

Abstract
Tectiviridae is a family of tailless bacteriophages with Gram-negative and Gram-positive hosts. The family model PRD1 and its close relatives all infect a broad range of enterobacteria by recognizing a plasmid-encoded conjugal transfer complex as a receptor. In contrast, tectiviruses with Gram-positive hosts are highly specific to only a few hosts within the same bacterial species. The cellular determinants that account for the observed specificity remain unknown. Here we present the genome sequence of Wip1, a tectivirus that infects the pathogen Bacillus anthracis. The Wip1 genome is related to other tectiviruses with Gram-positive hosts, notably, AP50, but displays some interesting differences in its genome organization. We identified Wip1 candidate genes for the viral spike complex, the structure located at the capsid vertices and involved in host receptor binding. Phage adsorption and inhibition tests were combined with immunofluorescence microscopy to show that the Wip1 gene product p23 is a receptor binding protein. His-p23 also formed a stable complex with p24, a Wip1 protein of unknown function, suggesting that the latter is involved with p23 in host cell recognition. The narrow host range of phage Wip1 and the identification of p23 as a receptor binding protein offer a new range of suitable tools for the rapid identification of B. anthracis.
INTRODUCTION
Bacteriophage-based diagnostics and therapeutics have been recognized as tools to combat bacterial infections for nearly a century. Wip1 (for worm intestinal phage 1) is a recently identified phage that infects the pathogen Bacillus anthracis and was isolated from the intestinal tracts of Eisenia fetida worms (1). It is a tailless, double-stranded DNA phage possessing an internal lipid membrane beneath an icosahedral protein coat (2). These features indicate that Wip1 belongs to the family Tectiviridae, a relatively rare phage group with surprising structural similarity to and a proposed evolutionary lineage in common with the mammalian adenovirus (3, 4). The Tectiviridae family consists of six isolates that infect Gram-negative bacteria, including PRD1 (5), and six that infect Gram-positive bacteria, including Bam35, Gil16, AP50, and Wip1 (1, 6–8). While PRD1 has been studied in detail, tectiviruses that infect Gram-positive bacteria are not as well characterized.
Wip1 phage exhibits a very narrow host range and is highly specific to B. anthracis (1), the notorious biothreat agent and Gram-positive bacterium that causes anthrax disease. The current gold standard for identifying suspected B. anthracis involves testing for γ phage sensitivity (9; http://www.bt.cdc.gov/agent/anthrax/faq/diagnosis.asp). However, using γ as a diagnostic tool can lead to false positives because of the susceptibility of several Bacillus cereus strains to infection with this phage (10, 11). Recent studies have shown that the host range of γ is less specific to B. anthracis than those of tectiviruses Wip1 and AP50 (1, 12). For example, B. cereus ATCC 4342 is sensitive to infection with γ phage but not to infection with either Wip1 or AP50. Additionally, the γ diagnostic phage yields plaques on B. anthracis ΔSterne only after 5 days, whereas Wip1 plaques can be detected at just 12 h postinfection (1). These advantages suggest that Wip1 may be superior to γ as a diagnostic tool.
Wip1's high specificity to B. anthracis is likely mediated by the initial recognition and binding of the virus to the host cell. Receptor binding proteins on the phage coat interact very specifically with receptors exposed on the surface of the bacterium (13). For tectiviruses, the receptor binding protein assembles with other phage proteins into a protruding complex that extends from each particle vertex (14). In the PRD1 spike complex, two elongated proteins, monomeric receptor binding protein P2 and trimeric spike protein P5, form two separate spikes that each protrude from penton base protein P31 (15–18).
Spike complex protein components have also been identified in Bam35, a tectivirus that infects Gram-positive Bacillus thuringiensis (19). By threading Bam35 gene products onto PRD1 X-ray structures, it was determined that gp28 is homologous to spike protein P5 and that gp29 is homologous to the C-terminal half of receptor binding protein P2 (20, 21). In addition, gp28 and gp29 were determined to reside on the surface of Bam35 by phage aggregation and neutralization assays with polyclonal antibodies (19). However, competitive binding assays with both recombinant and dissociated surface proteins were inconclusive and a Bam35 receptor binding protein could not be identified.
The goal of this study was to develop a better understanding of Wip1's highly specific tropism for B. anthracis. We started by imaging the morphological changes of Wip1 phage upon adsorption, expanding its host range analysis with adsorption studies, and sequencing its viral genome. On the basis of genomic analysis with other Gram-positive-bacterium-infecting tectiviruses, candidate gene products for the Wip1 spike complex were predicted and used to identify a Wip1 receptor binding protein that detects and exhibits specificity for B. anthracis.
MATERIALS AND METHODS
Bacterial strains and phages.
The majority of the bacterial strains used in the present study were previously described (1, 2, 11). All of the bacterial strains were grown in brain heart infusion (BHI) broth or on BHI agar plates at 30°C according to standard protocols. Bacteriophage Wip1 was isolated from the intestinal tracts of E. fetida worms from Pennsylvania. Phage propagation was performed on the B. anthracis ΔSterne strain.
Phage propagation.
High-titer phage stocks were obtained by infecting stationary-phase cell cultures (100 μl) with 100 μl of a series of diluted (1:100 to 1:1,000) Wip1 phage stocks. The phage-bacterium mixtures were incubated in a 37°C water bath for 15 min and then plated with molten top agar (0.8%) on BHI plates and incubated overnight at 30°C. When the viral plaques reached near confluence, the soft agar overlays were collected in conical tubes, incubated with 2 ml of 10 mM K phosphate per plate for 15 min at room temperature, and centrifuged at 4,000 rpm for 20 min at 4°C. The resulting supernatants were filtered (0.45-um-pore-size filter) and stored at 4°C.
TEM.
Wip1 phages were incubated with overnight cultures of B. anthracis ΔSterne at a multiplicity of infection (MOI) of 10 for 5 min at 37°C. After incubation, the mixtures were transferred to a new Eppendorf tube with solidified agar on the bottom to act as a cushion during the subsequent centrifugation at 6,000 rpm for 3 min. Supernatant was removed, and the pellet was resuspended in 1× glutaraldehyde fixative. Transmission electron microscopy (TEM) analyses were then performed at The Rockefeller University Bio-Imaging Resource Center as previously described (11).
