ml) was applied to an Ni+-nitrilotriacetic acid (NTA) agarose column (6.5 to 13 ml; Qiagen, Valencia, CA) equilibrated with buffer A (20 mM Tris-Cl [pH 8.0], 150 mM KCl) containing 25 mM imidazole. The column was washed with buffer A containing 25 mM imidazole (9 to 24 ml) followed by buffer A containing 70 mM imidazole (12 to 24 ml) at a flow rate of 1.0 ml min−1. The His10-tagged protein was eluted in buffer A containing 250 mM imidazole (9 to 10 ml). The active fraction was applied to a PD-10 desalting column (GE Healthcare, Piscataway, NJ) equilibrated with 20 mM Tris-Cl, pH 8.0, and eluted in the same buffer. Glycerol was added to a final concentration of 10%, and the protein was concentrated using the Vivaspin 20 ultrafiltration spin column (Sartorius Stedim, Göttingen, Germany) at 3,500 × g for 30 min (10°C). To the concentrated active fraction (6.5 to 9 ml), glycerol was further added to a final concentration of 20 to 30% and stored at −80°C.
Step 2: gel filtration.
An aliquot (0.2 ml) of the active fraction from step 1 was applied to a Superose 12 column (Amersham Pharmacia Biotech, Piscataway, NJ) equilibrated with 50 mM 4-morpholinepropanesulfonic acid (MOPS)-KOH (pH 7.5), 150 mM KCl, and 20% glycerol and eluted at a flow rate of 0.25 ml min−1 at 4°C for 120 min. The eluate was collected in 0.5-ml fractions and used immediately for enzyme assays.
Acrylyl-CoA reductase assay.
Enzyme activities were measured spectrophotometrically at 365 nm following the acrylyl-CoA- and protein-dependent oxidation of NAD(P)H [εNAD(P)H = 3,400 M−1 cm−1] in a cuvette with a path length of 1.0 cm at 30°C. Acrylyl-CoA was synthesized in situ using recombinant 3-hydroxypropionyl–CoA synthetase from S. tokodaii (28). The assay described below is a modification of that used by Teufel et al. (1). The reaction mixture (0.5 ml) contained 100 mM MOPS-KOH (pH 7.0), 10 mM MgCl2, 3 mM ATP, 0.1 mM CoA, 20 mM acrylate, 0.04 units (at 30°C) of recombinant S. tokodaii 3-hydroxypropionyl–CoA synthetase, and 0.35 mM NADPH. The mixture was incubated for 3 min, and the reaction was started by the addition of protein. The concentrations of 3-hydroxypropionyl–CoA synthetase were adjusted to ensure that it was not rate limiting. In determining the apparent Km value for acrylyl-CoA or NADPH, the concentration of one substrate was varied (CoA, 0.001 to 0.1 mM; NADPH, 0.005 to 0.35 mM), while the concentration of the other substrate was kept constant (CoA, 0.1 mM; NADPH, 0.35 mM). In determining the apparent Km value for NADH, the assays were carried out in a cuvette with a path length of 0.1 cm (0.2-ml reaction mixture); the concentration of NADH was varied from 0.2 to 4 mM, while the concentration of CoA (0.1 mM) was kept constant. The following buffers (all at a 0.1 M final concentration) were used to determine the pH optimum: 4-morpholineethanesulfonic acid (MES)-KOH (pH 6.0 to 6.5), MOPS-KOH (pH 6.5 to 7.5), 4-(2-hydroxyethyl)piperazine-1-ethanesulfonic acid (HEPES)-KOH (pH 7.5 to 8.0), N,N-Bis(2-hydroxyethyl)glycine (BICINE)-KOH (pH 8.0 to 9.0), 3-(cyclohexylamino)-1-propanesulfonic acid (CAPS)-KOH (pH 10.0). In determining the substrate specificity, chemically synthesized crotonyl-CoA (0.5 to 5 mM) was added to the reaction and 3-hydroxypropionyl–CoA was synthesized in situ by excess recombinant S. tokodaii 3-hydroxypropionyl–CoA synthetase in the assay mixture containing 3-hydroxypropionate instead of acrylate.
HPLC analysis.
An optimized reaction mixture (1.5 ml) was used to monitor product formation by acrylyl-CoA reductase (AcuI) following in situ acrylyl-CoA production by recombinant S. tokodaii 3-hydroxypropionyl–CoA synthetase. The assay mixture contained 100 mM Tris-Cl (pH 8.0), 10 mM MgCl2, 3 mM ATP, 0.12 units (at 30°C) of recombinant S. tokodaii 3-hydoxypropionyl–CoA synthetase, and 0.1 mM CoA. Acrylyl-CoA production was started by the addition of 20 mM acrylate. An aliquot (0.2 ml) of each reaction was removed and stopped by the addition of 4 μl 25% HCl at different time points (t = 0 min, 1 min, and 3 min). Subsequently, 1 mM NADPH was added to the reaction mixture from which 0.2 ml was withdrawn and mixed with 4 μl 25% HCl after 1 min. Acrylyl-CoA reduction was started by the addition of 0.3 μg of purified recombinant AcuI, and the reaction was stopped after 1 and 4 min by transferring 0.2 ml of the reaction mixture into 4 μl 25% HCl. The acidified samples were incubated on ice for 15 min, followed by centrifugation at 16,000 × g for 15 min to remove precipitated proteins. CoA-thioesters were analyzed by reverse-phase high-performance liquid chromatography (HPLC) using a Luna 5u C18 100A column (5 μm, 125 by 4.0 mm; Phenomenex, Torrance, CA). The standards and samples were eluted in a 30-min gradient from 2% (vol/vol) to 15% (vol/vol) acetonitrile in 50 mM potassium phosphate buffer (pH 6.7) at a flow rate of 0.6 ml min−1. All CoA-esters were detected at 260 nm. The retention times for standards were 12.8 min for methylmalonyl-CoA, 13.2 min for free CoA, 17 min for acetyl-CoA, 22 min for propionyl-CoA, and 25 min for crotonyl-CoA.
Protein analyses.
Protein concentration was determined by the method of Bradford (29) using bovine serum albumin as the standard. Protein fractions were analyzed by sodium dodecyl sulfate (SDS)-10% polyacrylamide gel electrophoresis (PAGE), and the protein bands were visualized by Coomassie brilliant blue R-250 stain. For AcuI, protein bands after PAGE were analyzed using in-gel trypsin digestion and liquid chromatography-mass spectrometry at the Mass Spectrometry and Proteomics Facility (The Ohio State University). Proteins were identified by a bioinformatic search using Mascot (Matrix Science, Boston, MA). Native molecular masses for all three recombinant proteins were determined on a Superose 12 gel filtration column calibrated with thyroglobulin (670 kDa), γ-globulin (158 kDa), ovalbumin (44 kDa), myoglobin (17 kDa), and vitamin B12 (1.35 kDa) (Gel Filtration Standard; Bio-Rad Laboratories, Hercules, CA).
