

Abstract
We performed whole-genome analyses of DNA methylation in Shewanella oneidensis MR-1 to examine its possible role in regulating gene expression and other cellular processes. Single-molecule real-time (SMRT) sequencing revealed extensive methylation of adenine (N6mA) throughout the genome. These methylated bases were located in five sequence motifs, including three novel targets for type I restriction/modification enzymes. The sequence motifs targeted by putative methyltranferases were determined via SMRT sequencing of gene knockout mutants. In addition, we found that S. oneidensis MR-1 cultures grown under various culture conditions displayed different DNA methylation patterns. However, the small number of differentially methylated sites could not be directly linked to the much larger number of differentially expressed genes under these conditions, suggesting that DNA methylation is not a major regulator of gene expression in S. oneidensis MR-1. The enrichment of methylated GATC motifs in the origin of replication indicates that DNA methylation may regulate genome replication in a manner similar to that seen in Escherichia coli. Furthermore, comparative analyses suggest that many Gammaproteobacteria, including all members of the Shewanellaceae family, may also utilize DNA methylation to regulate genome replication.
INTRODUCTION
DNA methylation plays a variety of functional roles in bacteria (1–3). For example, restriction-modification (R-M) systems use methylation patterns to identify and destroy foreign DNA during viral infections (4, 5). Bacteria also use DNA methylation to regulate genome replication (6), DNA mismatch repair (7), and gene expression (8–12). Methylation can even serve as an epigenetic modifier, influencing the expression patterns of daughter cells based on environmental conditions (13, 14). Because of these varied regulatory roles, DNA methylation should be incorporated into our emerging systems-level view of model microorganisms.
Despite the functional significance of DNA methylation, our understanding of its role in bacterial genetics and physiology remains incomplete due to methodological limitations. For example, bisulfite conversion can identify 5-methylcytosine modifications (15, 16), but there is no corresponding conversion assay for other common modifications in bacteria, such as N6-methyladenine (N6mA) or 4-methylcytosine (17). Methyl-sensitive restriction enzymes have been used to identify the methylation states of specific sequence motifs (18–20), but complete methylome analyses are not possible without prior knowledge of the entire set of methyltransferases and their sequence targets within a genome and access to methyl-sensitive restriction enzymes targeting these motifs. Single-molecule real-time (SMRT) sequencing overcomes these limitations and enables genome-wide analysis of DNA methylation with single base resolution (21). In this approach, modifications in the native-state DNA are revealed by deviations in the polymerase kinetics observed during sequencing. The specific type of DNA methylation can often be determined from the polymerase kinetics, e.g., N6-methyladenine or 4-methylcytosine. With SMRT sequencing, it is now possible to identify the complete set of methylated sequence motifs within a microbial genome and the methylation state for each instance of a motif (22–24). This represents a powerful tool for characterizing the functional roles of DNA methylation in a wide variety of bacteria.
Shewanella oneidensis MR-1 is a bacterial isolate belonging to the Shewanellaceae, a family distinguished by the wide variety of electron acceptors they can utilize (e.g., iron, manganese, uranium, chromium, and plutonium) (25–28). Because of their flexible respiratory pathways, Shewanella species are recognized as potential agents for bioremediation at sites contaminated with heavy metals and radionuclides (29). To better exploit its metabolic potential, S. oneidensis MR-1 has been characterized extensively, including analysis of gene expression (30, 31), identification of regulatory regions (32), and the determination of fitness levels for thousands of gene knockout mutants (33). However, the developing systems-level view of Shewanella does not yet incorporate DNA methylation and its potential regulatory roles. Genomic analyses reveal multiple putative methyltransferases in S. oneidensis MR-1 (34, 35), including several apparent “orphans” that lack corresponding restriction enzymes. It remains unclear what role these orphan methyltransferases might play.
Here we used SMRT sequencing to provide the first look at DNA methylation in S. oneidensis MR-1. We identified methylated sites throughout the genome, as well as the sequence motifs targeted by predicted methyltransferases. To determine if DNA methylation regulates gene expression, we examined whether changes in the expression level correspond with changes in the DNA methylation state when cultures are transferred from one set of growth conditions to another. Finally, we examined the finished genomes of all Gammaproteobacteria, including the Shewanellaceae, to determine which groups appear to use DNA methylation for regulating genome replication and DNA mismatch repair.
MATERIALS AND METHODS
Strains, culture conditions, and nucleic acid isolation.
S. oneidensis MR-1 was obtained from the American Type Culture Collection (catalog number 700550). The aerobic minimal medium contained, per liter, 1.5 g NH4Cl, 0.1 g KCl, 1.75 g NaCl, 0.61 g MgCl2-6H2O, 0.6 g NaH2PO4, 30 mM piperazine-N,N′-bis(2-ethanesulfonic acid) (PIPES) buffer, 20 mM dl-lactate, Wolfe's vitamins, and Wolfe's minerals (pH 7). Fumarate was added as an electron acceptor to anaerobic minimal medium (30 mM, final concentration). Anaerobic minimal medium experiments were set up in an anaerobic chamber (Coy) with a gas mix of 5% H2, 10% CO2, and 85% N2 and incubation in serum bottles closed with butyl rubber stopper at 200 rpm. Aerobic minimal medium experiments were conducted in 10-ml culture tubes or 250-ml culture flasks at 200 rpm. All cultures were incubated at 30°C.
To conduct the initial survey of DNA methylation, wild-type cells were collected during exponential growth in minimal medium (optical density at 600 nm [OD600] = 0.32). DNA and RNA were isolated with the DNeasy Blood & Tissue kit (Qiagen) and RNeasy minikit (Qiagen), respectively. Cells were treated with RNAprotect reagent (Qiagen) according to the manufacturer's instructions prior to RNA extraction, and total RNA was isolated after an on-column DNase treatment. To examine changes in methylation and gene expression in wild-type cells, we inoculated exponentially growing cells from aerobic minimal medium (OD600 = 0.32) into either rich medium (Luria-Bertani broth) or anaerobic minimal medium. DNA and RNA were collected after cultures reached exponential phase in the new medium (OD600 of 0.45 for rich medium, ∼5 population doublings post transfer; OD600 of 0.16 for anaerobic medium, ∼3 population doublings post transfer).
DNA methylation patterns were also determined for mutant strains whose putative methyltransferases were inactivated by transposon insertions (33). DNA from transposon mutants was collected from stationary-phase cultures growing in aerobic minimal medium (OD600 of ∼0.80). All mutants grew to the same density and did not display any substantial growth differences from the wild type.
DNA methylation detection.
Libraries of replicates of wild-type S. oneidensis MR-1 cultures were prepared for SMRT sequencing using a library construction protocol described previously (36). These libraries were sequenced to a mean genome coverage depth of 118× to 222× on the Pacific Biosciences RS instrument using C2 chemistry. One replicate grown in anaerobic minimal medium was excluded from further analysis due to poor coverage (<70×). Methyltransferase transposon mutants were sequenced to a depth of 72× to 113×. Average read lengths ranged from 2,296 to 2,889 bp in all libraries. Reads were mapped to the reference genome (RefSeq NC_004347.1) using the software program BLASR (37). Base modification and motif detection were performed using the Modification and Motif Detection protocol in the software program SMRTPipe v.1.3.3. Positions with coverage of >25× and kinetic scores of ≥QV 40 were considered modified. The QV is the −10 ∗ log (P value), where the P value was determined from a t test between the sample and the in silico model (http://pacb.com/applications/base_modification/index.html). Positions with coverage of <25× were excluded from analysis.
