

Abstract
Penicillin binding proteins (PBPs) are responsible for synthesizing and modifying the bacterial cell wall, and in Escherichia coli the loss of several nonessential low-molecular-weight PBPs gives rise to abnormalities in cell shape and division. To determine whether these proteins help connect the flagellar basal body to the peptidoglycan wall, we surveyed a set of PBP mutants and found that motility in an agar migration assay was compromised by the simultaneous absence of four enzymes: PBP4, PBP5, PBP7, and AmpH. A wild-type copy of any one of these restored migration, and complementation depended on the integrity of the PBP active-site serine. However, the migration defect was caused by the absence of flagella instead of improper flagellar assembly. Migration was restored if the flhDC genes were overexpressed or if the rcsB or cpxR genes were deleted. Thus, migration was inhibited because the Rcs and Cpx stress response systems were induced in the absence of these four specific PBPs. Furthermore, in this situation Rcs induction depended on the presence of CpxR. The results imply that small changes in peptidoglycan structure are sufficient to activate these stress responses, suggesting that a specific cell wall fragment may be the signal being sensed. The fact that four PBPs must be inactivated may explain why large perturbations to the envelope are required to induce stress responses.
INTRODUCTION
The peptidoglycan wall is vital to the bacterial cell. Situated between the inner and outer membranes of Gram-negative bacteria, this covalently linked scaffold of glycan chains and short peptides maintains cell shape and resists osmotic lysis (1). Peptidoglycan is synthesized and modified by a set of periplasmic penicillin binding proteins (PBPs), including the high-molecular-weight transglycosylase-transpeptidase PBPs, which polymerize and cross-link the glycan chains, and the low-molecular-weight (LMW) PBPs, which modify existing chains (2). Among the latter, the LMW PBPs 4 and 7 and AmpH are primarily endopeptidases that cleave peptide side chains and disconnect the glycan polymers (2), and PBPs 5 and 6 and DacD are d,d-carboxypeptidases that remove the terminal d-alanine from pentapeptide side chains (2). In Escherichia coli, the loss of several of these nonessential LMW PBPs affects the symmetry of cell division, slows daughter cell separation, and gives rise to abnormally shaped cells (3–5), but other than this, few physiological effects of the LMW PBPs are known. These proteins may modify peptidoglycan to accommodate cell growth or to allow large macromolecular structures, such as secretion systems or flagella, to assemble onto or across the cell wall (1, 6).
Because the wall is so important, it is not surprising that bacteria monitor its integrity and stability. In E. coli, the RcsBCD, CpxAR, BaeSR, and EnvZ-OmpR signal transduction systems are generally believed to sense and respond to stresses that damage or alter the cell envelope (7–11). For example, the Rcs multicomponent phosphorelay responds to stimuli such as osmotic shock and β-lactam exposure (10, 11) and modulates gene expression when one or more signals are sensed by the outer membrane lipoprotein RcsF or by the inner membrane sensor kinase RcsC (11–13). Upon activation, RcsC autophosphorylates a conserved histidine residue in its cytoplasmic domain; the phosphate moves to a neighboring aspartate, then to a histidine residue on the inner membrane protein RcsD, and finally to an aspartate residue on RcsB, the cytoplasmic response regulator (13, 14). A homodimer of phosphorylated RcsB or an RcsB-RcsA heterodimer modulates the expression of target genes, including those that encode the colanic acid synthesis pathway, the genes rprA, osmC, and ftsZ, and about 180 other genes (10). The Rcs phosphorelay also negatively regulates transcription of flhDC, the master regulator of flagella synthesis, thereby turning off motility when cells come under stress (15, 16). The CpxAR two-component system consists of the sensor kinase CpxA, the response regulator CpxR, and the periplasmic inhibitor CpxP (17, 18). This response is activated by surface attachment, misfolded pilus subunits, and changes in pH and osmolarity, among other stresses (18), and regulates the expression of bundle forming pili in enteropathogenic E. coli (19), the production of P pili in uropathogenic E. coli (20), and the attachment, invasion, and intracellular growth of Salmonella enterica serovar Typhimurium (21). Although the phosphotransfer pathways in these systems are well characterized, the molecular identities of the signals and the mechanisms by which they are sensed remain unknown.
Here, we hypothesized that the LMW PBPs might play a role in anchoring the flagellar basal body to the peptidoglycan cell wall, which is required for E. coli to be fully motile (22, 23). In fact, we found that the simultaneous removal of four specific PBPs (PBPs 4, 5, and 7 and AmpH) did inhibit bacterial motility, but this effect was not due to mis-assembly of flagella. Instead, these mutations induced the Rcs and Cpx stress responses, and the Rcs response inhibits motility by downregulating transcription of flagellar genes. The enzymatic activity of any one of these four PBPs was sufficient to rescue motility, strongly suggesting that these two stress responses respond to minor, possibly specific changes in peptidoglycan structure.
MATERIALS AND METHODS
Bacteria, plasmids, growth conditions, and migration assays.
E. coli strains and plasmids are listed in Table 1. The parental strain was E. coli CS109 (W1485F− rpoS rph-1 acnA) (24). Deletions marked with antibiotic resistance cassettes flanked by res sites were introduced by P1 transduction and cured by using the RP4 ParA resolvase (3). Deletions marked with antibiotic resistance cassettes flanked by FRT sites were introduced by P1 transduction and cured by using the FLP helper plasmid pCP20 (25). Bacteria were grown at 30 or 37°C in Luria-Bertani (LB) broth or agar (Difco) or at 30°C in tryptone broth (1% tryptone plus 0.25% NaCl) (Difco) or agar (tryptone broth plus 0.26% agar). When needed, kanamycin (50 μg/ml), chloramphenicol (20 μg/ml), or arabinose (0.05%) was added. For migration assays, bacteria were grown in LB broth overnight at 30°C, and 1.5 μl of this culture was spotted onto the surface of a migration plate (tryptone agar) and incubated at 30°C for 10 or 24 h.
Table 1.
E. coli strains and plasmids
| Strain or plasmid | Descriptiona | Source or reference |
|---|---|---|
| E. coli strains | ||
| CS109 | W1485 rph-1 katF (rpoS) | 3 |
| CS204-1 | CS109 dacA::res pbpG::res | 3 |
| CS206-3 | CS109 ampH::res pbpG::res | 3 |
| CS315-1 | CS109 dacA::res pbpG::res dacB::res | 3 |
| CS317-3 | CS109 ampH::res pbpG::res dacB::res | 3 |
| CS326-3 | CS109 ampH::res dacA::res dacB::res | 3 |
| CS345-3 | CS109 ampH::res dacA::res pbpG::res | 3 |
| CS448-3 | CS109 dacB::res dacA::res pbpG::res ampH::res | 3 |
| KE2 | CS109 ampG::kan | This study |
| KE8 | CS109 rcsB::tet | This study |
| KE9 | CS448-3 rcsB::tet | This study |
| KE11 | CS109 rcsF::cam | This study |
| KE12 | CS448-3 rcsF::cam | This study |
| KE14 | CS109 rcsC52::tet | This study |
| KE15 | CS448-3 rcsC52::tet | This study |
| KE18 | CS109 envZ::kan | This study |
| KE19 | CS109 ompR::kan | This study |
| KE20 | CS448-3 envZ::kan | This study |
| KE21 | CS448-3 ompR::kan | This study |
| KE36 | CS109 cpxR::kan | This study |
| KE37 | CS448-3 cpxR::kan | This study |
| KE39 | CS448-3 ampG::kan | This study |
| KE41 | CS109 fliC::kan | This study |
| KE42 | CS448-3 fliC::kan | This study |
| SKCS49-1 | CS109 flhA::kan | This study |
| Plasmids | ||
| pCP20 | cI857 repA(Ts) PR::flp, Ampr Camr | 25 |
| pDKR1 | colE1 PrprA::sfgfp; Kanr | 52 |
| pFlhDC | PBAD-flhDC; Camr | R. Harshey |
| pJW1 | pNLP10 with PcpxP::luxCDABE; Kanr | 33 |
| pKD46 | Lambda Red+ (Ampr pSC101 oriTS) | 25 |
| pLCM1 | Plac-ampH lacIq; Kanr | 53 |
| pLP8 | Plac lacIq, Kanr, pMLB1113 background | 54 |
| pLP405 | PBAD-dacB(S62A); Kanr | P. Potluri |
| pLP514 | Plac-dacA(S44G) lacIq; Kanr | 54 |
| pLP515 | Plac-dacA lacIq; Kanr | 54 |
| pLP706 | PBAD-pbpG(S67A); Kanr | P. Potluri |
| pLP18 | PBAD, Kanr | 55 |
| pLPKC403 | PBAD-dacB, Kanr | 55 |
| pLPKC704 | PBAD-pbpG, Kanr | 55 |
| pMEL1 | Plac-ampH(S87A) lacIq; Kanr | 53 |
| pVS182 | flhD::lacZ in pRS551; Ampr | 56 |
Camr, chloramphenicol resistance; Ampr, ampicillin resistance; Kanr, kanamycin resistance.
