

Abstract
The LytR-CpsA-Psr (LCP) proteins are thought to transfer bactoprenol-linked biosynthetic intermediates of wall teichoic acid (WTA) to the peptidoglycan of Gram-positive bacteria. In Bacillus subtilis, mutants lacking all three LCP enzymes do not deposit WTA in the envelope, while Staphylococcus aureus Δlcp mutants display impaired growth and reduced levels of envelope phosphate. We show here that the S. aureus Δlcp mutant synthesized WTA yet released ribitol phosphate polymers into the extracellular medium. Further, Δlcp mutant staphylococci no longer restricted the deposition of LysM-type murein hydrolases to cell division sites, which was associated with defects in cell shape and increased autolysis. Mutations in S. aureus WTA synthesis genes (tagB, tarF, or tarJ2) inhibit growth, which is attributed to the depletion of bactoprenol, an essential component of peptidoglycan synthesis (lipid II). The growth defect of S. aureus tagB and tarFJ mutants was alleviated by inhibition of WTA synthesis with tunicamycin, whereas the growth defect of the Δlcp mutant was not relieved by tunicamycin treatment or by mutation of tagO, whose product catalyzes the first committed step of WTA synthesis. Further, sortase A-mediated anchoring of proteins to peptidoglycan, which also involves bactoprenol and lipid II, was not impaired in the Δlcp mutant. We propose a model whereby the S. aureus Δlcp mutant, defective in tethering WTA to the cell wall, cleaves WTA synthesis intermediates, releasing ribitol phosphate into the medium and recycling bactoprenol for peptidoglycan synthesis.
INTRODUCTION
The peptidoglycan layer protects Gram-positive bacteria from osmotic lysis and serves as a barrier against compounds toxic to the membrane (1). Peptidoglycan also functions as a scaffold for the immobilization of capsular polysaccharides (2), wall teichoic acids (WTAs) (3), and proteins (4). Surface proteins are anchored by sortase A, a membrane-embedded transpeptidase that scans secreted polypeptides for the LPXTG motif of sorting signals (5). Sortase A cleaves the peptide bond between the threonine and the glycine of the LPXTG motif to form a thioester-linked intermediate between the carboxyl group of threonine at the C-terminal end of surface proteins and its active-site cysteine residue (5, 6). The sortase A acyl intermediate is resolved by the nucleophilic attack of the free amino group of the pentaglycine cross bridge within lipid II (7, 8), the substrate for peptidoglycan biosynthesis (9). Surface protein linked to lipid II is incorporated into the cell wall envelope via the transpeptidation and transglycosylation reactions of peptidoglycan synthesis (10–12). In Staphylococcus aureus, lipid II is comprised of C55-PP–MurNAc–[d-Ala–d-iGln-(NH2-Gly5)–l-Lys–d-Ala–d-Ala]–GlcNAc, i.e., a murein disaccharide-pentapeptide subunit linked to the membrane carrier bactoprenol (C55). In Gram-positive bacteria, bactoprenol/undecaprenyl is also used for the synthesis of extracellular polymers, such as WTA (13), teichuronic acid (14), and cell wall polysaccharides (15).
The S. aureus WTA is a polymer of 30 to 50 ribitol phosphate (Rbo-P) subunits connected via 1,5-phosphodiester bonds (16). Rbo-Pn is tethered to peptidoglycan via the murein linkage unit, GlcNAc-ManNAc-(glycerol phosphate [Gro-P])2-3 (17). Synthesis of the murein linkage unit is initiated by TagO (also referred to as TarO), which links UDP-GlcNAc and undecaprenyl phosphate to generate C55-PP–GlcNAc (14, 18, 19). The other WTA subunits are added to the undecaprenyl-linked intermediate via the enzymes TagA (ManNAc) (17, 20), TagBDF (Gro-P), and TarILJ (Rbo-P) (21–23). The product of this pathway, C55-PP–GlcNAc–ManNAc–(Gro-P)2-3–(Rbo-P)30-50, is presumably flipped across the plasma membrane by the TagGH transporter (24). Attachment of the WTA polymer to the C-6 hydroxyl of N-acetylmuramic acid within peptidoglycan [MurNAc-P–GlcNAc–ManNAc–(Gro-P)2-3–(Rbo-P)30-50] occurs during cell wall assembly (25). The first two genes of the WTA pathway (tagO, tagA) can be deleted without abolishing staphylococcal growth (20, 26, 27). In contrast, tagBDFGH and tarIJL cannot be deleted unless staphylococci carry inactivating mutations in tagA or tagO (28). This synthetic viable phenotype is explained by the limited availability of bactoprenol and its undecaprenyl phosphate derivatives (C55-PP and C55-P) for peptidoglycan cell wall biosynthesis (28). WTA synthesis is blocked by tunicamycin, an antibiotic from Streptomyces clavuligerus that inhibits TagO (29).
Kawai et al. proposed that a family of genes encoding the LytR-CpsA-Psr (LCP) proteins catalyzes attachment of the murein linkage unit of WTA to the peptidoglycan of Bacillus subtilis (30). B. subtilis encodes three lcp gene homologues, designated tagTUV, which are positioned within gene clusters for WTA biosynthetic enzymes (30). Simultaneous deletion of all three genes (tagTUV) is not compatible with bacterial growth unless bacilli lack the tagO gene. Blocking the expression of tagTUV causes a concomitant decrease in the synthesis of WTA (30). Further, X-ray crystallography identified polyprenyl phosphate bound to recombinant B. subtilis TagT or its Streptococcus pneumoniae Cps2A homologue, both of which also exert in vitro phosphatase activity (30, 31). On the basis of these observations, Kawai et al. proposed that LCP enzymes recognize WTA or capsular polysaccharide synthesis intermediates as the substrates for the formation of phosphodiester linkages formed between the C6-hydroxyl of MurNAc in peptidoglycan and GlcNAc-ManNAc murein linkage units (30). In agreement with this model, mutants (with the Δlcp mutation) lacking all of the three LCP homologues of S. aureus—lcpA (mrsR), lcpB (sa0908), and lcpC (sa2103)—do not harbor phosphate residues in the cell wall envelope (32) and cannot properly place cell division septa (33). Previous work left unresolved whether S. aureus Δlcp mutants are defective in the synthesis and/or the cell wall anchoring of WTA and whether their associated cell division defect is due to the sequestration of lipid II within the WTA pathway (32, 33). The present study was performed to address these questions.
MATERIALS AND METHODS
Bacterial strains, bacterial growth, and reagents.
S. aureus strains were grown in tryptic soy broth (TSB) or on tryptic soy agar (TSA) supplemented with appropriate antibiotics. Erythromycin (Erm) and chloramphenicol (Cm) were used at a concentration of 10 μg/ml, and tunicamycin (Tun), unless otherwise specified, was used at a concentration of 1 μg/ml. To examine the effect of tunicamycin on bacterial growth, overnight cultures grown in the absence of tunicamycin were diluted (1:100) into 100 μl fresh TSB with or without tunicamycin and growth at 37°C was monitored every 15 min for 12 h in a Synergy HT plate reader (BioTek) by measuring the absorbance at 600 nm (A600). To assess cell viability, overnight cultures of candidate strains were started by inoculating an isolated colony from a fresh plate into fresh medium. On the next day, a subculture was prepared by diluting an aliquot of the overnight culture in tubes containing fresh medium (1:100). The tubes were placed at 37°C with shaking. Sample aliquots were removed from the cultures after 3.5 h or 16 h, A600 values were recorded, and aliquots were serially diluted and plated on agar. The numbers of CFU were determined following incubation of the plates at 37°C overnight. The bacterial strains and plasmids utilized in this study are listed in Table 1. Mutations in the three LCP-encoding genes, msrR, sa0908, and sa2103, have been described earlier (32). For convenience, the genes are designated herein lcpA, lcpB, and lcpC, respectively. The nomenclature of the single, double, and triple mutants has been changed accordingly: each of the single or double mutants is referred to as the ΔlcpA (msrR), ΔlcpB (sa0908), ΔlcpC (sa2103), ΔlcpAB (msrR and sa0908), ΔlcpAC (msrR and sa2103), and ΔlcpBC (sa0908 and sa2103) mutants. The Δlcp mutant is the triple mutant lacking all three lcp genes (msrR, sa0908, sa2103). Deletion of tagO (ΔtagO) was achieved by allelic replacement using plasmid pKOR1 (34, 35). Transposon mutagenesis was performed using two plasmids, pFA545 and pBursa (36). Transposon mutants were isolated on agar plates containing tunicamycin, and candidate clones were rescreened for tunicamycin-dependent growth. To analyze the anchor structure of surface proteins, S. aureus strains were transformed with plasmid pHTT4 (10), which provides for the expression of the hybrid protein SEB-MH6-CWS, where SEB and CWS represent secreted staphylococcal enterotoxin B and the cell wall-sorting signal of protein A, respectively, separated by the engineered methionine–6-histidine (MH6) affinity tag for purification.
Table 1.