Chloroform sensitivity assay.
Wip1 phage samples (2 ml) were incubated with and without various volumes of chloroform (up to 80 μl) in capped glass tubes with gentle mixing at room temperature for 15 min. The titers of the mixtures were then determined on B. anthracis ΔSterne. W2 phage with and without chloroform plated on B. cereus ATCC 4342 was used as a control.
Phage adsorption assay.
Various bacterial strains were grown to stationary phase (approximately 2 × 108 CFU/ml), and 100 μl of bacteria was mixed with 100 μl of Wip1 at 2 × 107 PFU/ml. The phage-bacterium mixtures were incubated in a 37°C water bath for 20 min and then pelleted at 7,000 rpm for 3 min. The resulting supernatants were subsequently spin filtered (Millipore; 0.22-μm pore size), and titers were determined on plates of B. anthracis ΔSterne.
DNA manipulation and sequencing.
To obtain Wip1 DNA, Wip1 phage stocks (1 × 108 CFU/ml) were lysed as follows. A 25-μl volume of phage stock was suspended in 25 μl of 0.5 M NaOH (Sigma-Aldrich), incubated for 5 min at room temperature, neutralized with 50 μl of Tris (pH 8.0; Life Technologies), and diluted in 450 μl of distilled H2O. Wip1 DNA was processed as described previously (1), digested for 5 min at 65°C with 0.1 U of Tsp509I (New England BioLabs), ligated to EcoRI adaptors (GeneLink), PCR amplified with adaptor-specific primers, and cloned into the pBAD TOPO TA expression vector (Life Technologies). The resulting plasmid library was transformed into One Shot TOP10 Escherichia coli (Life Technologies), and random plasmid preparations were sequenced. Sequences derived from these transformed cells were confirmed by sequencing PCR products generated directly from Wip1 phage DNA. Primers were designed to sequence specific regions of the PCR products, and unknown sequence regions were determined by primer walking on the purified Wip1 genome.
Cloning of His-tagged Wip1 open reading frame 22 (ORF22), ORF23, and ORF24.
The PCR products containing the coding sequences for Wip1 gene products 22, 23, and 24 were separately amplified with specific primers. Each DNA fragment was inserted into a modified CDFDuet-1 plasmid between the SalI-NotI sites preceded by a T7lac promoter and ribosome binding site, as well as two His tag sequences. Clones were confirmed by sequencing with primers that flank the insert.
Purification of His-tagged Wip1 p22, p23, and p24.
Overnight cultures of E. coli DH5α cells carrying the cloned constructs were diluted 1:100 and grown in LB medium with spectinomycin (20 μg/ml) for 4 h while shaking at 37°C. After being moved to 16°C, the cultures were induced with isopropyl-β-d-thiogalactopyranoside (IPTG) at a final concentration of 0.25 mM and shaken for an additional 18 h. Bacterial pellets were collected by centrifugation (Sorvall SLC-6000 rotor, 7,200 rpm, 30 min, 4°C) and resuspended in a cold buffer (50 mM Tris [pH 8.0], 200 mM NaCl, 5 mM imidazole) at 1/100 of the original culture volume. Bacterial lysis was conducted by multiple passages through a French pressure cell (at ∼105 MPa) at 4°C. The cell debris was removed by centrifugation (Sorvall SS-34 rotor, 8,000 rpm, 20 min, 4°C), followed by filtration (Nalgene; 0.45-μm pore size).
The following purification steps were conducted at room temperature with buffers kept at 4°C. Twenty-five-milliliter columns were loaded with a 1.25-ml bed volume of Ni-nitrilotriacetic acid (NTA) agarose (Qiagen) and equilibrated with 2 column volumes of buffer (50 mM Tris [pH 8.0], 200 mM NaCl, 5 mM imidazole). The cell lysate from induced cultures was passed through the columns twice by gravity flow. The Ni-NTA agarose was then washed with 1 column volume of wash buffer A (50 mM Tris [pH 8.0], 500 mM NaCl, 30 mM imidazole) and 0.5 column volume of wash buffer B (50 mM Tris [pH 8.0], 500 mM NaCl, 60 mM imidazole). Finally, the eluate was collected by passing 5 bed volumes of elution buffer (50 mM Tris [pH 8.0], 500 mM NaCl, 250 mM imidazole) through the column.
In preparation for the next step of the purification process, the eluted proteins were dialyzed against buffer A (20 mM phosphate buffer, pH 7.4) at 4°C. Ion-exchange chromatography was then conducted with a 5-ml HiTrap Q HF column at a linear gradient of 100% buffer A targeting 50% buffer B (20 mM phosphate buffer, 1 M NaCl, pH 7.4). Fractions containing the purified proteins of interest were collected, analyzed by SDS-PAGE, pooled, and dialyzed against 1× phosphate-buffered saline (PBS) at 4°C (see Fig. S1 in the supplemental material).
Cloning and purification of coexpressed His-p23 and p24.
The cloning-and-purification schematic of His-p23 and p24 differs from that described for the individual recombinant proteins only in that the PCR insert at the multiple cloning site began with the start codon for ORF23 and ended with the last codon for ORF24. It should be noted that ORF24 was not cloned into a separate site with its own promoter and tag.
Inhibition of phage infection.
Overnight cultures of B. anthracis ΔSterne (12 μl) were mixed with molten soft agar (0.8%, 400 μl) and overlaid onto BHI agar plates measuring 35 by 10 mm. After allowing the bacterial soft agar to sit for 15 min, various dilutions of the purified His-tagged phage protein stocks (PBS dilutions, 100 μl) were evenly placed on top. The protein overlay was incubated atop the bacterial agar for 15 min before a final overlay of Wip1 phage (∼100 PFU) was evenly added. The resulting plaques were counted at 48 h postinfection. All steps were conducted at room temperature.
Inhibition of phage adsorption with polyclonal antisera against His-tagged Wip1 p23.