Enzyme assays and muropeptide analysis.
The β-galactosidase activity was assayed by growing bacteria in LB broth overnight at 30°C, diluting the culture 1:100 into 1% tryptone, and growing the cells until the optical density at 600 nm (OD600) of the culture reached 0.5 to 0.6. The enzyme activity was determined according to the procedure of Miller (26). The amounts of green fluorescent protein (GFP) and luciferase were determined by growing bacteria in LB broth as described above until the OD600 reached 0.5 to 0.6 and then transferring 100 μl (in triplicate) to wells of a 96-well microplate (Costar, catalog no. 3603; Corning, Inc.). Activity was expressed as the ratio of GFP fluorescence, 485 nm (excitation) and 528 nm (emission), or luminescence counts per second (cps) to OD630. For assays of cells from migration agar, a plug of cells and agar from 10- and 24-h migration plates was weighed and then sheared by passage through a 200-μl pipette tip. A 150-μl portion of 1% tryptone was added to the mixture, 100 μl of the sample was transferred in triplicate to a 96-well microplate, and the GFP fluorescence was measured as described above. The CFU were determined to ensure that equal numbers of cells were assayed.
Peptidoglycan was prepared and analyzed as described previously (27). Strains were grown overnight in LB medium at 30°C, diluted 1:100 into 150 ml of 1% tryptone, and grown at 30°C to an OD600 of 0.5 to 0.6. The cells were pelleted at 7,400 × g for 10 min at 4°C (Thermo Scientific Sorvall Legend XTR Centrifuge, F15 Fiberlite rotor). The cells were resuspended in 3 ml of LB medium and added dropwise to 6 ml of boiling 6% sodium dodecyl sulfate (SDS) in glass culture tubes. The samples were boiled and stirred for 1 h, then stirred at room temperature overnight. To isolate the peptidoglycan, the samples were boiled and stirred for an additional hour and pelleted by centrifugation at 65,000 rpm for 10 min at 30°C in an ultracentrifuge (Beckman-Optima TLX, TLA110 rotor). The pellet was resuspended in 3 ml of distilled H2O to wash the peptidoglycan. The wash step was repeated three to four times, and the pellet was resuspended in 1.5 ml of 10 mM Tris-HCl (pH 7.0), after which α-amylase was added to a final concentration of 100 μg/ml, and the sample was incubated at 37°C for 2 h. The samples were incubated with Pronase (100 µg/ml) at 60°C for 2 h and then boiled for 30 min in 1% SDS to destroy the Pronase activity. The samples were pelleted, washed three to four times with distilled H2O to remove the SDS, resuspended in 400 µl of NaPO4 (50 mM) buffer containing N-acetylmuramidase SG (catalog no. A0534; US Biologicals) (40 µg/ml), and incubated overnight at 37°C. The samples were then boiled for 5 min to destroy the muramidase activity and centrifuged at top speed for 10 min in a table-top microcentrifuge. The supernatant was transferred to a clean tube and vacuum dried for 2.5 h.
Western blotting.
Blots of whole cells were prepared as described previously (28). Cultures were grown overnight at 30°C in LB medium, diluted 1:100 into tryptone broth, and grown to an OD600 of 0.5 to 0.6, and 500 μl of the cells was pelleted by centrifugation and resuspended in 200 μl of double-distilled H2O. Next, 5 μl of this suspension was spotted onto a polyvinylidene difluoride (PVDF) membrane prewetted with methanol and allowed to absorb overnight at room temperature. Primary anti-FliC antibody (catalogue no. ab93713; Abcam, Cambridge, MA) was used at a 1:10,000 dilution. Secondary α-rabbit-HRP was used at a 1:25,000 dilution. Standard procedures were followed for Western blotting (29).
Flagellum staining.
Flagella were stained as described previously (30), with some modifications. All wash steps were carried out with extreme care, allowing up to 10 min for each wash. All centrifugation steps were 2,000 × g for 10 min in an Eppendorf 5415D tabletop centrifuge. Overnight cultures were grown in LB medium at 30°C, diluted 1:100 into 1% tryptone broth, and incubated until the OD600 reached 0.5 to 0.6. Motility was confirmed by microscopy before proceeding. The cell pellet was washed three times in equal volumes of phosphate buffer (pH 7.0) at room temperature, washed once in equal volumes of phosphate buffer (pH 7.5), and resuspended in 100 μl of Alexa Fluor 488 carboxylic acid succinimidyl ester (AF488, catalogue no. A20000; Invitrogen, Carlsbad, CA), which had been dissolved in phosphate buffer (pH 7.5). Then, 20 μl of 1 M NaHCO3 was added, and the dye-cell suspension was incubated at room temperature in the dark for 90 min, with gentle mixing on a rotator (Denville Scientific, South Plainfield, NJ). The stain was removed by centrifugation, and the cells were washed twice in 500 μl of phosphate buffer (pH 7.0) containing 0.002% Tween 20. This wash was carried out by gently swirling the liquid back and forth over the pellet. The pellet was resuspended in 500 μl of phosphate buffer (pH 7.0), containing 0.002% Tween 20, spotted onto a clean glass slide, and imaged using a wide-field epifluorescent Zeiss Axio Imager.Z1 microscope fitted with a 100× differential interference contrast objective (1.45 NA). Images were acquired with a Zeiss Axiocam MRm cold charge-coupled device camera, using appropriate filter cubes for fluorescence image acquisition.
RESULTS
Loss of four specific LMW PBPs inhibits motility.
We hypothesized that one or more of the LMW PBPs helps attach the flagellar machinery to the peptidoglycan cell wall. To test this, we screened 60 E. coli PBP mutants for defects in migration, expecting to find that nonmigratory strains had defects in flagellar assembly. Bacteria were inoculated into migration agar and incubated at 30°C for 10 h, and the diameters of their migration rings were measured. Of the 60, 8 did not migrate (Table 2), producing no ring extending beyond the original inoculum. All mutants lacking three or fewer PBPs migrated, as did all but one of those tested that lacked four (Table 2). The exception in this latter group was strain CS448-3 (Fig. 1A, row 2), from which the genes encoding PBP4, PBP5, PBP7, and AmpH had been deleted. Furthermore, from among strains lacking five to seven LMW PBPs, the only nonmigratory mutants were those from which these four PBPs were missing (Table 2). Tellingly, even strains lacking six LMW PBPs migrated as long as at least one of these four PBPs was intact (Table 2, strains with “CS6xx” prefixes). In no case did strains have growth defects that could account for these differences in migration (not shown). The absence of all four of these PBPs was necessary for the loss of motility, because mutants lacking any three of these four PBPs migrated (Fig. 1A, rows 3 to 6). As further verification, we complemented strain CS448-3 with plasmids carrying wild-type versions of each of these genes. Indeed, the presence of any one of these PBPs restored motility (Fig. 1B, rows 2, 4, 6, and 8). Finally, we determined whether loss of migration was caused by the absence of the proteins themselves or by the absence of their enzymatic activities. CS448-3 did not migrate when complemented with inactive versions of PBP 4, 5, or 7 or AmpH (Fig. 1B, rows 3, 5, 7, and 9) in which the active-site serine had been replaced with alanine or glycine. Thus, the enzymatic activity of these PBPs was responsible for the phenotype, not the physical presence or absence of the proteins. In sum, the results show that removing this one particular combination of four LMW PBPs was necessary and sufficient for inhibiting E. coli migration.
Table 2.