Strains and plasmids used in this study
| Strain or plasmid | Genotype or insert | Description | Reference or source |
|---|---|---|---|
| Strains | |||
| RN4220 | Wild type | S. aureus laboratory strain, a restriction-deficient derivative of NCTC 8325-4 | 56 |
| RN4220 ΔtagO | RN4220 tagO | RN4220 lacking tagO | 57 |
| RN4220 tagB::erm | RN4220 tagB | RN4220 with a bursa aurealis insertion in tagB | This work |
| RN4220 tarF::erm | RN4220 tarF | RN4220 with a bursa aurealis insertion in tarF | This work |
| RN4220 tarJ2::erm | RN4220 tarJ2 | RN4220 with a bursa aurealis insertion in tarJ2 | This work |
| MSSA 1112 | Wild type | MSSA clinical isolate | 58 |
| MSSA 1112 ΔlcpA | MSSA 1112 lcpA::erm | MSSA 1112 with ermB replacing lcpA (msrR) | 46 |
| MSSA 1112 ΔlcpB | MSSA 1112 lcpB | MSSA 1112 lacking lcpB (sa0908) | 33 |
| MSSA 1112 ΔlcpC | MSSA 1112 lcpC | MSSA 1112 lacking lcpC (sa2103) | 33 |
| MSSA 1112 ΔlcpAB | MSSA 1112 lcpA lcpB | MSSA 1112 lacking lcpA and lcpB | 33 |
| MSSA 1112 ΔlcpAC | MSSA 1112 lcpA lcpC | MSSA 1112 lacking lcpA and lcpC | 33 |
| MSSA 1112 ΔlcpBC | MSSA 1112 lcpB lcpC | MSSA 1112 lacking lcpB and lcpC | 33 |
| MSSA 1112 Δlcp | MSSA 1112 lcp | MSSA 1112 lacking all three lcp genes | 33 |
| MSSA 1112 Δlcp ΔtagO | MSSA 1112 lcp tagO | MSSA 1112 lacking all three lcp genes and tagO | This work |
| Plasmids | |||
| pΔtagO | DNA segments flanking tagO | pKOR1 carrying a 1-kbp DNA segment upstream and downstream of tagO, used for allelic replacement | This work |
| pHTT4 | SEB-MH6-CWS | pOS1 vector encoding SEB with the CWS motif of protein A | 10 |
| pBursa | Modified mariner transposon | Plasmid carrying the mariner-based transposon with erythromycin resistance (thermosensitive replicon) | 36 |
| pFA545 | Mariner transposase | Plasmid carrying the transposase (thermosensitive replicon) | 36 |
| plcpA | lcpA (msrR) | Plasmid pGC2 (pT194 based) encoding lcpA for complementation studies | 33 |
| plcpB | lcpB (sa0908) | Plasmid pGC2 (pT194 based) encoding lcpB for complementation studies | 33 |
| plcpC | lcpC (sa2103) | Plasmid pGC2 (pT194 based) encoding lcpC for complementation studies | 33 |
Purification of SEB-MH6-CWS and analysis of C-terminal anchor peptides.
Methicillin-sensitive S. aureus (MSSA) 1112 strains (wild type and Δlcp mutant) harboring pHTT4 (10) were grown to an A600 of 0.8. Cells were washed, suspended in 50 ml of 50 mM Tris-HCl buffer (pH 7.5) supplemented with 5 mM phenylmethylsulfonyl fluoride (PMSF), and broken in a BeadBeater instrument (Biospec Products Inc.). Crude lysates were centrifuged at 33,000 × g for 15 min, and sedimented material was suspended in 100 ml of 100 mM KH2PO4 (pH 7.5), containing 1% Triton X-100 and 1 mM PMSF. Samples were incubated for 3 h at 4°C with stirring to extract membrane lipids. Cell wall material containing SEB-MH6-CWS was sedimented by centrifugation at 33,000 × g for 15 min, washed three times with 100 ml of 100 mM sodium phosphate buffer (pH 6.0), and suspended in 30 ml of 100 mM Tris-HCl buffer (pH 7.5) supplemented with 1 mM PMSF. SEB-MH6-CWS was purified as described previously (10, 37). Briefly, the sample was incubated with 2 mg of lysostaphin for 16 h at 37°C and centrifuged at 33,000 × g for 15 min. The supernatant containing SEB-MH6-CWS was applied to gravity-flow columns packed with Ni-nitrilotriacetic acid (NTA) beads. The column was washed with 30 column volumes of buffer A (100 mM Tris-HCl buffer [pH 7.5], 150 mM NaCl) containing 10 mM imidazole. SEB-MH6-CWS was eluted in buffer A with 0.5 M imidazole. Protein in the eluate was precipitated with methanol-chloroform, dried under vacuum, and solubilized in 3 ml 70% formic acid prior to the addition of a cyanogen bromide crystal. Samples were incubated for 16 h at room temperature in the dark. Reaction products were washed twice with water, suspended in 1 ml of buffer B (10 mM Tris-HCl, 100 mM NaH2PO4, 6 M guanidine hydrochloride, pH 8.0), and applied to a gravity-flow column packed with Ni-NTA beads equilibrated in buffer B. The column was washed with 10 volumes each of buffer B, buffer C (8 M urea, 100 mM NaH2PO4, 10 mM Tris-HCl) at pH 8.0, and buffer C at pH 6.3. Anchor peptides including H6-CWS were eluted in buffer C containing 0.5 M imidazole at pH 8.0. For mass spectrometry analysis, peptides were desalted onto C18 matrix cartridges (Waters) that had been prewashed with 10 ml of acetonitrile (CH3CN) containing 0.1% trifluoroacetic acid (TFA) and 10 ml of 0.1% TFA in water. Peptides were eluted in 3 ml 60% CH3CN, 0.1% TFA, dried under vacuum, and suspended in 50 μl 30% CH3CN, 0.1% TFA. A 0.5-μl aliquot of this suspension was cospotted with 0.5 μl of matrix, α-cyano-4-hydroxycinnamic acid (10 mg/ml in 50% CH3CN, 0.1% TFA). Samples were subjected to matrix-assisted laser desorption ionization (MALDI)–time of flight (TOF) mass spectrometry (MS) using an Autoflex Speed Bruker MALDI instrument. Ions were detected in linear positive mode.
WTA preparations.
WTA was extracted from murein sacculi as described previously (38). Briefly, cells from a 30-ml culture of S. aureus grown in TSB at 37°C to an A600 of 1.0 were washed with 30 ml of buffer D [50 mM 2-(N-morpholino)ethanesulfonic acid (MES), pH 6.5] and suspended in 30 ml of buffer D containing 4% sodium dodecyl sulfate (SDS), followed by boiling for 1 h in a water bath and centrifugation for 10 min at 23,000 × g. The sediment was washed twice in 1 ml buffer D containing 4% SDS, once in buffer D containing 2% NaCl, and once in buffer D. Following each wash, murein sacculi were sedimented by centrifugation at 23,000 × g for 10 min, and after the final wash, the peptidoglycan was suspended in 1 ml of 20 mM Tris-HCl buffer at pH 8.0 containing 0.5% SDS and 20 μg proteinase K and the mixture was incubated at 50°C for 4 h. Protease-digested murein sacculi were washed once in buffer D containing 2% NaCl and thrice in water and suspended in 1 ml of 0.1 M NaOH for a 16-h incubation at room temperature with rotation, to hydrolyze WTA. Following sedimentation of NaOH-extracted murein sacculi at 23,000 × g for 10 min, the supernatant containing the released WTA was transferred to a new tube and neutralized with 50 μl of 0.1 M acetic acid and 100 mM Tris-HCl, pH 8.5, for subsequent analysis by polyacrylamide gel electrophoresis (PAGE).
WTA was also precipitated from 100-ml culture supernatants of S. aureus grown in TSB at 37°C to an A600 of 0.5 by adding 3 volumes of 95% ethanol and incubating at 4°C for 30 min. Precipitated material was sedimented by centrifugation (16,000 × g for 15 min), washed with 70% ethanol, air dried, and suspended in 5 ml of 100 mM Tris-HCl (pH 7.5) containing 5 mM CaCl2, 25 mM MgCl2, DNase (10 μg/ml), and RNase (50 μg/ml). Samples were incubated for 3 h at 37°C and subjected to methanol-chloroform extraction. The teichoic acid in the aqueous layer was dried under vacuum and solubilized in 1 ml of 20 mM Tris-HCl (pH 8.0).
Quantifying the phosphate content of the staphylococcal envelope.
Murein sacculi were prepared as described previously and stored in water (39). The phosphate content was determined by incubating 45 μl of a murein sacculus sample (A600, 1.0) with 5 μl of trichloroacetic acid at 80°C for 16 h. The inorganic phosphate released by this treatment was quantified with a colorimetric assay where a mix composed of 6 N H2SO4, water, 2.5% ammonium molybdate, and 10% ascorbic acid (in a ratio of 1:2:1:1) was added at 1:1 (vol/vol) to trichloroacetic acid-treated preparations, and the mixture was incubated at 37°C for 90 min. Product formation corresponding to free phosphate was measured in a spectrophotometer at 820 nm (A820), and the phosphate concentration in the samples was calculated from NaH2PO4 standards (concentration, 0 to 800 μM).
Gel electrophoresis and immunoblotting.
