

Abstract
NanR, one of >8,500 GntR superfamily helix-turn-helix transcriptional regulators, controls expression of the genes required for catabolism of sialic acids in Escherichia coli. It is predicted to do the same in related bacteria harboring orthologs of nanR. The sialic acids are a family of over 40 naturally occurring nine-carbon keto-sugar acids found mainly in the animal lineage, which includes starfish to humans in the deuterostome lineage. Sialic acids function in development, immunity, protein localization and stability, and homeostasis. They also serve as microbial carbon and nitrogen sources and ligands for cell recognition during host colonization. The importance of microbial sialic acid metabolism for host-microbe interactions has made it a target for therapeutic development. Exploiting this target depends on understanding sialometabolic pathways in a wide range of evolutionarily distinct bacteria. Here, we show by transcriptome, genetic, and biochemical analyses that the most common sialic acid, N-acetylneuraminate, induces the nanATEK-yhcH, yjhATS (nanCMS), and yjhBC operons by directly inactivating NanR, converting the predominantly dimeric form of the repressor to an inactive monomer of approximately 30-kDa. Additionally, other results identify critical amino acid residues and nucleotides in the regulator and operator, respectively. The combined results better define how sialic acids, acting through NanR, affect the metabolic flux of an important group of host-derived metabolites. Thus, E. coli serves as a valuable model for understanding sialocatabolic pathways in bacteria.
INTRODUCTION
Sialic acids (N-acylneuraminic acids) are nine-carbon amino sugars found in the deuterostome lineage, which includes starfish to humans. Many microbes that transiently or obligatorily colonize mammals and avian species catabolize sialic acids as sources of carbon and nitrogen (1, 2). The immediate by-products of sialic acid catabolism produced by the intracellular sialate aldolase (lyase) are pyruvate and a six-carbon amino sugar, N-acetylmannosamine (ManNAc), which is ultimately converted to fructose-6-phosphate. Given their most common positions as terminal nonreducing sugars attached to cell surface glycoproteins and glycolipids and serum glycoconjugates, the sialic acids are the first sugars an invading microorganism encounters upon entering an animal host. Mucins bathing mucosal surfaces are an especially rich source of these sugars (1). Recent bioinformatic surveys indicated that of 46 pathogenic or commensal species with presumptive or known N-acylneuraminate (nan) catabolic systems, 42 (33 pathogens and 9 commensals) colonize mammals (2, 3). Among these species, Escherichia coli has been considered a model system for understanding bacterial sialic acid catabolism since discovery of its nan system in 1985 (1, 2, 4, 5). In E. coli, the nanATEK-yhcH operon is positively controlled by cyclic AMP receptor protein (CRP) and negatively regulated by the upstream nanR-encoded transcriptional repressor, NanR (6–10). The combined enzymatic activities encoded by the operon ultimately connect catabolism of ManNAc, derived from exogenous sialic acids or internally as a product of de novo sialic acid synthesis, to fructose-6-phosphate in cooperation with the nagAB genes for N-acetylglucosamine (GlcNAc) catabolism. Although the most common sialic acid, N-acetylneuraminate (Neu5Ac), has been implicated as the inducer of the E. coli nan system (4, 5, 7, 9), there has been no direct evidence for its inducer function in any bacterium. In contrast, a case was made for ManNAc being the inducer of the Bacteroides fragilis nan system (11), while direct evidence has been published supporting glucosamine-6-phosphate (GlcN-6-P) as causing both activation and repression of sialometabolism in Haemophilus influenzae (12, 13), ManNAc-6-phosphate (ManNAc-6-P) as the nan-nag inducer in Vibrio vulnificus (14) and Staphylococcus aureus (15), and ManNAc or ManNAc-6-P as the nan inducer in Streptococcus pneumoniae (16). Each of the sialoregulators in these bacteria is homologous to RpiR and therefore distinct from E. coli NanR. Here we show that Neu5Ac converts NanR oligomers to monomers in solution and that Neu5Ac is the sole or at least primary inducer of the nan system in E. coli. Our results provide the first molecular characterization of any sialate repressor protein. Further, we identify some of the NanR amino acid residues and operator nucleotides critical for NanR-operator interactions firmly placing NanR in the GntR superfamily (17).
In addition to nan, Neu5Ac has been shown to increase expression of the divergent fim recombinase responsible for phase variation of type 1 pili and yjhA through the combined action of NanR and NagC (18). Despite uncertainty about the exact biological functions of the yjhAT gene products, yjhA (nanC) encodes an outer membrane diffusion channel (19), and yjhT (nanM) encodes a mutarotase that increases the conversion rate of the alpha-sialic acid isomer to the thermodynamically more stable beta form (20). Neither gene product is required for growth on Neu5Ac (2). We recently showed that YjhS (NanS) is an esterase required for growth of E. coli on O-acetylated sialic acids, a form of sialic acid that is prevalent in host mucin oligosaccharides (21). Thus, the primary function of the nanCMS, yjhBC, and yhcH genes could be to encode proteins that convert less common sialic acids to Neu5Ac (see Fig. 1A) (1), which is the preferred NanA (aldolase) substrate (2). Here we show that NanR regulates nanATEK-yhcH, nanCMS, and yjhBC operons by binding with similar affinities to a set of conserved operators with two or three exact, or nearly exact, repeats of the hexanucleotide sequence GGTATA, designated the Nan box (6, 7). Remarkably, all other bacteria with known or putative NanR-regulated nan genes lack the potential for both the coordinated regulation found in E. coli and, except for Shigella dysenteriae and some E. coli strains producing Shiga-like toxin, homologs of nanS (1). These findings make control of the nan genes in E. coli unique even among otherwise closely related enteric bacteria (1). The implications of these findings for a unified theory of bacterial sialometabolism have been described (1).
Fig 1.

Organization and expression of the sialoregulon. (A) Genetic organization of the sialoregulon, which is negatively controlled by NanR, with sizes (in kilobases) of nanA and operon transcripts shown above the arrows at the top. The lollypop structure between nanA and nanT indicates a potential stem-loop predicted from the DNA sequence. Bent arrows indicate promoter regions. Known or suspected functions of each gene are indicated, with the relative levels of expression from transcriptome analysis of the nanR-null mutant EV730 relative to the wild type shown in parentheses below the open reading frames (open arrows). (B) Triplicate Northern blot analysis of the wild type (lanes 1, 3, and 5) and strain EV730 (lanes 2, 4, and 6), which were grown in glycerol minimal medium. The mRNA was probed with full-length nanA DNA (top); estimated message sizes are given at right. (Bottom) Ethidium bromide-stained RNA. Positions of 16S and 23S rRNA are shown at left. (C and D) Lanes 1 show the labeled yjhBC and nanCMS (yjhATS) operators, respectively, with no NanR added. Lanes 2 through 9 represent incubations with 550, 55, 28, 14, 5.5, 2.8, 1.4, and 0.55 nM NanR, respectively. Lanes 10 contained 28 nM NanR incubated with the wild-type labeled operator (17). (E) Operator regions for the indicated yjh operons. Dissociation constants were determined as previously described (7), and the KD for the nan (nanATEK-yhcH) operator is taken from that reference. See the independent titration of the wild-type operator in Fig. S2 in the supplemental material.
Transcriptome analysis indicated that expression of a wider range of genes not directly controlled by NanR in E. coli is either increased or decreased by direct and probably indirect effects of sialic acid by-products. We suggest that the name “sialoregulon” be given to genes that are directly regulated by NanR, while the term “sialostimulon” should be used for genes affected by the by-products of sialic acid metabolism. We further suggest that regulation of the sialoregulon has been fine-tuned in different pathogens and commensals to maximize a potential for colonizing different hosts, with E. coli as the apotheosis of sialocatabolic efficiency (1). Therefore, denying bacteria their preferred niches by blocking sialometabolism or using sialo-prebiotics to stimulate growth of desired bacteria is leading to promising new therapeutic possibilities (1, 22, 23).
MATERIALS AND METHODS
Bacterial strains, plasmids, and growth conditions.
The bacterial strains and plasmids used in this study are listed in Table 1. Bacteria were routinely grown in LB broth (Lennox formulation) purchased from Thermo Fisher. For analyses of induction, bacteria were grown in minimal M63 medium (21) with the carbon sources indicated in the figure legends. Bacterial growth was at 37°C with vigorous aeration in a rotary shaker. Ampicillin (Ap), chloramphenicol, kanamycin (Km), and tetracycline (Tc) were used in media at 100, 20, 50, and 10 μg/ml, respectively.
Table 1.