Gene expression analyses.
Total RNA collected from biological replicates was sequenced on the Illumina platform to determine gene expression levels of S. oneidensis MR-1 grown under the three experimental conditions described above. rRNA was removed using the Ribo-Zero Meta-Bacteria kit (Epicentre) prior to creation of a strand-specific RNAseq library (38). Libraries were sequenced on the llumina HiSeq 2000 platform. Ten million mRNA reads randomly selected from each library were analyzed for differential expression using the DEseq (39), edgeR (40), and RankProd (41) software tools. Differentially expressed genes were those confidently identified by all three tests (Q < 0.005; false discovery rate [FDR] < 0.01) as having a fold change of >2.
Detecting dam, seqA, mutH, and GATC enrichment at the origin of replication.
We analyzed all 448 finished Gammaproteobacteria genomes available in the Joint Genome Institute's Integrated Microbial Genomes database (http://img.jgi.doe.gov/) (42) as of 6 March 2013. The numbers of genes assigned to COGs (clusters of orthologous groups) 0338, 3057, and 3066 were determined in order to quantify the number of dam, seqA, and mutH genes, respectively. The origin of replication was determined by in silico and/or in vivo predictions retrieved from the DoriC database v6.5 (http://tubic.tju.edu.cn/doric/index.php) (43, 44). For the small number of genomes missing from DoriC, the origin was found using the program Ori-Finder (http://tubic.tju.edu.cn/Ori-Finder) (45). The origin of replication was considered to be enriched in GATC motifs if the observed number of motifs with the origin was significantly greater than would be expected if GATC motifs were uniformly distributed throughout the genome (i.e., the total number of GATC sites divided by genome size). Significance was calculated using a binomial test with a P value threshold of 0.01.
In four of the genomes examined, we found that the origin annotated in DoriC was not enriched for GATC despite the presence of dam and seqA. The annotated origins shared the following characteristics: (i) the origin was predicted by in silico analysis, which considers both dnaA and gidA to be “indicator genes” of the origin; (ii) the proposed origin was located upstream of dnaA, whereas the predicted origin in other closely related strains was located upstream of gidA; and (iii) the region upstream of gidA was significantly enriched with GATC motifs. This suggested that the apparent anomalies were due to misannotation of the origin or replication. The origin was redefined around gidA in these cases.
Phylogeny of gammaproteobacteria.
16S rRNA sequences from finished Gammaproteobacteria genomes were aligned using the software program MAFFT v6.864b (46) and used to construct a maximum-likelihood phylogeny using the program RAxmL v7.2.6 (47), using the GTRGAMMA model and rapid bootstrap algorithm (1,000 iterations).
RESULTS AND DISCUSSION
Methylation profile of S. oneidensis MR-1.
To identify methylated sites within the genome of S. oneidensis MR-1, we performed SMRT sequencing on DNA extracted from triplicate exponential-phase cultures grown aerobically on minimal medium. Our analysis revealed 42,965 nucleotides (nt) that exhibited significant variations in polymerase kinetics that were diagnostic of DNA modification (21) (Fig. 1). Of those modified nucleotides, 41,853 were identified as N6-methyladenine (N6mA) based on their distinct kinetic fingerprint. The remaining kinetic variants included 396 cytosine, 301 guanine, and 415 thymine bases (see Table S1 in the supplemental material). While some of these were likely analytical artifacts, the agreement among all three biological replicates suggests additional, unidentified mechanisms for DNA modification at work in S. oneidensis MR-1, e.g., glucosylation, putrescinylation, and glutamylation (48, 49). The nature of these putative modifications could not be determined in this study but represent an interesting avenue for future investigation.
Fig 1.
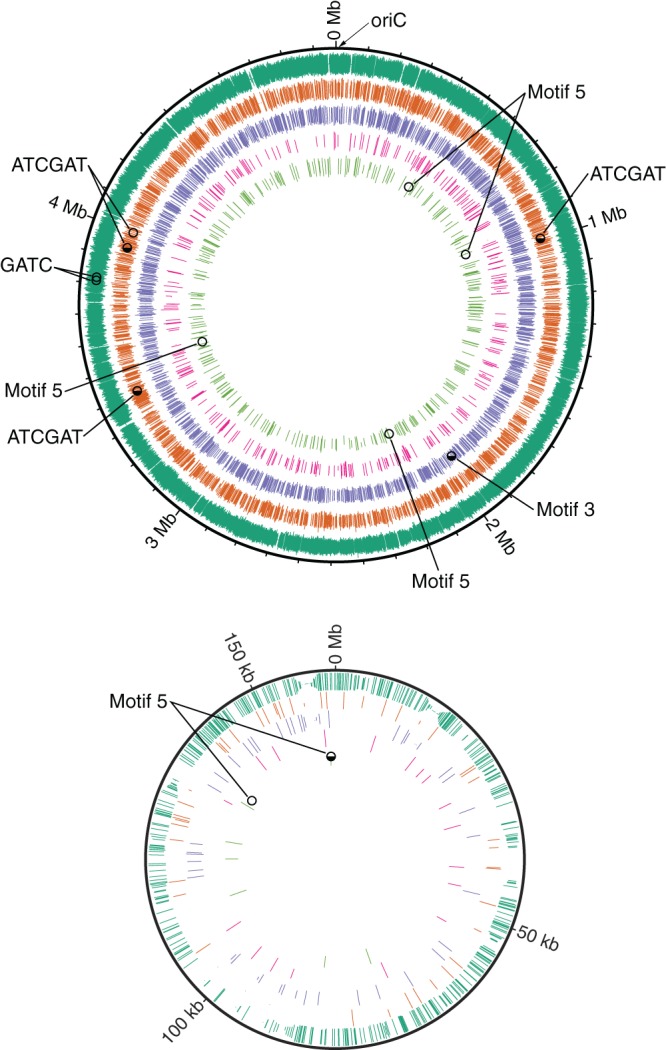
Locations of methylated sequence motifs in the chromosome and megaplasmid of S. oneidensis MR-1. Moving from the outer ring to the inner ring, the methylated motifs are as follows: GATC, ATCGAT, GCAN4GTC/GACN4TGC (motif 3), TACN6GTNGT/ACNACN6GTA (motif 4), and TGAYN6TGAC/GTCAN6RTCA (motif 5). The length of each methylation site marker is proportional to the confidence of methylation; all indicated sites have a P value of <0.0001. Hemimethylated (◒) and nonmethylated (○) sites in cultures grown aerobically in minimal medium are indicated. OriC indicates the position of the origin of replication.
Next, we analyzed the local sequence context of these N6mA bases to determine if they were located within specific sequence motifs. We identified two palindromic motifs (GATC and ATCGAT) and three bipartite motifs (GCAN4GTC/GACN4TGC, TACN6GTNGT/ACNACN6GTA, and TGAYN6TGAC/GTCAN6RTCA) enriched with N6mA bases (methylated bases are underlined) (Table 1). More than 99% of all N6mA residues could be assigned to one of these five sequence motifs, suggesting that methylation was performed by DNA methyltransferases targeting specific DNA sequences. Genome annotations of S. oneidensis MR-1 predicted three type I R-M systems with unknown target sequences (34). The discovery of three bipartite motifs is consistent with type I R-M systems (50), but the specific motif sequences we detected were novel R-M targets. The two methylated palindromes GATC and ATCGAT were also predicted in the restriction enzyme database REBASE (http://rebase.neb.com/rebase/rebase.html) based on sequence homology to methylases with known targets (35). However, S. oneidensis MR-1 has four putative type II methyltransferases, which is more than the number of methylated motifs detected once the three type-I-like bipartite motifs were excluded. All putative methyltransferases were expressed under these growth conditions, suggesting either redundancy among the methyltransferases in targeted motifs or possible misannotation of some genes.