WTA was separated on polyacrylamide gels using a Bio-Rad Protean II xi electrophoresis cell (20 cm by 16 cm by 1 mm) (40). Following separation of the extracts by electrophoresis, gels were incubated twice in 1 mg/ml alcian blue for 20 min, followed by two 30-min washes with deionized H2O and staining with a Silver Stain Plus kit (Bio-Rad). For comparative analyses of proteins, aliquots of the cultures used for WTA analysis were removed, treated with lysostaphin (4 μg), and incubated at 37°C for 10 min. Proteins in these lysates were precipitated with 7% trichloroacetic acid, washed once with acetone, dried, solubilized in 100 μl of 0.5 M Tris-HCl, pH 8.0, containing 4% SDS, and heated at 90°C for 10 min. Proteins were separated on 12% SDS-polyacrylamide gels and transferred to a polyvinylidene difluoride membrane (Millipore) for immunoblot analysis with appropriate polyclonal antibodies. Immunoreactive signals were revealed by using a secondary antibody coupled to IRDye 680 and visualized with a LI-COR Biosciences Odyssey imager.
Phenotypic characterization of mutants.
The ability of the S. aureus mutants to bind purified LysM-mCherry fluorescent protein was assessed as described previously (34, 41). Briefly, cells of logarithmically growing cultures were sedimented by centrifugation, washed in phosphate-buffered saline (PBS), and incubated with purified LysM-mCherry for 10 min. mCherry was used as a control. Following incubation, cells were washed twice in PBS and fixed in 4% paraformaldehyde in PBS prior to analysis by flow cytometry and fluorescence microscopy. Incubation with propidium iodide (Invitrogen) was used to evaluate the membrane integrity of the mutants compared to that of the wild type. Briefly, overnight cultures were diluted 1:100 into TSB containing 0 or 1 μg/ml tunicamycin and cultured at 37°C for 3.5 h. Following centrifugation of 1 ml culture, cells were washed twice with PBS, fixed for 20 min with 4% paraformaldehyde, and then blocked for 1 h in PBS with 1% bovine serum albumin. Cells were incubated for 15 min with 10 nM SYTO 9 (Invitrogen) and 2 μg/ml propidium iodide in PBS, washed twice, and suspended in PBS for flow cytometry measurements.
Flow cytometry and microscopy.
Flow cytometric analyses were performed using a BD LSR-II cytometer. mCherry fluorescence was quantified using the allophycocyanin (APC) parameter and gating on single cells using forward and side scatter. Staphylococci were gated using forward and side scatter, and SYTO 9-positive cells captured under the fluorescein isothiocyanate parameter were analyzed for propidium iodide staining in the peridinin chlorophyll protein A parameter. The parameters for negative propidium iodide staining were determined using an unstained control. For fluorescence microscopy, images were captured on a Leica TCS SP2 AOBS confocal laser scanning microscope with a ×100 objective using identical settings and exposure times between samples. For transmission electron microscopy, bacterial cells were washed twice with 50 mM Tris-HCl (pH 7.5), 150 mM NaCl, bathed in fixative (2% glutaraldehyde, 4% phosphonoformic acid, 0.1 M sodium cacodylate buffer) overnight at 4°C, and postfixed with 1% OsO4 in 0.1 M sodium cacodylate buffer for 60 min. Fixed samples were stained in 1% uranyl acetate in maleate buffer for 60 min, serially dehydrated with increasing concentrations of ethanol, embedded in Spurr resin for 48 h at 60°C, thin sectioned (90 nm) using a Reichert-Jung Ultracut device, and poststained in uranyl acetate and lead citrate. The samples were imaged on an FEI Tecnai F30 transmission electron microscope with a Gatan charge-coupled-device (CCD) digital micrograph.
RESULTS
The S. aureus Δlcp mutant releases WTA into the extracellular medium.
A S. aureus mutant with all three lcp genes deleted harbors very little phosphate in its cell wall envelope, suggesting that the mutant does not synthesize WTA (32). We wondered whether S. aureus lcp mutants may also be defective in the cell wall anchoring of WTA (30). To examine these possibilities, murein sacculi were isolated from wild-type S. aureus MSSA 1112 and its isogenic mutants lacking one lcp gene (the ΔlcpA, ΔlcpB, and ΔlcpC mutants), two lcp genes (the ΔlcpAB, ΔlcpAC, and ΔlcpBC mutants), or all three lcp genes (the Δlcp mutant). WTA was released from murein sacculi via alkaline hydrolysis; Rbo-P polymers of variable length were resolved by PAGE and visualized by staining with alcian blue-silver. As controls, the murein sacculi of wild-type S. aureus harbored WTA, whereas treatment of staphylococci with tunicamycin, an inhibitor of TagO, blocked the synthesis and incorporation of WTA into the cell wall (Fig. 1A, lanes 1 and 2). The MIC of tunicamycin required to block WTA synthesis in S. aureus MSSA 1112 was 1 μg/ml (see Fig. S1 in the supplemental material). WTA was detected in the murein sacculi of each single and double lcp mutant (Fig. 1A, lanes 3 to 8) but not in sacculi prepared from the Δlcp strain (Fig. 1A, lane 9). This WTA synthesis defect of the Δlcp mutant was restored by transformation with plasmids expressing any one of the three LCP homologues, lcpA, lcpB, or lcpC (Fig. 1B, lanes 11 to 13). We also determined the cell wall phosphate content of the wild-type versus mutant strains via colorimetric assay. Here, peptidoglycan was extracted and purified from exponentially growing bacteria, and the inorganic phosphate was liberated by incubation in 10% trichloroacetic acid. Staphylococci incubated with tunicamycin synthesized and incorporated very little phosphate into the cell wall envelope (Fig. 1C). As already noted by Dengler et al. (32), Rbo-P was not detected in cell wall extracts derived from the Δlcp mutant. Furthermore, the ΔlcpA and ΔlcpB single mutants but not the ΔlcpC single mutant contained reduced levels of phosphate compared to wild type, as did each of the double mutants (Fig. 1C). Nevertheless, loss of two lcp genes did not cause clear synergistic effects in reducing the phosphate content of staphylococcal cell walls (Fig. 1C).
Fig 1.
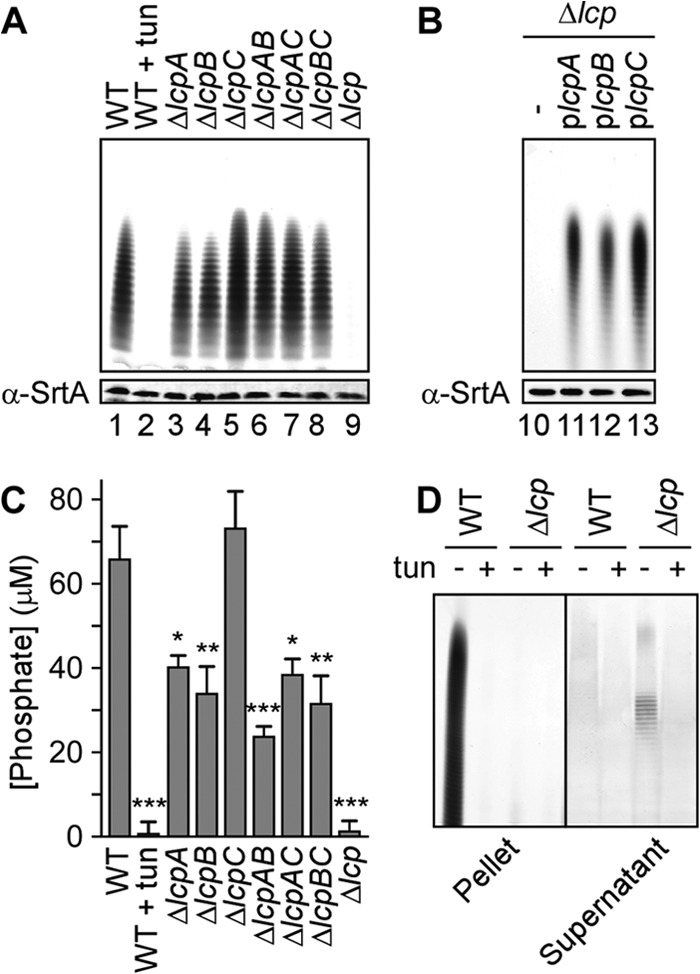
Δlcp mutant cells lack cell-associated wall teichoic acid. (A and B) Alcian blue- and silver-stained acrylamide gels of cell wall-associated WTA. (A) WTA was extracted from the S. aureus MSSA 1112 wild type (WT) grown without (lane 1) or with (lane 2) tunicamycin and from single mutant strains (the ΔlcpA, ΔlcpB, and ΔlcpC mutants), double mutant strains (the ΔlcpAB, ΔlcpAC, and ΔlcpBC mutants), and a triple mutant strain (the Δlcp mutant) (lanes 3 to 9, respectively). (B) WTA was extracted from the triple mutant strain (the Δlcp mutant) carrying an empty vector (lane 10) or complementing plasmid plcpA, plcpB, or plcpC (lanes 11 to 13, respectively). As a control for the number of staphylococci, cell extracts were immunoblotted with antibodies specific for sortase A. (C) Quantification of phosphate levels in purified peptidoglycan from the strains described for panel A. Average phosphate levels were derived from three independent experiments, and the standard errors of the means were calculated. Statistical significance between the wild type and each group was determined using one-way analysis of variance (ANOVA) with Bonferroni's multiple-comparison test. *, P < 0.05; **, P < 0.01; ***, P < 0.001. (D) Identification of teichoic acid in the supernatant of Δlcp cultures. Wild-type and Δlcp strains were grown in the absence (−) or presence (+) of tunicamycin, and teichoic acids in cell pellets (left) or culture supernatants (right) were resolved by PAGE and visualized by staining with alcian blue and silver.