RESULTS
Heterologous production of His10-tagged acrylyl-CoA reductases.
In order to test the possibility that RSP_1434 (acuI) from R. sphaeroides encodes an acrylyl-CoA reductase, a His10-tagged recombinant form of AcuI was heterologously produced using E. coli Rosetta2 (DE3). Cell extracts of E. coli producing AcuI showed acrylyl-CoA-dependent NADPH oxidation activity (12 μmol min−1 mg−1; Table 1), which was 1,000-fold greater than that detected in the cell extract of the nontransformed E. coli Rosetta2 (DE3) cells (0.013 μmol min−1 mg−1). This provided the first indication that AcuI indeed catalyzed the reduction of acrylyl-CoA. However, the activity in the cell extracts of E. coli was rapidly lost, similarly to what we observed for the overall conversion of 3-hydroxypropionate to propionyl-CoA in the cell extracts of R. sphaeroides grown with 3-hydroxypropionate (14). To this end, the addition of at least 10% (final concentration) glycerol to the cell extracts of E. coli stabilized the activity of acrylyl-CoA reduction. When glycerol was added to the cell extracts of R. sphaeroides, the activity for the overall conversion of 3-hydroxypropionate to propionyl-CoA also remained stable. This indicated that the previously observed rapid loss of the 3-hydroxypropionate conversion activity in the cell extracts of R. sphaeroides was due to the instability of AcuI.
Table 1.
Purification profiles of the heterologously produced histidine-tagged recombinant acrylyl-CoA reductases from Rhodobacter sphaeroides 2.4.1, Ruegeria pomeroyi DSS-3, and Escherichia coli K-12 substrain MG1655a
| Organism (gene locus) | Purification step | Vol (ml) | Total activity (μmol min−1) | Protein (mg) | Sp act (μmol min−1 mg−1) | Yield (%) | Purification (n-fold) |
|---|---|---|---|---|---|---|---|
| Rhodobacter sphaeroides 2.4.1 (acuI; RSP_1434) | Cell extract | 11 | 2,700 | 230 | 12 | 100 | 1 |
| Ni+ affinity chromatography | 12 | 620 | 11 | 56 | 23 | 5 | |
| Superose 12 gel filtration | 1.0 | 2.6 | 0.02 | 130 | NA | 11 | |
| Ruegeria pomeroyi DSS-3 (SPO_1914) | Cell extract | 12 | 7,100 | 310 | 23 | 100 | 1 |
| Ni+ affinity chromatography | 8.1 | 3,200 | 46 | 69 | 45 | 3 | |
| Superose 12 gel filtration | 1.5 | 32 | 0.33 | 98 | NA | 4 | |
| Escherichia coli K-12 substrain MG1655 (yhdH) | Cell extract | 12 | 3,200 | 260 | 12 | 100 | 1 |
| Ni+ affinity chromatography | 13 | 2,000 | 38 | 52 | 63 | 4 | |
| Superose 12 gel filtration | 2.0 | 14 | 0.20 | 72 | NA | 6 |
NA, not applicable, as only an aliquot was loaded onto the gel filtration column. In all cases, 15% (final concentration) glycerol was added to the cell extract. To the Superose 12 gel filtration column, 0.2 ml of the recombinant enzyme purified from the Ni+ affinity chromatography was applied.
In purifying the recombinant AcuI from the Ni+ affinity column, the His10-tagged enzyme (expected monomeric molecular mass, 37 kDa) coeluted with another protein sized approximately at 60 kDa on SDS-PAGE gels (Fig. 2A, lane 3). Protein fragment analyses identified the protein band at 37 kDa as AcuI (81% sequence coverage) and that at 60 kDa as the E. coli chaperone protein GroEL (GenBank accession number AAR21888; 81% coverage). Subsequent purification by gel filtration removed GroEL, and the recombinant AcuI was >95% pure as judged by electrophoresis and Coomassie staining (Fig. 2A, lane 4). The active form of the purified AcuI was recovered only when gel filtration was performed in the buffer containing 20% glycerol.
Fig 2.
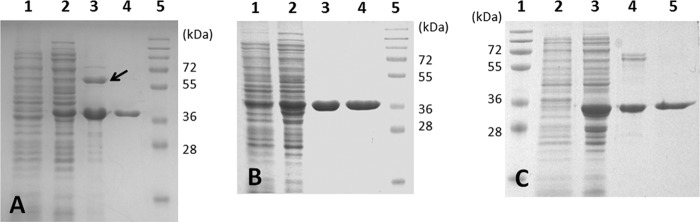
Purification of recombinant acrylyl-CoA reductases heterologously produced by E. coli Rosetta2 (DE3). Various fractions obtained during purification were separated on a 10% SDS-PAGE gel and stained with Coomassie blue. (A) Acrylyl-CoA reductase (AcuI) from Rhodobacter sphaeroides 2.4.1. Lane 1, whole cells before induction; lane 2, cell extract after 17 h of induction (20 μg); lane 3, recombinant AcuI eluted with E. coli GroEL (arrow) from the Ni+ affinity column (10 μg); lane 4, purified recombinant AcuI from the Superose 12 column (2 μg); lane 5, molecular mass standards. (B) Acrylyl-CoA reductase (product of SPO_1914) from Ruegeria pomeroyi DSS-3. Lane 1, whole cells before induction; lane 2, cell extract after 17 h of induction (20 μg); lane 3, recombinant product of SPO_1914 from the Ni+ affinity column (4 μg); lane 4, purified recombinant product of SPO_1914 from the Superose 12 column (4 μg); lane 5, molecular mass standards. (C) Acrylyl-CoA reductase (YhdH) from Escherichia coli K-12 substrain MG1655. Lane 1, molecular mass standards; lane 2, whole cells before induction; lane 3, cell extract after 18 h of induction (20 μg); lane 4, recombinant YhdH from the Ni+ affinity columns (4 μg); lane 5, purified recombinant YhdH from the Superose 12 column (2 μg).
Two other members of the MDR012 family, the product of SPO_1914 of Rg. pomeroyi and YhdH of E. coli, were also heterologously produced and purified to test for acrylyl-CoA reductase activity. SPO_1914 encodes a protein with 54% amino acid identity to AcuI (see Fig. S4 in the supplemental material) and is involved in DMSP metabolism by Rg. pomeroyi (21). YhdH is known as the founding member of the MDR012 family (23). While the activity of YhdH is yet unknown, it has been proposed to play a role in conferring resistance to acrylate (22). The amino acid sequence of YhdH is 54% identical to that of AcuI and 53% identical to that of the product of SPO_1914, indicating that neither protein is more or less closely related to YhdH (see Fig. S4). Thus, the activity of YhdH cannot be inferred solely on the basis of the sequence similarity. Unlike AcuI, the product of SPO_1914 or YhdH did not coelute with GroEL from an Ni+ affinity column (Fig. 2B and C). Both products were further purified by gel filtration (Table 1) and used in kinetic studies. All of the recombinant enzymes were stable for days in storage at −80°C.