Table 1.
Sequence motifs containing N6mA modifications (underlined bases) in S. oneidensis MR-1 grown aerobically on minimal mediuma
| Motif | No. of motifs |
No. (%) of motifs with methylation state |
|||||
|---|---|---|---|---|---|---|---|
| Total | CDS | Intergenic | Methylated | Hemi | Not | Conflict | |
| GATC | 16,376 | 14,328 | 2,048 | 16,281 (99.4) | 0 | 2 | 93 |
| ATCGAT | 2,342 | 2,052 | 290 | 2,292 (97.9) | 3 | 1 | 46 |
| GCAN4GTC/GACN4TGC | 1,803 | 1,543 | 260 | 1,793 (99.4) | 1 | 0 | 9 |
| TACN6GTNGT/ACNACN6GTA | 306 | 246 | 60 | 301 (98.4) | 0 | 0 | 5 |
| TGAYN6TGAC/GTCAN6RTCA | 291 | 243 | 48 | 282 (96.9) | 1 | 5 | 3 |
The total number of motifs and their locations in either coding (CDS) or intergenic regions were determined. The methylation state of motifs with <25× coverage could not be confidently determined, and these were excluded from these counts. “Methylated” motifs were modified on both strands, while “Hemi” motifs were modified on only one strand. Motifs with disagreements among replicates were in “Conflict.” Methylated bases occurring simultaneously in two motifs were counted toward each motif, e.g., the 225 instances of the methylated sequence ATCGATC were included in the tallies of both GATC and ATCGAT motifs.
To identify the specific target of each methyltransferase, we performed SMRT sequencing on methyltransferase transposon mutants. In these tests, a target motif was assigned to a predicted methyltransferase if the motif was not methylated in the transposon mutant. For example, the sequence motif ATCGAT was no longer methylated in a mutant lacking the putative methyltransferase SOA0004, thus indicating the target of this methyltransferase. The three bipartite motifs were also clearly assigned to the three predicted type I methyltransferases using this approach (Table 2). However, the protein(s) targeting GATC could not be identified unequivocally because this motif was always methylated in the individual transposon mutants. Interestingly, we were unable to generate a viable transposon mutant for predicted methyltransferase SO_0289, suggesting that this gene maybe required for viability. Two of the putative methyltransferase genes show homology to dam (SO_0289 and SO_0690) and were suspected to target GATC (Table 2). The gene SO_3004 does not show strong homology to dam but was also predicted in REBASE to target GATC. Thus, it appears that S. oneidensis MR-1 may use multiple genes to methylate GATC. Similar redundancies have been observed in Escherichia coli, which has three separate enzymes that methylate GATC (3, 22). In addition, some strains of Salmonella enterica may have up to five copies of dam genes (see Table S3 in the supplemental material). The potential redundancy of GATC-targeting methyltransferases and the lack of corresponding restriction enzymes suggest that S. oneidensis MR-1 may use GATC methylation in some regulatory capacity.
Table 2.
Assigning target motifs to putative methyltransferases using SMRT sequencing of gene knockout mutantsa
| Locus ID | R-M system | Predicted motif | Confirmed motif |
|---|---|---|---|
| SO_0383 | Type I | TGAYN6TGAC/GTCAN6RTCA | |
| SO_1457 | Type Ib | GCAN4GTC/GACN4TGC | |
| SO_4265 | Type I | TACN6GTNGT/ACNACN6GTA | |
| SO_0289 | Type II | GATC | ND |
| SO_0690 | Type II | ND | |
| SO_3004 | Type II | GATC | ND |
| SO_A0004 | Type II | ATCGAT | ATCGAT |
Underlined bases were methylated. Target motifs were predicted for three putative methyltransferase in REBASE. Loss of a methylated motif in mutants confirmed the target of the knocked-out gene. Motifs for three predicted methyltransferases were not detected (ND). No mutant was available for SO_0289, whereas no methylated motifs were lost for SO_0690 and SO_3004.
Type II according to REBASE but annotated as type I elsewhere (e.g., GenBank and COG).
To examine the efficiencies of methyltransferases in S. oneidensis MR-1, we determined the methylation state for each instance of the five sequence motifs throughout the genome. After excluding 418 motif locations with low sequence coverage, we found that 99.2% of the remaining 21,118 motif locations were methylated on both strands in all three replicates. The degree of methylation varied by motif, from as low as 96.9% to greater than 99.9% (Table 1). Interestingly, 5 sites were confidently hemimethylated, i.e., methylated on only one strand, in all three replicates, whereas 8 sites were not methylated on either strand (see Table S2 in the supplemental material). Five of the nonmethylated sites were in type I R-M motifs, two were located in GATC motifs, and one was found in an ATCGAT motif (Table 1; see also Table S2). Nonmethylated and hemimethylated sites have been detected in other bacteria (8, 20, 22, 23, 51, 52), and these sites are often protected from methyltransferases by DNA binding proteins.
Changes in DNA methylation can impact gene expression levels by altering the binding affinities of regulatory proteins. For example, the binding of leucine-responsive protein (Lrp), which regulates expression levels of the pap operon in E. coli, is controlled by the methylation state of two GATC sites in the Lrp binding site (8, 53). In S. oneidensis MR-1, GATC motifs are commonly found in the binding sites for the transcription factors Crp and Fnr (32). Indeed, 64 of the 174 Crp binding sites and 21 of 30 Fnr binding sites have one or more type II motifs (Table 3). This enrichment of GATC motifs in transcription factor binding sites presents an opportunity for methylation to impact gene expression on a large scale. That is, if DNA binding activities of Crp and Fnr are sensitive to the DNA methylation, then changes in the methylation of GATC sites could potentially influence gene expression in S. oneidensis MR-1.
Table 3.
Transcription factor binding sites containing type II motifsa
| Regulator | No. of sites |
No. of operons regulated | No. of genes regulated | No. of differentially expressed genes |
No. of differentially methylated sites |
|||
|---|---|---|---|---|---|---|---|---|
| Total | With motif | Rich medium | Anaerobic | Rich medium | Anaerobic | |||
| ArgR | 34 | 1 | 22 | 39 | 30 (4) | 13 (11) | 0 | 0 |
| Crp | 174 | 64 | 150 | 286 | 69 (48) | 12 (6) | 0 | 0 |
| Fnr | 30 | 21 | 26 | 73 | 34 (28) | 8 (6) | 0 | 0 |
| Fur | 32 | 1 | 32 | 58 | 12 (7) | 7 (2) | 1 | 0 |
| ModE | 2 | 2 | 2 | 4 | 0 | 0 | 0 | 0 |
| SO1578 | 2 | 2 | 2 | 3 | 0 | 0 | 0 | 0 |
| SO3385 | 1 | 1 | 1 | 3 | 0 | 0 | 0 | 0 |
| SO3393 | 6 | 2 | 2 | 2 | 1 (1) | 0 | 0 | 0 |
The numbers of genes and operons regulated by these transcription factors, as well as the number of genes differentially expressed when transferred to either rich medium or anaerobic minimal medium, are indicated. The numbers of upregulated genes are indicated in parentheses next to the total number of differentially expressed genes. Binding sites with different methylation states under different growth conditions are also displayed.