Migration of selected PBP mutants
| Strain | PBP(s) deleteda | Migrationb |
|---|---|---|
| BMCS20K-1 | 1b | + |
| BMCS04-1K | 1a | + |
| CS9-19 | 7 | + |
| CS12-7 | 5 | + |
| CS13-2 | 1a | + |
| CS14-2 | C | + |
| CS15-3 | H | + |
| CS16-1 | 1b | + |
| CS17-1 | 6 | + |
| CS206-3 | 7, H | + |
| CS211-2 | 5, 6 | + |
| CS212-3 | 6, H | + |
| CS221-3 | 4, H | + |
| CS224-2 | 1b, 5 | + |
| CS225-1 | 1b, 6 | + |
| CS226-3 | 1b, H | + |
| CS230-1K | 1a, H | + |
| CS236-1K | 4, dacD | + |
| CS315-1 | 4, 5, 7 | + |
| CS317-3 | 4, 7, H | + |
| CS320-1 | 4, C, H | + |
| CS322-1 | 4, 5, 6 | + |
| CS323-3 | 4, 6, H | + |
| CS326-3 | 4, 5, H | + |
| CS331-1 | 5, 6, 7 | + |
| CS332-3 | 6, 7, H | + |
| CS336-3 | 5, 6, H | + |
| CS345-3 | 5, 7, H | + |
| CS349-1 | 5, C, H | + |
| CS389-1K | 4, H, dacD | + |
| CS440-3 | 4, 6, 7, H | + |
| CS441-1 | 5, 6, C, H | + |
| CS442-3 | 4, 5, 6, H | + |
| CS443-3 | 5, 6, 7, H | + |
| CS444-1 | 5, 6, C, H | + |
| CS445-1 | 4, 5, 6, C | + |
| CS446-1 | 4, 5, 6, 7 | + |
| CS447-1 | 4, 6, 7, C | + |
| CS448-3 | 4, 5, 7, H | − |
| CS449-2 | 4, 5, 7, C | + |
| CS450-1 | 5, 6, 7, C | + |
| CS533-1 | 4, 5, 6, 7, C | + |
| CS534-1 | 4, 5, 7, C, H | − |
| CS535-1 | 4, 6, 7, C, H | + |
| CS536-1 | 5, 6, 7, C, H | + |
| CS539-1K | 4, 5, 6, C, H | + |
| CS607-4 | 1b, 4, 5, 6, 7, H | − |
| CS608-1 | 1b, 4, 5, 6, 7, C | + |
| CS609-1 | 1b, 4, 5, 7, C, H | − |
| CS610-1 | 1b, 4, 6, 7, C, H | + |
| CS611-1 | 1b, 5, 6, 7, C, H | + |
| CS612-1 | 4, 5, 6, 7, C, H | − |
| CS613-1 | 1b, 4, 5, 6, C, H | + |
| CS615-1K | 1a, 4, 5, 6, 7, C | + |
| CS616-1K | 1a, 4, 5, 7, C, H | − |
| CS617-1K | 1a, 4, 6, 7, C, H | + |
| CS618-1K | 1a, 5, 6, 7, C, H | + |
| CS619-1K | 1a, 4, 5, 6, C, H | + |
| CS702-1 | 1b, 4, 5, 6, 7, C, H | − |
| CS703-1K | 1a, 4, 5, 6, 7, C, H | − |
“C” and “H” refer to AmpC and AmpH, respectively.
“+” indicates the strain was as motile as the wild type after 10 h, and “−” indicates the strain was not motile after 10 h.
Fig 1.
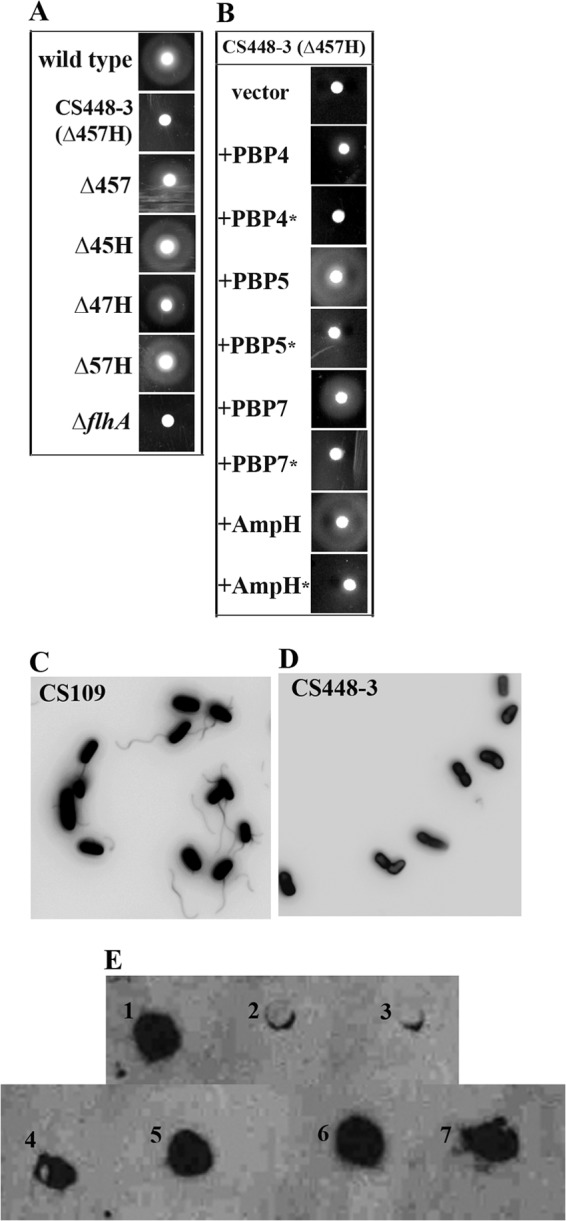
Loss of four specific PBPs inhibits migration by inhibiting flagellum production. The migration of each strain was assayed over 10 h on migration agar. (A) E. coli strain CS448-3, lacking PBPs 4, 5, and 7 and AmpH (Δ457H, row 2), is nonmotile after 10 h. Strains lacking any three of these PBPs remained motile: Δ457 (CS315-1), Δ45H (CS326-3), Δ47H (CS317-3), and Δ57H (CS345-3). A strain unable to make flagella, an ΔflhA mutant strain (SKCS49-1), served as a negative control. (B) Complementation with any one wild-type PBP gene restores the ability to migrate to E. coli CS448-3. CS448-3 was transformed with the following plasmids (the wild-type PBP that was expressed is noted in parentheses): pLPKC403 (PBP4, row 2), pLP515 (PBP5, row 4), pLPKC704 (PBP7, row 6), and pLCM1 (AmpH, row 8). No complementation occurred when PBPs with mutated active sites were expressed: pLP405 (PBP4*, row 3), pLP514 (PBP5*, row 5), pLP706 (PBP7*, row 7), and pMEL1 (AmpH*, row 9). CS448-3 transformed with plasmid vectors was nonmotile: CS448-3 pLP18 (top row) and CS448-3 pLP8 (data not shown). The parental strain containing plasmid vectors was motile (data not shown). (C and D) PBP loss inhibits flagellum production. E. coli CS109 (C) and CS448-3 (D) were stained with Alexa Fluor 488. Wild-type cells displayed at least one flagellum per cell (C), whereas CS448-3 lacked flagella (D). (E) Whole cells were spotted onto a PVDF membrane prewetted in methanol, and the FliC flagellar protein was detected by Western blotting. E1, CS109; E2, CS448-3; E3, CS109ΔflhA (SKCS49-1); E4, CS109ΔrcsB (KE8); E5, CS448-3ΔrcsB (KE9); E6, CS109/pFlhDC; E7, CS448-3/pFlhDC.
PBP loss antagonizes flagellum assembly.
Several scenarios could account for the inhibition of migration in these mutants: flagella might not be produced at all, the basal body might be so weakly anchored to peptidoglycan that the flagella become detached, or the flagella could be attached but not rotate properly. As a first step in distinguishing among these possibilities, we stained flagella with AF488 to determine whether flagella were present. The parent, E. coli CS109, had flagella (Fig. 1C), but the PBP mutant CS448-3 did not (Fig. 1D). To verify that flagella were not sheared off during the centrifugation steps, cells were spotted directly onto a PVDF membrane and probed with anti-FliC antibody. The parental strain produced cell surface FliC (Fig. 1, E1), but CS448-3 did not (Fig. 1, E2). Furthermore, flagellar proteins were not simply trapped inside CS448-3 cells, because Western blots of whole-cell lysates revealed that CS448-3 did not contain FliC (data not shown). We concluded that strain CS448-3 did not migrate simply because it did not make flagellar proteins.
PBP loss activates the Rcs phosphorelay.