Bacterial strains and plasmids used in this study
| Strain or plasmid | Genotype or relevant description | Source or reference |
|---|---|---|
| Strains | ||
| BL21 Star(DE3) | F− ompT hsdSB(rB− mB−) gal dcm rne131 (DE3) | Invitrogen |
| MG1655 | F− λ− ilvG rfb-5 rph-1 | 24 |
| BW30270 | rph+ derivative of MG1655 | E. coli Genetic Stock Center |
| 12-1 | JM101 Φ(nanA′-lac+)1(Hyb) | 7 |
| MC4100 | F− λ− araD139 Δ(argF-lac)U169 rpsL150 relA1 deoC rbsR fthD5301 fruA25 | Lab stock |
| EV730 | MC4100 ΔnanR::cat | This study |
| EV735 | BW30270 ΔnanS::kan | 21 |
| EV736 | EV735 ΔnanS; kan removed, leaving FRT site | This study |
| EV737 | BW30270 ΔyjhC::kan | This study |
| EV738 | EV737 ΔyjhC; kan removed, leaving FRT site | This study |
| EV741 | EV736 with Φ(nanS′-lac+) | This study |
| EV742 | EV738 with Φ(yjhC′-lac+) | This study |
| EV743 | EV738 with Φ(yjhC′-lac+)(Hyb) | This study |
| Plasmids | ||
| pAlter-1 | Site-directed mutagenesis vector, Tcr | Promega |
| pBAD24 | Arabinose inducible expression, Apr | 25 |
| pCE40 | Plasmid for construction of translational fusions to lacZ | 26 |
| pET-21C | T7 transcription vector, Apr, His tag | Novagen |
| pGEM-T Easy | PCR cloning vector | Promega |
| pGK137 | Plasmid with optimized ribosome-binding site for constructing lac+ operon fusions | J. Slauch 26 |
| pSX674 | nanR with 5′ NdeI and 3′ BamHI sites cloned into pET-21c, His tagged | 7 |
| pSX675 | nanR cloned into pBAD24 | 7 |
| pSX676 | pGEM-T Easy with 240-bp Nan box | 7 |
| pSX800 | Original stop codon reintroduced into pSX674 | This study |
| pSX801 | Double mutation in nanR by chemical mutagenesis of pSX675, R44CG65R | This study |
| pSX802 | Mutation in nanR by chemical mutagenesis of pSX675, G68L | This study |
| pSX803 | Double mutation in nanR by chemical mutagenesis of pSX675, S21AE71K | This study |
| pSX804 | Mutation in nanR by chemical mutagenesis of pSX675, R66C | This study |
| pSX805 | Site-directed mutagenesis of pSX675, R56A | This study |
| pSX806 | Site-directed mutagenesis of pSX675, R66A | This study |
| pSX807 | Site-directed mutagenesis of pSX675, R70A | This study |
| pSX808 | Site-directed mutagenesis of pSX675, N86A | This study |
| pSX809 | pAlter-1 with 246 bp nanR promoter in EcoRI site | This study |
| pSX810 | Site-directed mutagenesis of pSX809, GGTA to ATGG in first repeat | This study |
| pSX811 | Site-directed mutagenesis of pSX809, G to T in first repeat | This study |
| pSX812 | Site-directed mutagenesis of pSX809, G to T in second repeat and TAT to ATA in first | This study |
| pSX813 | Site-directed mutagenesis of pSX809, G to T in second repeat | This study |
| pSX814 | Site-directed mutagenesis of pSX809, G to T in third repeat | This study |
| pSX815 | Site-directed mutagenesis of pSX809, T to C in first repeat | This study |
| pSX816 | Site-directed mutagenesis of pSX809, C to A between first and second repeats | This study |
| pSX817 | Site-directed mutagenesis of pSX809, T to G 29 bp upstream of first repeat | This study |
| pSX818 | Site-directed mutagenesis of pSX809, G to T in first repeat and G to T in second repeat | This study |
| pSX819 | Site-directed mutagenesis of pSX809, G to T in second and G to T in third repeat | This study |
| pSX820 | Site-directed mutagenesis of pSX809, G to T in first repeat and G to T in third repeat | This study |
| pSX822 | pGEM-T Easy with 60 bp containing all repeats with an A added between repeats 1 and 2 | This study |
| pSX823 | pGEM-T Easy with 60 bp containing all repeats with an AC added between repeats 1 and 2 | This study |
| pSX824 | pGEM-T Easy with 60 bp containing all repeats with an ACA added between repeats 1 and 2 | This study |
The ΔyjhC strain EV737 (Table 1) was constructed as described previously for ΔnanS (21). The forward and reverse primers, respectively, used for the construction were 5′-GCCCGTGAATTGCAGTGTATCAATATGTGAAGCTTGGATATTCCGGGGATCCGTCGACC-3′ and 5′-GGCAGCATCAGCAGTAGAAATCGCCGACATGGCCGCCTCTGTAGGCTGGAGCTGCTTCG-3′, where underlining indicates homology to the target gene sequence removing 930 bp from the start to stop codons. Operon (pGK137) or protein (pCE40) fusions to nanS and yjhC deletions were constructed as previously described (23, 27), to yield strains EV741 and EV742, which have operon fusions of nanS′ and yjhC′ to lacZ, respectively, and strain EV743, which has yjhC′ fused to the lacZ open reading frame (Table 1). Strain 12-1 has a nanA′ protein fusion (including an intact nanA promoter) to lacZ inserted into the lambda att site (7). All fusion strains retain the intact nanR-nanETEK-yhcH region, where fusions to yjhC and nanS have nonpolar effects as the last genes in their respective operons (Fig. 1A). Strain EV730 harbors a nanR deletion marked with a chloramphenicol resistance cassette (cat) that was generated using methods described elsewhere (27). The forward and reverse primers for the constructions were, respectively, 5′-ATGAACGCATTTGATTCGCAAACCGAAGATTCTTGTGTAGGCTGGAGCTGCTTC-3′ and 5′-TTATTTCTTTTTGTTGGTGGTCTGACCGAAAGCCATATGAATATCCTCCTTAGTTCC-3′ and 5′-, where underlining indicates the regions of nanR homology.
Isolation and characterization of mutants with transdominant (dominant negative) nanR mutations.
Plasmid pSX675 (2 μg), with nanR expression under the control of the PBAD promoter, was mutagenized in 0.4 ml of 0.5 M potassium phosphate, pH 6.0, containing 5 mM EDTA, 0.8 ml of water, and 0.8 ml of 1 M hydroxylamine. Plasmid DNA was incubated at 37°C over a 5-h period, and samples were purified hourly using a Clean and Concentrator kit (Zymogen). Mutagenized plasmid from each time point was transformed into the nanA′ 12-1 reporter strain and plated on LB agar containing ampicillin and 30 to 40 μg/ml 5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside (X-Gal). Blue colonies were isolated; purified plasmids were backcrossed to confirm phenotypes. β-Galactosidase assays were carried out as previously described, with activities given in Miller units (21).
To confirm the dominant negative phenotype, four single mutations conferring R56A, R66A, R70A, and N86A alterations were made in plasmid pSX675 by site-directed mutagenesis using the Genetailor system (Invitrogen), resulting in plasmids pSX805 to pSX808, respectively. The primer pairs used for mutagenesis were the following, where underlining indicates the mutagenic changes: forward, 5′-GTGAACAATTACCGTCTGAAGCCGAACTGATGGC-3′, and reverse, 5′-TTCAGACGGTAATTGTTCACCTTCGCCAAAT-3′ (pSX805); forward, 5′-TGGCGTTCTTTAACGTGCGGGCTCCTTCGGTGCG-3′, and reverse, 5′-CCCGACGTTAAAGAACGCCATCAGTTCGCG-3′ (pSX806); forward, 5′-ACGTCGGGCGTCCTTCGGTGGCTGAAGCGCTGG-3′, and reverse, 5′-CACCGAAGGGCACCCGACGTTAAAGAACGC-3′ (pSX807); and forward, 5′-AAGGTCTGGTGCAAATAAACGCCGGCGAACGAG-3′, and reverse, 5′-GTTTATTTGCACCAGACCTTTGCGTTTTAA-3′ (pSX808). DNA sequencing of nanR verified or identified the mutations in the site-directed or chemically induced mutants, respectively.
Electrophoretic mobility gel shift analysis (EMSA) and stoichiometry of NanR-DNA complexes estimated by modified Ferguson analysis.
The DNA fragments for EMSA were purified using Elutatube (Fermentas) vials after amplification from pSX676 as described previously (7). After separation on a 5% polyacrylamide gel (Bio-Rad) in 0.5× TBE (45 mM Tris [pH 8.3], 45 mM sodium borate, 1 mM EDTA), the fragments were excised and eluted according to the manufacturer's instructions. Fragments were end labeled with 32P and purified using a Zymogen Clean and Concentrator kit, and EMSA was carried out as described previously (7). Binding was quantified using ImageQuant software after capturing signals using a Typhoon Imager (GE Healthcare). Forward and reverse primers for amplification and subsequent labeling of nanCMS and yjhBC were, respectively, 5′-CATATTCCTTTCAGACTG-3′, 5′-CCACCGCAAAGTGTTCGTCT-3′, 5′-CTTGGTTCTGGTGTTTGT-3′, and 5′-CCACCGCAAGTGTTCGTCT-3′.
Stoichiometries of NanR-DNA complexes were estimated by a modified Ferguson method (28, 29) using a 49-bp DNA operator containing all three direct Nan box repeats (7). DNA alone, NanR-DNA complexes, and nondenatured protein molecular weight markers (Sigma) were run on six gels ranging from 5 to 10% polyacrylamide concentrations. Dye fronts were marked on the gels prior to staining with Gradiflash (VWR) for 15 min, destaining in 6% acetic acid for 30 min, drying, and exposure to phosphorimaging. Analyses were carried out according to references 29 and 30.
Purification of native recombinant NanR.