Biochemical characterization of the recombinant AcuI of R. sphaeroides.
The properties of the recombinant AcuI are summarized in Table 2. The native molecular mass as determined by gel filtration was 64 kDa, suggesting that the enzyme in its native state formed a homodimer. The specific activity of NADPH-dependent acrylyl-CoA reduction was 130 ± 11 μmol min−1 mg−1, corresponding to a turnover number of 80 s−1 per subunit. In reverse-phase HPLC, the product of NADPH-dependent reduction of acrylyl-CoA was confirmed as propionyl-CoA (Fig. 3). After the addition of the CoA ligase, HPLC analysis indicated two additional peaks other than the one representing acrylyl-CoA (Fig. 3C). These two additional peaks constituted 25% of the total peak area of all three major peaks combined, and the earlier eluting compound is presumably due to adducts of acrylyl-CoA that formed as a result of a reaction between free CoA and acrylyl-CoA (30). Therefore, not all acrylyl-CoA formed by the ligase was available as a substrate for AcuI. In support of this account, we observed that for each mole of CoA added to the reaction, only 0.8 mol of NADPH was oxidized by AcuI in the coupled spectrophotometric assays. The time course HPLC profile showed that the adducts remained in the reaction mixture even after the addition of AcuI (Fig. 3E and F). This indicated that AcuI did not catalyze the reduction of those adducts. While it is still possible that AcuI may bind to these adducts, the possible inhibitory effect appears to be negligible given the high specific activity of acrylyl-CoA reduction observed in the reactions.
Table 2.
Molecular and catalytic properties of acrylyl-CoA reductases from Rhodobacter sphaeroides 2.4.1, Ruegeria pomeroyi DSS-3, Escherichia coli K-12 substrain MG1655, the crenarchaeon Sulfolobus tokodaii, and the Gram-positive bacterium Clostridium propionicumd
| Organism | Gene locus | Enzyme role in metabolism | Substrates | Products | Sp act (μmol min−1 mg−1) | Apparent Km for acrylyl-CoA (μM) | Apparent Km for NADPH (μM) | Turnover no. per subunit (s−1) | Native mol mass (kDa) | Calculated monomeric mol mass (kDa) | Composition | Specificity (%) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| MDR012 family | ||||||||||||
| Rhodobacter sphaeroides 2.4.1 | RSP_1434 (acuI) | 3-Hydroxypropionate assimilation | Acrylyl-CoA, NADPH, H+ | Propionyl-CoA, NADP+ | 130 ± 11 | ≤1.5 | 28 | 80 | 64 | 37 | Homodimer | Acrylyl-CoA, 100; crotonyl-CoA, <1; 3-OH–propionyl–CoA, <1; acrylate, <1; NADPH, 100; NADH, 7 |
| Ruegeria pomeroyi DSS-3 | SPO_1914 | Dimethylsulfoniopropionate metabolismb | Acrylyl-CoA, NADPH, H+ | Propionyl-CoA, NADP+ | 96 ± 4 | ≤2.8 | 18 | 60 | 62 | 37 | Homodimer | Acrylyl-CoA, 100; crotonyl-CoA, <1; 3-OH–propionyl–CoA, <1; acrylate, <1; NADPH, 100; NADH, 2 |
| Escherichia coli K-12 substrain MG1655 | yhdH | Unknown | Acrylyl-CoA, NADPH, H+ | Propionyl-CoA, NADP+ | 72 ± 6 | ≤1.1 | 33 | 45 | 56 | 37 | Homodimer | Acrylyl-CoA, 100; crotonyl-CoA, <1; 3-OH–propionyl–CoA, <1; acrylate, <1; NADPH, 100; NADH 10 |
| Sulfolobus tokodaii | ST0480 | Autotrophic CO2 fixation | Acrylyl-CoA, NADPH, H+ | Propionyl-CoA, NADP+ | 18.7 | 3 | 36 | 13 | 43 | 36 | Monomer | Acrylyl-CoA, 100; crotonyl-CoA, <1; NADPH, 100; NADH, <1 |
| Clostridium propionicum | Unidentified | Fermentation of alanine | Acrylyl-CoA, NADH, H+ | Propionyl-CoA, NAD+ | 0.79 | 2 ± 1 | ND, NADH, 8 μM | 4a | 600 ± 50 | 149c | Heterohexadecamer (α2βγ)4 | Acrylyl-CoA, 100; 3-buten–2-one, 0.7; crotonyl-CoA, <1 |
Turnover number per enzyme complex.
Physiological role of acrylyl-CoA reductase in Rg. pomeroyi DSS-3 was shown by Reisch et al. (21).
Experimentally derived molecular mass of α2βγ heterotetramer.
Fig 3.
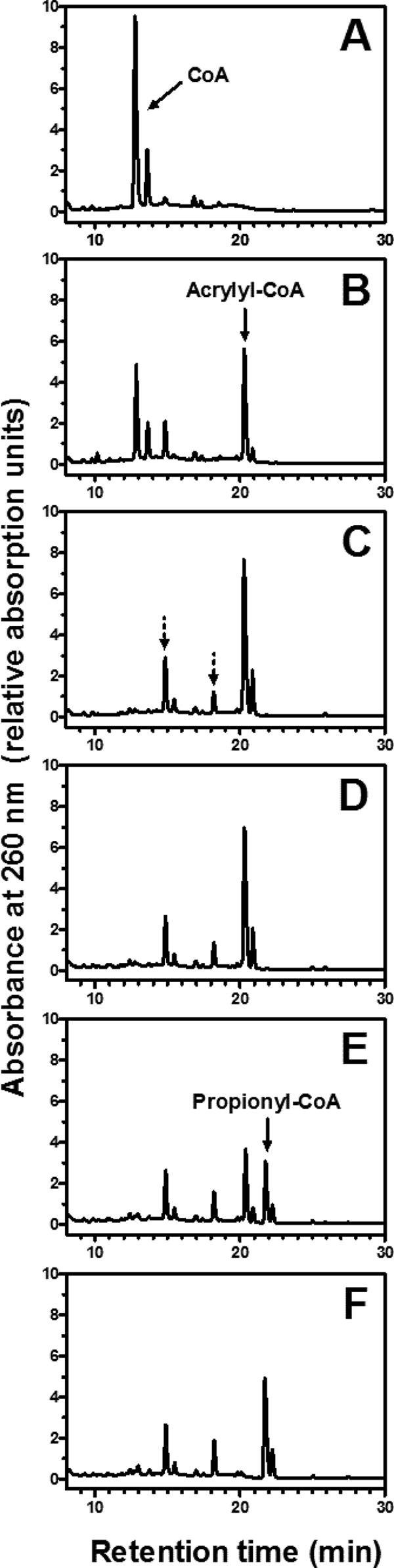
HPLC analysis of products formed by 3-hydroxypropionyl–CoA synthetase and acrylyl-CoA reductase (AcuI). Acrylyl-CoA was produced in situ by recombinant Sulfolobus tokodaii 3-hydroxypropionyl–CoA synthetase (A to C). (A) The reaction mixture contained 100 mM Tris-Cl (pH 8.0), 10 mM MgCl2, 3 mM ATP, recombinant S. tokodaii 3-hydroxypropionyl–CoA synthetase, and 0.1 mM CoA; (B) 1 min after the addition of 20 mM acrylate; (C) after 2 additional minutes of incubation; (D) 1 min after the addition of 1 mM NADPH; (E) 1 min after the addition of 0.3 μg of purified recombinant AcuI; (F) after 3 additional minutes of incubation. In panel C, the peaks denoted by the dotted arrows were considered the CoA adduct of acrylyl-CoA (30), which were not used as the substrates by AcuI. Free CoA and propionyl-CoA were identified by coelution with standard compounds (not shown).