To generate polyclonal antisera against His-tagged Wip1 p23, 500 μg of the purified protein was run on SDS-PAGE and excised bands were used as the antigen to immunize two New Zealand White rabbits. Preimmune serum was collected before the first immunization in complete Freund's adjuvant and for the second, third, and fourth immunizations in incomplete Freund's adjuvant. Immunization and production blood samples were collected at 3-week intervals for a total of 21 weeks. An indirect enzyme-linked immunosorbent assay (ELISA) was used to determine the titer of the polyclonal antisera against His-p23. The titer was found to be 2,000 (the highest dilution to yield an absorbance of 1 in 15 min).
The antisera were then tested for the ability to inhibit Wip1 phage binding activity. Various dilutions of serum (in BHI medium) were mixed with Wip1 phage stock (∼800 PFU) and incubated in a 37°C water bath for 30 min. After the addition of 100 μl of overnight cultures of B. anthracis ΔSterne, the serum-phage-bacterium mixtures were incubated at room temperature for 30 min. Bacterial pellets were collected via centrifugation at 12,500 rpm for 30 s, washed, and resuspended in 10 mM K phosphate buffer. Bacterium-bound phage were plated onto BHI and proceeded to form plaques overnight. The inhibition rate was measured as the percent reduction of Wip1 PFU compared to a no-serum PBS control.
Fluorescence microscopy.
The following protocol is a modified version of one previously described (22). Overnight cultures of various bacterial strains were inoculated at a 1:100 dilution into BHI medium and grown to mid-exponential phase (unless otherwise noted) by shaking for 3 h at 30°C. The cell cultures were then pelleted by centrifugation (Eppendorf 5810R, 4,000 rpm, 5 min, 4°C), washed, and then resuspended in PBS. To fix the cells, paraformaldehyde and NaPO4, pH 7.4, were added to the suspension at final concentrations of 2.6% and 30 mM, respectively. The cells were incubated for 15 min at room temperature, followed by 30 min on ice; washed with PBS; and then attached to polylysine-coated glass coverslips. The fixed cells attached to glass were then washed with PBS and blocked with PBS containing 1% bovine serum albumin for 15 min. The cells subsequently underwent a series of three labeling steps, first with purified, His-tagged Wip1 protein, then with anti-His mouse antibodies, and finally with anti-mouse rhodamine dye and 4′,6-diamidino-2-phenylindole (DAPI) stain. Each labeling step consisted of incubating the cells with the labeling mixture for 45 min in a moist chamber and was followed by thorough washing with PBS. To reduce bleaching, the slides were mounted with 50% glycerol and 0.1% p-phenylenediamine in PBS, pH 8.0. Images were captured with a Delta-Vision image restoration microscope (Applied Precision/Olympus) equipped with a CoolSnap QE cooled charge-coupled device camera (Photometrics). An Olympus 100× oil immersion objective was used in conjunction with a 1.5× Optovar magnification changer.
Nucleotide sequence accession number.
The complete sequence of the Wip1 bacteriophage was submitted to GenBank and assigned accession number KF188458.
RESULTS
Wip1 is morphologically similar to other tectiviruses.
Wip1 phage is able to produce clear plaques on the B. anthracis Sterne and ΔSterne strains. Samples of the viral particles with and without the bacterium B. anthracis ΔSterne were analyzed by TEM. Wip1 phage alone are tailless, bilayer, icosahedral particles with a vertex-to-vertex diameter of approximately 60 nm and are similar in morphology to other tectiviral phages (2). Treatment with chloroform inhibited Wip1 activity, which is consistent with Wip1 possessing an inner lipid membrane (data not shown).
Upon incubation with its host B. anthracis ΔSterne, Wip1 displayed a tube-like structure (Fig. 1) that has been described in other tectiviruses such as Bam35 and PRD1 (6, 20, 23). In PRD1, interaction of protein P2 with the host receptor leads to a conformational change that results in the dissociation of the spike complex proteins from the virion (24). This is followed by the transformation of the spherical internal membrane into a tubular channel structure for the delivery of viral DNA into the host (25, 26). In some cases, the phage tails were seen to interact in a conventional way with the bacterial cell surface. However, interestingly, TEM images also captured several Wip1 phages with tube-like structures facing away from the bacterial surface (Fig. 1) instead of directly toward the host, a phenomenon not reported in other tectiviruses (D. H. Bamford, personal communication, 2006). This could indicate that the labile vertex undergoing tubular transformation may not be the same as the vertex that initially binds the receptor. This could also be explained by reversible binding between the Wip1 tube-like structure and its host.
Fig 1.

TEM of the Wip1 phage on B. anthracis. Wip1 was incubated with B. anthracis ΔSterne for 5 min at an MOI of 10 before being fixed for TEM imaging. Scale bars = 0.1 μm. The arrow in panel B indicates a tubular channel facing away from the bacterial surface instead of directly towards the host.
Wip1 infection and adsorption are highly specific to B. anthracis.
Previous studies determined that Wip1 infectivity is more specific to B. anthracis than is the standard diagnostic tool γ phage (1). We decided to expand the host range study to include adsorption and infection of Wip1 virions to the surface of different bacteria. Adsorption assays showed that Wip1 binding also exhibited a high specificity to B. anthracis that corresponded to its narrow infectivity host range (Table 1). This observation further supported the model of Wip1 tropism being mediated by the receptor binding proteins on its surface.
Table 1.
Wip1 infectivity and adsorption rangea
| Bacterial strain | Phage infectivity (PFU/ml of stock) |
% Wip1 adsorption | |
|---|---|---|---|
| γ | Wip1 | ||
| Bacillus anthracis ΔSterne | 3.0E+09 | 6.0E+09 | 100 |
| Bacillus cereus | |||
| ATCC 4342 | 1.0E+05 | <10 | <5 |
| CDC32805 | 4.0E+07 | 3.0E+07 | 94 |
| CDC13100 | <10 | <10 | <5 |
| CDC13140 | <10 | <10 | <5 |
| ATCC 10987 | <10 | <10 | <5 |
| NRL 569 | <10 | <10 | <5 |
| ATCC 14579 | <10 | <10 | <5 |
| ATCC 13472 | <10 | <10 | <5 |
| ATCC 11980 | <10 | <10 | <5 |
| RTS100 | <10 | <10 | <5 |
| Bacillus thuringiensis | |||
| HD1 | <10 | <10 | <5 |
| HD73 | <10 | <10 | <5 |
| Bacillus subtilis SL4 | <10 | <10 | <5 |
| Bacillus pumilus SL4680 | <10 | <10 | <5 |
| Sporosarcina ureae | <10 | <10 | <5 |
| Bacillus megaterium WH32 | <10 | <10 | <5 |
| Brevibacillus laterosporus | <10 | <10 | <5 |
Various bacterial strains were tested for Wip1 infectivity and adsorption. The lower limit of detection of infectivity is indicated by a value of <10, while the lower limit of detection of adsorption is indicated by a value of <5. The Wip1 host range is highly specific for B. anthracis in terms of both infectivity and adsorption.