The simplest explanation for the lack of flagella was that transcription or translation of these genes was inhibited in CS448-3. The Rcs phosphorelay negatively regulates motility by inhibiting transcription of flhDC, the master transcriptional activator of flagella genes (15). Indeed, expression of a flhD::lacZ reporter gene was decreased 4-fold in CS448-3 relative to the wild type (not shown). To determine whether the Rcs phosphorelay inhibited motility and thus migration, we removed the response regulator RcsB from CS448-3 (Fig. 2, A5) and found that this mutant was as capable of migration as the wild type at 10 h (Fig. 2, A1). Removing RcsC, the phosphorelay sensor histidine kinase, also restored the ability to migrate to CS448-3 at 10 h (Fig. 2, A7). If the Rcs system was inhibiting the transcription of flhDC in CS448-3, then overexpressing flhDC should restore migration. In fact, ectopic expression of flhDC in the parent strain produced a much enlarged migration ring (Fig. 2, A8), and ectopic expression of flhDC in CS448-3 also restored the ability to migrate (Fig. 2, A9). This latter ring was greater than that exhibited by the parental strain without an ectopic source of FlhDC (Fig. 2, A1). At the cellular level, removing RcsB (Fig. 1, E5) or overexpressing flhDC (Fig. 1, E7) restored cell surface flagellin to CS448-3, and these cells were also hyperflagellated (Fig. 2B, lower panel), which is the likely explanation for the abnormally large migration rings in these strains (15). Finally, transcription of the rprA gene is positively regulated by RcsB and is a surrogate for activation of the Rcs pathway (31). Expression of a PrprA::sfgfp reporter gene was significantly greater in CS448-3 than in the parental strain, and deleting RcsB diminished reporter expression (Fig. 2C). Reporter expression in CS315 and additional PBP mutants was intermediate between wild type and CS448-3 (Fig. 2D), suggesting that Rcs was moderately activated in these strains, although migration was not affected (Fig. 1A). Overall, the results established that CS448-3 was nonmigratory at 10 h because the Rcs phosphorelay was activated in this mutant and inhibited the expression of flagellum genes.
Fig 2.
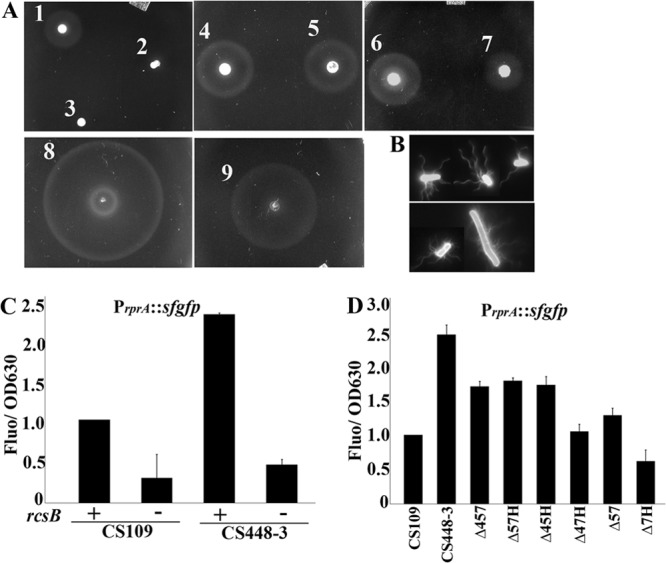
PBP loss activates the Rcs phosphorelay. (A) Migration assays. A1, E. coli CS109; A2, CS448-3; A3, CS109 ΔflhA (SKCS49-1); A4, CS109 ΔrcsB (KE8); A5, CS448-3 ΔrcsB (KE9); A6, CS109 ΔrcsC (KE17); A7, CS448-3 ΔrcsC (KE15); A8, CS109/pFlhDC; A9, CS448-3/pFlhDC. The flhDC genes were expressed from plasmid pFlhDC by supplementing the plates with arabinose (0.05%). (B) Overexpressing flhDC produces hyperflagellated cells. Cells were stained with Alexa Fluor 488. CS109/pFlhDC (upper panel) and CS448-3/pFlhDC (lower panel) exhibited many more flagella than did CS109 (Fig. 1D). Some cells appear enlarged because of the intensity of the fluorescence. (C) The absence of four PBPs increases the relative amount of PrprA::sfgfp expression in an RcsB-dependent manner in E. coli CS448-3. “+”, Wild-type rcsB present; “−”, rcsB is deleted. (D) Rcs reporter expression in double and triple PBP mutants: Δ457 (CS315-1), Δ57H (CS345-3), Δ45H (CS326-3), Δ47H (CS317-3), Δ57 (CS204-1), Δ7H (CS206-3). The strains in panels C and D contain plasmid pDKR1, which carries the PrprA::sfgfp reporter gene. In panels C and D, data from three experiments were normalized relative to CS109.
CS448-3 recovers motility after prolonged incubation.
Curiously, though strain CS448-3 did not migrate at 10 h, it exhibited near-wild-type migration after 24 h of incubation (Fig. 3A, bottom right). This delay was not due to poor or slow growth because CS448-3 did not show significant growth defects (not shown). Because the loss of these PBPs produces aberrantly shaped cells (3), it was possible that migration was hindered or slowed because of a nonoptimum shape. However, this was not the case because CS448-3 remained misshapen even after motility was restored at 24 h (data not shown). Also, the loss of RcsB, although it restored the ability to migrate to CS448-3, did not improve the shape of this mutant (not shown). Furthermore, mutants lacking other combinations of PBPs 4, 5, and 7 or AmpH migrated (Fig. 1A, rows 3 to 6) but were aberrantly shaped (not shown). These results indicated that neither growth nor morphology accounted for the delay in migration. Another potential explanation for migration recovery at 24 h was that CS448-3 had accumulated compensatory suppressor mutations during this time. If so, then cells isolated from the extremities of the migration ring should be motile at 10 h in subsequent assays. However, when such cells were transferred to LB broth, grown overnight at 30°C and inoculated onto a migration plate, none migrated after 10 h (not shown), indicating that migration at 24 h was not caused by the accumulation of suppressor mutants.
Fig 3.
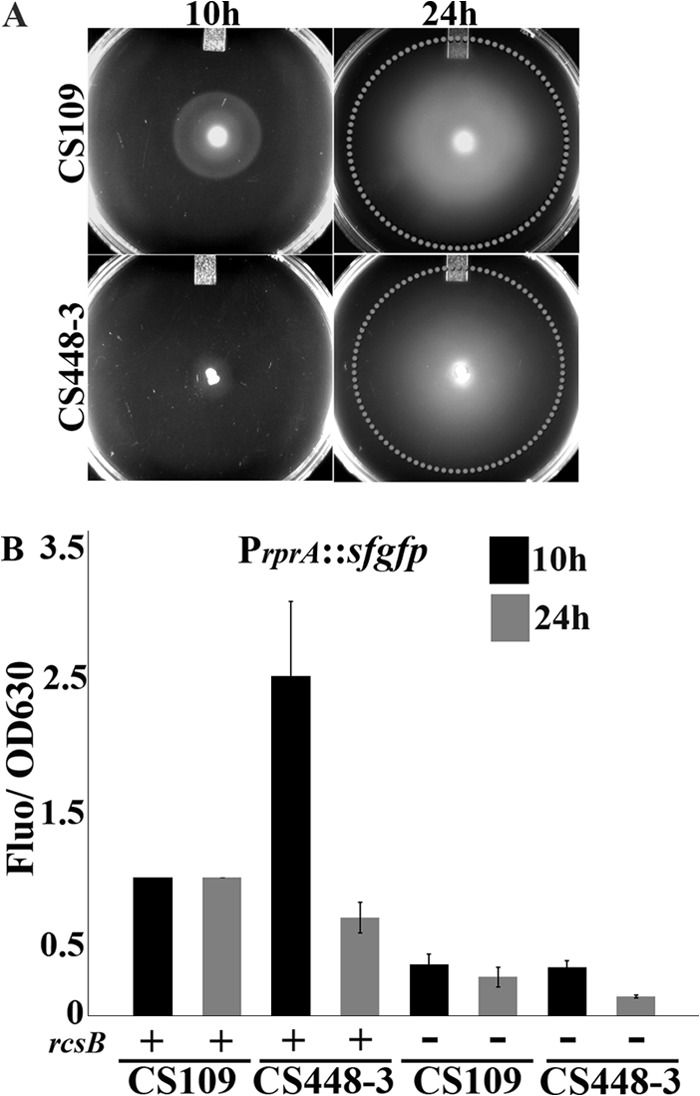
CS448-3 exhibits wild-type migration and an attenuated Rcs response after 24 h of incubation. (A) Strains were assayed for migration at 10 and 24 h. At 10 h, CS109 migrates (top left), but CS448-3 does not migrate (bottom left). CS448-3 recovers wild-type migration after 24 h (bottom right, white dotted circle indicates migration ring). (B) Cells were removed from the migration plate and assayed for PrprA::sfgfp expression (black bars, 10 h; gray bars, 24 h). Each strain contained plasmid pDKR1, which contains the PrprA::sfgfp reporter. Reporter expression in CS448-3 pDKR1 returns to wild-type levels at 24 h. “+”, Wild type rcsB is present; “−”, rcsB is deleted. The data from three experiments were normalized relative to CS109.
