

Abstract
Pasteurella multocida is a Gram-negative multispecies pathogen and the causative agent of fowl cholera, a serious disease of poultry which can present in both acute and chronic forms. The major outer membrane component lipopolysaccharide (LPS) is both an important virulence factor and a major immunogen. Our previous studies determined the LPS structures expressed by different P. multocida strains and revealed that a number of strains belonging to different serovars contain the same LPS biosynthesis locus but express different LPS structures due to mutations within glycosyltransferase genes. In this study, we report the full LPS structure of the serovar 4 type strain, P1662, and reveal that it shares the same LPS outer core biosynthesis locus, L3, with the serovar 3 strains P1059 and Pm70. Using directed mutagenesis, the role of each glycosyltransferase gene in LPS outer core assembly was determined. LPS structural analysis of 23 Australian field isolates that contain the L3 locus revealed that at least six different LPS outer core structures can be produced as a result of mutations within the LPS glycosyltransferase genes. Moreover, some field isolates produce multiple but related LPS glycoforms simultaneously, and three LPS outer core structures are remarkably similar to the globo series of vertebrate glycosphingolipids. Our in-depth analysis showing the genetics and full range of P. multocida lipopolysaccharide structures will facilitate the improvement of typing systems and the prediction of the protective efficacy of vaccines.
INTRODUCTION
Pasteurella multocida is a Gram-negative pathogen that causes a range of diseases in wild and domestic animals (1). These diseases include fowl cholera in poultry (2), hemorrhagic septicemia and shipping fever in ruminants (3), and atrophic rhinitis and respiratory disease in pigs (4), all of which cause significant economic losses to primary industries worldwide. P. multocida can also cause human infections following animal bites or close contact with domestic animals (5). Current classification of P. multocida strains combines Heddleston lipopolysaccharide (LPS) typing with Carter capsular typing, either by traditional serological methods or by multiplex PCR (6–8). P. multocida strains are currently classified into 5 capsular serogroups (A, B, D, E, and F) and 16 Heddleston LPS serovars (6, 8).
Protective immunity against P. multocida is generally considered to be mediated humorally, with protective antibodies directed primarily, but not exclusively, against the LPS antigen located on the surface of the P. multocida cell. Although both live and killed vaccines are available for protection against P. multocida infection, few afford good levels of protection against strains expressing different LPS (9, 10). Therefore, elucidating the full range of LPS structures expressed by P. multocida strains, including field isolates, and understanding the role of LPS in protective immunity are crucial for the formulation of effective, cross-protective vaccines.
The LPS structures produced by P. multocida type strains representing the Heddleston serovars 1, 2, 5, 8, 9, 13, and 14 have been determined, as well as the structure of the LPS expressed by the genome-sequenced serovar 3 strain Pm70 (11–17). Some P. multocida strains can produce two LPS inner core glycoforms simultaneously, termed glycoform A and glycoform B. The inner core glycoform A is produced by all strains examined to date and contains a single phosphorylated 3-deoxy-d-manno-octulosonic acid (Kdo) residue that is substituted with a phosphoethanolamine (PEtn) residue (16). This glycoform also contains a second glucose residue (Glc II) attached to the 6 position of the first heptose (Hep I). The inner core glycoform B contains two Kdo residues and does not have the additional Glc on Hep I. LPS structural analysis of a range of P. multocida strains representing seven serovars has revealed that the most variable part of the molecule is the outer core (structure beyond Glc I). Analysis of the corresponding LPS outer core biosynthesis locus in each strain revealed that although some shared a nearly identical locus, they expressed different LPS molecules due to point mutations or deletions within LPS biosynthesis genes (11, 13). In this study, we report that the LPS outer core biosynthesis locus, first identified in Pm70 (17, 18) and named L3 in this study, is found in the serovar 3 and serovar 4 type strains (P1059 and P1662) and in 23 Australian field isolates. Each of these strains express the same conserved inner core structure, but the strains display significant variability in the length of the LPS outer core and in the number of LPS glycoforms produced simultaneously. We also determined the role of each of the L3 LPS outer core glycosyltransferase genes in the assembly of the LPS outer core.
MATERIALS AND METHODS
Bacterial strains, plasmids, media, and growth conditions.
The bacterial strains and plasmids used in this study are listed in Table 1. Escherichia coli was grown routinely in Luria-Bertani broth. P. multocida was grown in nutrient broth, brain heart infusion (BHI), or heart infusion (HI) broth. Solid media were obtained by the addition of 1.5% (wt/vol) agar. When required, the media were supplemented with kanamycin (50 μg/ml), spectinomycin (50 μg/ml), or tetracycline (2.5 μg/ml). To isolate LPS from the P. multocida serovar 4 type strain (P1662), 2 liters of an early-log-phase culture of P. multocida (grown in BHI at 37°C with shaking at 200 rpm) was used to inoculate 24 liters of BHI in a 28-liter NBS fermenter. The culture was grown for 18 h at 37°C with shaking at 200 rpm, with aeration at 24 liters min−1 and 20% O2 saturation. Following treatment with hyaluronidase for 1 h (1 g; Sigma) to remove capsular material, phenol (2% final concentration) was added and the culture incubated at room temperature for 4 h to kill bacteria, which were then harvested with a Sharples continuous-flow centrifuge. For compositional analysis of LPS produced by the Heddleston serovar 3 type strain (P1059) and the Australian field isolates, small quantities of LPS were isolated from plate-grown cells as described previously (19).
Table 1.