To test whether AcuI could catalyze the reductive carboxylation of acrylyl-CoA to form (2S)-methylmalonyl–CoA, a coupled acrylyl-CoA reduction assay was carried out in the presence of 33 mM NaHCO3, and the products were analyzed by HPLC. This was tested because crotonyl-CoA carboxylase/reductase, an enzyme of the MDR050 family (23) related to AcuI (the MDR012 family), has been shown to catalyze reductive carboxylation of acrylyl-CoA (25). For AcuI, the HPLC profile of acrylyl-CoA reduction performed in the presence of NaHCO3 showed only propionyl-CoA as the product, indicating that AcuI did not catalyze the reductive carboxylation of acrylyl-CoA (data not shown).
NADPH-dependent reduction of acrylyl-CoA by AcuI followed Michaelis-Menten kinetics with an apparent Km for acrylyl-CoA of 1.5 μM and that for NADPH of 28 μM. Although NADH could substitute NADPH, the catalytic efficiency for the holoenzyme with NADH as the electron donor (kcat/Km = 0.42 μM−1 s−1) was 14-fold lower than that with NADPH (5.7 μM−1 s−1). NADPH-dependent reduction of crotonyl-CoA, a C4 analog of acrylyl-CoA, was not observed. AcuI did not catalyze reduction of acrylate or dehydration/reduction of 3-hydroxypropionyl–CoA. Specific activity of acrylyl-CoA reduction was unchanged at pH 6 to 9. Overall, the results confirmed that AcuI was an acrylyl-CoA reductase. Hence, acuI was annotated to encode an acrylyl-CoA reductase, and the sequence was deposited as GenBank accession number JQ582462 in the NCBI database.
Biochemical characterization of the recombinant enzymes encoded by SPO_1914 and yhdH.
The products of SPO_1914 and yhdH catalyzed NADPH-dependent reduction of acrylyl-CoA, confirming that all three enzymes of the MDR012 family examined in this study were indeed acrylyl-CoA reductase (Table 2). For the product of SPO_1914, the turnover number (60 s−1 per subunit) was slightly lower than that observed with AcuI (80 s−1 per subunit). The turnover number of acrylyl-CoA reduction by YhdH (45 s−1 per subunit) was the slowest of the three acrylyl-CoA reductases and only half of that of AcuI (Table 2).
As was the case with AcuI, NADH could substitute for NADPH in acrylyl-CoA reduction, albeit at low catalytic efficiency. For the holoenzyme encoded by SPO_1914, the catalytic efficiency with NADH (0.1 μM−1 s−1) was 70-fold lower than that with NADPH (6.7 μM−1 s−1). Likewise, for the holoenzyme of YhdH, the catalytic efficiency with NADH (0.27 μM−1 s−1) was 10-fold lower than that with NADPH (2.7 μM−1 s−1).
In contrast to AcuI, there was low but detectable activity for NADPH-dependent crotonyl-CoA reduction by the product of SPO_1914 and YhdH. In the presence of 1 mM crotonyl-CoA, the product of SPO_1914 catalyzed crotonyl-CoA reduction at 1 μmol min−1 mg−1. Given that this specific activity was 100-fold less than that of acrylyl-CoA reduction, further assays to determine the Km for crotonyl-CoA were not pursued. For YhdH, which exhibited slightly higher specific activity for crotonyl-CoA reduction than the product of SPO_1914, the kinetics of NADPH-dependent crotonyl-CoA reduction were determined. The activity followed Michaelis-Menten kinetics, with the apparent Km for crotonyl-CoA of 0.8 mM and Vmax of 6 μmol min−1 mg−1. Thus, the catalytic efficiencies for crotonyl-CoA and acrylyl-CoA were 0.009 and 83 μM−1s−1, respectively. This corresponds to a mere 0.01% specificity for crotonyl-CoA compared to acrylyl-CoA. Therefore, the results suggest that crotonyl-CoA reduction by the three acrylyl-CoA reductases examined in this study is likely to be physiologically insignificant.
Heterologous complementation of the acuI-null mutant by genes encoding acrylyl-CoA reductases (SPO_1914, yhdH) and crotonyl-CoA carboxylase/reductase (ccr).
Previously, we demonstrated that the R. sphaeroides ΔacuI::kan mutant was compromised for growth with 3-hydroxypropionate as a sole carbon source and hence concluded that acuI was involved in 3-hydroxypropionate assimilation by R. sphaeroides (14). Here, we conducted a similar growth experiment, in which the R. sphaeroides ΔacuI::kan mutant complemented with acuI, SPO_1914, yhdH, or ccr (RSP_0960) was tested for growth in minimal medium containing 3-hydroxypropionate or succinate (positive control) as a sole carbon source (Fig. 4). This was performed to test if an alternative gene, which encodes an enzyme that can act on acrylyl-CoA, could complement acuI to rescue the mutant for growth with 3-hydroxypropionate. Based on the biochemical analysis of the purified recombinant proteins, it was clear that both SPO_1914 and yhdH encoded NADPH-dependent acrylyl-CoA reductases. Therefore, we hypothesized that these genes could complement acuI in vivo. For optimal expression in R. sphaeroides, both SPO_1914 and yhdH introduced into the mutant were codon optimized (see Fig. S2 and S3 in the supplemental material) and placed under the control of a constitutive promoter. The ccr gene encodes the enzyme crotonyl-CoA carboxylase/reductase and can act on acrylyl-CoA as an alternative substrate (25). Reductive carboxylation of acrylyl-CoA by Ccr results in the product (2S)-methylmalonyl–CoA. Because methylmalonyl-CoA is an intermediate of the methylmalonyl-CoA pathway required for assimilation of propionyl-CoA (31, 32) and 3-hydroxypropionate (14), we hypothesized that ccr could also complement acuI for growth on 3-hydroxypropionate.