We next examined whether the Rbo-P polymer can be detected in the culture medium of Δlcp staphylococci. Following growth in the presence or absence of tunicamycin, carbohydrates and teichoic acid polymers in the supernatant of centrifuged cultures from wild-type MSSA 1112 or its Δlcp variant were precipitated with ethanol. Teichoic acids were sedimented by centrifugation, and contaminating nucleic acids were digested with DNase and RNase. Samples were extracted with methanol and chloroform to remove proteins. Teichoic acids in the aqueous layer were concentrated by lyophilization, suspended in water, subjected to PAGE, and revealed by alcian blue-silver staining. As controls, murein sacculi of wild-type MSSA 1112 harbored Rbo-P WTA, whose synthesis was blocked by tunicamycin treatment (Fig. 1D). Of note, the Rbo-P polymer was detected in the extracellular medium of cultures of the Δlcp mutant but not those of wild-type S. aureus (Fig. 1D). Tunicamycin treatment of Δlcp mutant cultures abolished the release of the Rbo-P polymer into the culture medium (Fig. 1D). To ascertain that Rbo-P polymer release did not result from cell lysis or aberrant peptidoglycan turnover, we asked whether the Δlcp mutant also released cytoplasmic proteins or other cell wall polymers, such as sortase-anchored proteins, into the medium. We showed that a hybrid protein with the cell wall-sorting signal of protein A, SEB-MH6-CWS, is properly tethered to the murein sacculus of the Δlcp mutant. Immunoblotting analyses revealed that the hybrid is not aberrantly released into the extracellular milieu of Δlcp mutant cultures compared to cultures of wild-type staphylococci (see Fig. S2 in the supplemental material). The cytoplasmic protein L6 was not detected in extracts of spent culture medium, ruling out bacterial lysis as a mechanism for surface protein release into the culture medium (see Fig. S2 in the supplemental material). Thus, we conclude that the Δlcp mutant fails to deposit WTA in the cell wall envelope and instead releases Rbo-P into the extracellular medium. Taken together, these data suggest that LcpA, LcpB, and LcpC display overlapping functions in depositing WTA in the cell wall envelope, presumably by forming a phosphodiester bond between the C-6 hydroxyl of MurNAc and the murein linkage unit tethered to Rbo-Pn.
Cell division and envelope defects of Δlcp and Δlcp ΔtagO mutant staphylococci.
In B. subtilis, deletion of all three lcp genes is not compatible with bacterial growth, unless the mutant cells also harbor a deletion of the tagO gene (30). In contrast, the S. aureus Δlcp mutant, which also lacks all three lcp genes, continues to grow (A600), albeit at a lower rate than the wild type (see Fig. S3 in the supplemental material). To assess the viability of staphylococcal cells, culture aliquots were removed, serially diluted, and plated after 3.5 h to enumerate the number of viable CFU. We observed a 3-log-unit reduction in plating efficiency for the Δlcp mutant compared to wild-type MSSA 1112 (Fig. 2A). Introduction of the ΔtagO allele into the Δlcp mutant neither restored the plating efficiency to wild-type levels nor enhanced the growth of the mutant at the earlier 3.5-h time point (Fig. 2A), when the LCP enzymes are predominantly expressed (42). Further, when growth was measured via determination of the absorbance of liquid cultures (A600), the Δlcp ΔtagO mutant replicated at a rate similar to that for the Δlcp mutant but not at the rate of its wild-type parent (see Fig. S3 in the supplemental material). To measure the number of nonviable staphylococci, culture aliquots of the three strains were stained with propidium iodide to identify lysed bacteria with membrane damage via flow cytometry (Fig. 2B). These experiments revealed that 30.2% of Δlcp mutant cells and 18.7% of Δlcp ΔtagO mutant cells were positive for propidium iodide, whereas only 1.68% of wild-type cells were positive for propidium iodide (Fig. 2B). Wild-type and mutant staphylococci were fixed, thin sectioned, and viewed by transmission electron microscopy. As expected, wild-type strain MSSA 1112 formed round cells with a thick cell wall envelope and cross-wall septa positioned at midcell, perpendicular to previous cell division planes (Fig. 2C). Uranyl acetate staining revealed the uniform deposition of WTA as electron-dense deposits in the envelope of wild-type staphylococci (Fig. 2C) (43). In contrast, the Δlcp and Δlcp ΔtagO variants generated deformed cells with thin, irregular envelopes lacking the WTA staining of wild-type staphylococci (Fig. 2C). Of note, the Δlcp and Δlcp ΔtagO variants generated cross-wall septa parallel to the previous division planes, indicating that the physiological mechanisms for the selection of cell division planes had been abolished in the Δlcp and Δlcp ΔtagO mutants (Fig. 2C). Interestingly, this phenotype was also observed for the isolated tagO and lcpA mutants, the latter of which still produces WTA (33, 43). These data suggest that both WTA synthesis and assembly are important for physiological cell division (43). In conclusion, the growth delay observed in the Δlcp mutant is not a direct result of the continued synthesis of WTA and cannot be restored by preventing WTA synthesis.
Fig 2.
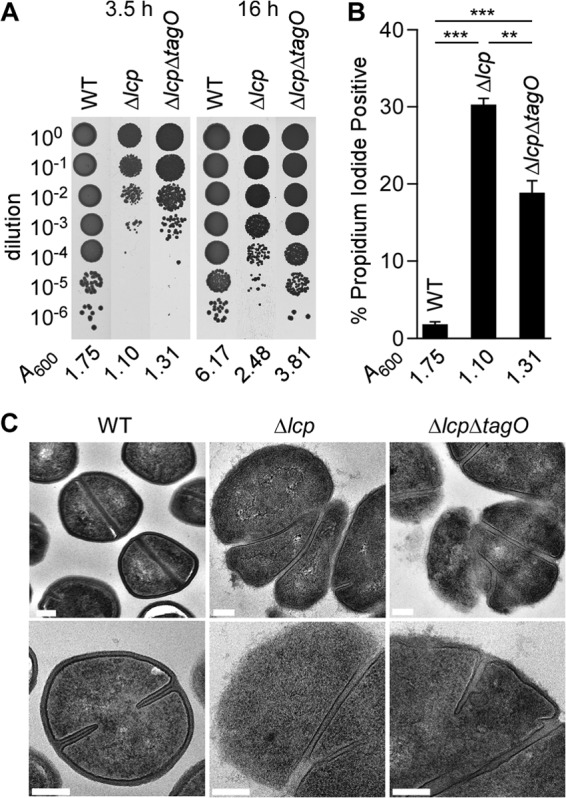
Deletion of tagO does not improve growth of the Δlcp mutant. (A) Staphylococcal viability was examined by plating serial dilutions (0- to 6-fold) of culture aliquots of the wild type and the Δlcp and Δlcp ΔtagO mutants that had been grown for 3.5 and 16 h. Images of the agar plates are shown along with the A600 values of the cultures at the time of plating. (B) Membrane integrity of staphylococci was assessed with propidium iodide staining. Culture aliquots of the wild type and the Δlcp and Δlcp ΔtagO mutants that had been grown for 3.5 h were fixed with paraformaldehyde and stained with SYTO 9 (total cells) and propidium iodide. SYTO 9-positive cells were analyzed for propidium iodide staining using flow cytometry. Combined data from two independent experiments with triplicate analyses of 20,000 cells per sample are presented. Statistical significance was determined using the Student t test. **, P < 0.01; ***, P < 0.001. (C) Transmission electron micrographs of wild-type, Δlcp, and Δlcp ΔtagO bacteria. Cells were cultured for 3.5 h, as described for panel A. Bars, 200 nm.
Effect of tunicamycin on the generation of nonviable daughter cells by Δlcp mutant staphylococci.