Wip1 is related to other tectiviruses, with notable differences.
The Wip1 genome was determined to be a linear molecule of DNA measuring 14,319 bp. Detailed analysis of the Wip1 sequence revealed the existence of 27 putative ORFs, as shown in Fig. 2. Wip1 exhibits sequence similarities to Gram-positive infecting tectiviruses Bam35c and Gil16c. Wip1 is most closely related to AP50, which also has a narrow host range highly specific to B. anthracis (12). The genome of Wip1 from ORF5 through ORF25 is strikingly similar to the section of AP50 from ORF10 through ORF30 in ORF size, sequence, and organization (Table 2 and Fig. 2). Of the total of 27 putative Wip1 ORFs, 19 show a sequence identity of at least 50% to other tectiviral proteins and 14 show a high sequence identity of at least 75% to AP50 proteins. We noted that the Wip1 genome GC content at both extremities is lower than the percentage observed in the central section of the genome. The GC content is approximately the same in all of the Wip1 ORFs as in their corresponding AP50 homologs (12).
Fig 2.

Alignment of Bam35c, Gil16c, AP50, and Wip1 genome maps. Predicted genes are represented as block arrows, and the color key in the bottom right indicates postulated functions. Shaded regions pair sequence segments conserved between phages and amino acid identity (id.). Percentage ranges are shown in the lower left corner. HVR, highly variable region.
Table 2.
Comparison of genes in Wip-1 with those of other members of the family Tectiviridaea
| Wip1 ORF | No. of residues (genome coordinates) | Strand | % G+C | % Identity |
ORF (no. of residues) |
PRD1 protein (no. of residues) [reference(s) or source] | Postulated function | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| AP50 | GIL16c | Bam35c | AP50 | GIL16c | Bam35c | ||||||
| 1 | 57 (396–569) | + | 29.5 | ||||||||
| 2 | 196 (598–1188) | + | 30.7 | Transcription factor | |||||||
| 3 | 112 (1219–1557) | + | 25.7 | LexA-type repressor | |||||||
| 4 | 91 (1690–1965) | + | 25.7 | ||||||||
| 5 | 67 (1958–2161) | + | 27.9 | 55.1 | 10 (57) | ||||||
| 6 | 116 (2121–2471) | + | 43.9 | 69.6 | 62.5 | 45.7 | 11 (114) | 9 (125) | 10 (145) | P6 (166) [31] | DNA packaging/unique vertex |
| 7 | 236 (2309–3019) | + | 46.8 | 78.4 | 42.2 | 44.0 | 12 (235) | 10 (248) | 11 (252) | P10 (203) [32, 33] | Assembly |
| 8 | 83 (2865–3116) | + | 46.4 | 94.0 | 53.2 | 53.2 | 13 (83) | 11 (80) | 12 (80) | ||
| 9 | 212 (3125–3763) | + | 42.4 | 93.9 | 59.9 | 63.7 | 14 (212) | 13 (212) | 14 (212) | P9 (227) [34] | DNA packaging ATPase |
| 10 | 54 (3760–3924) | + | 36.4 | 89.1 | 60.9 | 60.9 | 15 (46) | 14 (46) | 15 (46) | ||
| 11 | 47 (3938–4081) | + | 33.6 | 80.9 | 48.8 | 48.8 | 16 (49) | 16 (46) | 16 (46) | P20 (42) [uniprot.org/uniprot/P27587] | DNA packaging/unique vertex |
| 12 | 353 (4081–5142) | + | 40.7 | 83.8 | 63.1 | 63.9 | 17 (354) | 18 (356) | 18 (356) | P3 (395) [35] | Major capsid protein |
| 13 | 74 (5191–5415) | + | 38.2 | 78.4 | 35.7 | 34.3 | 18 (74) | 19 (76) | 19 (76) | P22 (47) [uniprot.org/uniprot/P27388] | DNA packaging/unique vertex |
| 14 | 59 (5421–5600) | + | 32.8 | 34.4 | 46.8 | 39.4 | 19 (56) | 20 (52) | 20 (68) | ||
| 15 | 157 (5677–6150) | + | 38.8 | 86.6 | 37.2 | 37.8 | 20 (157) | 22 (143) | 21 (143) | ||
| 16 | 61 (6138–6323) | + | 36.0 | 79.3 | 60.7 | 60.7 | 21 (58) | 23 (58) | 22 (58) | ||
| 17 | 48 (6323–6469) | + | 36.1 | 77.1 | 54.2 | 54.2 | 22 (48) | 24 (48) | 23 (48) | ||
| 18 | 91 (6482–6757) | + | 43.1 | 82.4 | 35.6 | 33.3 | 23 (91) | 25 (91) | 24 (91) | ||
| 19 | 213 (6761–7402) | + | 44.8 | 85.1 | 23.5 | 25.9 | 24 (210) | 26 (204) | 25 (204) | P11 (207) [36] | DNA delivery |
| 20 | 218 (7402–8058) | + | 44.1 | 91.7 | 66.4 | 65.4 | 25 (218) | 27 (250) | 26 (250) | P7 (265) | Lysin |
| 21 | 175 (8055–8582) | + | 46.4 | 84.6 | 62.2 | 63.4 | 26 (175) | 28 (170) | 27 (170) | ||
| 22 | 291 (8593–9468) | + | 44.7 | 63.9 | 43.1 | 40.3 | 27 (304) | 29 (297) | 28 (304) | P5 (340) [37] | Trimeric spike protein |
| 23 | 117 (9472–9825) | + | 39.8 | ||||||||
| 24 | 118 (9838–10194) | + | 36.7 | 50.8 | 29 (118) | ||||||
| 25 | 48 (10196–10342) | + | 33.3 | 71.7 | 30 (49) | ||||||
| 26 | 213 (10344–10985) | + | 42.3 | Lysin | |||||||
| 27 | 898 (11342–14038) | − | 29.7 | DNA polymerase | |||||||
ORFs were predicted by GeneMark (http://exon.biology.gatech.edu/heuristic_hmm2.cgi), and G+C percentages were determined by the GC calculator at http://www.sciencebuddies.org. Protein identities were determined by the Pairwise Sequence Alignment program Water available from EMBOSS at the EMBL-EBI node (http://www.ebi.ac.uk/Tools/psa/emboss_water/). A summary of PRD1 gene functions can be found in reference 38, and PRD1 homologs in Bam35c were determined in reference 21.