To overproduce and purify native recombinant NanR, plasmid pSX674, generated previously to express a C-terminal histidine-tagged polypeptide (7), was mutagenized with using Genetailor (Invitrogen) to reintroduce the original nanR stop codon. Plasmid pSX674 was methylated with S-adenosylmethionine and amplified with overlapping forward (5′-AGACCACCAACAAAAAGAAATAGGATCCGAATTC-3′ [reengineered stop codon underlined]) and reverse (5′-TTTCTTTTTCTTGGTGGTCTGACCGAAAGC-3′) primers using Platinum high-fidelity Taq polymerase. Mutagenized plasmid was transformed into strain DH5α, where linear, unmethylated product was recircularized. Plasmids obtained from transformants were sequenced to verify the mutation; the resulting plasmid was designated pSX800. This plasmid was transformed into strain BL21(DE3) Star, grown to an A600 of 0.4 in 1 liter of Terrific broth, and induced with 1 mM isopropyl thio-β-d-galactoside (IPTG) for 1 h with aeration. The cell pellet was resuspended in 10 mM Tris-HCl (pH 8.0), 100 mM NaCl, and protease inhibitors at 1 ml per 10 g (wet weight). Cells were lysed by French pressure cell disruption at 10,000 lb/in2, and debris was removed by centrifugation at 23,000 × g for 20 min. The supernatant was brought to 20% saturation with ammonium sulfate, and NanR in the resulting supernatant was precipitated by saturation to 40%. The pellet was resuspended in 10 mM Tris-HCl (pH 8.0), 50 mM NaCl (Q buffer) and dialyzed against three changes of the same buffer over 12 h at 4°C. The dialyzed sample was added to a column with a bed volume of 40 ml Q-Sepharose (Sigma) resin equilibrated with Q buffer. Protein was eluted with a linear gradient of 50 mM to 1 M NaCl; NanR was identified by gel electrophoresis and staining with Coomassie blue. Fractions containing NanR were pooled and dialyzed against H buffer (20 mM Tris-HCl [pH 8.0], 1 mM EDTA, 5% glycerol, 20 mM β-mercaptoethanol, and 100 mM NaCl) for 12 h. Dialyzed protein was applied to a heparin column (Amersham) equilibrated with H buffer, and then washed with 300 ml of H buffer. NanR was eluted with a linear gradient of 0.1 to 1.1 M NaCl. Fractions containing NanR were pooled and biological activity confirmed by EMSA. Some preparations were subjected to an additional step involving size fractionation through Sephacryl S-200. Protein was stored in 50% glycerol at −20°C. Protein concentration was determined by Pierce Micro BCA using bovine serum albumin as standard. Figure S1 in the supplemental material shows an example of a typical purification result.
Operator mutations generated by site-directed mutagenesis.
Plasmid pSX676 harboring the 246-bp DNA fragment, including the Nan box (7) was digested with EcoRI, and the resulting fragment was subcloned into pAlter-1 (Altered Sites; Promega). Cloning was confirmed by DNA sequencing. pAlter-1 carries genes for both ampicillin and tetracycline resistance, but the ampicillin resistance gene has been inactivated. The resulting tetracycline-resistant plasmid, pSX809, was then used as the platform for generating the desired operator mutations. The protocol utilized three primers per alteration: a phosphorylated ampicillin repair primer (5′-pGTTGCCATTGCTGCAGGCATCGTGGTG-3′), a tetracycline resistance knockout primer (5′-pGCCGGGCCTCTTGCGGGCGTCCATTCC-3′), and primers specific for each mutation as given in Results. Mutagenesis was carried out by denaturing pSX809 at 95°C for 10 min followed by lowering to 75°C for 5 min and cooling a degree per min to 45°C before rapid cooling to room temperature. After annealing, the new mutant strand was synthesized with T4 DNA polymerase and strands were ligated. The reaction mixtures were then transformed into strain BMH 71-18 mutS cells (Promega) and cultured with ampicillin selection. Plasmids were purified with a Wizard Plus SV miniprep kit (Promega) and transformed into strain JM109. Mutations were confirmed by DNA sequencing. Prior to isolation of operator fragments for binding studies, the plasmids were treated with plasmid-safe DNase (Clontech) to remove traces of chromosomal DNA that could interfere with the analyses. Amplified fragments were labeled and subjected to EMSA analysis as previously described (7). Mutant operators were cloned into pGEM-T Easy and transformed into the 12-1 nanA′ reporter strain prior to β-galactosidase assays, thereby allowing estimates of the relative competition between wild-type chromosomal and episomal operators for NanR binding, as measured by the promoter titration effects (7).
To introduce nucleotide extensions between the first two Nan box GGTATA repeats, the oligonucleotides shown in Results were synthesized and annealed to their complements as described previously (7). The resulting double-stranded operators were cloned into pGEM-T Easy and used as described above for promoter titration assays or EMSA as indicated in Results. Each mutant operator generated by synthesis or mutagenesis was given a numerical designation as given in the text and accompanying table and figure.
Chemical cross-linking.
To determine the effect of Neu5Ac on NanR oligomerization, purified protein from the heparin sulfate stage was pretreated with ethylene glyco-bis succinimidylsuccinate (EGS) prior to sodium dodecyl sulfate (SDS)-polyacrylamide gel electrophoresis (PAGE) and visualization of polypeptides with Coomassie blue. EGS purchased from Pierce was dissolved in dimethyl sulfoxide to a final concentration of 10 mM. Various concentrations of purified NanR preincubated with or without Neu5Ac or ManNAc were treated with 1 mM EGS for 1 h at room temperature; reactions were quenched by adding glycine to a 20 mM final concentration before SDS-PAGE using 4 to 20% gels purchased from BioExpress or Bio-Rad. Unless indicated otherwise, polypeptides were visualized by staining with Coomassie blue.
Antibody production and Western blot analysis.
Monospecific polyclonal antibody against NanR was produced by the Immunological Resource Center at the University of Illinois. A 9-week-old female New Zealand White rabbit was injected with 0.5 mg of antigen emulsified with Titermax (Sigma) adjuvant for the primary immunization. Subsequent immunizations were carried out at 3-week intervals with antigen (0.25 mg) emulsified with incomplete Freund's adjuvant (Difco). The antiserum from the first bleed relative to the third injection detected a NanR-specific band in Western blotting when used at a 1:10,000 dilution. Preimmune serum was nonreactive. Antibody was used for Western blot analysis at a dilution of 1:10,000. After denaturing gel electrophoresis, gels were blotted to nitrocellulose and blocked with SuperBlock (Pierce) for 1 h. Blots were incubated with primary antibody overnight at 4°C and then washed three times with 20 mM Tris, 150 mM NaCl, and 0.1% Tween 20, pH 7.5. Horseradish peroxidase (HRP)-conjugated anti-rabbit IgG secondary antibody (Amersham) was incubated for 1 h, washed, and reacted with Supersignal West Pico reagent (Pierce) for detection of NanR signal.
Northern blot analysis.
The prototrophic E. coli strain MC4100 and its nanR derivative were grown overnight with orbital shaking in M63 minimal medium containing 0.4% glycerol. The culture was diluted 50-fold into the same medium and incubated with shaking at 37° for 2 h. A portion (2 ml) of each culture was added to 4 ml of RNA Protect Bacteria (Qiagen) and incubated for 5 min before pelleting cells. RNA was purified from the cell pellets using a Qiagen RNeasy kit according to the manufacturer's instructions, including treatment with DNase. RNA integrity was verified by electrophoresis of a 3-μg sample on an agarose gel. For the Northern blot analysis, 5 and 10 μg samples were separated in a 0.8% agarose–2.2 M formaldehyde–morpholinepropanesulfonic acid (MOPS) (40 mM MOPS, 10 mM sodium acetate, 1 mM EDTA [pH 7.0]) gel. After electrophoresis, RNA was blotted overnight to Nytran. The RNA was immobilized by treating with UV light in a Stratagene UV Stratalinker. The nanA gene was amplified with the primers 5′-ATGGCAACGAATTTACGTGGCGTA-3′ and 5′-TCACCCGCGCTCTTGCATCAACTG-3′ and labeled by random prime labeling using the Megaprime DNA labeling system (Amersham Biosciences). Briefly, 25 ng of DNA was denatured at 95°C in the presence of random hexamer primers. This was added to a Tris buffer containing dGTP, dATP, dTTP, α-[32P]dCTP, and the Klenow fragment of DNA polymerase I. After incubation at 37° EDTA was added to a final concentration of 20 mM. Hybridization and radiographic detection were carried out as previously described (31).
Transcriptome analysis.
The prototrophic Escherichia coli K-12 laboratory strain MC4100 was grown with orbital shaking in M63 minimal medium containing 0.4% glycerol as the sole carbon source at 37°C. When the bacteria reached an A600 of 0.2, the culture was split into two equal volumes. Neu5Ac (100 μg/ml, final concentration) was added to one culture, and both cultures were allowed to grow for an additional 40 min. The flasks were removed from the incubator and 10 ml of each culture was added to 20 ml of RNA Protect Bacteria (Qiagen). RNA was purified using the Qiagen RNeasy kit according to the manufacturer's instructions, including treatment with DNase. Running a 5-μg sample on an agarose gel and staining with ethidium bromide (EtBr) verified RNA integrity. RNA from each sample (1 μg) was mixed with 4 μl of E. coli cDNA labeling primers (C5603; Sigma Genosys). This mixture was heated to 94°C for 2 min and then ramped to 42°C over 20 min in a thermal cycler to allow primer annealing. cDNA was prepared by the addition of dATP, dGTP, dTTP (333 μM), [α-33P]dCTP (20 mCi, with a specific activity of 2,000 to 3,000 Ci/mmol), 50 U of avian myeloblastosis virus (AMV) reverse transcriptase (Sigma Genosys), and reverse transcriptase buffer followed by incubation for 3 h at 42°C. Unincorporated nucleotides were removed by purification over a Sephadex G-25 spin column (PRSP0001; Sigma Genosys). Two Panorama E. coli gene array membranes (Sigma catalog no. G1666) were prepared by rinsing in 2× SSPE for 5 min. The membranes were prehybridized with hybridization solution (PR485) at 65°C for 2 h. The labeled cDNA was mixed with 3 ml hybridization solution and heated to 95°C for 10 min to denature the cDNA. The prehybridization solution was removed and replaced with the labeled cDNA in hybridization solution. Hybridization was carried out at 65°C with mixing overnight. The membranes were washed (0.5× SSPE, 0.2% SDS) as directed in the Panorama protocol. Duplicate arrays were exposed to a phosphorimager screen for 2 days and scanned on an Amersham Typhoon imager. The data were averaged and analyzed using ArrayVision software from Imaging Research. The growth, isolation of RNA, preparation of cDNA, and transcriptome analysis of wild-type Escherichia coli strain MC4100 relative to EV730, a nanR-null mutant, were carried out as described above, with the exception that both strains were grown in M63 minimal medium containing 0.4% glycerol as the sole carbon source without addition of sialic acid. The complete list of results for both analyses is available at www.cvm.uiuc.edu/path/sialobiology.