Dengler et al. (32) reported that tunicamycin treatment could partially alleviate the growth defect of Δlcp mutant staphylococci, a phenotype that was also observed in our experiments (Fig. 3A). We wondered whether tunicamycin treatment also suppressed the phenotype of the Δlcp mutant of nonviable daughter cells. Bacterial cultures were grown without or with 1 μg/ml of tunicamycin for 3.5 h. Culture aliquots were serially diluted and plated on TSA for colony formation (Fig. 3A). As expected, the plating efficiency of wild-type MSSA 1112 was slightly reduced when the strain was grown in the presence of tunicamycin. In contrast, the addition of tunicamycin did not affect the plating efficiency of the Δlcp strain (Fig. 3A), a phenotype that is in stark contrast to that of strains with mutational lesions in genes of the WTA biosynthetic pathway. For example, disruption of S. aureus tagB, tarF, or tarJ2 via bursa aurealis transposon insertion was conditional for the presence of tunicamycin in agar medium, whereas the plating efficiency of the ΔtagO mutant was not (see Fig. S3 in the supplemental material). The plating efficiency of the Δlcp strain and its sensitivity to tunicamycin were almost restored to wild-type levels upon transformation with plasmids that provide for the expression of any one of the three lcp genes (Fig. 3A). Of note, the reduction in plating efficiency was not always correlated with the bacterial growth measured for wild-type and Δlcp cultures via increases in the A600 (Fig. 3A). As before, this difference can be explained by the generation of nonviable daughter cells, revealed by the increase of propidium iodide-positive cells in the Δlcp mutant culture. While incubation of cultures with tunicamycin led to further increases in propidium iodide-positive Δlcp cells, tunicamycin treatment did not affect the membrane integrity of wild-type staphylococci (Fig. 3B). Transmission electron microscopy revealed that tunicamycin treatment did not affect the overall morphology and cell division septum formation in wild-type cells; as expected, the outer electron-dense layer of WTA was abolished when staphylococci were grown in the presence of the antibiotic (Fig. 3C). Further, tunicamycin treatment of Δlcp staphylococci did not change the aberrant gross morphology and septation defect of the mutant cells (Fig. 3C). These data show that treatment with tunicamycin, an inhibitor of WTA synthesis, suppresses neither the cell division defect of Δlcp staphylococci nor its generation of nonviable daughter cells.
Fig 3.
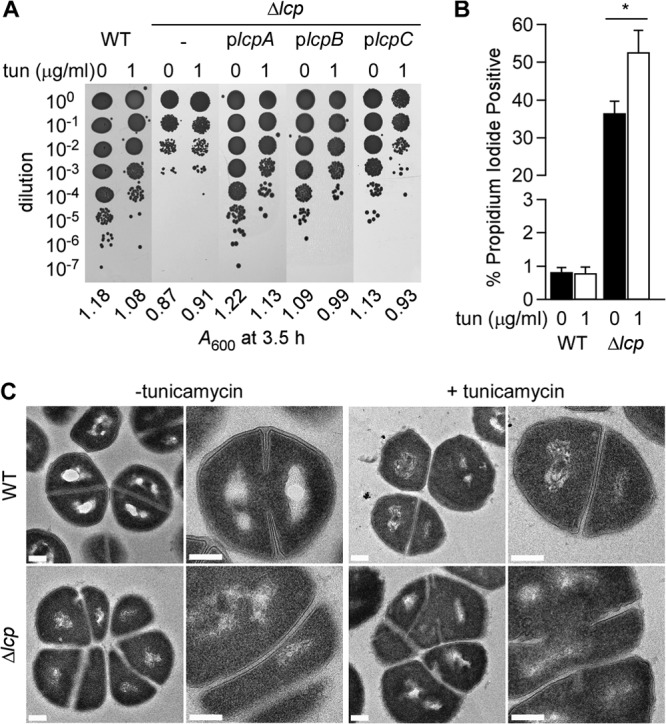
Inhibition of TagO with tunicamycin does not suppress the growth defects of the Δlcp mutant. (A) Bacterial growth was examined by plating serial dilutions of cultures grown for 3.5 h without or with tunicamycin (0 or 1 μg/ml). Culture aliquots of the wild type and the Δlcp mutant without or with plcpA, plcpB, or plcpC were spotted on agar plates. A600 values were recorded at the time of plating and are reported under the images of the agar plates. (B) Membrane integrity of staphylococci assessed with propidium iodide staining. Culture aliquots of the wild type and the Δlcp mutant grown for 3.5 h without or with tunicamycin (0 or 1 μg/ml) were fixed and stained as described in the legend to Fig. 2B. The data were analyzed as described in the legend to Fig. 2B. *, P < 0.05). (C) Transmission electron micrographs of wild-type and Δlcp mutant cells from cultures grown for 3.5 h without or with tunicamycin (0 or 1 μg/ml). Bars, 200 nm.
The lcp genes are not required for surface protein anchoring to the staphylococcal cell wall.
Previous work identified lipid II to be the peptidoglycan substrate of the sortase A-catalyzed transpeptidation reaction of surface protein anchoring to the cell wall envelope (8). Consistent with this model, surface protein anchoring can be blocked by treating staphylococci with inhibitors of lipid II polymerization (vancomycin or moenomycin) or bactoprenol metabolism (nisin) (8). LCP proteins have been proposed to transfer WTA precursors [C55-PP–GlcNAc–ManNAc–(Gro-P)2-3–(Rbo-P)30-50] onto the glycan strands of peptidoglycan (30). Since both reactions—surface protein and WTA anchoring—require bactoprenol-linked intermediates, we wondered whether sortase A-mediated protein anchoring is altered in S. aureus strains lacking the three LCP phosphotransferases (Δlcp strain). To test this possibility, the wild-type parent strain MSSA 1112 and the Δlcp mutant were transformed with plasmid pHTT4, which provides for the expression of SEB-MH6-CWS, an engineered surface protein consisting of the N-terminal signal peptide and staphylococcal enterotoxin B (SEB) fused to the C-terminal cell wall-sorting signal (CWS) of protein A (Fig. 4A). Peptidoglycan preparations from both MSSA 1112 strains (wild type/pHTT4 and Δlcp mutant/pHTT4) were treated with lysostaphin to solubilize anchored SEB-MH6-CWS for purification via metal-chelating affinity chromatography (Fig. 4B). Purified proteins were subjected to cyanogen bromide (CNBr) cleavage at methionine residues. C-terminal anchor peptides with their N-terminal six-histidine (H6) tag were again purified by affinity chromatography and analyzed by MALDI MS (Fig. 4CD). Ion signals labeled with m/z 1,665 (compound 1) and m/z 1,722 (compound 3) correspond to surface protein anchor peptides with two or three glycine residues [H2N-H6AQALPETGG(G)], which are released from the cell wall via lysostaphin cleavage (Fig. 4C and D; Table 2). Ion signals with m/z 1,693 (compound 2) and m/z 1,750 (compound 4) represent formylated peptides of compounds 1 and 3, respectively (Table 2). Compounds with m/z 3,826 (compound 5), m/z 3,854 (compound 6), and m/z 3,903 (compound 7) represent formylated or carbamylated anchor peptides with two or three glycine residues, also liberated via lysostaphin cleavage from the cell wall [NH2-VDSKDVKIEVYLTTKKGTMH6AQALPETG(G)]; these peptides were generated via incomplete CNBr cleavage of SEB-MH6-CWS at methionyl residues (Table 2). Thus, SEB-MH6-CWS anchor peptides with similar structures and relative abundances were liberated via lysostaphin treatment from the cell wall envelope of wild-type and Δlcp mutant staphylococci. These data indicate that the absence of LCP enzymes does not affect sortase A-mediated anchoring of surface proteins in S. aureus.
Fig 4.
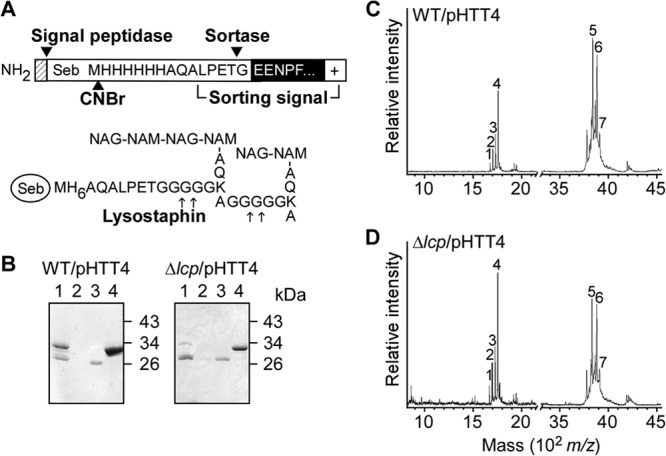
Sortase A anchoring of proteins to the cell wall is not affected in the Δlcp mutant. (A) Diagram of recombinant SEB-MH6-CWS encoded by the pHTT4 plasmid. Cleavage sites of the signal peptidase, sortase A, and CNBr are indicated. (B) Coomassie brilliant blue-stained SDS-polyacrylamide gel of SEB-MH6-CWS released with lysostaphin from the cell wall of either wild-type (left) or Δlcp mutant (right) staphylococci. Solubilized cell wall preparations (lanes 1) were subjected to affinity chromatography on Ni-NTA beads. Material not retained on the column is shown in lanes 2. The beads were washed (lanes 3), and SEB-MH6-CWS was eluted with buffer containing 0.5 M imidazole (lanes 4). Molecular mass markers are indicated on the right side of the gel. (C and D) Mass spectrometry of CNBr-cleaved SEB-MH6-CWS purified from the wild type (C) and the Δlcp mutant (D). Proteins eluted in lanes 4 of panel B were digested with CNBr, and fragments were purified once more over Ni-NTA, eluted, desalted, and subjected to MALDI-TOF MS. The analysis of the ion signals labeled 1 to 7 and structural predictions are listed in Table 2.
Table 2.