Dynamics of DNA methylation and gene expression.
To explore changes in genome-wide methylation patterns and their possible impact on gene expression, we measured DNA methylation and mRNA levels in exponential-phase S. oneidensis MR-1 cultures grown under various conditions. More specifically, triplicate cultures grown aerobically in minimal medium were first analyzed by SMRT sequencing and RNAseq and then analyzed again after transfer to either aerobic rich medium or anaerobic minimal medium. Not surprisingly, gene expression varied from one condition to another, with 426 genes differentially expressed between minimal and rich media (201 upregulated and 225 downregulated) and 99 genes differentially expressed between aerobic and anaerobic conditions (51 upregulated and 48 downregulated) (Fig. 2; see also Tables S4 and S5 in the supplemental material).
Fig 2.
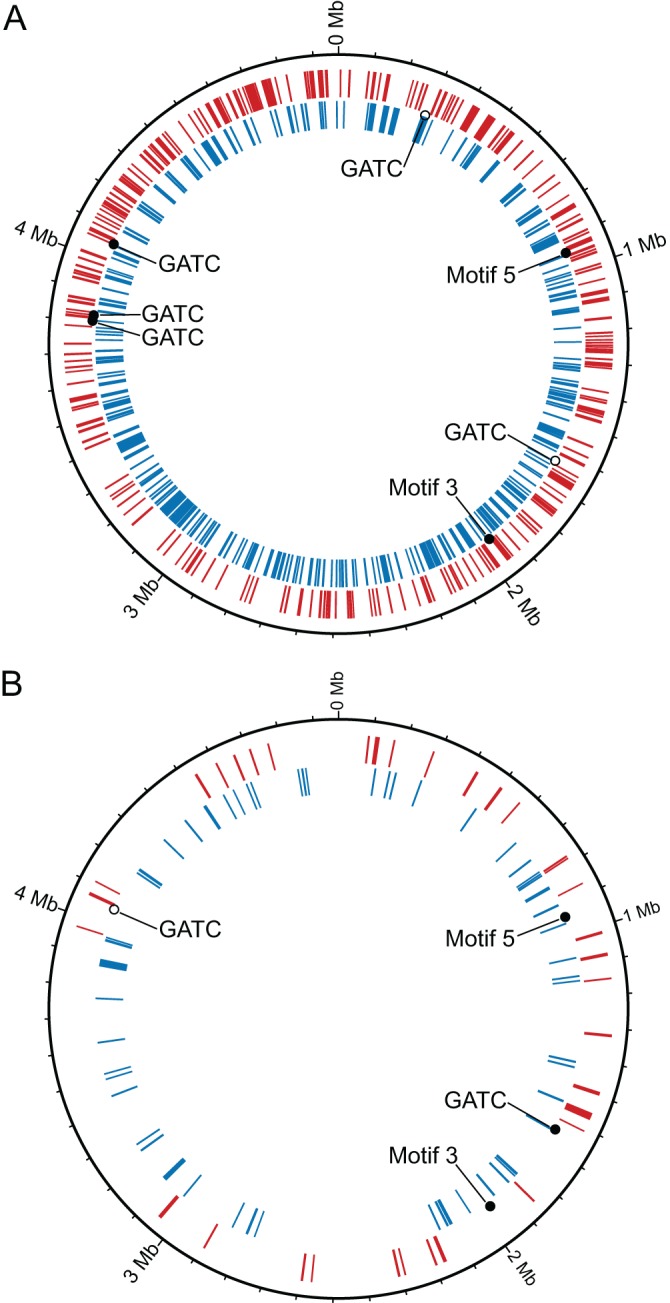
Locations of differentially expressed genes (red and blue rings) and differentially methylated motifs in S. oneidensis MR-1 cultures transferred from aerobic minimal medium to rich medium (A) or anaerobic minimal medium (B). Upregulated genes are red, and downregulated genes are blue. Motif locations that became methylated (●), hemimethylated (◒), or nonmethylated (○) after transfer to different culture conditions are indicated. The methylated motifs are as follows: GCAN4GTC/GACN4TGC (motif 3), TACN6GTNGT/ACNACN6GTA (motif 4), and TGAYN6TGAC/GTCAN6RTCA (motif 5).
We did not observe widespread changes in methylation in cultures growing under different conditions. However, reproducible differences in methylation state were observed at 8 sites (Table 4; see also Table S2 in the supplemental material). Of these sites, 5 were GATC motifs located within intergenic regions. One of these GATC sites (genome position 4,061,174) was located within the binding site of a ferric uptake regulator protein (Fur) transcription factor (Table 4), although neither gene regulated by this transcription factor was differentially expressed. The methylation state of two other GATC motifs flanking another Fur binding site (genome positions 3,823,765 and 3,823,792) also differed between minimal and rich media. One of the nine genes (SO_3667) regulated by this binding site was upregulated when cultures were transferred to rich medium (see Table S4). Finally, a differentially methylated GATC motif was detected 215 bp upstream of argC (SO_0275), a gene involved in arginine synthesis. The expression of argC and four other genes in the same operon was repressed when cultures were transferred from minimal medium lacking arginine into rich medium containing arginine (see Table S4). The latter two cases were suggestive of a possible connection between DNA methylation and gene expression, although the nature of the connection remains unclear. That is, did changes in the methylation state influence expression levels, or did binding of regulatory proteins inadvertently protect these sites from methylation? Establishing a direct causal link was not possible with the current data set and will require additional future investigations.
Table 4.
Genomic contexts of motifs with differences in methylation state among conditionsa
| Position (+/−) | Motif | Methylation stateb |
Genomic context |
||||
|---|---|---|---|---|---|---|---|
| Min medium, aerobic | Rich medium | Min medium, anaerobic | Left flanking gene | Within gene | Right flanking gene | ||
| 280309/280310 | GATC | ● | ○ | ● | ← SO_0274 | SO_0275 → | |
| 1642124/1642125 | GATC | ○ | ● | ← SO_1563 | SO_1565 → | ||
| 3823765/3823766 | GATC | ○ | ● | ○ | ← SO_3669 | SO_3670 → | |
| 3823792/3823793 | GATC | ○ | ● | ○ | ← SO_3669 | SO_3670 → | |
| 4061174/4061175* | GATC | ● | ○ | ← SO_3914 | SO_3915 → | ||
| 1965318/1965324 | GCAN4GTC/GACN4TGC | ◒ | ● | ● | → SO_1871 | SO_1872 ← | |
| 938801/938809 | TGAYN6TGAC/GTCAN6RTCA | ○ | ● | ● | SO_0912 | ||
| 160490/160498 P | TGAYN6TGAC/GTCAN6RTCA | ◒ | ● | SO_A0172 | |||
Left and right flanking genes, as well as their orientation, are provided for motifs found within intergenic regions. Motifs were upstream of a left flanking gene if the arrow points to the left and upstream of right flanking genes if the arrow points to the right. “P” indicates a motif located in the plasmid, while “∗” indicates a motif in a Fur transcription factor binding site.
Min, minimal; ●, methylated; ○, nonmethylated; ◒, hemimethylated.