RESULTS
Identification of the E. coli sialoregulon and sialostimulon.
To identify loci directly regulated by NanR, we compared the relative gene expression of an E. coli K-12 ΔnanR strain (EV730) with that of its wild-type MC4100 parent, both grown in M63 with glycerol and no Neu5Ac, as well as MC4100 grown in glycerol minimal medium either with or without Neu5Ac (see Materials and Methods). As shown in Fig. 1A, three operons comprising 10 genes of 4,290 open reading frames examined expressed more messages in the deletion strain under the two conditions used for transcriptome analyses (see Materials and Methods). The yjhBC and yjhATS operons were apparently controlled by upstream DNA sequences that are similar or identical to the previously identified Nan box controlling nanATEK-yhcH expression (7), supporting the conclusion that NanR regulates expression of the sialoregulon by binding to identical or nearly identical operators. This idea was confirmed by direct EMSA analyses (Fig. 1C to E). Figure 1E shows the Nan box (nan operator) identified previously by DNase I foot printing (7). NanR bound to all coregulated operons with similar efficiencies (Fig. 1C to E). The 10 genes organized into three operons shown in Fig. 1A were the only ones overexpressed under both conditions used for the transcriptome analyses. We refer to genes directly regulated by NanR as the sialoregulon. As shown in Fig. 1B, message from the first gene (nanA) of the nanATEK-yhcH operon was the most prevalent transcript in the deletion strain, suggesting that the stem-loop structure located between nanA and nanT coding sequences attenuates the previously noted expression of downstream genes (4).
Thirteen other genes with at least 4-fold-increased expression were detected when cells were grown with Neu5Ac (see Table S1 in the supplemental material). The addition of Neu5Ac also resulted in ≥4-fold-decreased expression of 29 other genes, known or predicted to be involved in synthetic or catabolic processes. Because expression of the genes listed in Table S1 was unaltered by deletion of nanR in EV730, we propose that by-products of sialic acid metabolism, like pyruvate and ManNAc, either directly or indirectly affect cell physiology and the set of NanR-independent genes referred to as the sialostimulon.
To confirm that Neu5Ac influenced expression of nanCMS and yjhBC operons, we measured the β-galactosidase activities of reporter operon or translational fusions of lacZ to nanA as a control (7), yjhC, or nanS. As shown in Fig. 2, cells containing the in-frame polypeptide fusions to nanA and yjhC in strains 12-1 and EV743, respectively, produced 10-fold or greater activity when Neu5Ac was added to the growth medium relative to cells grown on glycerol alone. The much greater activity of the nanA reporter in strain 12-1 is consistent with the increased nanA transcription detected by Northern blotting (Fig. 1B) and previous enzymatic and mRNA runoff experiments (4, 7). The induction of the nanS reporter, the last gene of the nanCMS operon, was comparable to expression of a fusion to nanC (19), consistent with the genetic organization shown in Fig. 1A. The approximately 20-fold induction of the nanS reporter fusion in strain EV741 was confirmed by quantitative PCR results (data not shown). Addition of Neu5,9Ac2, a substrate of NanS, increased expression of the nanA and yjhC fusions in strains 12-1 and EV742 but not in strain EV741 lacking the sialate O-acetyl esterase (Fig. 2). This result indicates that either Neu5,9Ac2 is not transported by NanT or removal of the acetyl group is necessary for induction.
Fig 2.
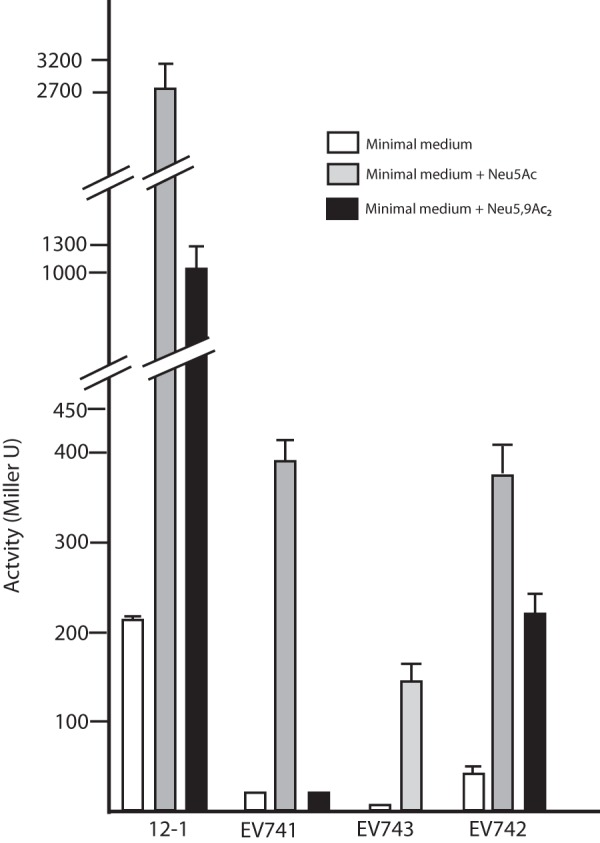
Effects of two different sialic acids on expression of reporter lacZ transcriptional or translational reporter fusions to nanA (strain 12-1), nanS (strain EV741), or yjhC (strains EV742 and EV743). The indicated bacterial strains were grown in glycerol minimal medium or medium supplemented with 100 μg/ml Neu5Ac or Neu5,9Ac2 for 1 h and assayed for reporter β-galactosidase activity. The results are the averages from three independent experiments; error bars show standard deviations (SD). A lack of visible error bars means that the SD was less than the width of the bar outline.
Crystallization of native NanR.
Purified NanR was crystallized using robotic Emerald Wizard crystallization (Jena Bioscience) and a Hampton Research grid screen and Cryo Crystal screen in 2-methyl-2,4-pentanediol. A total of 384 experiments were conducted at 10 mg/ml NanR with half the experiments including 15 mM Neu5Ac. Despite repeated attempts to crystallize NanR from the heparin sulfate or S-200 steps (see Materials and Methods), we were able to obtain crystals diffracting to no better than 5 to 10 Å, and then only when Neu5Ac was included in the crystallization buffer. Failure to obtain crystals in the absence of Neu5Ac suggests that purified NanR is polydisperse, and that Neu5Ac functions by converting NanR oligomers to monomers while yielding crystals that were not suitable for structural determination. Our inability to solve the structure of NanR and the lack of an effect of Neu5Ac or ManNAc addition during EMSA despite in vivo results indicating these as the only two inducer candidates (4, 6, 7) led us to pursue a combination of biochemical and genetic approaches.
Analysis of NanR oligomerization by chemical cross-linking.
In the absence of Neu5Ac or EGS, NanR migrated in SDS-PAGE (see Materials and Methods) with its expected size of about 30 kDa (Fig. 3A and B, lanes 1). When Neu5Ac was added to NanR in concentrations ranging from 1 to 10 mM, the majority of repressor migrated as the monomer, but with the appearance of a minor band that was slightly larger than the homodimer (Fig. 3A, lane 2; Fig. 3B, lanes 2 to 5; and Fig. 3C, lanes 2 and 4). This band's formation was dependent on Neu5Ac concentration (Fig. 3B, lanes 1 to 5), suggesting that Neu5Ac alters NanR conformation such that a fraction of molecules form an SDS-insoluble complex of an undefined nature. If this conclusion is correct, it might explain why low-quality crystals were obtained, because a fraction of molecules were in a disordered state. In the presence of EGS and NanR (5.4 to 9 μM), homodimers predominated together with a concentration-dependent increase in oligomers with estimated masses consistent with 3 or more cross-linked monomers or a combination of monomers and dimers (Fig. 3A, lanes 3 to 5). Clearly, the cross-linking results confirm that NanR exists primarily as a homodimer but that it also might produce higher-molecular-weight forms depending on concentration, despite the ambiguity in the exact oligomerization state of molecules larger than dimers. When 10 mM Neu5Ac was added to NanR prior to EGS, most of the NanR molecules migrated as monomers, together with the minor band noted above (Fig. 3A, lanes 6 and 7) and some molecules forming very high-molecular-weight complexes that were resistant to Neu5Ac-induced dissociation or otherwise entrained at the origin (Fig. 3A, lane 8). Figure 3B (lanes 6 to 9) confirms the effect of Neu5Ac on NanR dissociation. Although we do not know the intracellular concentration of NanR, the results shown in Fig. 3 indicate that the repressor can exist in a variety of structures ranging from the monomer to multioligomeric forms. Multioligomerization of regulatory proteins solely through protein-protein interactions is not an unprecedented finding (32).
Fig 3.