Sortase A-catalyzed surface protein anchoring to the cell wall is not altered in the Δlcp mutanta
| Ion | Observed m/z |
Calculated m/z | Proposed structure | |
|---|---|---|---|---|
| Wild type/pHTT4 | Δlcp/pHTT4 | |||
| 1 | 1,665.941 | 1,665.811 | 1,665.729 | NH2-H6AQALPET-Gly2-CO2H |
| 2 | 1,693.893 | 1,693.830 | 1,693.739 | HCO-NH-H6AQALPET-Gly2-CO2H |
| 3 | 1,722.888 | 1,722.827 | 1,722.781 | NH2-H6AQALPET-Gly3-CO2H |
| 4 | 1,750.956 | 1,750.867 | 1,750.791 | HCO-NH-H6AQALPET-Gly3-CO2H |
| 5 | 3,826.572 | 3,826.436 | 3,826.215 | NH2-VDSKDVKIEVYLTTKKGTMH6AQALPET-Gly2-CO2H, Na+ |
| 6 | 3,854.116 | 3,854.111 | 3,854.225 | HCO-NH-VDSKDVKIEVYLTTKKGTMH6AQALPET-Gly2-CO2H, Na+ |
| 7 | 3,901.934 | 3,902.469 | 3,903.301 | H2NCO-NH-VDSKDVKIEVYLTTKKGTMH6AQALPET-Gly3-CO2H |
Cyanogen bromide cleavage products of cell wall-anchored SEB-MH6-CWS liberated by lysostaphin treatment and purified by Ni-NTA chromatography (Fig. 4) were analyzed by MALDI-TOF MS in linear positive mode. The program CS ChemDraw was used for m/z calculations.
Deposition of LysM murein hydrolases in the envelope of Δlcp mutant staphylococci.
The decoration of the staphylococcal peptidoglycan with WTA restricts the binding of secreted murein hydrolases to the cell wall and limits the autolytic activity of these enzymes to the cross-wall compartment of dividing staphylococci (44). For example, the deposition of the LytN and Sle1 murein hydrolases is restricted to the cross wall (34, 41). Each of these enzymes harbors LysM domains at their N termini (34). LysM domains are both necessary and sufficient for the targeting of Sle1 to the cross wall (34). Inhibition of WTA synthesis, for example, by treatment of staphylococci with tunicamycin or deletion of tagO, abolishes the specificity of LysM domain targeting to the bacterial envelope (34). In a confocal fluorescence microscopy experiment, an abundance of the LysM domain of LytN (LysMLytN)-mCherry and the LysM domain of Sle1 (LysMSle1)-mCherry was found to be deposited uniformly throughout the envelope of tagO mutant but not wild-type staphylococci (34) (Fig. 5A). As a control, mCherry alone did not bind to the staphylococcal envelope. We wondered whether the Δlcp mutant, which fails to deposit WTA in the staphylococcal envelope, is also defective for the targeting of LysM-type murein hydrolases. Similar strong binding activities of LysMLytN-mCherry and LysMSle1-mCherry were observed for the Δlcp mutant strain but not for its MSSA 1112 parent (Fig. 5A).
Fig 5.

The LysM domains of LytN and Sle1 bind uniformly to the envelope of the Δlcp mutant. Purified mCherry, LysMLytN-mCherry, or LysMSle1-mCherry was incubated with wild-type parent (strains RN4220 and MSSA 1112), ΔtagO mutant, or Δlcp mutant staphylococci. Binding of hybrid mCherry to the bacterial envelope was analyzed by fluorescence microscopy (A) and flow cytometry (B). (A) (Left) Differential inference contrast (DIC) images of staphylococcal cells analyzed by fluorescence microscopy (middle); (right) merged images derived from both data sets.
The deposition of mCherry, LysMLytN-mCherry, and LysMSle1-mCherry on the staphylococcal surface was quantified by measuring bacterial fluorescence in a flow cytometry experiment (Fig. 5B). Ten- and 100-fold increases in LysMLytN-mCherry and LysMSle1-mCherry fluorescence, respectively, were measured with Δlcp mutant staphylococci relative to that measured with wild-type strain MSSA 1112. These differences were comparable to those observed between wild-type and ΔtagO mutant staphylococci (Fig. 5B). As a control, mCherry did not bind to the envelope of wild-type, ΔtagO mutant, or Δlcp mutant staphylococci (Fig. 5A and B). These data suggest that the WTA deposition defect of the Δlcp mutant causes the unrestricted deposition of murein hydrolases in the bacterial envelope, a phenotype that likely contributes to the decreased viability of Δlcp daughter cells generated during cell division.
DISCUSSION
Recent reports implicated a family of genes encoding the LCP proteins in WTA synthesis and in the attachment of secondary cell wall polymers to bacterial peptidoglycan (30, 31). LCP-encoding genes are present in virtually all Gram-positive bacteria, and often, numerous homologues are present within a single genome (45). The LCP proteins share a similar predicted secondary structure, consisting of a short N-terminal cytoplasmic tail, a single transmembrane domain, and an extracellular C-terminal region encompassing a mixed α-helical/β-sheet LCP domain (30, 45). While the biochemical activity of these proteins is still not known, variants carrying defective alleles of lcp genes display pleiotropic phenotypes. For example, in both S. aureus and S. pneumoniae, loss of one or more of these genes resulted in aberrant septum formation (33, 46, 47), increased susceptibility to β-lactam antibiotics (33, 46), autolysis (33), aberrant biofilm formation (46), induction of cell wall envelope stress responses (32), and reduced cell wall phosphate (32). B. subtilis contains three homologues of the LCP genes, tagTUV, which cluster with genes of the WTA synthesis pathway (30). Deletion of all three genes is not compatible with growth of the mutant bacilli (30). Similar to late-stage WTA mutants, this phenotype can be rescued through the additional deletion of tagO (28); furthermore, tagTUV mutants cannot assemble WTA in the bacterial cell wall (30). In S. pneumoniae, mutants with deletions of one or two of the LCP homologues (Cps2A, LytR, and Psr) synthesize reduced amounts of capsular polysaccharide and release some of this material into the extracellular medium (31, 48, 49). The crystal structures of recombinant B. subtilis TagT and S. pneumoniae Cps2A revealed the presence of a polyprenyl phosphate lipid bound to the purified protein (30, 31). These findings have led to the hypothesis that LCP enzymes transfer secondary cell wall polymers from their undecaprenyl phosphate precursors and link them to the bacterial peptidoglycan (30). Specifically, LCP enzymes are proposed to tether the murein linkage unit of WTA or other secondary cell wall polysaccharides to the C-6 hydroxyl of MurNAc in the peptidoglycan of Gram-positive bacteria (30).
Although several layers of evidence support the hypothesis of Kawai and colleagues (30), there are also arguments against it. For example, S. pneumoniae is not known to express functional tagO and tagA homologues or synthesize murein linkage units (C55-PP–GlcNAc–ManNAc); pneumococci are thought to anchor their teichoic acid polymers and capsular polysaccharides to the C-6 hydroxyl of MurNAc via phosphodiester bonds with other carbohydrate structures (50). Further, if B. subtilis TagTUV or S. aureus LcpABC were to tether the murein linkage units of WTA to the cell wall, one would expect that the corresponding mutant strains either harbor WTA precursors [C55-PP–GlcNAc–ManNAc–Gro2-3–(Rbo-Pn)] in the envelope or release unanchored products into the extracellular medium. Although this was tested, the B. subtilis tagTUV mutant did not release WTA into the medium and also did not deposit WTA in the envelope (30).
We are interested in sortase-mediated anchoring of surface proteins to the cell wall envelope (5), a biosynthetic pathway that also utilizes an undecaprenyl precursor, lipid II, to immobilize polypeptides in the bacterial envelope (7, 8, 51). We presumed that the S. aureus Δlcp mutant may accumulate undecaprenyl-linked intermediates of the WTA pathway, thereby causing a block in surface protein anchoring. This, however, was not observed; instead, the S. aureus Δlcp mutant synthesized WTA and released the Rbo-P polymers into the extracellular medium. We surmise that the observed release of WTA may be catalyzed by undecaprenyl pyrophosphate phosphatases (UppPs), which are otherwise involved in the recycling of translocated lipid II (52). If so, this could explain why undecaprenyl recycling occurs in the Δlcp variant but not in tagB, tarF, or tarJ2 mutants, which require mutations in tagO or tagA or inhibition of WTA synthesis with tunicamycin for growth. We entertain a model whereby the S. aureus Δlcp mutant translocates its WTA precursors via TagGH for subsequent hydrolysis via UppPs, thereby enabling the return of bactoprenol into the cytoplasm. This model could also explain why other WTA biosynthetic intermediates, those accumulating in tagB, tarF, or tarJ2 mutants (without substrate properties for TagGH), either deplete staphylococci of the lipid carrier or accumulate toxic intermediates. It seems plausible that UppPs do not have access to cytoplasmic intermediates of the WTA pathway and therefore cannot relieve the growth-inhibitory attributes of tagB, tarF, or tarJ2 mutations. Our model offers insights into the development of new drugs that target key steps of WTA synthesis on the bacterial surface. At least for S. aureus, the causative agent of human infections with antibiotic-resistant strains (53), small-molecule inhibitors of LCPs would not be expected to cause bactericidal activity, whereas the combined inhibition of LCPs and UppPs may be associated with antibiotic activity. In contrast, the B. subtilis tagTUV mutant appears to sequester WTA synthesis intermediates, as these mutations inhibit the growth of the mutant, unless WTA synthesis is blocked via tagO mutation (30). Thus, B. subtilis may lack the relevant UppP activity and cannot recycle WTA synthesis intermediates. A combined literature and GenBank search suggests that S. aureus encodes three genes with undecaprenyl pyrophosphate phosphatase activity. The canonical bacA-uppP gene has been described and is dispensable for growth (54). S. aureus also encodes two predicted phosphatases with a PAP2 domain reminiscent of ybjG and pgpB, two genes that have been shown to encode redundant UppP activities E. coli (52). A similar search performed for B. subtilis suggests that this organism encodes only two undecaprenyl pyrophosphate phosphatases, bacA (formerly yubB) and bcrC (formerly ywoA) (55).