The most remarkable distinction of the Wip1 genome is the placement of the putative DNA polymerase, ORF27, at the 3′ end of the genome on the negative strand. All other tectiviral DNA polymerases are encoded in the first 5,000 bp of their genomes on the positive strand. While several polymerase motifs were identified in the Wip1 ORF27 sequence, the unusual Wip1 ORF27 gene product does not share any significant homology with any other proteins in the NCBI database.
Another notable section of the genome includes Wip1 ORF22, ORF23, and ORF24. Wip1 ORF22 is predicted to be a putative spike complex protein, as it shares 40.3% sequence identity with Bam35 gp28, a homolog of PRD1 trimeric spike protein P5. Bam35 gp28 is followed by gp29, a 293-amino-acid protein that also resides on the phage surface (21). If the Wip1 and AP50 genomes both align with that of Bam35c, then a putative spike complex protein should follow. Interestingly, in Wip1 and AP50, the genes downstream of the P5 homolog are located in a highly variable region and do not share sequence identity with any Bam35 ORFs (Fig. 2). Despite the lack of homology, we predicted that Wip1 gene products 23 and 24 (totaling 235 amino acids together) were putative spike complex proteins based on strikingly similar gene cassette alignment both upstream and downstream of this region. Finally, we were intrigued by the observation that while Wip1 ORF24 shares 51% sequence identity with AP50 ORF29, Wip1 ORF23 is a unique gene that does not share sequence homology with any other known genes, tectiviral or otherwise.
Wip1 p23 and p24 form a stable complex.
Genomic analysis predicted three Wip1 proteins, p22, p23, and p24, to be possible candidates for the Wip1 spike complex. We developed expression-and-purification schemes for the His-tagged constructs of all three viral proteins. However, the separately expressed His-p23 and His-p24 constructs resulted in extremely low yields of soluble protein because of the formation of inclusion bodies (data not shown). Curiously, when His-p23 and p24 were coexpressed, significantly higher yields of soluble protein were generated, suggesting that His-p23 and p24 assist each other in proper folding when expressed together. We used this coexpressed complex to purify the two molecules apart from each other. However, when the His-p23–p24 complex was eluted from a stringently washed Ni-NTA column, both proteins eluted together despite the fact that p24 was not His tagged (Fig. 3). When we used this Ni-NTA eluate to separate the two proteins by ion-exchange chromatography, they were again observed together in the same fractions despite the fact that His-p23 and p24 exhibit drastically different pIs of 8.4 and 4.9, respectively (Fig. 3). This observation strongly suggests that p23 and p24 form a stable complex.
Fig 3.

Purified proteins. The Ni-NTA eluate containing both His-p23 and p24 (leftmost lane) was further purified by charge by ion-exchange chromatography. The distinct peaks were evaluated by SDS-PAGE, which revealed the presence of His-p23 alone in peak 1 and the presence of His-p23 and p24 together in peak 2. The values to the left of the gel are molecular sizes in kilodaltons. OD, optical density; FPLC, fast protein liquid chromatography.
Wip1 p23 is a receptor-binding protein.
The purified recombinant proteins were subsequently used in the Wip1 activity inhibition assay, which consisted of overlaying the viral proteins on top of B. anthracis ΔSterne growing in soft agar before adding a final overlay of infectious Wip1 phage. His-p23 was shown to competitively inhibit Wip1 infectivity up to 100% in a dose-dependent manner, while His-p22 and His-p24 had no effect on phage infectivity (Fig. 4A). This finding suggests that Wip1 p23 is a receptor binding protein. We also tested the His-p23–p24 complex in this assay at a 2× total concentration to account for the presence of the two proteins that make up each protein complex. The His-p23–p24 complex exhibited higher inhibition levels than His-p23 alone, a finding that may be explained by either increased protein stability or enhanced activity of the receptor binding complex. The possibility that recombinant His-p22 and His-p24 do not possess their full biological activity because of the linked histidine should be noted. However, the issues of potential misfolding or activity interference caused by a histidine tag are partially addressed for p24 with the coexpressed His-p23–p24 complex.
Fig 4.

Wip1 inhibition of infectivity by recombinant Wip1 proteins. (A) Wip1 phage was assayed for inactivation by recombinant viral proteins at various concentrations. Concentrations are indicated per protein. His-p23 and the His-p23–p24 complex inactivated Wip1 infectivity by up to 100% in a dose-dependent manner. His-p22 and His-p24 did not affect phage activity. Bars represent the standard errors of a minimum of three experiments. (B) Anti-His-p23 antibody neutralization of Wip1 activity. Polyclonal antibodies were generated against His-p23 and tested for neutralization of Wip1 activity as described in Materials and Methods. After preincubation with phage, anti-His-p23 serum inhibited Wip1 adsorption to B. anthracis ΔSterne by up to 90% in a dose-dependent manner. Prebleed serum did not affect Wip1 adsorption activity. Bars represent the standard errors of at least three experiments.