Effects of Neu5Ac and ManNAc on oligomerization of NanR. Purified native NanR from the heparin sulfate stage was used in all experiments. The concentration of sialic acid (Neu5Ac) (Sia), when present (+), was 10 mM. In other lanes, Neu5Ac concentrations of ≤10 mM are specified. ManNAc was used at a 10 mM final concentration. Cross-linking results with EGS are defined in the text. Unnumbered lanes show molecular mass markers, with sizes (in kDa) at left. A. Lanes 1 through 3 and 6 contained 5.4 μM NanR, while lanes 4 and 7 contained 7.2 μM and lanes 5 and 8 contained 9 μM NanR. Numbers beside the arrows show estimates of three different superoligomeric sizes (in kDa). (B) NanR was included at a 2.7 μM final concentration in lanes 1 through 9. (C) NanR was used at a 35 μM final concentration in lanes 1 through 5. NanR concentrations are based on the monomeric form. Asterisks indicate the apparent SDS-resistant oligomer discussed in the text.
Previous analyses of nan induction from within by a polysialic acid capsule-deficient E. coli K1 mutant that accumulates free Neu5Ac from biosynthesis (4, 5) or addition of various sugars to the reporter strain 12-1 suggested that Neu5Ac was the natural inducer (7). However, to determine if ManNAc had inducer activity, which could not be previously tested (7, 8), we investigated whether it affected NanR oligomerization. Figure 3C (lane 1) shows the expected NanR monomer after electrophoresis (Fig. 3C, lane 2). Preincubation with 10 mM ManNAc resulted in a profile identical to that of NanR incubated without sugar (Fig. 3C, compare lane 3 with lane 1). Note that ManNAc did not result in formation of the minor band described above. This finding adds to the evidence that Neu5Ac induces the aberrant form in a small fraction of NanR molecules. As shown in Fig. 3A and B, addition of EGS after incubation with Neu5Ac resulted in mostly monomer, while treatment with ManNAc prior to EGS did not cause the dissociation of NanR dimers or higher-order oligomers (Fig. 3C, lane 5). These results confirm the polydispersity of NanR in solution and establish Neu5Ac as the physiological inducer of sialic acid catabolism in E. coli.
Characterization of nanR mutations conferring a transdominant phenotype.
Nucleotide changes in plasmid pSX675, which expresses arabinose-inducible nanR+, were generated either by hydroxylamine or site-directed mutagenesis. Transformation of the nanA reporter strain 12-1 with randomly mutagenized plasmid yielded 24 of 1,800 colonies with deep blue color on medium with X-Gal and arabinose compared to the pale blue or white color of colonies expressing active NanR. The dominant negative or transdominant phenotype of the blue colonies was confirmed by backcrossing plasmids into strain 12-1 on X-Gal with arabinose and observing the same phenotype. The nanR genes from four different mutants chosen at random were sequenced to identify the mutation(s) responsible for transdominance. The remaining 20 mutants were not investigated further, because the results described below are consistent with defects in the N-terminal DNA recognition domain common to the GntR superfamily. This region in NanR was previously hypothesized to be the recognition domain on the basis of its similarity to FadR (7, 33).
In the absence of sialic acid, the reporter expressed 22 ± 5 units, while the uninduced strain harboring pSX675 produced 5.8 ± 2.3 units and <1 unit (0.6 ± 0.02) when induced with 0.2% arabinose, indicating the range of reporter values above which dominant negative mutants would be expected to have a detectable phenotype. As shown in Fig. 4, all mutants expressed full-length NanR relative to induced wild type harboring pSX675 (lane 1) or purified NanR (lane 2), as detected by Western blotting. As expected, the mutants obtained by chemical mutagenesis produced more activity than the reporter expressing unmutagenized pSX675 (Fig. 4, compare expression values in lanes 3 to 6 to that in lane 1). On the basis of its predicted structural similarity with the FadR N terminus (6, 7) and overall similarity to the GntR superfamily (6, 17), the G65L mutant is expected to disrupt the tertiary structure of the predicted recognition helix (α3) shown in Fig. 5B. This glycine residue is likely to have a function similar to that of FadR G42 (Fig. 5A) in orienting the α2 and α3 helices. The R66C alteration is predicted to directly affect α3 by removing the positive charge necessary for interacting with DNA (Fig. 5A and B). This helix was shown to be within contact distance of the DNA binding site in the FadR-DNA cocrystal (33), as well as being predicted to be the recognition domain for the entire GntR superfamily (17). It is not possible without reproducing the single mutations in pSX801 and pSX803 to determine whether one or both changes are needed for the transdominant phenotype, although G65 was one of the residues altered in pSX801 (Fig. 4). However, characterization of R66A and R70A mutants after site-directed mutagenesis both supports our conclusion about the R66C phenotype and strengthens the overall conclusion that dominant negative mutants result from effects on the NanR N terminus. A site-directed mutation causing the N86A change outside α3 had a lesser effect on reporter expression (Fig. 4, lanes 1 and 7), as would be expected of a region not as critical for direct protein-DNA interactions (Fig. 5B). However, the lack of an effect by the R56A alteration suggests FadR is only a loose model for interpreting the effects of mutations on NanR function, which as shown below is consistent with a different mechanism of NanR binding to its operator than FadR's. Taken together, the isolation and characterization of dominant negative nanR mutants supports the conclusion that NanR functions as an oligomer (Fig. 3) and that it shares at least some structural features with FadR and other members of the GntR superfamily (Fig. 5), confirming earlier predictions based on molecular modeling and phylogenetic analyses (6, 7, 17).
Fig 4.
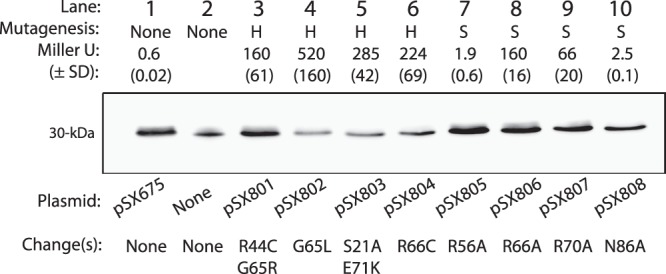
Effects of hydroxylamine-induced (H) and site-directed (S) nanR mutations in a nanA′ reporter, nanR+ background. The indicated plasmids expressing wild-type or mutant nanR were induced with arabinose, and extracts were subjected to Western blot analysis against anti-NanR antibody, as indicated by the expected 30-kDa bands shown in lane 1 and lanes 3 through 10. Lane 2 is a positive control containing 0.2 μg of purified NanR as the antigen. Amino acid changes in the mutated NanR are indicated. Effects of the unmutagenized and mutant NanR expressed by the plasmids on reporter fusion activity are indicated by the average activities obtained from three separate experiments, with the SD in parentheses.
Fig 5.
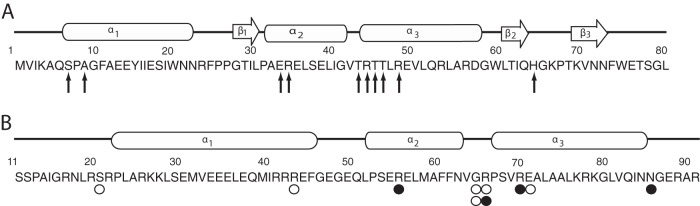
Functional comparison of FadR and NanR N-terminal DNA binding domains. Amino acid residues are numbered from the N terminus of FadR (A) or NanR (B) and are shown below the known or predicted regions of α-helical, β-helical, or random helical structures. Arrows indicate residues known to contact operator DNA nucleotides. Open circles indicate residues found altered in transdominant mutants isolated after random mutagenesis with hydroxylamine (H), while closed circles represent alterations resulting from site-directed mutagenesis (S) as described in the text.
Stoichiometries of operator-NanR complexes.
As shown previously (7) and in Fig. 1C and D, NanR binding to the 256-bp promoter results in three concentration-dependent complexes detected by EMSA. Previous evidence suggested that these complexes are not conformational isomers (6, 7), indicating that they might represent increasing numbers of NanR molecules bound per operator; the complex with the lowest mobility represents the most NanR bound per DNA molecule. Therefore, we estimated the stoichiometry of NanR binding to a 49-bp operator (7) by modified Ferguson analysis (28, 29).
Figure 6A (lane 2) shows an example of the EMSA results obtained from a 9% gel with NanR binding (complexes C1 to C3) to the synthetic 49-bp operator with a calculated mass of 30.3 kDa (Fig. 6A, lane 1). When the relative migration of the indicated molecular weight markers (Fig. 6B) and the three DNA-NanR complexes (Fig. 6C) were plotted against acrylamide concentration, the set of curves obtained by plotting the negative slopes against molecular weights of the markers on a double log scale yielded a straight line, from which the size of each complex was estimated by extrapolation or interpolation (Fig. 6D). After subtraction of the calculated mass of the operator (30.3 kDa), which was within 10% of the estimated mass of 33.5 kDa (Fig. 6D), thus providing an internal verification of the method, and assuming one mole of operator per complex, masses of 92, 128, and 188 kDa were obtained for C1, C2, and C3, respectively (Fig. 6D). These sizes are most consistent with 3, 4, and 6 NanR monomers bound per respective complex. However, because NanR monomers appear to be the inactive form (Fig. 3), the results of Ferguson analysis suggest that either a trimer binds initially (C1) or a dimer followed by a monomer binds, perhaps cooperatively. Then, either the monomer dissociates followed by addition of one (C2) or two (C3) additional dimers, or another monomer (C2) followed by one dimer or two more monomers (C3). Each possible binding scenario leads to a total of six monomers bound to produce C3. Of course, C1 mass might be an overestimate that actually represents the homodimer, in which case each complex would be formed by the simple addition of homodimers (7). However, the molecular masses of C2 and C3 are extrapolated values, while that of C1 is very close to the size of the bovine serum albumin (BSA) dimer (132 kDa), suggesting that the C1 mass estimation should be the most accurate of the estimations for the three complexes. Indeed, the exact 29,221 molecular weight of a NanR monomer implies a homotrimer of 87.6 kDa, which is much closer to the estimated size of C1 (92 kDa) than the 58,441 calculated for the homodimer. On the basis of previous EMSA results (7), C1 appears to have the highest affinity for NanR (7) (Fig. 1D and E), which might be achieved through binding of a homotrimer or a dimer followed by one monomer. The footprint of C1 is consistent with NanR protecting the two most 5′ GGTATA repeats (6), clearly supporting the conclusion that the fully protected footprint (7) requires more molecules of bound NanR than that just those required for formation of C1. We conclude that the NanR operator is composed of overlapping binding sites accommodating the various numbers of bound NanR molecules within three turns of the DNA helix, or about 30 bp, as detected by DNase I protection assay showing coverage of all three repeats (7). Full coverage requires six NanR monomers, and as shown below, all three GGTATA repeats are important for binding. Note that oligomerization solely through protein-protein interactions even with regulator bound to DNA cannot be excluded (32). Thus, NanR polydispersity and disorganization in the presence of sialic acid (Fig. 3), an overlapping DNA binding site consisting of three weak consensus GntR superfamily palindromes (6, 7, 17, 33) (Fig. 7A), and unusual binding stoichiometry likely contributed to our inability to crystallize otherwise exceptionally pure native NanR (see Fig. S1B, lane 4, in the supplemental material).