Fig 4.
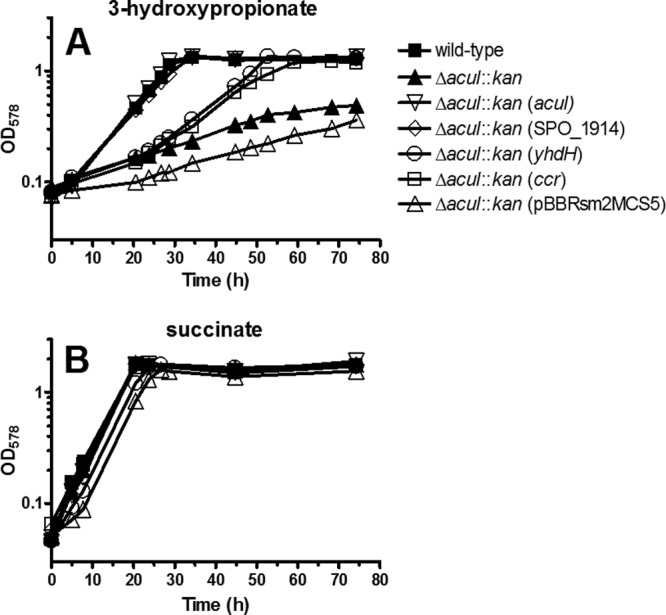
Photoheterotrophic (anoxic/light) growth of Rhodobacter sphaeroides 2.4.1. R. sphaeroides wild-type, ΔacuI::kan mutant, ΔacuI::kan mutant complemented with constitutively expressing acuI, SPO_1914, yhdH, or ccr from the plasmid (indicated in parentheses), and ΔacuI::kan mutant carrying the null vector pBBRsm2MCS5 were grown in the minimal medium containing 3-hydroxypropionate (10 mM) (A) or succinate (10 mM) (B) as a sole carbon source. See Materials and Methods for the names of the plasmids used in the study.
The results showed that the doubling time of the wild-type R. sphaeroides grown on 3-hydroxypropionate was 7 h, whereas that of the ΔacuI::kan mutant or the mutant carrying the vector pBBRsm2MCS5 was 20 to 30 h. The latter clearly indicated that acuI was involved in 3-hydroxypropionate assimilation. Doubling times of the acuI- or SPO_1914-complemented mutant grown on 3-hydroxypropionate was 7 h. This demonstrated that these genes restored the doubling time to the level comparable to that of the wild type. Doubling time of the yhdH-complemented mutant grown on 3-hydroxypropionate was 11 h. This was 4 h longer than that observed with the wild-type-, acuI-, or SPO_1914-complemented mutant. However, the yhdH-complemented mutant grew on 3-hydroxypropionate at a growth rate nearly twice as fast as that of the ΔacuI::kan mutant, indicating that yhdH was able to replace the function of acuI to some extent. The doubling time of the ccr-complemented mutant grown on 3-hydroxypropionate was 11 h, equivalent to that observed with the yhdH-complemented mutant. Therefore, this also indicated that ccr could at least partially replace the function of acuI.
DISCUSSION
AcuI defines a new class of acrylyl-CoA reductases.
Here, we present AcuI from R. sphaeroides as the first member of the MDR012 family of enzymes for which a biochemical activity has been demonstrated. Catalytic properties of the purified recombinant products of acuI and two additional members of the MDR012 family, the products of SPO_1914 and yhdH, clearly show that these enzymes are NADPH-dependent acrylyl-CoA reductases and exhibit similar kinetic properties. The crystal structure of YhdH had been solved almost a decade ago; however, its catalytic property has remained unknown until now (33).
Interestingly, the acrylyl-CoA reductase from crenarchaea (e.g., Sulfolobus) is also a member of the medium-chain dehydrogenase/reductase superfamily (1). However, its precise phylogenetic position based on the bioinformatic algorithm of the MDR superfamily (23) remains unidentified. While Sulfolobus acrylyl-CoA reductase is not a member of the MDR012 family, some of its properties do resemble those of the MDR012 family. As shown in Table 2, the enzymes of both families use NADPH as a reductant. They are also similar in size as monomers. Based on the amino acid sequence alignment of AcuI and Sulfolobus acrylyl-CoA reductase, the N-terminal half of the two proteins shares 25% identical amino acids (14) (see Fig. S4 in the supplemental material). On the other hand, the enzymes of the two families differ in their native composition. While Sulfolobus acrylyl-CoA reductase in its native state exists as a monomer (1), the enzymes of the MDR012 family function as homodimers. Furthermore, Sulfolobus acrylyl-CoA reductase appears to contain one or two Zn2+ ions (1), whereas the enzymes of the MDR012 family appear to contain none (23, 33), which is supported by the bioinformatic analysis (see Fig. S4).
The C-terminal acrylyl-CoA reductase domain of propionyl-CoA synthase from C. aurantiacus is also considered a member of the MDR superfamily (2, 14); however, its affiliation to a specific family within the MDR superfamily remains unclear (23). Although a partially digested propionyl-CoA synthase lacking the C-terminal domain has been shown as catalytically active in converting 3-hydroxypropionate to acrylyl-CoA (2), the catalytic activity by the standalone C-terminal acrylyl-CoA reductase domain has yet to be evidenced. Therefore, its kinetic comparison with other acrylyl-CoA reductases is currently not feasible.
Acrylyl-CoA reductase from C. propionicum is clearly distinct from the other above-described acrylyl-CoA reductases. Clostridial acrylyl-CoA reductase is not a member of the MDR superfamily. Instead, it is a multisubunit enzyme composed of a flavin-containing propionyl-CoA dehydrogenase and electron-transferring flavoprotein components. The latter is required for shuttling electrons from NADH to the flavin of the propionyl-CoA dehydrogenase component (3). Notably, acrylyl-CoA reductase activity by this clostridial enzyme complex can be measured only under anoxic conditions. This is because the NADH oxidase activity is linked to an electron-transferring flavoprotein. Under aerobic conditions, the reduced form (i.e., FADH2) readily reacts with molecular oxygen and produces H2O2. The clostridial enzyme complex has also been shown to catalyze the oxidation of propionyl-CoA using the artificial electron acceptor ferrocenium hexafluorophosphate (3). This activity is likely to be associated with the propionyl-CoA dehydrogenase component of the complex, as the transfer of electrons from an acyl-CoA to an artificial electron acceptor with a positive standard redox potential is typical for acyl-CoA dehydrogenases (34). In contrast, the enzymes of the MDR012 family or Sulfolobus acrylyl-CoA reductase would not catalyze the oxidation of propionyl-CoA. This is because these enzymes do not contain a flavin cofactor, and therefore the oxidation of propionyl-CoA would require transfer of electrons from propionyl-CoA directly onto NADP+. The latter is not feasible given the difference in standard redox potential of the NADP+-NADPH couple (E0′ = −0.32 V) and that of the acrylyl-CoA–propionyl-CoA couple (E0′ = +69 mV) (35).