Finally, CS448-3 might have regained motility and thus the ability to migrate if Rcs phosphorelay activity was attenuated after 24 h. To test this, the parent strain and CS448-3 carrying a PrprA::sfgfp reporter gene (on plasmid pDKR1) were incubated on migration plates, agar plugs containing cells from the inoculation site extending to the periphery of the migration ring were collected, and the amount of sfGFP in these cells at 10 and 24 h was determined. There was some expression of the PrprA::sfgfp gene in the parental strain CS109 (Fig. 3B), which was probably nonspecific background caused by leaky transcriptional control. This level was the same in CS109 at both 10 and 24 h (Fig. 3B). In contrast, PrprA::sfgfp expression was elevated significantly in CS448-3 at 10 h (Fig. 3B, black bar). However, after 24 h, PrprA::sfgfp expression in CS448-3 returned to wild-type levels (Fig. 3B, gray bar). Therefore, it appears that Rcs activity in CS448-3 was attenuated after prolonged incubation, which would account for the restored motility. Such a scenario further corroborates the conclusion that in this mutant the Rcs response delays motility and thus the ability to migrate.
The Cpx stress response is activated in CS448-3.
Signal transduction systems other than the Rcs pathway also affect flagellar motility. In the EnvZ-OmpR two-component system, OmpR, like RcsAB, negatively regulates flhDC transcription (32). To determine whether the EnvZ-OmpR response was activated in CS448-3, we removed either EnvZ, the sensor kinase, or OmpR, the response regulator. The loss of neither restored the ability to migrate to CS448-3 at 10 h (not shown). Similarly, the mdtABCD::luxCDABE reporter gene, which is induced by the BaeSR envelope stress response, was not expressed in CS448-3 (not shown), indicating that the BaeSR system was not active under these conditions. In contrast, inactivation of the Cpx two-component system in CS448-3 restored its ability to migrate (Fig. 4B5). This system affects motility in E. coli via CpxR-mediated transcriptional repression of the motAB genes (33). Also, expression of the PcpxP::luxCDABE reporter, indicating Cpx activation, increased in CS448-3 relative to the wild type (Fig. 4C). Strains lacking different combinations of PBP 4, 5, or 7 or AmpH expressed the reporter at levels that fell in between those of wild type and CS448-3 (Fig. 4D). This indicated that Cpx was moderately activated in these mutants, although migration was not affected. Thus, the Cpx stress response was induced in CS448-3, as well as the Rcs response. However, PcpxP::luxCDABE expression remained high in CS448-3 mutants lacking rcsB (not shown) and rcsC (Fig. 4C), indicating that activation of the Cpx pathway was independent of the ability to mount an Rcs response. Surprisingly, however, PrprA::sfGFP expression returned to a basal level when cpxR was deleted from E. coli CS448-3 (Fig. 4E), indicating that under these conditions Rcs induction depended on the presence of CpxR. The simultaneous activity of both of these pathways was required to fully inhibit motility and thus migration in CS448-3, because removing either RcsB (Fig. 2A5) or CpxR (Fig. 4B5) restored migration to CS448-3 at 10 h. The results suggest that the two responses must function cooperatively to antagonize motility.
Fig 4.
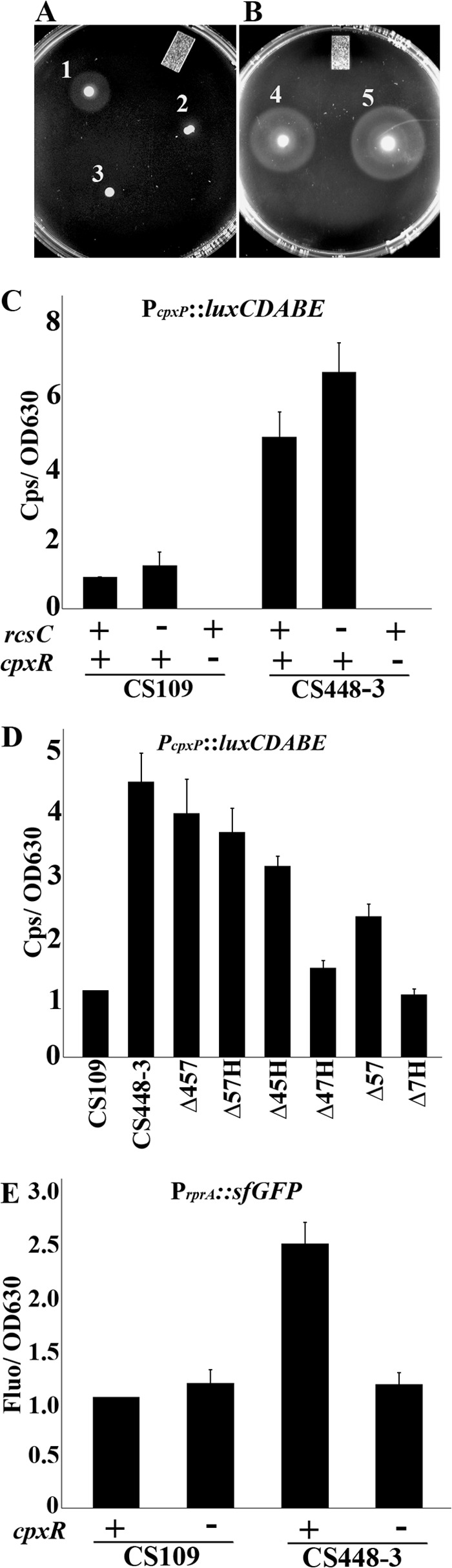
The Cpx stress response is activated in E. coli CS448-3. (A) Migration assays: A1, CS109; A2, CS448-3; A3, CS109 ΔflhA (SKCS49-1). (B) Deleting cpxR restores the ability to migrate to CS448-3: B4, CS109 ΔcpxR (KE36); B5, CS448 ΔcpxR (KE37). (C) Cpx reporter activity is increased in CS448-3 and is independent of the Rcs phosphorelay. (D) Cpx reporter activity in double and triple PBP mutants. Strains are the same as in Fig. 2D. (E) Rcs reporter activity depends on CpxR. The experimental details are the same as in the legend to Fig. 2. The strains in panels C and D contain plasmid pJW1, which carries the PcpxP::luxCDABE reporter. The strains in panel E contain plasmid pDKR1, which carries the PrprA::sfgfp reporter. “+”, rcsC or cpxR are present; “−”, the gene is deleted. In panels C, D, and E, the data from three experiments were normalized relative to CS109.
Peptidoglycan composition of PBP mutants.
Because the loss of PBPs 4, 5, and 7 and AmpH induced the Rcs and Cpx systems, we hypothesized that the signal(s) triggering these responses must be a type of modified peptidoglycan. To determine whether CS448-3 contained unique peptidoglycan modifications, sacculi from CS448-3 and mutants deleted for PBP4, PBP5, PBP7, or AmpH were isolated and analyzed by HPLC. Sacculi from CS448-3 contained high levels of monomer (M5) and dimer pentapeptides (D45), as well as having a greater number of cross-linked muropeptides and lower amounts of lipoprotein-containing muropeptides (Table 3).
Table 3.