Fig 6.
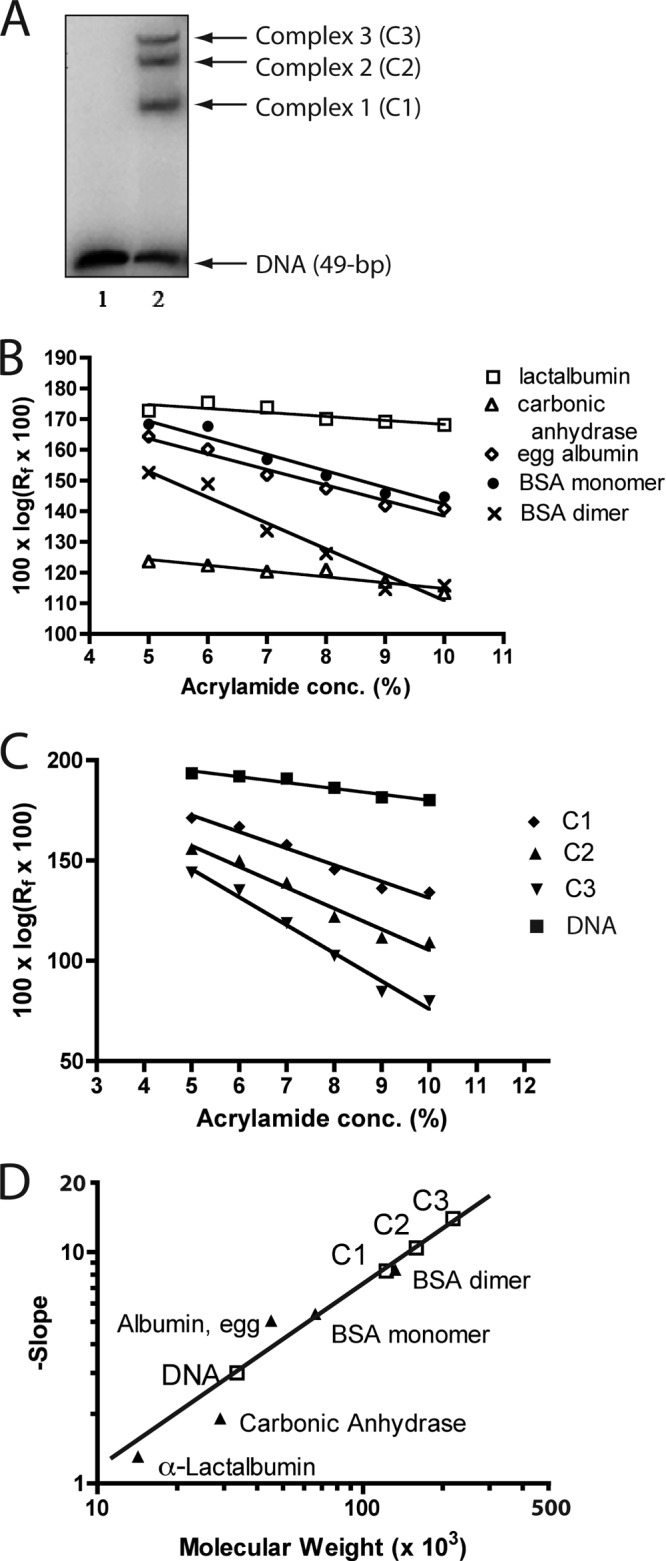
Stoichiometries of NanR binding to synthetic operator. (A) Example of the three NanR-DNA complexes detected by EMSA in a 9% gel with 49 bp synthetic operator DNA. (B) Migrations of the indicated polypeptide standards in polyacrylamide gels of differing porosities. (C) Migrations of the indicated radiolabeled NanR-DNA complexes, DNA alone, and stained polypeptide standards with estimated molecular weights. (D) The negative slopes of migration for C1 to C3 and operator DNA are plotted against the migrations and molecular weights of standards (solid triangles). The estimated molecular weights of complexes C2 and C3 are extrapolated values based on the slope of the standard curve.
Fig 7.

Nan boxes in E. coli and selected bacteria predicted to encode orthologs of NanR. (A) One strand of the 49-bp oligonucleotide used for modified Ferguson analysis is depicted, with boxed nucleotides indicating the conserved regions in the nanATEK-yhcH, nanCMS, and yjhBC operators (Fig. 1E). Nucleotides above the box indicate overlapping repeats of the GntR superfamily consensus sequence, where lowercase letters signify deviations from the consensus and “n” denotes any nucleotide. Brackets indicate nucleotides deviating from the Nan box sequence. Nucleotides below the box indicate the overlapping FadR consensus sequence (41). Underlined nucleotides indicate the hexanucleotide repeats typically found in Nan boxes. (B) Nan boxes in bacteria with NanR orthologs. Sd, Shigella dysenteriae; Ck, Citrobacter koseri; St, Salmonella enterica serovar Typhimurium; Es, Enterobacter sakazakii; Et, Edwardsiella tarda; Sp, Shewanella pealeana; Ph, Pseudoalteromonas haloplanktis; Cf, Citrobacter freundii.
Effects of operator mutations on DNA-NanR interactions.
To confirm the importance of the GGTATA repeats to NanR binding, we used site-directed mutagenesis of pSX809 and the promoter titration assay described elsewhere (7). In this assay, plasmids bearing subcloned wild-type or mutated operators are transformed into reporter strain 12-1 such that increased β-galactosidase activity indicates in vivo titration of wild-type NanR. Table 2 shows the relative induction of wild-type and mutant operators. The wild type and mutants 7 and 8 had the same induction values within about a factor of two, which we assume reflects the deviation in assays comparing different plasmids. Mutant operators 1, 2, and 4 to 6 with defects in any one of the three GGTATA repeats had decreased induction in the titration assay and worse binding (except for mutant 5), reflected by increased dissociation constant, indicating that all three repeats are important for full repression by NanR (Table 2). In general, there was agreement between the in vitro binding and in vivo titration assays. However, these assays may not precisely measure identical phenomena, which could explain why mutant 5 had reduced binding yet a wild-type dissociation constant. Note that its induction was the least affected among all mutants examined, suggesting that the third repeat is less critical than the first two for NanR binding. This suggestion is consistent with deviation of the third repeat in the consensus NanR binding site (Fig. 7B). As expected for binding of an oligomeric regulator, spacing between the first two repeats is important (Table 2, mutants 13 to 15). At least two contiguous repeats are needed for repression, since mutants 3, 9, 10, and 11 (Table 2) with defects in two repeats had the most severe phenotypes, which further supports the importance of the complete operator for wild type repression.
Table 2.
Effects of nan operator mutations on NanR binding and promoter titration

Mutations are shaded in gray. Underlining indicates the repeated operator hexanucleotides.
The activity of strain 12-1 harboring the nanA reporter fusion was 36 ± 13 Miller units; its induction was set as 1.
WT, wild type.
Figure S2 in the supplemental material gives the EMSA results from which dissociation constants of wild-type and selected mutant operators were determined. The results document loss of binding except at a high NanR concentration in mutants with severe in vivo phenotypes (Table 2, mutants 3, 13, and 9 to 11). Interpreting apparent effects of different mutants on formation of C1 to C3 is not straightforward because of the relatively high NanR concentrations needed to detect a gel-shifted complex. However, mutants 1 and 2 with defects in the first repeat show reduced C1 (see Fig. S2). Therefore, if complexes C2 and C3 represent 4 and 6 NanR monomers bound, respectively (Fig. 6), some oligomerization must occur through solely protein-protein interactions with bound NanR, as observed for NtrC (32). The results in Table 2 and Fig. S2 support the overall importance of the Nan box for NanR binding and repression of the sialoregulon using a repressor in the FadR family of helix-turn-helix (HTH) regulators with, however, atypical responses to inducer, its oligomerization state, and binding mechanism. The comparisons shown in Fig. 7 allow us to propose a consensus sequence for the Nan box, which reveals a unique overlapping extension of the FadR/Gnt consensus operator sequences wherein the second and third overlaps have increasingly more deviations from the consensus. This unusual operator structure may have evolved to permit tight regulation of nan genes while avoiding interference with other GntR-like regulators.