In a second phage inhibition assay, we used polyclonal antisera against Wip1 His-p23 to inhibit phage binding activity. B. anthracis ΔSterne bacteria were added to preincubated mixtures of Wip1 phage and antiserum at multiple dilutions. The cells were pelleted, and the supernatant plated to reveal the titer of unbound phage. The results revealed that anti-His-p23 antibodies reduced Wip1 adsorption by up to 90% in a dose-dependent manner. Prebleed serum did not have an effect on Wip1 activity (Fig. 4B). This suggests that Wip1 p23 protein resides on the phage surface and is consistent with the finding that it is a receptor-binding molecule.
Wip1 p23 binding is specific to B. anthracis.
To further understand the interaction between Wip1 proteins and bacterial surfaces, the purified His-tagged viral proteins were tested for surface labeling of selected bacterial strains by indirect immunofluorescence microscopy. Figure 5 shows that both His-p23 and the His-p23–p24 complex bind specifically to the surface of B. anthracis ΔSterne. Surprisingly, the Wip1 proteins bound all mid-log-phase ΔSterne bacteria uniformly but bound only a subpopulation of stationary-phase ΔSterne bacteria. His-p22 and His-p24 were unable to bind either growth phase of ΔSterne, further suggesting that Wip1 p22 and p24 are not involved in phage adsorption. The lesser ability of Wip1 to bind to stationary-phase bacteria is probably due to the replacement of surface array protein (Sap) with EA1 protein as the bacteria transition from log phase to stationary phase (27); Sap is believed to be the AP50 tectivirus receptor (28) and may be important for Wip1 attachment as well.
Fig 5.

Indirect immunofluorescence microscopy of His-tagged Wip1 proteins. ×1,000 magnification.
In Fig. 6, the specificity of the binding of His-p23 and the His-p23–p24 complex to B. anthracis is shown by indirect immunofluorescence microscopy. Neither of the protein constructs was able to bind the surface of B. cereus ATCC 4342, CDC13100, or CDC13140, the three strains that are susceptible to γ infection but not to Wip1 infection. As expected, they were also unable to bind B. cereus strains ATCC 10987 and NRL 569, as well as B. thuringiensis strains HD1 and HD73, none of which can be infected by either γ or Wip1 phage. His-p23 and the His-p23–p24 complex bound positively to only B. anthracis Sterne and ΔSterne and B. cereus CDC32805, the only B. cereus strain in our host range analysis that supported Wip1 replication (see Table S1 in the supplemental material). These findings suggest that His-p23 does not bind to just any Gram-positive bacterial surface but binds very specifically to the bacterial hosts that support Wip1 infection, including B. anthracis ΔSterne.
Fig 6.
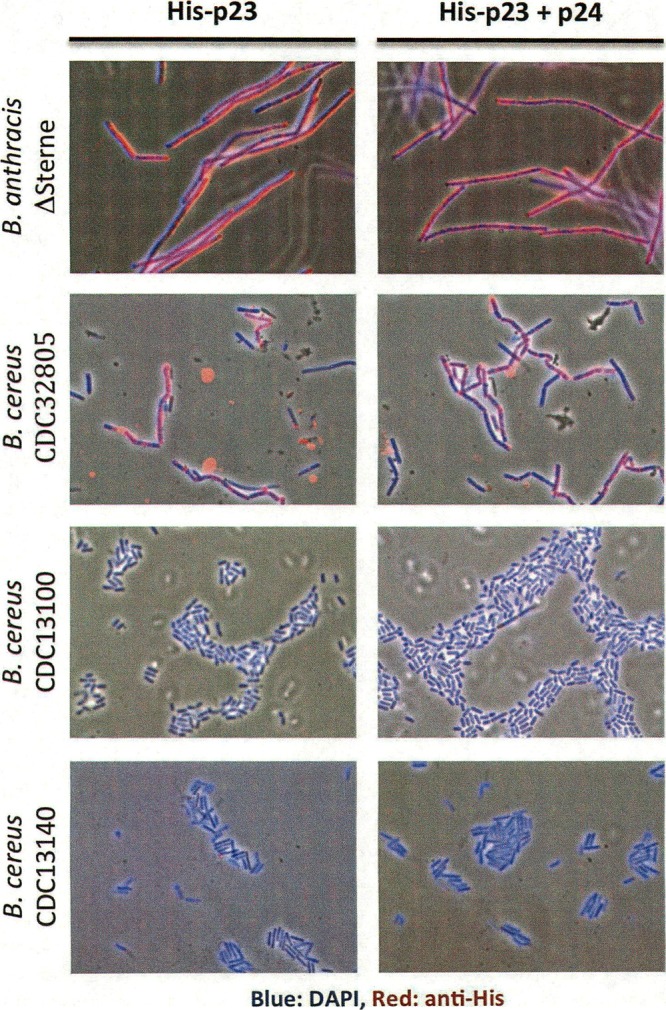
Indirect immunofluorescence microscopy of His-p23 and the His-p23–p24 complex. ×1,000 magnification.
DISCUSSION
In the present study, we characterized the Wip1 phage and its genome to develop the tools to identify Wip1 gene product 23 as a receptor binding protein. Wip1 tropism was previously shown to be highly specific to B. anthracis. Here, we determined by adsorption assays that specificity to B. anthracis is mediated by Wip1's receptor binding. Indeed, receptor binding protein p23 was demonstrated to bind very specifically to bacterial strains that correspond to Wip1's narrow host range.
The identification of Wip1 p23 as a receptor binding protein is noteworthy because it is a unique protein with no homology to any other known proteins. It is particularly surprising because ORF23 shares no sequence identity with AP50, a tectivirus with a similar host range that is also highly specific to B. anthracis. Genomic analysis showed that the overall Wip1 genome shows significant ORF size, sequence, and organization similarities to the AP50 genome. In fact, the genes neighboring ORF23 display this conservation. ORF22 (291 residues) shares 64% sequence identity with AP50 ORF27 (304 residues), and ORF24 (118 residues) shares 51% sequence identity with AP50 ORF29 (118 residues).