For all acrylyl-CoA reductases thus far studied, the catalytic efficiency toward crotonyl-CoA, a C4-analog of acrylyl-CoA, is less than 1% (Table 2). Recently, a bona fide crotonyl-CoA reductase from the spirochete Treponema denticola has been characterized (36, 37). The enzyme is NADH dependent and belongs to the family of trans-2-enoyl–CoA reductase, which is clearly distinct from the MDR superfamily. Some members of the trans-2-enoyl–CoA reductase family have been shown to act on longer-chain enoyl-CoA substrates (e.g., hexenoyl-CoA) (38). However, for all the members of the trans-2-enoyl–CoA reductase family, the specificity toward acrylyl-CoA has, to our knowledge, not been tested and remains unknown.
A striking feature common to all acrylyl-CoA reductases is the extremely low Km values for acrylyl-CoA (<3 μM) (Table 2). Herrmann and colleagues (39) indicate that acrylyl-CoA is a strong electrophile and can react readily with essential biomolecules and may cause substantial damage to the cells. However, cells that rely on primary carbon and/or energy metabolism involving acrylyl-CoA as an intermediate face a steady flux of acrylyl-CoA. In the case of Sulfolobus and C. aurantiacus, acrylyl-CoA is an intermediate during autotrophic CO2 fixation (see Fig. S1 in the supplemental material). For Clostridium, acrylyl-CoA is an intermediate in energy metabolism (i.e., fermentation) during growth with alanine (see Fig. S1). In principle, acrylyl-CoA reductases with an extremely low Km for acrylyl-CoA would minimize the accumulation of acrylyl-CoA in these cells.
Metabolic role of AcuI.
We have previously shown that acuI is involved in 3-hydroxypropionate assimilation by R. sphaeroides (14). This was demonstrated by in vivo growth analysis in which an acuI mutant was compromised for growth with 3-hydroxypropionate. Although it has been recently proposed that the role of AcuI is merely to detoxify acrylyl-CoA (22), we must emphasize that the role of AcuI is in the assimilation of 3-hydroxypropionate. This is because AcuI catalyzes the reduction of acrylyl-CoA to propionyl-CoA, which is then converted to succinyl-CoA through the reactions of the methylmalonyl-CoA pathway by propionyl-CoA carboxylase, methylmalonyl-CoA/ethylmalonyl-CoA epimerase, and (2R)-methylmalonyl–CoA mutase (Fig. 1). Because succinyl-CoA is an intermediate of the tricarboxylic acid cycle, the pathway described here essentially functions as an anaplerotic pathway for R. sphaeroides growing with 3-hydroxypropionate.
In the past, much of the studies on acuI have been focused on DMSP. However, a major caveat of the studies concerning the metabolism of DMSP in R. sphaeroides is that this compound does not serve as a sole carbon source for growth by R. sphaeroides (15). In addition, it is quite possible that dddL, which is located immediately downstream of and cotranscribed with acuI (16), may not encode a bona fide DMSP lyase. This is because the reported activity of DMSP cleavage by R. sphaeroides is extremely low (15). This suggests that DMSP is probably not the primary substrate for catalysis by DddL. Thus, DddL may actually act on an unknown substrate whose chemical structure somewhat resembles DMSP (40).
Metabolic role of the SPO_1914 product.
Unlike R. sphaeroides, Rg. pomeroyi is capable of growing with DMSP or acrylate as a sole carbon and energy source (17). An SPO_1914 mutant of Rg. pomeroyi was unable to grow with DMSP or acrylate (21, 22), indicating that acrylyl-CoA reductase encoded by SPO_1914 is involved in DMSP and acrylate metabolism by Rg. pomeroyi.
The marine proteobacteria Halomonas sp. HTNK1 (22, 41) and Alcaligenes faecalis M3A (22, 42), both of which can use DMSP as a sole carbon and energy source, have the acuI-like genes present in large gene clusters, including genes involved in DMSP metabolism. This strongly suggests that these acuI-like genes encode functional acrylyl-CoA reductases that function in DMSP metabolism.
The metabolic role of YhdH is still unknown.
Based on our analysis, it is not possible to assign a metabolic role to YhdH of E. coli. Because E. coli K-12 substrain MG1655 was unable to grow aerobically in M63 minimal medium containing 3-hydroxypropionate (10 to 20 mM) or acrylate (5 to 10 mM) as a sole carbon and energy source (M. Asao, unpublished data), we deduced that YhdH had no role in the metabolism of these substrates. While many species of Enterobacteriaceae also carry yhdH, which is highly conserved among them, the gene is found in various genomic contexts with no obvious indication of its metabolic role. The proposed role of YhdH in providing acrylate tolerance remains unclear, as the fate of propionyl-CoA, the also potentially toxic product of acrylyl-CoA reduction, remains unexplained (22).
Heterologous genes can replace acuI during growth of R. sphaeroides with 3-hydroxypropionate.
As expected based on the biochemical data that demonstrated efficient acrylyl-CoA reduction, SPO_1914 from Rg. pomeroyi or yhdH from E. coli could rescue an acuI mutant of R. sphaeroides for growth with 3-hydroxypropionate. The gene ccr from R. sphaeroides could also, at least partially, restore the acuI mutant for growth with 3-hydroxypropionate. The result is likely due to the reductive carboxylation of acrylyl-CoA, a side activity of crotonyl-CoA carboxylase/reductase (Ccr) (25). This would imply that the product (2S)-methylmalonyl–CoA is assimilated via the methylmalonyl-CoA pathway by R. sphaeroides growing with 3-hydroxypropionate. The partial rescue of the 3-hydroxypropionate phenotype of the acuI mutant by ccr is remarkable, as Ccr has an at least 300-fold-higher Km value for acrylyl-CoA than AcuI (25) (Table 2). Ccr has no inherent role in 3-hydroxypropionate assimilation by wild-type R. sphaeroides (14); however, Ccr activity was detectable in cell extracts of the noncomplemented acuI mutant during growth with 3-hydroxypropionate (unpublished results). The upregulation of Ccr, therefore, may account for the slow growth of the acuI mutant. The finding that ccr can rescue an acuI mutant for growth with 3-hydroxypropionate has significant implications in interpreting the results of in vivo complementation. For one, because ccr is required for acetate assimilation but not for 3-hydroxypropionate metabolism, it is clear that the complementation does not necessarily identify genes serving the same native physiological function or role. Two, because it was not the primary catalytic activity (crotonyl-CoA carboxylation/reduction) but the side activity of Ccr (acrylyl-CoA carboxylation/reduction) that was responsible for the complementation, it becomes clear that substrate specificity of an enzyme could be easily misidentified without further analysis. Indeed, it may actually be the case that some DMSP lyases have been misidentified based on complementation in E. coli that results in production of dimethylsulfide from DMSP (40). Because at least some of these lyases identified in this manner are likely to act on DMSP as an alternative substrate, they might not be involved in DMSP metabolism in their native system.