Peptidoglycan composition of E. coli PBP mutants
| Muropeptide | Avg value (mol%) for various E. coli strains (PBPs deleted)a |
|||||
|---|---|---|---|---|---|---|
| CS109 (wt) | CS315-1 (4, 5, 7) | CS317-3 (4, 7, AmpH) | CS326-3 (4, 5, AmpH) | CS345-3 (5, 7, AmpH) | CS448-3 (4, 5, 7, AmpH) | |
| M3 | 9.73 | 5.58 | 7.30 | 7.50 | 5.33 | 3.11 |
| M3G | 2.22 | 1.15 | 1.56 | 1.38 | 1.20 | 0.46 |
| M4 | 42.32 | 30.91 | 33.67 | 38.48 | 34.44 | 18.01 |
| M4G | 0.00 | 1.89 | 0.03 | 0.44 | 1.64 | 2.57 |
| M2 | 3.60 | 2.21 | 4.03 | 2.39 | 2.41 | 2.57 |
| M5 | 0.00 | 13.43 | 0.34 | 7.91 | 9.23 | 25.13 |
| M3L | 10.01 | 6.13 | 7.79 | 6.54 | 4.83 | 1.56 |
| D33D | 0.22 | 0.00 | 0.08 | 0.06 | 0.03 | 0.00 |
| D34D | 1.30 | 0.43 | 0.89 | 1.03 | 0.89 | 0.25 |
| D43 | 4.19 | 1.38 | 4.07 | 1.86 | 1.89 | 1.20 |
| D44G | 0.00 | 2.70 | 0.45 | 0.62 | 2.68 | 3.52 |
| D44 | 18.76 | 14.64 | 29.44 | 19.27 | 20.19 | 14.44 |
| D45 | 0.21 | 13.13 | 0.47 | 7.00 | 8.31 | 19.66 |
| T443 | 0.25 | 0.09 | 0.13 | 0.00 | 0.05 | 0.06 |
| T444 | 1.51 | 1.15 | 2.21 | 1.17 | 1.60 | 1.30 |
| D43L | 2.97 | 2.21 | 4.12 | 2.15 | 2.55 | 1.60 |
| T445 | 0.00 | 0.83 | 0.00 | 0.34 | 0.49 | 1.10 |
| D43N | 0.37 | 0.22 | 0.59 | 0.21 | 0.24 | 0.21 |
| D44N | 1.16 | 0.64 | 1.30 | 0.65 | 0.93 | 0.67 |
| D45N | 0.09 | 0.69 | 0.00 | 0.49 | 0.47 | 1.38 |
| T444N | 0.34 | 0.15 | 0.42 | 0.22 | 0.22 | 0.18 |
| T445N | 0.05 | 0.18 | 0.00 | 0.11 | 0.09 | 0.32 |
| D44NN | 0.36 | 0.23 | 0.37 | 0.18 | 0.18 | 0.16 |
| Monomers | 68.19 | 61.30 | 55.24 | 64.62 | 58.98 | 53.40 |
| Dimers | 29.67 | 36.31 | 42.01 | 33.55 | 38.58 | 43.40 |
| Trimers | 2.14 | 2.39 | 2.76 | 1.85 | 2.44 | 3.20 |
| Lipoprotein | 13.04 | 8.323 | 11.90 | 8.69 | 7.38 | 3.41 |
| Anhydro | 3.02 | 2.39 | 3.70 | 2.09 | 2.45 | 3.15 |
| Dap-dap | 1.58 | 0.49 | 1.21 | 1.15 | 1.16 | 0.33 |
| Pentapeptide | 0.33 | 28.25 | 0.81 | 15.84 | 18.58 | 47.83 |
| Penta-glycine | 0 | 4.59 | 0.48 | 1.05 | 4.33 | 6.09 |
| Cross-linkage | 33.98 | 41.08 | 47.50 | 37.25 | 43.45 | 49.78 |
| Avg length | 33.68 | 41.95 | 27.10 | 48.60 | 40.85 | 32.68 |
Values (given as the mole percentage) are averages of four (CS109, CS315-1, and CS448-3) or three (CS317-3, CS326-3, and CS345-3) HPLC analyses of peptidoglycan composition. The PBP status (i.e., wild type [wt] or PBPs deleted) is indicated in parentheses after each strain.
The relevant signal could be the increase of a peptidoglycan component that activates the Rcs or Cpx systems. Alternately, the presence of a particular muropeptide in wild-type cells might inhibit these stress responses, in which case a decrease in the concentration of this inhibitor would activate these systems. With this in mind, we compared the abundance of specific muropeptides with Rcs and Cpx reporter expression in the respective strains. If the accumulation of a specific muropeptide activated the Rcs and Cpx pathways, then that muropeptide should be abundant in strains with high Rcs or Cpx reporter expression and low in strains with low reporter expression. The patterns of Rcs and Cpx induction were similar (Fig. 2D and Fig. 4D). In both cases, reporter genes were expressed in increasing amounts according to the following relationship: CS317-3 < CS326-3 ≤ CS345-3, CS315-1 < CS448-3 (Fig. 2D and Fig. 4D). A candidate inducer should increase accordingly, and the abundance of the following muropeptides followed this trend: M5, M4G, D45, D44G, T445, and T445N (Table 3). Note that the overall frequency of pentapeptide- and pentaglycine-containing muropeptides increased in concert with this predicted pattern (Table 3). If the signal is a negative regulator, then the candidate inhibitory muropeptide should decrease in these strains as follows: CS317-3 > CS326-3 ≥ CS345-3, CS315-1 > CS448-3. Muropeptides that followed this trend included: M3, M3G, D43, D44NN, and those with attached lipoprotein (Table 3).
Finally, we examined the possibility that the peptidoglycan signaling compound might be transported to the cytoplasm by the muropeptide recycling pathway (34). If so, then this could account for the attenuated Rcs response observed after 24 h (Fig. 3B). However, deleting the gene encoding the inner membrane permease AmpG did not prevent Rcs induction in strain CS448-3, nor did it prevent the resumption of migration at 24 h (not shown). Thus, the peptidoglycan signal did not have to enter the cytoplasm via the recycling pathway, nor was Rcs attenuated because the inducing signal was depleted in this way.
DISCUSSION
Envelope stress responses are activated by peptidoglycan modification.
E. coli encodes multiple envelope stress responses, but the specific signals that trigger these different responses have not been described. Thus far, the inducing treatments that have been identified affect the envelope in multiple ways. For example, lysozyme (35) and EDTA (36) significantly damage the peptidoglycan and outer membrane, respectively. Exposure to alcohols (37), acid stress (38), and changes in osmolarity (39, 40) likewise probably damage numerous envelope proteins or seriously alter overall membrane integrity. These gross effects make it nearly impossible to identify specific molecular stimuli that activate any one envelope stress response. Here, we demonstrate that the loss of four LMW PBPs (PBP4, PBP5, PBP7, and AmpH) activates the Rcs and Cpx envelope stress responses. Importantly, the pathways are activated by the loss of PBP enzyme activity, not because the proteins themselves are absent, strongly suggesting that a peptidoglycan compound is the activating signal. The results imply that the Rcs and Cpx pathways detect changes among peptidoglycan species in the cell wall.
Rcs and Cpx sensor components in the periplasm and on the periplasmic side of the cytoplasmic membrane are conveniently located to respond to such a cell wall-associated stimulus, and the responses could be activated either because an inducer accumulates or because an inhibitory compound disappears. Thus, PBPs 4, 5, and 7 and AmpH may normally degrade or modify an activator (which would then accumulate in their absence), or they may synthesize an inhibitor (which would then disappear in their absence). Whatever its identity and mode of action, the activating compound may be sensed directly by Rcs or Cpx receptors or may induce these stress responses indirectly via a downstream event (e.g., alterations in periplasmic protein folding or changes in another envelope characteristic). In any case, the critical concentration of the signal(s) will depend on the activities of PBPs 4, 5, and 7 and AmpH.
Although we cannot yet identify the specific peptidoglycan species responsible, we can draw some conclusions about its nature. If the signal is an activator, then the fact that PBP5 must be inactivated indicates the compound probably contains a pentapeptide. PBP4 and AmpH also exhibit low levels of d,d-carboxypeptidase activity, and their loss may also contribute to the accumulation of such a pentapeptide. However, because PBPs 4 and 7 and AmpH are primarily endopeptidases (2), the fact that the stress responses are induced in their absence implies that the activator probably includes a cross-linked muropeptide. In fact, E. coli CS448-3 contains much higher amounts of pentapeptide and cross-linked muropeptides (Table 3). More specifically, the concentrations of two monomers (M5 and M4G), two dimers (D45 and D44G), and two trimers (T445 and T445N) rise in parallel with the degree to which the Rcs and Cpx responses become activated, making these good candidates for behaving as the activator. Interestingly, two of these muropeptides contain glycine as the terminal amino acid in the peptide side chains, making it possible that Rcs and Cpx sense these termini, which are not abundant under normal circumstances. On the other hand, if the signal involves the disappearance of an inhibitor, then by inverting the above considerations the inhibitory compound would most likely contain a tetrapeptide or tripeptide and would either be a monomer or dimer that appears frequently in wild-type E. coli but not in CS448-3. Finally, the proximal inducing signal might not be a specific peptidoglycan species but rather some downstream alteration that affects the cell envelope or its properties. At the moment, the most obvious possibility along this line is that E. coli CS448-3 has many fewer muropeptides that are covalently connected to Braun's lipoprotein (Table 3), and this alteration in envelope structure might destabilize the outer membrane (although there is no evidence of this as yet).
Although a small change in peptidoglycan structure may trigger the Rcs and Cpx responses, other types of envelope damage may induce these responses by independent routes or signals. Alternately, all stressors might eventually generate a common signal. For example, the Cpx response can be induced by overexpressing the outer membrane lipoprotein NlpE (41), which has no obvious relationship to peptidoglycan synthesis. However, it is possible that accumulation of periplasmic NlpE could affect the synthesis and composition of peptidoglycan, which would then be the proximal signal. Conversely, and even more theoretically, extensive peptidoglycan modification might affect the folding of periplasmic proteins, which might themselves act as the proximal stress-inducing signal. In either case, the involvement of a specific set of LMW PBPs offers a new avenue to explore these questions.