DISCUSSION
In the absence of a NanR crystal structure, we investigated interactions between nan repressor, inducer, and operator using genetic and biochemical approaches. The combined results provide a better understanding of the mechanisms regulating sialic acid catabolism in E. coli and the first direct evidence explaining how Neu5Ac derepresses nanATEK-yhcH, nanCMS, and yjhBC. We postulate that this coregulated set of 10 genes (the sialoregulon) evolved to metabolize the diversity of sialic acid structures naturally existing at animal mucosal surfaces (21, 34, 35), leading to a unified theory of bacterial sialocatabolism (1). Additionally, we identified a potentially larger group of genes, designated the sialostimulon, which is likely to be affected positively or negatively by downstream metabolites of sialic acid catabolism and probable alterations in cellular physiology. In contrast, increased nag and fim gene expression previously shown to be affected by Neu5Ac or its downstream metabolites (8, 18) was not detected by transcriptome analyses, suggesting that altered expression of these genes was below the level of our detection method. Finally, by providing an analysis of NanR amino acid residues and operator nucleotides critical for regulation, our results explain how tight control of the nan system is achieved through interactions of multiple NanR molecules with overlapping binding sites. These conclusions should be true for all bacteria with NanR-responsive operators (Fig. 7).
Physiological functions of the sialoregulon.
When E. coli is growing in glucose minimal medium, nanA expression is barely detectable due to inactivation of CRP and repression by NanR (4, 7). However, with CRP activated during growth on a non-catabolite-repressing substrate like glycerol, nanA expression increases, presumably by RNA polymerase blocking repressor binding (14), followed by at least another 10-fold increase resulting from inactivating NanR with Neu5Ac (Fig. 2). Northern blot analysis comparing a nanR-null mutant to its wild-type parent strain (Fig. 1B) is consistent with the previously described magnitude of induction (4, 7), suggesting that quantitative differences between transcriptome values and those of reporter fusions, Northern blotting, and previous enzymatic assays were caused by priming effects (4–8). Therefore, the combined results indicate no qualitative differences between the various approaches, all of which demonstrate induction of the sialoregulon by derepressing (inactivating) NanR. When this result is considered together with the genetic and biochemical characterization of the repressor, it appears that full induction of nan occurs when the inducer (Neu5Ac) destabilizes NanR oligomers upon binding to the C-terminal domain, which is the common domain for regulation of all characterized members of the GntR superfamily (17). The C-terminal domain of NanR has predicted secondary structure most similar to FadR, placing it in the FadR subfamily of the GntR superfamily (6, 17). In addition to NanA and the other enzymes needed for dissimilating Neu5Ac, the nan system includes genes encoding a putative permease(s), porin, mutarotase, esterase(s), oxidoreductase, epimerase, and dehydrogenase that are either known or presumed to convert structurally distinct sialic acids or sialic acid-like molecules to Neu5Ac (1, 2, 23). We suggest that the sialoregulon's primary function is to funnel the diverse chemical forms of sialic acids originating from the host mucosal surface to NanA-digestible Neu5Ac. As shown in this and a previous communication (7), CRP and especially NanR control the overall activity of the E. coli sialoregulon. This regulon is conspicuously absent in all other bacteria examined to date, even closely related enterics (1).
Because the sialic acids are synthesized almost exclusively by deuterostomes, it is unsurprising that a nanAT deletion might be deficient for colonization in a rodent model (36, 37), though this phenotype might only be true for commensals and extraintestinal E. coli, while a transient such as E. coli O157 seems to be less dependent on sialometabolism (38, 39). In addition to nutritional aspects of sialometabolism during colonization, intracellular accumulation of Neu5Ac inhibits cell growth (4). E. coli depends on NanA eliminating toxic intracellular Neu5Ac (4, 9), which is the likely reason why nanA expression is greater than that of the downstream nanTEK-yhcH genes. The dual function of NanA connecting metabolism to detoxification presumably accounts for the potential stem-loop structure in the nanAT intergenic region attenuating downstream gene expression (Fig. 1A). The multiple NanR binding sites might fine-tune nanA expression in response to changing sialic acid concentrations through graded reduction of occupied Nan box-binding sites.
Mechanism of NanR binding.
As discussed elsewhere (7), the lac operon achieves a high level of repression by a looping mechanism involving multiple operator-repressor interactions. In contrast, induction of the tightly regulated nan operon depends on freeing multiple overlapping sites composed of three copies of the loose palindrome recognized by members of the GntR superfamily, with the first site conforming almost exactly to the superfamily consensus sequence and to that of the FadR subfamily (Fig. 7A). The length of these overlapping sites corresponds to the protected footprint described previously (7). The C1 footprint indicates overlap with the second GGTATA repeat (6), an expected result for overlapping binding sites and multiple stoichiometries. The NanR binding mechanism presumably reflects the need to tightly control the dual function of NanA in nutrition and detoxification.
Winged HTH (wHTH) regulators were first identified in eukaryotes in which the secondary structures of their DNA binding domains are organized from the N termini as α1-β1-α2-α3-β2-w1-β3-w2 secondary structural units (40). The α3 helix binds the DNA major groove, with stability and specificity being provided by the wings contacting the flanking minor grooves (40). This binding structure explains how some monomeric wHTH regulators achieve specificity. In contrast, the bacterial wHTH regulator FadR binds as a dimer after undergoing a structural change caused by effector binding to the C terminus such that both recognition helices of the homodimer interact with a single major groove, with one wing of each monomer contacting the flanking minor grooves. Figure 5A shows the FadR amino acid residues within contact distance of the DNA binding site observed in the DNA-FadR cocrystal (33). The predicted secondary structure of the NanR DNA-binding domain (Fig. 5B) provides scant evidence for a β structure, while the three N-terminal α-helices typical of GntR superfamily regulators (17) are clearly present. Though not shown in Fig. 5, the C-terminal NanR effector binding domain is predicted to be composed of either turns or α-helical secondary structural units (6), which is topologically identical to FadR and clearly places NanR in the FadR subfamily of bacterial HTH regulators despite the fact that NanR uses overlapping binding sites instead of wings for specificity. There are currently >8,500 members of the GntR superfamily, which is composed of seven subfamilies, but with additional subfamilies likely to be identified (17). Despite its similarity to FadR, our results further indicate a distinct binding mechanism for NanR, with Neu5Ac causing a conformational change leading to dissociation of oligomers (Fig. 3). We also showed that ManNAc has no activity in this system, eliminating the only other remaining likely inducer candidate (7). Therefore, NanR is distinct from FadR and probably most other GntR regulators in undergoing oligomeric dissociation in response to the conformational change(s) triggered upon Neu5Ac binding to the C-terminal domain. Chemical cross-linking is thus a useful method for assessing the effects of various ligands on sialoregulators such as NanR when EMSA is unsuccessful (7).
Regulation of sialocatabolism in organisms with predicted NanR orthologs.
To obtain the information summarized in Fig. 7B, we searched for at least two GGTATA repeats between orthologs of nanR and the start of operons potentially encoding NanA and containing the other canonical genes needed to convert Neu5Ac to GlcNAc-6-phosphate (1). Interestingly, when we extended this analysis to other homologous genes of the E. coli sialoregulon (Fig. 1A), we often found them, with the notable exception of nanS, organized completely differently than in E. coli, including having locations near a putative transporter(s) in the sodium solute symporter family (1). One of these symporters was recently cloned from Salmonella enterica serovar Typhimurium and shown to complement an E. coli nanT mutant (41). These observations indicate that unlike E. coli, other enteric organisms are unable to metabolize Neu5,9Ac2 (1) but have evolved sialic acid transporters that are functional only in the presence of physiological concentrations of sodium. Furthermore, almost all of the noncanonical sialocatabolic genes in organisms other than E. coli lack upstream Nan boxes (1), indicating that coordinate regulation of the sialoregulon by NanR is unique to E. coli even among closely related enteric species. These observations are consistent with the previous suggestion that E. coli attained its position as the predominant facultative anaerobic commensal and pathogen by its efficiency for scavenging the diversity of sialic acids found in animal hosts (1). Finally, despite only weak similarity to NanS, most mucosal pathogens with known or predicted sialocatabolic systems include predicted orthologs of nanS, indicating that an ability to metabolize alternative sialic acids, and to regulate this process, might be an essential trait in a wide range of evolutionarily distinct bacteria sharing otherwise common mucosal niches (1). Therefore, understanding E. coli sialocatabolism should continue to guide both basic and applied research on some likely very important nutritional aspects of the host-microbe interaction.
Supplementary Material
ACKNOWLEDGMENTS
K.A.K. was supported in part by an American Heart Association Predoctoral Fellowship. E.R.V. gratefully acknowledges NIH grant AI042015 for continued support.
We thank Jacqueline Plumbridge for reading and commenting on a preliminary version of the manuscript; some comments were incorporated into the submitted copy. James Slauch kindly provided the plasmids pGK137 and pCE40 used for converting nanS and yjhC deletions to lac+ operon or protein fusions. We are grateful to Eric Sixmister for many fruitful conversations. We thank Eric L. Deszo for help quantifying phosphorimages and analysis of transcriptome data. We thank Jamie L. Jirik for the β-galactosidase assays, representing in part the results of her undergraduate research project supporting the University of Illinois B.A. degree with distinction. Finally, we are especially grateful for Elspeth Garman's and Daan M. F. van Aalten's heroically independent attempts to crystallize NanR.
Footnotes
Published ahead of print 9 August 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JB.00692-13.