Supplementary Material
ACKNOWLEDGMENTS
We thank F. Robert Tabita (The Ohio State University) for the valuable advice in stabilizing the enzyme activity. We also thank Sriram Satagopan and Chris Rocco (both of The Ohio State University) for their expertise in assisting M.A. with conducting gel filtration and HPLC, respectively. We thank Michael Carter and Patrice Hamel for critical review of the manuscript.
This work was supported by grant MCB0842892 from the National Science Foundation.
Footnotes
Published ahead of print 16 August 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JB.00685-13.
REFERENCES
- 1.Teufel R, Kung JW, Kockelkorn D, Alber BE, Fuchs G. 2009. 3-Hydroxypropionyl-coenzyme A dehydratase and acryloyl-coenzyme A reductase, enzymes of the autotrophic 3-hydroxypropionate/4-hydroxybutyrate cycle in the Sulfolobales. J. Bacteriol. 191:4572 4581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Alber BE, Fuchs G. 2002. Propionyl-coenzyme A synthase from Chloroflexus aurantiacus, a key enzyme of the 3-hydroxypropionate cycle for autotrophic CO2 fixation. J. Biol. Chem. 277:12137–12143 [DOI] [PubMed] [Google Scholar]
- 3.Hetzel M, Brock M, Selmer T, Pierik AJ, Golding BT, Buckel W. 2003. Acryloyl-CoA reductase from Clostridium propionicum. An enzyme complex of propionyl-CoA dehydrogenase and electron-transferring flavoprotein. Eur. J. Biochem. 270:902–910 [DOI] [PubMed] [Google Scholar]
- 4.Berg IA, Kockelkorn D, Buckel W, Fuchs G. 2007. A 3-hydroxypropionate/4-hydroxybutyrate autotrophic carbon dioxide assimilation pathway in archaea. Science 318:1782–1786 [DOI] [PubMed] [Google Scholar]
- 5.Berg IA, Kockelkorn D, Ramos-Vera WH, Say RF, Zarzycki J, Hügler M, Alber BE, Fuchs G. 2010. Autotrophic carbon fixation in archaea. Nat. Rev. Microbiol. 8:447–460 [DOI] [PubMed] [Google Scholar]
- 6.Herter S, Farfsing J, Gad'on N, Rieder C, Eisenreich W, Bacher A, Fuchs G. 2001. Autotrophic CO2 fixation by Chloroflexus aurantiacus: study of glyoxylate formation and assimilation via the 3-hydroxypropionate cycle. J. Bacteriol. 183:4305–4316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Strauss G, Fuchs G. 1993. Enzymes of a novel autotrophic CO2 fixation pathway in the phototrophic bacterium Chloroflexus aurantiacus, the 3-hydroxypropionate cycle. Eur. J. Biochem. 215:633–643 [DOI] [PubMed] [Google Scholar]
- 8.Zarzycki J, Brecht V, Müller M, Fuchs G. 2009. Identifying the missing steps of the autotrophic 3-hydroxypropionate CO2 fixation cycle in Chloroflexus aurantiacus. Proc. Natl. Acad. Sci. U. S. A. 106:21317–21322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cardon BP, Barker HA. 1946. Two new amino-acid-fermenting bacteria, Clostridium propionicum and Diplococcus glycinophilus. J. Bacteriol. 52:629–634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schweiger G, Buckel W. 1984. On the dehydration of (R)-lactate in the fermentation of alanine to propionate by Clostridium propionicum. FEBS Lett. 171:79–84 [DOI] [PubMed] [Google Scholar]
- 11.Schweiger G, Buckel W. 1985. Identification of acrylate, the product of the dehydration of (R)-lactate catalyzed by cell-free extracts from Clostridium propionicum. FEBS Lett. 185:253–256 [DOI] [PubMed] [Google Scholar]
- 12.Brockman HL, Wood WA. 1975. Electron-transferring flavoprotein of Peptostreptococcus elsdenii that functions in the reduction of acrylyl-coenzyme A. J. Bacteriol. 124:1447–1453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Marx H, Graf AB, Tatto NE, Thallinger GG, Mattanovich D, Sauer M. 2011. Genome sequence of the ruminal bacterium Megasphaera elsdenii. J. Bacteriol. 193:5578–5579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schneider K, Asao M, Carter MS, Alber BE. 2012. Rhodobacter sphaeroides uses a reductive route via propionyl coenzyme A to assimilate 3-hydroxypropionate. J. Bacteriol. 194:225–232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Curson ARJ, Rogers R, Todd JD, Brearley CA, Johnston AWB. 2008. Molecular genetic analysis of a dimethylsulfoniopropionate lyase that liberates the climate-changing gas dimethylsulfide in several marine α-proteobacteria and Rhodobacter sphaeroides. Environ. Microbiol. 10:757–767 [DOI] [PubMed] [Google Scholar]
- 16.Sullivan MJ, Curson ARJ, Shearer N, Todd JD, Green RT, Johnston AWB. 2011. Unusual regulation of a leaderless operon involved in the catabolism of dimethylsulfoniopropionate in Rhodobacter sphaeroides. PLoS One 6:e15972. 10.1371/journal.pone.0015972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gonzáles JM, Covert JS, Whitman WB, Henriksen JR, Mayer F, Scharf B, Schmitt R, Buchan A, Fuhrman JA, Kiene RP, Moran MA. 2003. Silicibacter pomeroyi sp. nov. and Roseovarius nubinhibens sp. nov., dimethylsulfoniopropionate-demethylating bacteria from marine environments. Int. J. Syst. Evol. Microbiol. 53:1261–1269 [DOI] [PubMed] [Google Scholar]
- 18.Howard EC, Henriksen JR, Buchan A, Reisch CR, Bürgmann H, Welsh R, Ye W, González J, Mace K, Joye SB, Kiene RP, Whitman WB, Moran MA. 2006. Bacterial taxa that limit sulfur flux from the ocean. Science 314:649–652 [DOI] [PubMed] [Google Scholar]