An interesting genomic observation is that AP50 ORF28, which is located in the genomic position corresponding to that of Wip1 ORF23, is the gene that harbors one of two sequence mutations that differentiate isolate AP50t, which produces turbid plaques, from isolate AP50c, which produces clear plaques. Generally, clear plaques are formed when the host is completely susceptible to the phage while turbid plaques are formed if the host is partially resistant to the phage (e.g., if 10% of the cells survive infection). Furthermore, AP50 ORF28 and Wip1 ORF23 are located in a highly variable region of their respective genomes. On the one hand, it is not surprising that the Eip1 receptor-binding domain ORF23 exhibits sequence diversity. On the other hand, it is rather unusual that two closely related tectiviruses with similar host ranges have evolved uniquely different receptor binding proteins.
Another notable observation is that the His-p23–p24 complex exhibited higher competitive inhibition than His-p23 alone, suggesting that p24 complemented or enhanced His-p23 binding activity. This enhancement could result from simply protecting His-p23 proteins from degradation. However, we were careful to use fresh protein stocks in all assays. It is also possible that p24 plays a secondary but complementary role in Wip1 binding. The adsorption of phage to the Gram-positive bacterial surface has been suggested to occur in two stages. The first step involves reversible binding to general recognition molecules in the cell wall and is followed by a subsequent irreversible step involving a more specific factor (29, 30). In fact, the seahorse-like structure of PRD1 receptor binding protein P2 consists of multiple domains with different purported functions. In one P2 model, the fin-shaped domain is proposed to make initial contacts by scanning the host surface in order to bring the head domain closer to its receptor (18). It is possible that Wip1 p24 is such a spike complex domain with nonspecific, reversible surface-scanning properties.
Polyclonal antibodies against His-p23 were also able to inactivate Wip1 binding activity. It should be noted, however, that the neutralizing effect of anti-His-p23 serum on Wip1 activity was much weaker than that of anti-gp28 or anti-gp29 serum on Bam35 activity (19). At a mere 20× dilution, anti-His-p23 antibody inactivation measured <20%. In contrast, anti-gp28 and anti-29 antibodies did not demonstrate inactivation rates of <20% until they reached dilutions of 5,000× and 100,000×, respectively. One explanation for this difference is the possibility that p23 is a poor antigen that generates a weak immunogenic response. However, an indirect ELISA determined that the polyclonal antisera against His-p23 exhibited reasonable immunogenic strength (absorbance reading of 1 at a 1:2,000 dilution).
The narrow host infectivity-and-adsorption range of Wip1 indicates that its receptor is unique to the exposed surface of B. anthracis and B. cereus CDC32805 in a manner that is accessible to phage. Indirect immunofluorescence microscopy demonstrated that p23 and the p23-p24 complex detect and label B. anthracis with a specificity that seems to match its narrow host range. This activity makes both the Wip1 phage and its receptor molecules potentially useful diagnostic tools for B. anthracis with better specificity than the reagents currently being used.
Supplementary Material
ACKNOWLEDGMENTS
This study was supported by USPHS grant AI057472 to V.A.F.
We thank Erec Stebbins for the Duet Vector.
Footnotes
Published ahead of print 26 July 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JB.00655-13.
REFERENCES
- 1.Schuch R, Pelzek A, Fischetti VA. 2010. Prevalence of Bacillus anthracis-like organisms and bacteriophages in the intestinal tract of the earthworm Eisenia fetida. Appl. Environ. Microbiol. 76:2286–2294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schuch R, Fischetti VA. 2009. The secret life of the anthrax agent Bacillus anthracis: bacteriophage-mediated ecological adaptations. PLoS One 4:e6532. 10.1371/journal.pone.0006532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Merckel MC, Huiskonen JT, Bamford DH, Goldman A, Tuma R. 2005. The structure of the bacteriophage PRD1 spike sheds light on the evolution of viral capsid architecture. Mol. Cell 18:161–170 [DOI] [PubMed] [Google Scholar]
- 4.Bamford D. 2002. Evolution of viral structure. Theor. Popul. Biol. 61:461–470 [DOI] [PubMed] [Google Scholar]
- 5.Olsen RH, Siak J, Gray RH. 1974. Characteristics of PRD1, a plasmid-dependent broad host range DNA bacteriophage. J. Virol. 14:689–699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ackermann HW, Roy R, Martin M, Murthy MR, Smirnoff WA. 1978. Partial characterization of a cubic Bacillus phage. Can. J. Microbiol. 24:986–993 [DOI] [PubMed] [Google Scholar]
- 7.Verheust C, Fornelos N, Mahillon J. 2005. GIL16, a new Gram-positive tectiviral phage related to the Bacillus thuringiensis GIL01 and the Bacillus cereus pBClin15 elements. J. Bacteriol. 187:1966–1973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nagy E, Prágai B, Ivánovics G. 1976. Characteristics of phage AP50, an RNA phage containing phospholipids. J. Gen. Virol. 32:129–132 [DOI] [PubMed] [Google Scholar]
- 9.Abshire TG, Brown JE, Ezzell JW. 2005. Production and validation of the use of gamma phage for identification of Bacillus anthracis. J. Clin. Microbiol. 43:4780–4788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schuch R, Fischetti VA. 2006. Detailed genomic analysis of the Wbeta and gamma phages infecting Bacillus anthracis: implications for evolution of environmental fitness and antibiotic resistance. J. Bacteriol. 188:3037–3051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schuch R, Nelson D, Fischetti VA. 2002. A bacteriolytic agent that detects and kills Bacillus anthracis. Nature 418:884–889 [DOI] [PubMed] [Google Scholar]