LMW PBPs perform different physiological roles.
It is a mystery why E. coli and other bacteria contain so many PBPs with similar or overlapping enzymatic activities. Here, we show that the loss of one specific combination of LMW PBPs activates two stress responses, which suggests that these and other ostensibly redundant PBPs may have somewhat more individual physiological roles than is now appreciated. Specifically, the Rcs and Cpx responses are triggered by removing one d,d-carboxypeptidase (PBP5) and three of the four E. coli endopeptidases. No other combination of PBP deletions reproduced this phenotype, indicating that these enzymes exhibit a great deal of selectivity as they interact with their substrates. For example, we conclude that in vivo, PBP4, PBP7, AmpH, and MepA must have small but physiologically relevant differences in endopeptidase activity or substrate specificity, so that these enzymes may not overlap in function or be as superfluous as has been assumed.
Another question is why multiple LMW PBPs should have to be inactivated before the Rcs and Cpx responses are highly induced. Because many β-lactam antibiotics inhibit multiple PBPs, deleting four PBPs may reproduce the cell wall modifications that accompany exposure to β-lactams. We speculate that this requirement may allow E. coli to monitor cell wall damage in order to activate stress responses only when the damage becomes sufficiently severe, as reflected by the simultaneous inactivation of multiple PBPs.
Finally, why might the Rcs and Cpx responses be activated by PBP inactivation? Other than the antilysozyme proteins Ivy and MliC (35), no known Rcs-induced proteins are related to peptidoglycan synthesis or modification (10). Thus, it is not clear that the Rcs system is set up to repair damage to the peptidoglycan or whether this kind of damage serves only as a convenient signal. On the other hand, the Cpx response stimulates the expression of two peptidoglycan amidase genes (amiA and amiC) (42), as well as that of at least four other cell wall-associated genes (dacC, slt, ycbB, and ygaU) (43), so this response may actively repair cell wall damage.
The Rcs and Cpx stress responses synergistically inhibit motility.
Removing the Rcs response did not appreciably affect activation of the Cpx pathway in E. coli CS448-3, but removing CpxR returned Rcs reporter expression to its basal level, indicating that Rcs activity depends on CpxR in this mutant. This phenotype has not been reported previously, and we are characterizing it further. One possibility is that expression of a Cpx-regulated gene is required to activate Rcs; alternately, although unlikely, in the absence of CpxR, CpxA may dephosphorylate RcsB and thus inhibit the Rcs response. The latter scenario might reflect a natural mechanism that keeps the level of phosphorylated RcsB in check, or it may simply be an artifact caused by removing CpxR. In any case, both the Rcs and Cpx responses must be activated before migration is inhibited completely; that is, the CS448-3 mutant can migrate if only one or the other response is disabled. The simplest explanation is that neither of these inhibitory events is fully effective on its own. Such regulation is not unprecedented. For example, a high level of spy gene expression requires that both the Cpx and the Bae stress responses be activated (44). Thus, E. coli is able to fine-tune transcription levels and cellular physiology based on environmental stimuli, the degree of stress-induced damage, and the specific stress responses that become activated.
Conclusion.
Peptidoglycan or its modified components can serve as biological signals in several different species and systems. For example, peptidoglycan fragments induce sporulation in Bacillus subtilis (45, 46), alert the innate immune system (47, 48), act as virulence factors in Bordetella pertussis (49), and induce the production of β-lactamase (50, 51). We now report that peptidoglycan modifications activate the Rcs and Cpx stress responses in a system that may help integrate and improve our understanding of how cell envelope stress is sensed and conveyed to the requisite genetic regulators.
ACKNOWLEDGMENTS
We thank Tracy Raivio, Rasika Harshey, and Prasad Potluri for plasmids used in this study.
This study was supported by the National Institute of General Medical Sciences of the National Institutes of Health under award R01-GM61019 and by the Arkansas Biosciences Institute, the major research component of the Arkansas Tobacco Settlement Proceeds Act of 2000.
Footnotes
Published ahead of print 26 July 2013
REFERENCES
- 1.Holtje JV. 1998. Growth of the stress-bearing and shape-maintaining murein sacculus of Escherichia coli. Microbiol. Mol. Biol. Rev. 62:181–203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Typas A, Banzhaf M, Gross CA, Vollmer W. 2012. From the regulation of peptidoglycan synthesis to bacterial growth and morphology. Nat. Rev. Microbiol. 10:123–136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Denome SA, Elf PK, Henderson TA, Nelson DE, Young KD. 1999. Escherichia coli mutants lacking all possible combinations of eight penicillin binding proteins: viability, characteristics, and implications for peptidoglycan synthesis. J. Bacteriol. 181:3981–3993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nelson DE, Young KD. 2001. Contributions of PBP 5 and dd-carboxypeptidase penicillin binding proteins to maintenance of cell shape in Escherichia coli. J. Bacteriol. 183:3055–3064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Meberg BM, Paulson AL, Priyadarshini R, Young KD. 2004. Endopeptidase penicillin-binding proteins 4 and 7 play auxiliary roles in determining uniform morphology of Escherichia coli. J. Bacteriol. 186:8326–8336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Scheurwater EM, Burrows LL. 2011. Maintaining network security: how macromolecular structures cross the peptidoglycan layer. FEMS Microbiol. Lett. 318:1–9 [DOI] [PubMed] [Google Scholar]
- 7.Vogt SL, Raivio TL. 2012. Just scratching the surface: an expanding view of the Cpx envelope stress response. FEMS Microbiol. Lett. 326:2–11 [DOI] [PubMed] [Google Scholar]
- 8.Leblanc SK, Oates CW, Raivio TL. 2011. Characterization of the induction and cellular role of the BaeSR two-component envelope stress response of Escherichia coli. J. Bacteriol. 193:3367–3375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cai SJ, Inouye M. 2002. EnvZ-OmpR interaction and osmoregulation in Escherichia coli. J. Biol. Chem. 277:24155–24161 [DOI] [PubMed] [Google Scholar]
- 10.Laubacher ME, Ades SE. 2008. The Rcs phosphorelay is a cell envelope stress response activated by peptidoglycan stress and contributes to intrinsic antibiotic resistance. J. Bacteriol. 190:2065–2074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Majdalani N, Gottesman S. 2005. The Rcs phosphorelay: a complex signal transduction system. Annu. Rev. Microbiol. 59:379–405 [DOI] [PubMed] [Google Scholar]
- 12.Majdalani N, Heck M, Stout V, Gottesman S. 2005. Role of RcsF in signaling to the Rcs phosphorelay pathway in Escherichia coli. J. Bacteriol. 187:6770–6778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Castanie-Cornet MP, Cam K, Jacq A. 2006. RcsF is an outer membrane lipoprotein involved in the RcsCDB phosphorelay signaling pathway in Escherichia coli. J. Bacteriol. 188:4264–4270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Takeda S, Fujisawa Y, Matsubara M, Aiba H, Mizuno T. 2001. A novel feature of the multistep phosphorelay in Escherichia coli: a revised model of the RcsC → YojN → RcsB signaling pathway implicated in capsular synthesis and swarming behaviour. Mol. Microbiol. 40:440–450 [DOI] [PubMed] [Google Scholar]
- 15.Francez-Charlot A, Laugel B, Van Gemert A, Dubarry N, Wiorowski F, Castanie-Cornet MP, Gutierrez C, Cam K. 2003. RcsCDB His-Asp phosphorelay system negatively regulates the flhDC operon in Escherichia coli. Mol. Microbiol. 49:823–832 [DOI] [PubMed] [Google Scholar]
- 16.Soutourina OA, Bertin PN. 2003. Regulation cascade of flagellar expression in Gram-negative bacteria. FEMS Microbiol. Rev. 27:505–523 [DOI] [PubMed] [Google Scholar]
- 17.Danese PN, Snyder WB, Cosma CL, Davis LJ, Silhavy TJ. 1995. The Cpx two-component signal transduction pathway of Escherichia coli regulates transcription of the gene specifying the stress-inducible periplasmic protease, DegP. Genes Dev. 9:387–398 [DOI] [PubMed] [Google Scholar]
- 18.Hunke S, Keller R, Muller VS. 2012. Signal integration by the Cpx-envelope stress system. FEMS Microbiol. Lett. 326:12–22 [DOI] [PubMed] [Google Scholar]