- 19.Reisch CR, Moran MA, Whitman WB. 2008. Dimethylsulfoniopropionate-dependent demethylase (DmdA) from Pelagibacter ubique and Silicibacter pomeroyi. J. Bacteriol. 190:8018–8024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bürgmann H, Howard EC, Ye W, Sun F, Sun S, Napierala S, Moran MA. 2007. Transcriptional response of Silicibacter pomeroyi DSS-3 to dimethylsulfoniopropionate (DMSP). Environ. Microbiol. 9:2742–2755 [DOI] [PubMed] [Google Scholar]
- 21.Reisch CR, Crabb WM, Gifford SM, Teng Q, Stoudemayer MJ, Moran MA, Whitman WB. 2013. Metabolism of dimethylsulphoniopropionate by Ruegeria pomeroyi DSS-3. Mol. Microbiol. 89:774–791 [DOI] [PubMed] [Google Scholar]
- 22.Todd JD, Curson ARJ, Sullivan MJ, Kirkwood M, Johnston AWB. 2012. The Ruegeria pomeroyi acuI gene has a role in DMSP catabolism and resembles yhdH of E. coli and other bacteria in conferring resistance to acrylate. PLoS One 7:e35947. 10.1371/journal.pone.0035947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hedlund J, Jörnvall H, Persson B. 2010. Subdivision of the MDR superfamily of medium-chain dehydrogenases/reductases through iterative hidden Markov model refinement. BMC Bioinformatics 11:534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Simon EJ, Shemin D. 1953. The preparation of S-succinyl coenzyme A. J. Am. Chem. Soc. 75:2520 [Google Scholar]
- 25.Erb TJ, Brecht V, Fuchs G, Müller M, Alber BE. 2009. Carboxylation mechanism and stereochemistry of crotonyl-CoA carboxylase/reductase, a carboxylating enoyl-thioester reductase. Proc. Natl. Acad. Sci. U. S. A. 106:8871–8876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Alber BE, Spanheimer R, Ebenau-Jehle C, Fuchs G. 2006. Study of an alternate glyoxylate cycle for acetate assimilation by Rhodobacter sphaeroides. Mol. Microbiol. 61:297–309 [DOI] [PubMed] [Google Scholar]
- 27.Larsen RA, Wilson MM, Guss AM, Metcalf WW. 2002. Genetic analysis of pigment biosynthesis in Xanthobacter autotrophicus Py2 using a new, highly efficient transposon mutagenesis system that is functional in a wide variety of bacteria. Arch. Microbiol. 178:193–201 [DOI] [PubMed] [Google Scholar]
- 28.Alber BE, Kung JW, Fuchs G. 2008. 3-Hydroxypropionyl-coenzyme A synthetase from Metallospaera sedula, an enzyme involved in autotrophic CO2 fixation. J. Bacteriol. 190:1383–1389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bradford MM. 1976. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72:248–254 [DOI] [PubMed] [Google Scholar]
- 30.Patel SS, Walt DR. 1988. Acetyl coenzyme A synthetase catalyzed reactions of coenzyme A with α,β-unsaturated carboxylic acids. Anal. Biochem. 170:355–360 [DOI] [PubMed] [Google Scholar]
- 31.Maruyama K, Kitamura H. 1975. Some effects of propionate on the growth of Rhodopseudomonas spheroides S. Agr. Biol. Chem. 39:1521–1526 [Google Scholar]
- 32.Maruyama K. 1979. The metabolic pathway of propionate in Rhodopseudomonas sphaeroides S. Agric. Biol. Chem. 43:2385–2386 [Google Scholar]
- 33.Sulzenbacher G, Roig-Zamboni V, Salomoni A, Valencia C, Campanacci V, Vincentelli R, Tegoni M, Eklund H, Cambillau C. 2004. Structure of Escherichia coli YhdH, a putative quinone oxidoreductase. Acta Crystallogr. D Biol. Crystallogr. 60:1855–1862 [DOI] [PubMed] [Google Scholar]
- 34.Lehman TC, Thorpe C. 1990. Alternate electron acceptors for medium-chain acyl-CoA dehydrogenase: use of ferrecenium salts. Biochemistry 29:10594–10602 [DOI] [PubMed] [Google Scholar]
- 35.Sato K, Nishina Y, Setoyama C, Miura R, Shiga K. 1999. Unusually high standard redox potential of acrylyl-CoA/propionyl-CoA couple among enoyl-CoA/acyl-CoA couples: a reason for the distinct metabolic pathway of propionyl-CoA from longer acyl-CoAs. J. Biochem. 126:668–675 [DOI] [PubMed] [Google Scholar]
- 36.Bond-Watts BB, Weeks AM, Chang MCY. 2012. Biochemical and structural characterization of the trans-enoyl-CoA reductase from Treponema denticola. Biochemistry 51:6827–6837 [DOI] [PubMed] [Google Scholar]
- 37.Tucci S, Martin W. 2007. A novel prokaryotic trans-enoyl-CoA reductase from the spirochete Treponema denticola. FEBS Lett. 581:1561–1566 [DOI] [PubMed] [Google Scholar]
- 38.Hoffmeister M, Piotrowski M, Nowitzki U, Martin W. 2005. Mitochondrial trans-2-enoyl-CoA reductase of wax ester fermentation from Euglena gracilis defines a new family of enzymes involved in lipid synthesis. J. Biol. Chem. 280:4329–4338 [DOI] [PubMed] [Google Scholar]
- 39.Herrmann G, Selmer T, Jessen HJ, Gokarn RR, Selifonova O, Gort SJ, Buckel W. 2005. Two beta-alanyl-CoA:ammonia lyases in Clostridium propionicum. FEBS J. 272:813–821 [DOI] [PubMed] [Google Scholar]
- 40.Johnston AWB, Todd JD, Curson ARJ. 2012. Microbial origins and consequences of dimethyl sulfide. Microbe 7:181–185 [Google Scholar]
- 41.Todd JD, Curson ARJ, Nikolaidou-Katsaraidou N, Brearley CA, Watmough N, Chan Y, Page PCB, Sun L, Johnston AWB. 2010. Molecular dissection of bacterial acrylate catabolism—unexpected links with dimethlsulfoniopropionate catabolism and dimethyl sulfide production. Environ. Micrbiol. 12:327–343 [DOI] [PubMed] [Google Scholar]
- 42.Ansede JH, Pellechia PJ, Yoch DC. 1999. Metabolism of acrylate to β-hydroxypropionate and its role in dimethylsulfoniopropionate lyase induction by a salt marsh sediment bacterium, Alcaligenes faecalis M3A. Appl. Environ. Microbiol. 65:5075–5081 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.