- 12.Sozhamannan S, McKinstry M, Lentz SM, Jalasvouri M, McAfee F, Smith A, Dabbs J, Ackermann HW, Bamford JKH, Mateczun A, Read TD. 2008. Molecular characterization of a variant of Bacillus anthracis-specific phage AP50 with improved bacteriolytic activity. Appl. Environ. Microbiol. 74:6792–6796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Haywood AM. 1994. Virus receptors: binding, adhesion strengthening, and changes in viral structure. J. Virol. 68:1–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sokolova A, Malfois M, Caldentey J, Svergun DI, Koch MH, Bamford DH, Tuma R. 2001. Solution structure of bacteriophage PRD1 vertex complex. J. Biol. Chem. 276:46187–46195 [DOI] [PubMed] [Google Scholar]
- 15.Bamford JK, Bamford DH. 2000. A new mutant class, made by targeted mutagenesis, of phage PRD1 reveals that protein P5 connects the receptor binding protein to the vertex. J. Virol. 74:7781–7786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Huiskonen JT, Manole V, Butcher SJ. 2007. Tale of two spikes in bacteriophage PRD1. Proc. Natl. Acad. Sci. U. S. A. 104:6666–6671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mindich L, Bamford D, McGraw T, Mackenzie G. 1982. Assembly of bacteriophage PRD1: particle formation with wild-type and mutant viruses. J. Virol. 44:1021–1030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Xu LB SD, Butcher SJ, Bamford DH, Burnett RM. 2003. The receptor binding protein P2 of PRD1, a virus targeting antibiotic-resistant bacteria, has a novel fold suggesting multiple functions. Structure 11:309–322 [DOI] [PubMed] [Google Scholar]
- 19.Gaidelyte A, Cvirkaite-Krupovic V, Daugelavicius R, Bamford JK, Bamford DH. 2006. The entry mechanism of membrane-containing phage Bam35 infecting Bacillus thuringiensis. J. Bacteriol. 188:5925–5934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Laurinmäki PA, Huiskonen JT, Bamford DH, Butcher SJ. 2005. Membrane proteins modulate the bilayer curvature in the bacterial virus Bam35. Structure 13:1819–1828 [DOI] [PubMed] [Google Scholar]
- 21.Ravantti JJG A, Bamford DH, Bamford JK. 2003. Comparative analysis of bacterial viruses Bam35, infecting a gram-positive host, and PRD1, infecting gram-negative hosts, demonstrates a viral lineage. Virology 313:401–414 [DOI] [PubMed] [Google Scholar]
- 22.Raz A, Fischetti VA. 2008. Sortase A localizes to distinct foci on the Streptococcus pyogenes membrane. Proc. Natl. Acad. Sci. U. S. A. 105:18549–18554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rydman PS, Caldentey J, Butcher SJ, Fuller SD, Rutten T, Bamford DH. 1999. Bacteriophage PRD1 contains a labile receptor-binding structure at each vertex. J. Mol. Biol. 291:575–587 [DOI] [PubMed] [Google Scholar]
- 24.Grahn AM, Caldentey J, Bamford JK, Bamford DH. 1999. Stable packaging of phage PRD1 DNA requires adsorption protein P2, which binds to the IncP plasmid-Encoded conjugative transfer complex. J. Bacteriol. 181:6689–6696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Daugelavicius R, Bamford JK, Bamford DH. 1997. Changes in host cell energetics in response to bacteriophage PRD1 DNA entry. J. Bacteriol. 179:5203–5210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Grahn AM, Daugelavicius R, Bamford DH. 2002. Sequential model of phage PRD1 DNA delivery: active involvement of the viral membrane. Mol. Microbiol. 46:1199–1209 [DOI] [PubMed] [Google Scholar]
- 27.Mignot T, Mesnage S, Couture-Tosi E, Mock M, Fouet A. 2002. Developmental switch of S-layer protein synthesis in Bacillus anthracis. Mol. Microbiol. 43:1615–1627 [DOI] [PubMed] [Google Scholar]
- 28.Bishop-Lilly KA, Plaut RD, Chen PE, Akmal A, Willner KM, Butani A, Dorsey S, Mokashi V, Mateczun AJ, Chapman C, George M, Luu T, Read TD, Calendar R, Stibitz S, Sozhamannan S. 2012. Whole genome sequencing of phage resistant Bacillus anthracis mutants reveals an essential role for cell surface anchoring protein CsaB in phage AP50c adsorption. Virol. J. 9:246. 10.1186/1743-422X-9-246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jacobson ED, Lind OE. 1977. Adsorption of bacteriophages phi 29 and 22a to protoplasts of Bacillus subtilis. J. Virol. 21:1223–1227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Monteville MR, Ardestani B, Geller BL. 1994. Lactococcal bacteriophages require a host cell wall carbohydrate and a plasma membrane protein for adsorption and ejection of DNA. Appl. Environ. Microbiol. 60:3204–3211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Stromsten NJ, Benson SD, Burnett RM, Bamford DH, Bamford JK. 2003. The Bacillus thuringiensis linear double-stranded DNA phage Bam35, which is highly similar to the Bacillus cereus linear plasmid pBClin15, has a prophage state. J. Bacteriol. 185:6985–6989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mindich L, Bamford D, McGraw T, Mackenzie G. 1982. Assembly of bacteriophage PRD1: particle formation with wild-type and mutant viruses. J. Virol. 44:1021–1030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rydman PS, Bamford JK, Bamford DH. 2001. A minor capsid protein P30 is essential for bacteriophage PRD1 capsid assembly. J. Mol. Biol. 313:785–795 [DOI] [PubMed] [Google Scholar]
- 34.Bamford JK, Hanninen AL, Pakula TM, Ojala PM, Kalkkinen N, Frilander M, Bamford DH. 1991. Genome organization of membrane-containing bacteriophage PRD1. Virology 183:658–676 [DOI] [PubMed] [Google Scholar]
- 35.Benson SD, Bamford JK, Bamford DH, Burnett RM. 2002. The X-ray crystal structure of P3, the major coat protein of the lipid-containing bacteriophage PRD1, at 1.65 A resolution. Acta. Crystallogr. D Biol. Crystallogr. 58:39–59 [DOI] [PubMed] [Google Scholar]
- 36.Bamford JK, Bamford DH. 1991. Large-scale purification of membrane-containing bacteriophage PRD1 and its subviral particles. Virology 181:348–352 [DOI] [PubMed] [Google Scholar]
- 37.Bamford JK, Bamford DH. 1990. Capsomer proteins of bacteriophage PRD1, a bacterial virus with a membrane. Virology 177:445–451 [DOI] [PubMed] [Google Scholar]
- 38.Bamford JKH, Cockburn JJB, Diprose J, Grimes JM, Sutton G, Stuart DI, Bamford DH. 2002. Diffraction quality crystals of PRD1, a 66-MDa dsDNA virus with an internal membrane. J. Struct. Biol. 139:103–112 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.