While these methylation dynamics are intriguing, it is clear that widespread changes in gene expression were not accompanied by equally widespread changes in DNA methylation (Fig. 2). Nor was there an obvious relationship between methylation at known transcription factor binding sites and expression. For example, 69 Crp-regulated genes were differentially expressed when cultures were transferred from minimal medium to rich medium, but none of the Crp binding sites were differentially methylated (Table 3; see also Tables S4 and S5 in the supplemental material). The only differentially methylated transcription factor binding site we observed did not exhibit a significant difference in expression for the corresponding genes. It is worth noting that Shewanella cultures were not synchronized, and it is conceivable that short-term changes in methylation state and gene expression within a small subset of the culture could have gone undetected. Still, while methylation may impact the expression of a few genes, our data suggest that methylation does not play a large and direct role in regulating gene expression in S. oneidensis MR-1, at least not under the conditions we tested.
To our knowledge, this is the first study to directly measure the genome-wide dynamics of methylation and gene expression in a wild-type bacterium. Three previous studies examined gene expression in E. coli mutants lacking dam (Dam−), and in each case the expression patterns of the mutants differed from those of the wild-type strain (54–56). However, support for a direct regulatory role for methylation was inconsistent among studies. For example, Oshima et al. 54) found that a disproportionately large number of the differentially expressed genes in Dam− mutants were regulated by transcription factors with GATC motifs within their binding sites. In contrast, Lobner-Olesen et al. (55) and Robbins-Menke et al. (56) found that the regulatory regions of differentially expressed genes were not enriched with GATC for both Dam− and Dam-overproducing mutants. While there are clear cases where methylation directly regulates gene expression in E. coli (57), the last two studies suggest that most differential gene expression in Dam mutants results from indirect effects of variable Dam concentrations on cell physiology, not from direct regulation of expression via methylation. Similarly, >30% of genes in E. coli C227-11 were differentially expressed after deleting the RM.EcoGIII methyltransferase, yet there were no clear connections to RM.EcoGIII methylation sites for most of these genes (22). These studies highlight the challenges of interpreting a direct regulatory roles for DNA methylation by examining methyltransferase knockout mutants. Monitoring methylation dynamics in wild-type bacteria, which is now possible using SMRT sequencing, should provide a clearer picture of the regulatory roles of DNA methylation in bacteria.
Roles for DNA methylation in genome replication and DNA mismatch repair.
If DNA methylation is not a major regulator of gene expression in S. oneidensis MR-1, then why does its genome encode orphan methyltransferases? One possibility is that DNA methylation plays a critical role in DNA mismatch repair and/or genome replication. For example, S. oneidensis MR-1 contains the DNA mismatch repair gene mutH, which in E. coli nicks the unmethylated strand near hemimethylated GATC sites when mismatches occur during genome replication. This enables removal of the unmethylated strand and resynthesis of the correct sequence from the methylated template (7). In addition, the nonuniform distribution of GATC sites throughout the genome suggests that S. oneidensis MR-1 may use DNA methylation to regulate genome replication in a manner similar to that of E. coli. Specifically, the origin of replication (oriC) is enriched with GATC sites in both organisms (Fig. 3; see also Fig. S1 in the supplemental material), and in E. coli the methylation state of these sites plays an important role in regulating genome replication (58–61). Briefly, genome replication in E. coli is initiated in part by the binding of DnaA to the origin of replication. Shortly after replication begins, SeqA binds to newly formed hemimethylated GATC sites in the origin, thus preventing additional binding of DnaA and the reinitiation of replication (58, 62). SeqA also binds to hemimethylated GATC sites in the promoter region of dnaA and reduces transcription of dnaA once the replication fork has passed (6, 63), thereby decreasing the chance of replication reinitiation. We hypothesize that S. oneidensis MR-1 uses similar mechanisms to control DNA replication based on the presence of dam and seqA in the genome as well as enrichment of GATC sites in the oriC.
Fig 3.
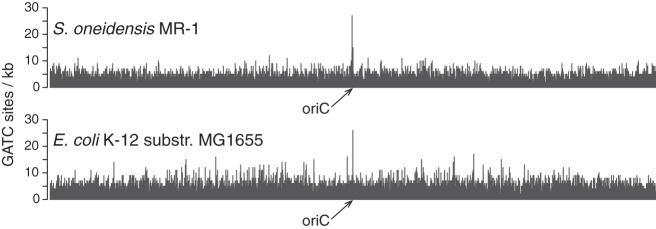
Distribution of GATC motifs throughout the genomes of S. oneidensis MR-1 and E. coli. The arrow indicates the origin of replication (oriC).
To determine if DNA methylation regulates genome replication and mismatch repair in S. oneidensis MR-1, we attempted to construct dam and seqA mutants. DNA replication and cell division are not synchronized in Δdam and ΔseqA mutants of E. coli (63, 64), and we wished to observe if the same was true for S. oneidensis MR-1. However, both dam (locus tag SO_0289) and seqA appear to be essential for viability as determined by high-throughput transposon mutagenesis and sequencing (TnSEQ) (65) (K. M. Wetmore, M. Blow, and A. Deutschbauer, unpublished data), so mutants could not be recovered. dam is also essential for fellow Gammaproteobacteria members Vibrio cholerae and Yersinia pseudotuberculosis, where it plays a role in regulating genome replication (66, 67). Interestingly, mutants were viable for the two other S. oneidensis MR-1 genes that either showed some homology to dam (SO_0690) or were predicted to methylate GATC by REBASE (SO_3004). Both genes were expressed in wild-type S. oneidensis MR-1, indicating that they were functional but not completely redundant to SO_0289. The nonviability of both dam and seqA mutants suggests that methylation of GATC motifs plays a critical functional role in S. oneidensis MR-1, mostly likely in regulating genome replication.
Next, we asked if other Shewanella species might utilize methylation to regulate genome replication or mismatch repair by searching their genomes for dam, seqA, mutH, and GATC enrichment around the origin of replication. In addition to S. oneidensis MR-1, there are 21 finished Shewanella genomes isolated from diverse environments, including marine and freshwater sediments, hydrothermal vents, and the nidamental gland of a squid. Nineteen isolates encoded the mismatch repair protein MutH, whereas all 21 isolates were positive for dam, seqA, and GATC enrichment around oriC (Fig. 4; see also Table S3 in the supplemental material), the same pattern observed in S. oneidensis MR-1 and E. coli. This suggests that regulation of genome replication through DNA methylation might be a universal feature of the Shewanellaceae family.
Fig 4.

Phylogenetic patterns of DNA methylation usage in the Gammaproteobacteria. The presence of dam, seqA, and GATC enrichment at the origin of replication is illustrated on a phylogenetic tree constructed from the 16S sequences of 448 finished genomes. The scale bar represents the fraction of substitutions per site. The star indicates a node beyond which all but 24 genomes encoded dam and seqA, and were enriched for GATC motifs in the origin of replication. Although this node does not have strong bootstrap support based on 16S rRNA sequences, this node was supported by a rare indel in RpoB (70, 71).