In sum, data reported here support the model of Kawai and colleagues, whereby LCP enzymes catalyze the transfer of WTA synthesis intermediates to the cell wall peptidoglycan (30). The observed pleiotropic phenotypes of the Δlcp mutant are likely due to its WTA synthesis and WTA cell wall deposition defects. Without WTA, staphylococci display aberrant cell size, septum formation, autolysis, and susceptibility to antibiotics and defects in biofilm formation (32, 33, 45, 46). The molecular basis of the observed phenotypes is likely due to defects in the positioning and functional coordination of the peptidoglycan biosynthesis and cell wall separation machines, which involve a wide spectrum of different enzymes.
Supplementary Material
ACKNOWLEDGMENTS
We thank Yimei Chen for assistance with transmission electron microscopy, Andrea C. Dedent for technical advice, and members of the Olaf Schneewind and Dominique Missiakas laboratories for discussions.
Yvonne G. Y. Chan acknowledges support from the American Heart Association (award 13POST16980091). This work was supported by NIAID grant RO1 AI38897. We acknowledge membership within and support from the Region V Great Lakes Regional Center of Excellence in Biodefense and Emerging Infectious Diseases Consortium (GLRCE; NIAID award 1-U54-AI-057153).
Footnotes
Published ahead of print 9 August 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JB.00544-13.
REFERENCES
- 1.Shockman GD, Barrett JF. 1983. Structure, function, and assembly of cell walls of gram-positive bacteria. Annu. Rev. Microbiol. 37:501–527 [DOI] [PubMed] [Google Scholar]
- 2.Munoz E, Ghuysen JM, Heymann H. 1967. Cell walls of Streptococcus pyogenes, type 14. C polysaccharide-peptidoglycan and G polysaccharide-peptidoglycan complexes. Biochemistry 6:3659–3670 [DOI] [PubMed] [Google Scholar]
- 3.Coley J, Archibald AR, Baddiley J. 1976. A linkage unit joining peptidoglycan to teichoic acid in Staphylococcus aureus H. FEBS Lett. 61:240–242 [DOI] [PubMed] [Google Scholar]
- 4.Schneewind O, Fowler A, Faull KF. 1995. Structure of the cell wall anchor of surface proteins in Staphylococcus aureus. Science 268:103–106 [DOI] [PubMed] [Google Scholar]
- 5.Mazmanian SK, Liu G, Ton-That H, Schneewind O. 1999. Staphylococcus aureus sortase, an enzyme that anchors surface proteins to the cell wall. Science 285:760–763 [DOI] [PubMed] [Google Scholar]
- 6.Navarre WW, Schneewind O. 1994. Proteolytic cleavage and cell wall anchoring at the LPXTG motif of surface proteins in gram-positive bacteria. Mol. Microbiol. 14:115–121 [DOI] [PubMed] [Google Scholar]
- 7.Ruzin A, Severin A, Ritacco F, Tabei K, Singh G, Bradford PA, Siegel MM, Projan SJ, Shlaes DM. 2002. Further evidence that a cell wall precursor [C(55)-MurNAc-(peptide)-GlcNAc] serves as an acceptor in a sorting reaction. J. Bacteriol. 184:2141–2147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Perry AM, Ton-That H, Mazmanian SK, Schneewind O. 2002. Anchoring of surface proteins to the cell wall of Staphylococcus aureus. III. Lipid II is an in vivo peptidoglycan substrate for sortase-catalyzed surface protein anchoring. J. Biol. Chem. 277:16241–16248 [DOI] [PubMed] [Google Scholar]
- 9.Higashi Y, Strominger JL, Sweeley CC. 1967. Structure of a lipid intermediate in cell wall peptidoglycan synthesis: a derivative of C55 isoprenoid alcohol. Proc. Natl. Acad. Sci. U. S. A. 57:1878–1884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ton-That H, Faull KF, Schneewind O. 1997. Anchor structure of staphylococcal surface proteins. A branched peptide that links the carboxyl terminus of proteins to the cell wall. J. Biol. Chem. 272:22285–22292 [DOI] [PubMed] [Google Scholar]
- 11.Ton-That H, Liu G, Mazmanian SK, Faull KF, Schneewind O. 1999. Purification and characterization of sortase, the transpeptidase that cleaves surface proteins of Staphylococcus aureus at the LPXTG motif. Proc. Natl. Acad. Sci. U. S. A. 96:12424–12429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Strominger JL. 1968-1969. Penicillin-sensitive enzymatic reactions in bacterial cell wall synthesis. Harvey Lect. 64:179–213 [PubMed] [Google Scholar]
- 13.Yokoyama K, Mizuguchi H, Araki Y, Kaya S, Ito E. 1989. Biosynthesis of linkage units for teichoic acids in gram-positive bacteria: distribution of related enzymes and their specificities for UDP-sugars and lipid-linked intermediates. J. Bacteriol. 171:940–946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Soldo B, Lazarevic V, Karamata D. 2002. tagO is involved in the synthesis of all anionic cell-wall polymers in Bacillus subtilis 168. Microbiology 148:2079–2087 [DOI] [PubMed] [Google Scholar]
- 15.Kern J, Ryan C, Faull K, Schneewind O. 2010. Bacillus anthracis surface-layer proteins assemble by binding to the secondary cell wall polysaccharide in a manner that requires csaB and tagO. J. Mol. Biol. 401:757–775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Armstrong JJ, Baddiley J, Buchanan JG. 1959. Structure of teichoic acid from the walls of Bacillus subtilis. Nature 184:248–249 [DOI] [PubMed] [Google Scholar]
- 17.Yokoyama K, Miyashita T, Arakai Y, Ito E. 1986. Structure and functions of linkage unit intermediates in the biosynthesis of ribitol teichoic acids in Staphylococcus aureus H and Bacillus subtilis W23. Eur. J. Biochem. 161:479–489 [DOI] [PubMed] [Google Scholar]
- 18.Wyke AW, Ward JB. 1977. Biosynthesis of wall polymers in Bacillus subtilis. J. Bacteriol. 130:155–163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Xia G, Kohler T, Peschel A. 2010. The wall teichoic acid and lipoteichoic acid polymers of Staphylococcus aureus. Int. J. Med. Microbiol. 300:148–154 [DOI] [PubMed] [Google Scholar]
- 20.D'Elia MA, Henderson JA, Beveridge TJ, Heinrichs DE, Brown ED. 2009. The N-acetylmannosamine transferase catalyzes the first committed step of teichoic acid assembly in Bacillus subtilis and Staphylococcus aureus. J. Bacteriol. 191:4030–4034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mauel C, Young M, Margot P, Karamata D. 1989. The essential nature of teichoic acids in Bacillus subtilis as revealed by insertional mutagenesis. Mol. Gen. Genet. 215:388–394 [DOI] [PubMed] [Google Scholar]
- 22.Pooley HM, Abellan FX, Karamata D. 1991. A conditional-lethal mutant of Bacillus subtilis 168 with a thermosensitive glycerol-3-phosphate cytidylyltransferase, an enzyme specific for the synthesis of the major wall teichoic acid. J. Gen. Microbiol. 137:921–928 [DOI] [PubMed] [Google Scholar]
- 23.Lazarevic V, Abellan FX, Möller SB, Karamata D, Mauël C. 2002. Comparison of ribitol and glycerol teichoic acid genes in Bacillus subtilis W23 and 168: identical function, similar divergent organization, but different regulation. Microbiology 148:815–824 [DOI] [PubMed] [Google Scholar]
- 24.Lazarevic V, Karamata D. 1995. The tagGH operon of Bacillus subtilis 168 encodes a two-component ABC transporter involved in the metabolism of two wall teichoic acids. Mol. Microbiol. 16:345–355 [DOI] [PubMed] [Google Scholar]
- 25.Kojima N, Araki Y, Ito E. 1983. Structure of linkage region between ribitol teichoic acid and peptidoglycan in cell walls of Staphylococcus aureus H. J. Biol. Chem. 258:9043–9045 [PubMed] [Google Scholar]
- 26.Weidenmaier C, Kokai-Kun JF, Kristian SA, Chanturiya T, Kalbacher H, Gross M, Nicholson G, Neumeister B, Mond JJ, Peschel A. 2004. Role of teichoic acids in Staphylococcus aureus nasal colonization, a major risk factor in nosocomial infections. Nat. Med. 10:243–245 [DOI] [PubMed] [Google Scholar]
- 27.D'Elia MA, Millar KE, Beveridge TJ, Brown ED. 2006. Wall teichoic acid polymers are dispensable for cell viability in Bacillus subtilis. J. Bacteriol. 188:8313–8316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.D'Elia MA, Pereira MP, Chung YS, Zhao W, Chau A, Kenney TJ, Sulavik MC, Black TA, Brown ED. 2006. Lesions in teichoic acid biosynthesis in Staphylococcus aureus lead to a lethal gain of function in the otherwise dispensable pathway. J. Bacteriol. 188:4183–4189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hancock IC, Wiseman G, Baddiley J. 1976. Biosynthesis of the unit that links teichoic acid to the bacterial wall: inhibition by tunicamycin. FEBS Lett. 69:75–80 [DOI] [PubMed] [Google Scholar]