- 19.Vogt SL, Nevesinjac AZ, Humphries RM, Donnenberg MS, Armstrong GD, Raivio TL. 2010. The Cpx envelope stress response both facilitates and inhibits elaboration of the enteropathogenic Escherichia coli bundle-forming pilus. Mol. Microbiol. 76:1095–1110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hung DL, Raivio TL, Jones CH, Silhavy TJ, Hultgren SJ. 2001. Cpx signaling pathway monitors biogenesis and affects assembly and expression of P pili. EMBO J. 20:1508–1518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Humphreys S, Rowley G, Stevenson A, Anjum MF, Woodward MJ, Gilbert S, Kormanec J, Roberts M. 2004. Role of the two-component regulator CpxAR in the virulence of Salmonella enterica serotype Typhimurium. Infect. Immun. 72:4654–4661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chevance FF, Hughes KT. 2008. Coordinating assembly of a bacterial macromolecular machine. Nat. Rev. Microbiol. 6:455–465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.DePamphilis ML, Adler J. 1971. Attachment of flagellar basal bodies to the cell envelope: specific attachment to the outer, lipopolysaccharide membrane and the cytoplasmic membrane. J. Bacteriol. 105:396–407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schnaitman CA, McDonald GA. 1984. Regulation of outer membrane protein synthesis in Escherichia coli K-12: deletion of ompC affects expression of the OmpF protein. J. Bacteriol. 159:555–563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Datsenko KA, Wanner BL. 2000. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. U. S. A. 97:6640–6645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Miller J. 1992. A short course in bacterial genetics: a laboratory manual and handbook for Escherichia coli and related bacteria. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 27.Li SY, Holtje JV, Young KD. 2004. Comparison of high-performance liquid chromatography and fluorophore-assisted carbohydrate electrophoresis methods for analyzing peptidoglycan composition of Escherichia coli. Anal. Biochem. 326:1–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Alaniz RC, Cummings LA, Bergman MA, Rassoulian-Barrett SL, Cookson BT. 2006. Salmonella typhimurium coordinately regulates FliC location and reduces dendritic cell activation and antigen presentation to CD4+ T cells. J. Immunol. 177:3983–3993 [DOI] [PubMed] [Google Scholar]
- 29.Sambrook J, Fritsch EF, Maniatis T. 1989. Molecular cloning: a laboratory manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 30.Turner L, Ryu WS, Berg HC. 2000. Real-time imaging of fluorescent flagellar filaments. J. Bacteriol. 182:2793–2801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Majdalani N, Chen S, Murrow J, St John K, Gottesman S. 2001. Regulation of RpoS by a novel small RNA: the characterization of RprA. Mol. Microbiol. 39:1382–1394 [DOI] [PubMed] [Google Scholar]
- 32.Prüss BM, Besemann C, Denton A, Wolfe AJ. 2006. A complex transcription network controls the early stages of biofilm development by Escherichia coli. J. Bacteriol. 188:3731–3739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Price NL, Raivio TL. 2009. Characterization of the Cpx regulon in Escherichia coli strain MC4100. J. Bacteriol. 191:1798–1815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cheng Q, Park JT. 2002. Substrate specificity of the AmpG permease required for recycling of cell wall anhydro-muropeptides. J. Bacteriol. 184:6434–6436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Callewaert L, Vanoirbeek KG, Lurquin I, Michiels CW, Aertsen A. 2009. The Rcs two-component system regulates expression of lysozyme inhibitors and is induced by exposure to lysozyme. J. Bacteriol. 191:1979–1981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bury-Mone S, Nomane Y, Reymond N, Barbet R, Jacquet E, Imbeaud S, Jacq A, Bouloc P. 2009. Global analysis of extracytoplasmic stress signaling in Escherichia coli. PLoS Genet. 5:e1000651. 10.1371/journal.pgen.10000651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Clarke EJ, Voigt CA. 2011. Characterization of combinatorial patterns generated by multiple two-component sensors in Escherichia coli that respond to many stimuli. Biotechnol. Bioeng. 108:666–675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Castanie-Cornet MP, Cam K, Bastiat B, Cros A, Bordes P, Gutierrez C. 2010. Acid stress response in Escherichia coli: mechanism of regulation of gadA transcription by RcsB and GadE. Nucleic Acids Res. 38:3546–3554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Raivio TL, Silhavy TJ. 1997. Transduction of envelope stress in Escherichia coli by the Cpx two-component system. J. Bacteriol. 179:7724–7733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mileykovskaya E, Dowhan W. 1997. The Cpx two-component signal transduction pathway is activated in Escherichia coli mutant strains lacking phosphatidylethanolamine. J. Bacteriol. 179:1029–1034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Snyder WB, Davis LJ, Danese PN, Cosma CL, Silhavy TJ. 1995. Overproduction of NlpE, a new outer membrane lipoprotein, suppresses the toxicity of periplasmic LacZ by activation of the Cpx signal transduction pathway. J. Bacteriol. 177:4216–4223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Weatherspoon-Griffin N, Zhao G, Kong W, Kong Y, Morigen Andrews-Polymenis H, McClelland M, Shi Y. 2011. The CpxR/CpxA two-component system up-regulates two Tat-dependent peptidoglycan amidases to confer bacterial resistance to antimicrobial peptide. J. Biol. Chem. 286:5529–5539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Raivio TL, Leblanc SK, Price NL. 2013. The Escherichia coli Cpx envelope stress response regulates genes of diverse function that impact antibiotic resistance and membrane integrity. J. Bacteriol. 195:2755–2767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rosner JL, Martin RG. 2013. Reduction of cellular stress by TolC-dependent efflux pumps in Escherichia coli indicated by BaeSR and CpxARP activation of spy in efflux mutants. J. Bacteriol. 195:1042–1050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dworkin J, Shah IM. 2010. Exit from dormancy in microbial organisms. Nat. Rev. Microbiol. 8:890–896 [DOI] [PubMed] [Google Scholar]
- 46.Lee M, Hesek D, Shah IM, Oliver AG, Dworkin J, Mobashery S. 2010. Synthetic peptidoglycan motifs for germination of bacterial spores. Chembiochem 11:2525–2529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Girardin SE, Boneca IG, Carneiro LA, Antignac A, Jehanno M, Viala J, Tedin K, Taha MK, Labigne A, Zahringer U, Coyle AJ, DiStefano PS, Bertin J, Sansonetti PJ, Philpott DJ. 2003. Nod1 detects a unique muropeptide from gram-negative bacterial peptidoglycan. Science 300:1584–1587 [DOI] [PubMed] [Google Scholar]
- 48.Clarke TB, Davis KM, Lysenko ES, Zhou AY, Yu Y, Weiser JN. 2010. Recognition of peptidoglycan from the microbiota by Nod1 enhances systemic innate immunity. Nat. Med. 16:228–231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cookson BT, Tyler AN, Goldman WE. 1989. Primary structure of the peptidoglycan-derived tracheal cytotoxin of Bordetella pertussis. Biochemistry 28:1744–1749 [DOI] [PubMed] [Google Scholar]
- 50.Tayler AE, Ayala JA, Niumsup P, Westphal K, Baker JA, Zhang L, Walsh TR, Wiedemann B, Bennett PM, Avison MB. 2010. Induction of β-lactamase production in Aeromonas hydrophila is responsive to beta-lactam-mediated changes in peptidoglycan composition. Microbiology 156:2327–2335 [DOI] [PubMed] [Google Scholar]
- 51.Moya B, Dotsch A, Juan C, Blazquez J, Zamorano L, Haussler S, Oliver A. 2009. β-lactam resistance response triggered by inactivation of a nonessential penicillin-binding protein. PLoS Pathog. 5:e1000353. 10.1371/journal.ppat.1000353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ranjit DK, Young KD. 2013. The Rcs stress response and accessory envelope proteins are required for de novo generation of cell shape in Escherichia coli. J. Bacteriol. 195:2452–2462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Laubacher ME, Melquist AL, Chandramohan L, Young KD. 2013. Cell sorting enriches Escherichia coli mutants that rely on peptidoglycan endopeptidases to suppress highly aberrant morphologies. J. Bacteriol. 195:855–866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Potluri L, Karczmarek A, Verheul J, Piette A, Wilkin JM, Werth N, Banzhaf M, Vollmer W, Young KD, Nguyen-Disteche M, den Blaauwen T. 2010. Septal and lateral wall localization of PBP5, the major d,d-carboxypeptidase of Escherichia coli, requires substrate recognition and membrane attachment. Mol. Microbiol. 77:300–323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Potluri LP, de Pedro MA, Young KD. 2012. Escherichia coli low-molecular-weight penicillin-binding proteins help orient septal FtsZ, and their absence leads to asymmetric cell division and branching. Mol. Microbiol. 84:203–224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sperandio V, Torres AG, Giron JA, Kaper JB. 2001. Quorum sensing is a global regulatory mechanism in enterohemorrhagic Escherichia coli O157:H7. J. Bacteriol. 183:5187–5197 [DOI] [PMC free article] [PubMed] [Google Scholar]