Looking beyond the Shewanellaceae, we searched all finished Gammaproteobacteria genomes for the presence of dam, mutH, seqA, and GATC enrichment in oriC. Of the 448 finished Gammaproteobacteria genomes in the Integrated Microbial Genomes database (42), 331 encoded one or more putative dam genes, while 266 encoded seqA and 278 encoded mutH (Fig. 4; see also Fig. S2 and Table S3 in the supplemental material). Interestingly, virtually all Gammaproteobacteria arising after a single evolutionary branch point were positive for dam, mutH, seqA, and GATC enrichment at oriC (Fig. 4; see also Fig. S2). There were 24 exceptions beyond this branch point that lacked seqA and GATC enrichment, and of these, 23 were endosymbionts with massively reduced genomes (see Table S3). Genome reduction, including loss of regulatory elements, is a common feature of endosymbionts (68), and it appears that both dam and seqA were lost during these reductions. Glaciecola nitratireducens FR1064 is not an endosymbiont, but its genome is >1Mbp smaller than those of the two other sequenced members of the genus Glaciecola (69), suggesting it too may have lost seqA during genome reduction. While some basal lineages within the Gammaproteobacteria encoded dam and/or mutH genes, none encoded seqA or showed enrichment of GATC sites at the origin of replication (Fig. 4). Using the limited number of genomes available at the time, Lobner-Olesen et al. (3) identified a “DamMT clade” within the Gammaproteobacteria. Our more comprehensive analysis generally agrees with this earlier report, although it would be more appropriate to discuss a “seqA/GATC-oriC clade” since dam and mutH were not exclusive to one clade of the Gammaproteobacteria (Fig. 4).
The development of epigenetic regulation of genome replication appears to be a key evolutionary event within the Gammaproteobacteria. The phylogenetic pattern of dam, seqA, and GATC enrichment at the origin indicates that this mechanism for regulating chromosome replication via DNA methylation was acquired and maintained by more recent lineages, not lost by the more basal groups within the clade. Moreover, this mechanism has been maintained throughout the evolution of multiple families, even as these groups have diversified and expanded into a wide range of different aquatic, terrestrial, and host-associated environments. Dam has even become essential for viability of some Gammaproteobacteria, such as S. oneidensis MR-1 (this study), Vibrio cholerae, and Yersina pseudotuberculosis (66). The maintenance of dam, seqA, and GATC enrichment at oriC throughout their evolutionary history suggests that many members of the Gammaproteobacteria, regardless of their environment, experience strong selective pressure to synchronize genome replication with cell division and that more recently evolved lineages do so with DNA methylation. Presumably, the more basal lineages of Gammaproteobacteria lacking seqA and GATC enrichment at oriC use alternative strategies. Interestingly, many of these basal gammaproteobacteria lineages encode dam, but its functional role remains a mystery since they do not appear to use methylation for DNA mismatch repair or genome replication. Further analyses with SMRT sequencing will shed additional light on the role of DNA methylation in these bacteria.
Conclusions.
DNA methylation appears to serve a variety of functions in S. oneidensis MR-1, including restriction/modification, DNA mismatch repair, and regulation of genome replication. Methylation may also regulate expression of a few genes, but it does not appear to be a major regulator of gene expression. This connection to gene expression is one of the more interesting, but poorly understood, facets of DNA methylation, and it remains to be seen if it is a major regulator of expression in any bacterium. However, with the exception of E. coli and Caulobacter crescentus, our understanding of DNA methylation is extremely limited for the vast majority of prokaryotes. The further application of SMRT sequencing will dramatically expand our understanding of DNA methylation in these understudied clades. Large-scale surveys of diverse microbial groups using SMRT sequencing will help provide new insights into the scope and variety of DNA methylation in various phylogenetic groups, whereas analyses of wild-type and knockout mutants will help uncover and experimentally verify the functional roles of methylation.
Supplementary Material
ACKNOWLEDGMENT
The DOE Joint Genome Institute was supported by the US Department of Energy Office of Science under contract number DE-AC02-05CH11231.
Footnotes
Published ahead of print 30 August 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JB.00935-13.
REFERENCES
- 1.Casadesus J, Low D. 2006. Epigenetic gene regulation in the bacterial world. Microbiol. Mol. Biol. Rev. 70:830–856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Low DA, Casadesus J. 2008. Clocks and switches: bacterial gene regulation by DNA adenine methylation. Curr. Opin. Microbiol. 11:106–112 [DOI] [PubMed] [Google Scholar]
- 3.Lobner-Olesen A, Skovgaard O, Marinus MG. 2005. Dam methylation: coordinating cellular processes. Curr. Opin. Microbiol. 8:154–160 [DOI] [PubMed] [Google Scholar]
- 4.Bickle TA, Kruger DH. 1993. Biology of DNA Restriction. Microbiol. Rev. 57:434–450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vasu K, Nagaraja V. 2013. Diverse functions of restriction-modification systems in addition to cellular defense. Microbiol. Mol. Biol. Rev. 77:53–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Katayama T, Ozaki S, Keyamura K, Fujimitsu K. 2010. Regulation of the replication cycle: conserved and diverse regulatory systems for DnaA and oriC. Nat. Rev. Microbiol. 8:163–170 [DOI] [PubMed] [Google Scholar]
- 7.Modrich P, Lahue R. 1996. Mismatch repair in replication fidelity, genetic recombination, and cancer biology. Annu. Rev. Biochem. 65:101–133 [DOI] [PubMed] [Google Scholar]
- 8.Braaten BA, Platko JV, Vanderwoude MW, Simons BH, Degraaf FK, Calvo JM, Low DA. 1992. Leucine-responsive regulatory protein controls the expression of both the pap and fan pili operons in Escherichia coli. Proc. Natl. Acad. Sci. U. S. A. 89:4250–4254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Braun RE, Wright A. 1986. DNA methylation differentially enhances the expression of one of the 2 Escherichia coli dnaA promoters in vivo and in vitro. Mol. Gen. Genet. 202:246–250 [DOI] [PubMed] [Google Scholar]
- 10.Brunet YR, Bernard CS, Gavioli M, Lloubes R, Cascales E. 2011. An epigenetic switch involving overlapping fur and DNA methylation optimizes expression of a type VI secretion gene cluster. Plos Genet. 7:e1002205. 10.1371/journal.pgen.1002205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Camacho EM, Casadesus J. 2005. Regulation of traJ transcription in Salmonella virulence plasmid by strand-specific DNA adenine hemimethylation. Mol. Microbiol. 57:1700–1718 [DOI] [PubMed] [Google Scholar]
- 12.Srikhanta YN, Fox KL, Jennings MP. 2010. The phasevarion: phase variation of type III DNA methyltransferases controls coordinated switching in multiple genes. Nat. Rev. Microbiol. 8:196–206 [DOI] [PubMed] [Google Scholar]