- 30.Kawai Y, Marles-Wright J, Cleverley RM, Emmins R, Ishikawa S, Kuwano M, Heinz N, Bui NK, Hoyland CN, Ogasawara N, Lewis RJ, Vollmer W, Daniel RA, Errington J. 2011. A widespread family of bacterial cell wall assembly proteins. EMBO J. 30:4931–4941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Eberhardt A, Hoyland CN, Vollmer D, Bisle S, Cleverley RM, Johnsborg O, Havarstein LS, Lewis RJ, Vollmer W. 2012. Attachment of capsular polysaccharide to the cell wall in Streptococcus pneumoniae. Microb. Drug Resist. 18:240–255 [DOI] [PubMed] [Google Scholar]
- 32.Dengler V, Meier PS, Heusser R, Kupferschmied P, Fazekas J, Friebe S, Staufer SB, Majcherczyk PA, Moreillon P, Berger-Bachi B, McCallum N. 2012. Deletion of hypothetical wall teichoic acid ligases in Staphylococcus aureus activates the cell wall stress response. FEMS Microbiol. Lett. 333:109–120 [DOI] [PubMed] [Google Scholar]
- 33.Over B, Heusser R, McCallum N, Schulthess B, Kupferschmied P, Gaiani JM, Sifri CD, Berger-Bachi B, Stutzmann Meier P. 2011. LytR-CpsA-Psr proteins in Staphylococcus aureus display partial functional redundancy and the deletion of all three severely impairs septum placement and cell separation. FEMS Microbiol. Lett. 320:142–151 [DOI] [PubMed] [Google Scholar]
- 34.Frankel MB, Schneewind O. 2012. Determinants of murein hydrolase targeting to cross-wall of Staphylococcus aureus peptidoglycan. J. Biol. Chem. 287:10460–10471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bae T, Schneewind O. 2006. Allelic replacement in Staphylococcus aureus with inducible counter-selection. Plasmid 55:58–63 [DOI] [PubMed] [Google Scholar]
- 36.Bae T, Glass EM, Schneewind O, Missiakas D. 2008. Generating a collection of insertion mutations in the Staphylococcus aureus genome using bursa aurealis. Methods Mol. Biol. 416:103–116 [DOI] [PubMed] [Google Scholar]
- 37.Marraffini LA, Schneewind O. 2005. Anchor structure of staphylococcal surface proteins. V. Anchor structure of the sortase B substrate IsdC. J. Biol. Chem. 280:16263–16271 [DOI] [PubMed] [Google Scholar]
- 38.Meredith TC, Swoboda JG, Walker S. 2008. Late-stage polyribitol phosphate wall teichoic acid biosynthesis in Staphylococcus aureus. J. Bacteriol. 190:3046–3056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.de Jonge BL, Chang YS, Gage D, Tomasz A. 1992. Peptidoglycan composition of a highly methicillin-resistant Staphylococcus aureus strain. The role of penicillin binding protein 2A. J. Biol. Chem. 267:11248–11254 [PubMed] [Google Scholar]
- 40.Pollack JH, Neuhaus FC. 1994. Changes in wall teichoic acid during the rod-sphere transition of Bacillus subtilis 168. J. Bacteriol. 176:7252–7259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Frankel MB, Hendrickx AP, Missiakas DM, Schneewind O. 2011. LytN, a murein hydrolase in the cross-wall compartment of Staphylococcus aureus, is involved in proper bacterial growth and envelope assembly. J. Biol. Chem. 286:32593–32605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rossi J, Bischoff M, Wada A, Berger-Bachi B. 2003. MsrR, a putative cell envelope-associated element involved in Staphylococcus aureus sarA attenuation. Antimicrob. Agents Chemother. 47:2558–2564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Campbell J, Singh AK, Santa Maria JP, Jr, Kim Y, Brown S, Swoboda JG, Mylonakis E, Wilkinson BJ, Walker S. 2011. Synthetic lethal compound combinations reveal a fundamental connection between wall teichoic acid and peptidoglycan biosyntheses in Staphylococcus aureus. ACS Chem. Biol. 6:106–116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schlag M, Biswas R, Krismer B, Kohler T, Zoll S, Yu W, Schwarz H, Peschel A, Gotz F. 2010. Role of staphylococcal wall teichoic acid in targeting the major autolysin Atl. Mol. Microbiol. 75:864–873 [DOI] [PubMed] [Google Scholar]
- 45.Hubscher J, Luthy L, Berger-Bachi B, Stutzmann Meier P. 2008. Phylogenetic distribution and membrane topology of the LytR-CpsA-Psr protein family. BMC Genomics 9:617. 10.1186/1471-2164-9-617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hubscher J, McCallum N, Sifri CD, Majcherczyk PA, Entenza JM, Heusser R, Berger-Bachi B, Stutzmann Meier P. 2009. MsrR contributes to cell surface characteristics and virulence in Staphylococcus aureus. FEMS Microbiol. Lett. 295:251–260 [DOI] [PubMed] [Google Scholar]
- 47.Johnsborg O, Havarstein LS. 2009. Pneumococcal LytR, a protein from the LytR-CpsA-Psr family, is essential for normal septum formation in Streptococcus pneumoniae. J. Bacteriol. 191:5859–5864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cieslewicz MJ, Kasper DL, Wang Y, Wessels MR. 2001. Functional analysis in type Ia group B Streptococcus of a cluster of genes involved in extracellular polysaccharide production by diverse species of streptococci. J. Biol. Chem. 276:139–146 [DOI] [PubMed] [Google Scholar]
- 49.Morona JK, Morona R, Paton JC. 2006. Attachment of capsular polysaccharide to the cell wall of Streptococcus pneumoniae type 2 is required for invasive disease. Proc. Natl. Acad. Sci. U. S. A. 103:8505–8510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Denapaite D, Bruckner R, Hakenbeck R, Vollmer W. 2012. Biosynthesis of teichoic acids in Streptococcus pneumoniae and closely related species: lessons from genomes. Microb. Drug Resist. 18:344–358 [DOI] [PubMed] [Google Scholar]
- 51.Marraffini LA, Dedent AC, Schneewind O. 2006. Sortases and the art of anchoring proteins to the envelopes of gram-positive bacteria. Microbiol. Mol. Biol. Rev. 70:192–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.El Ghachi M, Derbise A, Bouhss A, Mengin-Lecreulx D. 2005. Identification of multiple genes encoding membrane proteins with undecaprenyl pyrophosphate phosphatase (UppP) activity in Escherichia coli. J. Biol. Chem. 280:18689–18695 [DOI] [PubMed] [Google Scholar]
- 53.Klevens RM, Morrison MA, Nadle J, Petit S, Gershman K, Ray S, Harrison LH, Lynfield R, Dumyati G, Townes JM, Craig AS, Zell ER, Fosheim GE, McDougal LK, Carey RB, Fridkin SK, Active Bacterial Core Surveillance (ABCs) MRSA Investigators 2007. Invasive methicillin-resistant Staphylococcus aureus infections in the United States. JAMA 298:1763–1771 [DOI] [PubMed] [Google Scholar]
- 54.Chalker AF, Ingraham KA, Lunsford RD, Bryant AP, Bryant J, Wallis NG, Broskey JP, Pearson SC, Holmes DJ. 2000. The bacA gene, which determines bacitracin susceptibility in Streptococcus pneumoniae and Staphylococcus aureus, is also required for virulence. Microbiology 146(Pt 7):1547–1553 [DOI] [PubMed] [Google Scholar]
- 55.Bernard R, El Ghachi M, Mengin-Lecreulx D, Chippaux M, Denizot F. 2005. BcrC from Bacillus subtilis acts as an undecaprenyl pyrophosphate phosphatase in bacitracin resistance. J. Biol. Chem. 280:28852–28857 [DOI] [PubMed] [Google Scholar]
- 56.Kreiswirth BN, Lofdahl S, Betley MJ, O'Reilly M, Schlievert PM, Bergdoll MS, Novick RP. 1983. The toxic shock syndrome exotoxin structural gene is not detectably transmitted by a prophage. Nature 305:709–712 [DOI] [PubMed] [Google Scholar]
- 57.Grundling A, Missiakas DM, Schneewind O. 2006. Staphylococcus aureus mutants with increased lysostaphin resistance. J. Bacteriol. 188:6286–6297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Entenza JM, Vouillamoz J, Glauser MP, Moreillon P. 1997. Levofloxacin versus ciprofloxacin, flucloxacillin, or vancomycin for treatment of experimental endocarditis due to methicillin-susceptible or -resistant Staphylococcus aureus. Antimicrob. Agents Chemother. 41:1662–1667 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.