REFERENCES
- 1.Vimr ER. 2013. Unified theory of bacterial sialometabolism: how and why bacteria metabolize host sialic acids. ISRN Microbiol. 2013:816713. 10.1155/816713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Vimr ER, Kalivoda KA, Deszo EL, Steenbergen SM. 2004. Diversity of microbial sialic acid metabolism. Microbiol. Mol. Biol. Rev. 68:132–153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Almagro-Moreno S, Boyd EF. 2009. Insights into the evolution of sialic acid catabolism among bacteria. BMC Evol. Biol. 9:118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Vimr ER, Troy FA. 1985. Identification of an inducible catabolic system for sialic acids (nan) in Escherichia coli. J. Bacteriol. 164:845–853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vimr ER, Troy FA. 1985. Regulation of sialic acid metabolism in Escherichia coli: role of N-acylneuraminate pyruvate-lyase. J. Bacteriol. 164:854–860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kalivoda KA. 2004. Ph.D thesis University of Illinois at Urbana-Champaign, Urbana, IL [Google Scholar]
- 7.Kalivoda KA, Steenbergen SM, Vimr ER, Plumbridge J. 2003. Regulation of sialic acid catabolism by the DNA binding protein NanR in Escherichia coli. J. Bacteriol. 185:4806–4815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Plumbridge J, Vimr E. 1999. Convergent pathways for utilization of the amino sugars N-acetylglucosamine, N-acetylmannosamine, and N-acetylneuraminic acid by Escherichia coli. J. Bacteriol. 181:47–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ringenberg M, Lichtensteiger C, Vimr E. 2001. Redirection of sialic acid metabolism in genetically engineered Escherichia coli. Glycobiology 11:533–539 [DOI] [PubMed] [Google Scholar]
- 10.Steenbergen SM, Lee YC, Vann WF, Vionnet J, Wright LF, Vimr ER. 2006. Separate pathways for O acetylation of polymeric and monomeric sialic acids and identification of sialyl O-acetyl esterase in Escherichia coli K1. J. Bacteriol. 188:6195–6206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Brigham C, Caughlan R, Gallegos R, Dallas MB, Godoy VG, Malamy MH. 2009. Sialic acid (N-acetyl neuraminic acid) utilization by Bacteroides fragilis requires a novel N-acetyl mannosamine epimerase. J. Bacteriol. 191:3629–3638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Johnston JW, Zaleski A, Allen S, Mootz JM, Armbruster D, Gibson BW, Apicella MA, Munson RS., Jr2007. Regulation of sialic acid transport and catabolism in Haemophilus influenzae. Mol. Microbiol. 66:26–39 [DOI] [PubMed] [Google Scholar]
- 13.Johnston JW, Shamsulddin H, Miller A-F, Apicella MA. 2010. Sialic acid transport and catabolism are cooperatively regulated by SiaR and CRP in nontypeable Haemophilus influenzae. BMC Microbiol. 10:240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kim BS, Hwang J, Kim MH, Choi SH. 2011. Cooperative regulation of the Vibrio vulnificus nan gene cluster by NanR protein, CAMP receptor protein, and N-acetylmannosamine 6-phosphate. J. Biol. Chem. 286:40889–40899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Olson ME, King JM, Yahr TL, Horswill AR. 2013. Sialic acid catabolism in Staphylococcus aureus. J. Bacteriol. 195:1779–1788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gualdi L, Hayre JK, Gerlini A, Bidossi A, Colomba L, Trappetti C, Pozzi G, Docquier J-D, Andrew P, Ricci S, Oggioni MR. 2012. Regulation of neuraminidase expression in Streptococcus pneumoniae. BMC Microbiol. 12:200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hoskisson PA, Rigali S. 2009. Chapter 1. Variation in form and function: the helix-turn-helix regulators of the GntR superfamily. Adv. Appl. Microbiol. 69:1–22 [DOI] [PubMed] [Google Scholar]
- 18.Sohanpal BK, El-Labany S, Lahooti M, Plumbridge JA, Blomfield IC. 2004. Integrated regulatory responses of fimB to N-acetylneuraminic (sialic) acid and GlcNAc in Escherichia coli K-12. Proc. Natl. Acad. Sci. U. S. A. 101:16322–16327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Condemine G, Berrier C, Plumbridge J, Ghazi A. 2005. Function and expression of an N-acetylneuraminic acid-inducible outer membrane channel in Escherichia coli. J. Bacteriol. 187:1959–1965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Severi E, Muller A, Potts JR, Leech A, Williamson D, Wilson KS, Thomas GH. 2008. Sialic acid mutarotation is catalyzed by the Escherichia coli beta-propeller protein YjhT. J. Biol. Chem. 283:4841–4849 [DOI] [PubMed] [Google Scholar]
- 21.Steenbergen SM, Jirik JL, Vimr ER. 2009. YjhS (NanS) is required for Escherichia coli to grow on 9-O-acetylated N-acetylneuraminic acid. J. Bacteriol. 191:7134–7139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Izard T, Lawrence MC, Malby RL, Lilley GG, Colman PM. 1994. The three-dimensional structure of N-acetylneuraminate lyase from Escherichia coli. Structure 2:361–369 [DOI] [PubMed] [Google Scholar]
- 23.Vimr ER, Steenbergen SM. 2006. Targeting microbial sialic acid metabolism for new drug development, p 125–150, In Bewley CA. (ed), Protein-carbohydrate interactions in infectious disease. Royal Society of Chemistry, London, United Kingdom [Google Scholar]
- 24.Blattner FR, Plunkett G, III, Block CA, Perna NT, Burland V, Riley M, Collado-Vides J, Glasner JD, Rode CK, Mayhew GF, Gregor J, Davis NW, Kirkpatrick HA, Goeden MA, Rose DJ, Mau B, Shao Y. 1997. The complete genome sequence of Escherichia coli K-12. Science 277:1453–1474 [DOI] [PubMed] [Google Scholar]
- 25.Guzman LM, Belin D, Carson MH, Beckwith J. 1995. Tight regulation, modulation, and high-level expression by vectors containing the arabinose pBAD promoter. J. Bacteriol. 177:4121–4130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ellermeier CD, Janakiraman A, Slauch JM. 2002. Construction of targeted single copy lac fusions using lambda Red and FLP-mediated site-specific recombination in bacteria. Gene 290:153–161 [DOI] [PubMed] [Google Scholar]
- 27.Ringenberg MA, Steenbergen SM, Vimr ER. 2003. The first committed step in the biosynthesis of sialic acid by Escherichia coli K1 does not involve a phosphorylated N-acetylmannosamine intermediate. Mol. Microbiol. 50:961–975 [DOI] [PubMed] [Google Scholar]
- 28.Lavrrar JL, McIntosh MA. 2003. Architecture of a fur binding site: a comparative analysis. J. Bacteriol. 185:2194–2202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Orchard K, May GE. 1993. An EMSA-based method for determining the molecular weight of a protein-DNA complex. Nucleic Acids Res. 21:3335–3336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sigma Chemical Co 1986. Sigma technical bulletin MKR-137. Sigma Chemical Co., St. Louis, MO [Google Scholar]
- 31.Steenbergen S, Vimr ER. 1990. Mechanism of polysialic acid chain elongation in Escherichia coli K1. Mol. Microbiol. 4:603–611 [DOI] [PubMed] [Google Scholar]
- 32.Wyman C, Rombel I, North AK, Bustamante C, Kustu S. 1997. Unusual oligomerization required for activity of NtrC, a bacterial enhancer-binding protein. Science 275:1658–1661 [DOI] [PubMed] [Google Scholar]
- 33.Xu Y, Heath RJ, Li Z, Rock CO, White SW. 2001. The FadR · DNA complex. Transcriptional control of fatty acid metabolism in Escherichia coli. J. Biol. Chem. 276:17373–17379 [DOI] [PubMed] [Google Scholar]
- 34.Robbe C, Capon C, Maes E, Rousset M, Zweibaum A, Zanetta J-P, Michalski J-C. 2003. Evidence of regio-specific glycosylation in human intestinal mucins. J. Biol. Chem. 278:46337–46348 [DOI] [PubMed] [Google Scholar]
- 35.Robbe-Masselot C, Maes E, Rousset M, Michalski J-C, Capon C. 2009. Glycosylation of human fetal mucins: a similar repertoire of O-glycans along the intestinal tract. Gycoconj. J. 26:397–413 [DOI] [PubMed] [Google Scholar]
- 36.Chang DE, Smalley DJ, Tucker DL, Leatham MP, Norris WE, Stevenson SJ, Anderson AB, Grissom JE, Laux DC, Cohen PS, Conway T. 2004. Carbon nutrition of Escherichia coli in the mouse intestine. Proc. Natl. Acad. Sci. U. S. A. 101:7427–7432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fabich AJ, Jones SA, Chowdhury FZ, Cernosek A, Anderson A, Smalley D, McHargue JW, Hightower GA, Smith JT, Autieri SM, Leatham MP, Lins JJ, Allen RL, Laux DC, Cohen PS, Conway T. 2008. Comparison of carbon nutrition for pathogenic and commensal Escherichia coli strains in the mouse intestine. Infect. Immun. 76:1143–1152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jacobsen L, Durso L, Conway T, Nickerson KW. 2009. Escherichia coli O157:H7 and other E. coli strains share physiological properties associated with intestinal colonization. Appl. Environ. Microbiol. 75:4633–4635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Snider TA, Fabich AJ, Conway T, Clinkenbeard KD. 2009. E. coli O157:H7 catabolism of intestinal mucin-derived carbohydrates and colonization. Vet. Microbiol. 136:150–154 [DOI] [PubMed] [Google Scholar]
- 40.Gajiwala KS, Burley SK. 2000. Winged helix proteins. Curr. Opin. Struct. Biol. 10:110–116 [DOI] [PubMed] [Google Scholar]
- 41.Severi E, Hosie AH, Hawkhead JA, Thomas GH. 2010. Characterization of a novel sialic acid transporter of the sodium solute symporter (SSS) family and in vivo comparison with known bacterial sialic acid transporters. FEMS Microbiol. Lett. 304:47–54 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.