- 13.Hernday AD, Braaten BA, Low DA. 2003. The mechanism by which DNA adenine methylase and PapI activate the pap epigenetic switch. Mol. Cell 12:947–957 [DOI] [PubMed] [Google Scholar]
- 14.Lim HN, van Oudenaarden A. 2007. A multistep epigenetic switch enables the stable inheritance of DNA methylation states. Nat. Genet. 39:269–275 [DOI] [PubMed] [Google Scholar]
- 15.Kahramanoglou C, Prieto AI, Khedkar S, Haase B, Gupta A, Benes V, Fraser GM, Luscombe NM, Seshasayee AS. 2012. Genomics of DNA cytosine methylation in Escherichia coli reveals its role in stationary phase transcription. Nat. Commun. 3:886. 10.1038/ncomms1878 [DOI] [PubMed] [Google Scholar]
- 16.Bormann Chung CA, Boyd VL, McKernan KJ, Fu Y, Monighetti C, Peckham HE, Barker M. 2010. Whole methylome analysis by ultra-deep sequencing using two-base encoding. PLoS One 5:e9320. 10.1371/journal.pone.0009320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ehrlich M, Wilson GG, Kuo KC, Gehrke CW. 1987. N4-methylcytosine as a minor base in bacterial DNA. J. Bacteriol. 169:939–943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang MX, Church GM. 1992. A whole genome approach to in vivo DNA-protein interactions in Escherichia coli. Nature 360:606–610 [DOI] [PubMed] [Google Scholar]
- 19.Ringquist S, Smith CL. 1992. The Escherichia coli chromosome contains specific, unmethylated dam and dcm sites. Proc. Natl. Acad. Sci. U. S. A. 89:4539–4543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hale WB, Vanderwoude MW, Low DA. 1994. Analysis of nonmethylated GATC sites in the Escherichia coli chromosome and identification of sites that are differentially methylated in response to environmental stimuli. J. Bacteriol. 176:3438–3441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Flusberg BA, Webster DR, Lee JH, Travers KJ, Olivares EC, Clark TA, Korlach J, Turner SW. 2010. Direct detection of DNA methylation during single-molecule, real-time sequencing. Nat. Methods 7:461–465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fang G, Munera D, Friedman DI, Mandlik A, Chao MC, Banerjee O, Feng Z, Losic B, Mahajan MC, Jabado OJ, Deikus G, Clark TA, Luong K, Murray IA, Davis BM, Keren-Paz A, Chess A, Roberts RJ, Korlach J, Turner SW, Kumar V, Waldor MK, Schadt EE. 2012. Genome-wide mapping of methylated adenine residues in pathogenic Escherichia coli using single-molecule real-time sequencing. Nat. Biotechnol. 30:1232–1239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Clark TA, Murray IA, Morgan RD, Kislyuk AO, Spittle KE, Boitano M, Fomenkov A, Roberts RJ, Korlach J. 2012. Characterization of DNA methyltransferase specificities using single-molecule, real-time DNA sequencing. Nucleic Acids Res. 40:e29. 10.1093/nar/gkr1146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Murray IA, Clark TA, Morgan RD, Boitano M, Anton BP, Luong K, Fomenkov A, Turner SW, Korlach J, Roberts RJ. 2012. The methylomes of six bacteria. Nucleic Acids Res. 40:11450–11462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Boukhalfa H, Icopini GA, Reilly SD, Neu MP. 2007. Plutonium(IV) reduction by the metal-reducing bacteria Geobacter metallireducens GS15 and Shewanella oneidensis MR1. Appl. Environ. Microbiol. 73:5897–5903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fredrickson JK, Romine MF, Beliaev AS, Auchtung JM, Driscoll ME, Gardner TS, Nealson KH, Osterman AL, Pinchuk G, Reed JL, Rodionov DA, Rodrigues JLM, Saffarini DA, Serres MH, Spormann AM, Zhulin IB, Tiedje JM. 2008. Towards environmental systems biology of Shewanella. Nat. Rev. Microbiol. 6:592–603 [DOI] [PubMed] [Google Scholar]
- 27.Myers CR, Nealson KH. 1988. Bacterial manganese reduction and growth with manganese oxide as the sole electron acceptor. Science (New York, NY) 240:1319–1321 [DOI] [PubMed] [Google Scholar]
- 28.Caccavo F, Blakemore RP, Lovley DR. 1992. A hydrogen-oxidizing, Fe(Iii)-reducing microorganism from the Great Bay Estuary, New Hampshire. Appl. Environ. Microbiol. 58:3211–3216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hau HH, Gralnick JA. 2007. Ecology and biotechnology of the genus Shewanella. Annu. Rev. Microbiol. 61:237–258 [DOI] [PubMed] [Google Scholar]
- 30.Kolker E, Picone AF, Galperin MY, Romine MF, Higdon R, Makarova KS, Kolker N, Anderson GA, Qiu X, Auberry KJ, Babnigg G, Beliaev AS, Edlefsen P, Elias DA, Gorby YA, Holzman T, Klappenbach JA, Konstantinidis KT, Land ML, Lipton MS, McCue L-A, Monroe M, Pasa-Tolic L, Pinchuk G, Purvine S, Serres MH, Tsapin S, Zakrajsek BA, Zhu W, Zhou J, Larimer FW, Lawrence CE, Riley M, Collart FR, Yates JR, Smith RD, Giometti CS, Nealson KH, Fredrickson JK, Tiedje JM. 2005. Global profiling of Shewanella oneidensis MR-1: expression of hypothetical genes and improved functional annotations. Proc. Natl. Acad. Sci. U. S. A. 102:2099–2104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Price MN, Deutschbauer AM, Skerker JM, Wetmore KM, Ruths T, Mar JS, Kuehl JV, Shao WJ, Arkin AP. 2013. Indirect and suboptimal control of gene expression is widespread in bacteria. Mol. Syst. Biol. 9:660. 10.1038/msb.2013.16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Novichkov PS, Laikova ON, Novichkova ES, Gelfand MS, Arkin AP, Dubchak I, Rodionov DA. 2010. RegPrecise: a database of curated genomic inferences of transcriptional regulatory interactions in prokaryotes. Nucleic Acids Res. 38:D111–D118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Deutschbauer A, Price MN, Wetmore KM, Shao W, Baumohl JK, Xu Z, Nguyen M, Tamse R, Davis RW, Arkin AP. 2011. Evidence-based annotation of gene function in Shewanella oneidensis MR-1 using genome-wide fitness profiling across 121 conditions. Plos Gen. 7:e1002385. 10.1371/journal.pgen.1002385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Heidelberg JF, Paulsen IT, Nelson KE, Gaidos EJ, Nelson WC, Read TD, Eisen JA, Seshadri R, Ward N, Methe B, Clayton RA, Meyer T, Tsapin A, Scott J, Beanan M, Brinkac L, Daugherty S, DeBoy RT, Dodson RJ, Durkin AS, Haft DH, Kolonay JF, Madupu R, Peterson JD, Umayam LA, White O, Wolf AM, Vamathevan J, Weidman J, Impraim M, Lee K, Berry K, Lee C, Mueller J, Khouri H, Gill J, Utterback TR, McDonald LA, Feldblyum TV, Smith HO, Venter JC, Nealson KH, Fraser CM. 2002. Genome sequence of the dissimilatory metal ion-reducing bacterium Shewanella oneidensis. Nat. Biotechnol. 20:1118–1123 [DOI] [PubMed] [Google Scholar]
- 35.Roberts RJ, Vincze T, Posfai J, Macelis D. 2010. REBASE—a database for DNA restriction and modification: enzymes, genes and genomes. Nucleic Acids Res. 38:D234–D236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Travers KJ, Chin CS, Rank DR, Eid JS, Turner SW. 2010. A flexible and efficient template format for circular consensus sequencing and SNP detection. Nucleic Acids Res. 38:e159. 10.1093/nar/gkq543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chaisson MJ, Tesler G. 2012. Mapping single molecule sequencing reads using basic local alignment with successive refinement (BLASR): application and theory. BMC Bioinformatics 13:238. 10.1186/1471-2105-13-238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Parkhomchuk D, Borodina T, Amstislavskiy V, Banaru M, Hallen L, Krobitsch S, Lehrach H, Soldatov A. 2009. Transcriptome analysis by strand-specific sequencing of complementary DNA. Nucleic Acids Res. 37:e123. 10.1093/nar/gkp596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Anders S, Huber W. 2010. Differential expression analysis for sequence count data. Genome Biol. 11:R106. 10.1186/gb-2010-11-10-r106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Robinson MD, McCarthy DJ, Smyth GK. 2010. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26:139–140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hong F, Breitling R, McEntee CW, Wittner BS, Nemhauser JL, Chory J. 2006. RankProd: a bioconductor package for detecting differentially expressed genes in meta-analysis. Bioinformatics 22:2825–2827 [DOI] [PubMed] [Google Scholar]
- 42.Markowitz VM, Chen IM, Palaniappan K, Chu K, Szeto E, Grechkin Y, Ratner A, Jacob B, Huang J, Williams P, Huntemann M, Anderson I, Mavromatis K, Ivanova NN, Kyrpides NC. 2012. IMG: the Integrated Microbial Genomes database and comparative analysis system. Nucleic Acids Res. 40:D115–D122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gao F, Luo H, Zhang CT. 2013. DoriC 5.0: an updated database of oriC regions in both bacterial and archaeal genomes. Nucleic Acids Res. 41:D90–D93. 10.1093/nar/gks990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gao F, Zhang CT. 2007. DoriC: a database of oriC regions in bacterial genomes. Bioinformatics 23:1866–1867 [DOI] [PubMed] [Google Scholar]
- 45.Gao F, Zhang CT. 2008. Ori-Finder: a web-based system for finding oriCs in unannotated bacterial genomes. BMC Bioinformatics 9:79. 10.1186/1471-2105-9-79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Katoh K, Toh H. 2008. Recent developments in the MAFFT multiple sequence alignment program. Brief. Bioinform. 9:286–298 [DOI] [PubMed] [Google Scholar]
- 47.Stamatakis A. 2006. RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 22:2688–2690 [DOI] [PubMed] [Google Scholar]
- 48.Gommers-Ampt JH, Borst P. 1995. Hypermodified bases in DNA. FASEB J. 9:1034–1042 [DOI] [PubMed] [Google Scholar]
- 49.Iyer LM, Zhang D, Maxwell Burroughs A, Aravind L. 2013. Computational identification of novel biochemical systems involved in oxidation, glycosylation and other complex modifications of bases in DNA. Nucleic Acids Res. 2013:1–21. 10.1093/nar/gkt573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dryden DTF, Murray NE, Rao DN. 2001. Nucleoside triphosphate-dependent restriction enzymes. Nucleic Acids Res. 29:3728–3741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lluch-Senar M, Luong K, Llorens-Rico V, Delgado J, Fang G, Spittle K, Clark TA, Schadt E, Turner SW, Korlach J, Serrano L. 2013. Comprehensive methylome characterization of Mycoplasma genitalium and Mycoplasma pneumoniae at single-base resolution. Plos Genet. 9:e1003191. 10.1371/journal.pgen.1003191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tavazoie S, Church GM. 1998. Quantitative whole-genome analysis of DNA-protein interactions by in vivo methylase protection in E. coli. Nat. Biotechnol. 16:566–571 [DOI] [PubMed] [Google Scholar]
- 53.Low DA, Weyand NJ, Mahan MJ. 2001. Roles of DNA adenine methylation in regulating bacterial gene expression and virulence. Infect. Immun. 69:7197–7204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Oshima T, Wada C, Kawagoe Y, Ara T, Maeda M, Masuda Y, Hiraga S, Mori H. 2002. Genome-wide analysis of deoxyadenosine methyltransferase-mediated control of gene expression in Escherichia coli. Mol. Microbiol. 45:673–695 [DOI] [PubMed] [Google Scholar]
- 55.Lobner-Olesen A, Marinus MG, Hansen FG. 2003. Role of SeqA and Dam in Escherichia coli gene expression: a global/microarray analysis. Proc. Natl. Acad. Sci. U. S. A. 100:4672–4677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Robbins-Manke JL, Zdraveski ZZ, Marinus M, Essigmann JM. 2005. Analysis of global gene expression and double-strand-break formation in DNA adenine methyltransferase- and mismatch repair-deficient Escherichia coli. J. Bacteriol. 187:7027–7037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Casadesus J, Low DA. 2013. Programmed heterogeneity: epigenetic mechanisms in bacteria. J. Biol. Chem. 288:13929–13935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Slater S, Wold S, Lu M, Boye E, Skarstad K, Kleckner N. 1995. E. coli SeqA protein binds oriC in two different methyl-modulated reactions appropriate to its roles in DNA replication initiation and origin sequestration. Cell 82:927–936 [DOI] [PubMed] [Google Scholar]
- 59.Boye E, Lobner-Olesen A, Skarstad K. 2000. Limiting DNA replication to once and only once. EMBO Rep. 1:479–483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Oka A, Sugimoto K, Takanami M, Hirota Y. 1980. Replication origin of the Escherichia coli K-12 chromosome—the size and structure of the minimum DNA segment carrying the information for autonomous replication. Mol. Gen. Genet. 178:9–20 [DOI] [PubMed] [Google Scholar]
- 61.Ogden GB, Pratt MJ, Schaechter M. 1988. The replicative origin of the Escherichia coli chromosome binds to cell membranes only when hemimethylated. Cell 54:127–135 [DOI] [PubMed] [Google Scholar]
- 62.von Freiesleben U, Rasmussen KV, Schaechter M. 1994. Seqa limits DnaA activity in replication from oriC in Escherichia coli. Mol. Microbiol. 14:763–772 [DOI] [PubMed] [Google Scholar]
- 63.Lu M, Campbell JL, Boye E, Kleckner N. 1994. SeqA—a negative modulator of replication initiation in Escherichia coli. Cell 77:413–426 [DOI] [PubMed] [Google Scholar]
- 64.Boye E, Lobner-Olesen A. 1990. The role of dam methyltransferase in the control of DNA replication in E. coli. Cell 62:981–989 [DOI] [PubMed] [Google Scholar]
- 65.van Opijnen T, Bodi KL, Camilli A. 2009. Tn-seq: high-throughput parallel sequencing for fitness and genetic interaction studies in microorganisms. Nat. Methods 6:767–772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Julio SM, Heithoff DM, Provenzano D, Klose KE, Sinsheimer RL, Low DA, Mahan MJ. 2001. DNA adenine methylase is essential for viability and plays a role in the pathogenesis of Yersinia pseudotuberculosis and Vibrio cholerae. Infect. Immun. 69:7610–7615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Demarre Gl, Chattoraj DK. 2010. DNA adenine methylation is required to replicate both Vibrio cholerae chromosomes once per cell cycle. Plos Genetics 6:e1000939. 10.1371/journal.pgen.1000939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Moran NA. 2002. Microbial minimalism: genome reduction in bacterial pathogens. Cell 108:583–586 [DOI] [PubMed] [Google Scholar]
- 69.Bian F, Qin Q-L, Xie B-B, Shu Y-L, Zhang X-Y, Yu Y, Chen B, Chen X-L, Zhou B-C, Zhang Y-Z. 2011. Complete genome sequence of seawater bacterium Glaciecola nitratireducens FR1064(T). J. Bacteriol. 193:7006–7007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Gao B, Mohan R, Gupta RS. 2009. Phylogenomics and protein signatures elucidating the evolutionary relationships among the Gammaproteobacteria. Int. J. Syst. Evol. Microbiol. 59(Part 2):234–247 [DOI] [PubMed] [Google Scholar]
- 71.Williams KP, Gillespie JJ, Sobral BW, Nordberg EK, Snyder EE, Shallom JM, Dickerman AW. 2010. Phylogeny of gammaproteobacteria. J. Bacteriol. 192:2305–2314 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.