Bacterial strains and plasmids used in this study
| Strain or plasmid | Relevant description | Source or referencea |
|---|---|---|
| Strains | ||
| E. coli DH5α | deoR endA1 gyrA96 hsdR17(rK− mK+) recA1 relA1 supE44 thi-1 Δ(lacZYA-argF)U169 ϕ80dlacZΔM15 F− | Bethesda Research Laboratories |
| P. multocida strains | ||
| AL523 | P. multocida VP161 strain containing Tn916 carrying tetM | This study |
| AL539 | VP161 insertional gatA mutant generated using pAL290; no expression of hptE due to polar effects of the gatA insertion | This study |
| AL806 | VP161 gatA mutant (AL539) containing pAL446 | |
| AL2116 | P1059 gatF (pm1141) TargeTron mutant of P1059 | This study |
| AL2117 | P1059 natB (pm1140) TargeTron mutant of P1059 | This study |
| AL2155 | P1059 gatG (pm1139) TargeTron mutant of P1059 | This study |
| AL2192 | P1059 gatG mutant containing pAL1022 | This study |
| AL2297 | P1059 gatF mutant containing pAL1046 | This study |
| AL2299 | P1059 natB mutant containing pAL1045 | This study |
| AL2354 | P1059 natB mutant containing vector pAL99T | This study |
| AL2355 | P1059 gatF mutant containing vector pAL99T | This study |
| AL2395 | P1059 gatG mutant containing vector pAL99S | This study |
| P1059 | Heddleston serovar 3 type strain | 32 |
| P1662 | Heddleston serovar 4 type strain | 32 |
| PM1 | P. multocida turkey isolate | 33 |
| PM3 | P. multocida turkey isolate | 33 |
| PM8 | P. multocida turkey isolate | 33 |
| PM18 | P. multocida chicken isolate | 33 |
| PM48 | P. multocida chicken isolate | 33 |
| PM72 | P. multocida chicken isolate | 33 |
| PM146 | P. multocida chicken isolate | 34 |
| PM1075 | P. multocida field isolate | MRG culture collection |
| PM1098 | P. multocida field isolate | MRG culture collection |
| PM1099 | P. multocida field isolate | MRG culture collection |
| PM1103 | P. multocida field isolate | MRG culture collection |
| PM1120 | P. multocida chicken isolate | MRG culture collection |
| PM1153 | P. multocida chicken isolate | MRG culture collection |
| PM1205 | P. multocida emu isolate | MRG culture collection |
| PM1258 | P. multocida chicken isolate | MRG culture collection |
| PM1268 | P. multocida chicken isolate | MRG culture collection |
| PM1320 | P. multocida chicken isolate | MRG culture collection |
| PM1369 | P. multocida chicken isolate | MRG culture collection |
| PM1434 | P. multocida chicken isolate | MRG culture collection |
| PM1439 | P. multocida chicken isolate | MRG culture collection |
| PM1441 | P. multocida turkey isolate | MRG culture collection |
| PM1470 | P. multocida turkey isolate | MRG culture collection |
| PM1474 | P. multocida duck isolate | MRG culture collection |
| Plasmids | ||
| pAL99 | P. multocida expression plasmid (Kanr) which contains the P. multocida constitutive Ptpi promoter | 35 |
| pAL99S | P. multocida expression plasmid (Specr); derivative of pAL99 | This study |
| pAL99T | P. multocida expression plasmid (Tetr); derivative of pAL99 | This study |
| pAL290 | pUA826 derivative containing an internal fragment of gatA | This study |
| pAL446 | Complete gctC and hptE genes from Pm70 cloned into pAL99 | This study |
| pAL692 | P. multocida plasmid (Specr) containing TargeTron group II intron | 20 |
| pAL953 | pAL692 with aph3 (Kanr) inserted into TargeTron group II intron | This study |
| pAL1003 | pAL953 retargeted to natB by using primers BAP6851/6852/6853 | This study |
| pAL1004 | pAL953 retargeted to gatG by using primers BAP6854/6855/6856 | This study |
| pAL1006 | pAL953 retargeted to gatF by using primers BAP6864/6865/6866 | This study |
| pAL1022 | Complete gatG gene cloned into pAL99S | This study |
| pAL1045 | Complete natB gene cloned into pAL99T | This study |
| pAL1046 | Complete gatF gene cloned into pAL99T | This study |
| pUA826 | Mob+ R6K replicon; single-crossover mutagenesis vector; Apr Spcr | 36 |
MRG, Microbiology Research Group, Queensland Alliance for Agriculture and Food Innovation, University of Queensland, Australia.
DNA manipulations.
Restriction digests and ligations were performed using enzymes and buffers obtained from NEB or Roche Diagnostics GmbH. Plasmid DNA and genomic DNA were prepared using a plasmid minikit from Qiagen and a genomic DNA extraction kit from RBC, respectively. PCR amplification of DNA was performed using Taq DNA polymerase or the Expand High Fidelity PCR system (Roche Diagnostics), and amplified fragments were purified using a Qiagen PCR purification kit. The oligonucleotides used in this study were synthesized by Sigma, Australia, and are listed in Table S1 in the supplemental material. Preparation of electrocompetent P. multocida and electroporation conditions were as described previously (20). DNA sequencing reactions and cycle conditions using either plasmid DNA or PCR products as templates were performed using BigDye Terminator, version 3.1 (Applied Biosystems), per the manufacturer's instructions, with the following modifications: 1.5 μl of 10× Taq PCR buffer (Roche) and 1 μl of BigDye Terminator, version 3.1, were used in a 15-μl (total volume) reaction mixture. For direct sequencing of P. multocida genomic DNA, reaction and cycle conditions were as described previously (21), with the following modifications: reactions were performed in a 40-μl volume with 3 μl 10× Taq PCR buffer (Roche), 13 pmol of oligonucleotide, and 4 μl of BigDye Terminator, version 3.1 (Applied Biosystems). All DNA sequences were determined on a capillary-platform genetic analyzer (model 3730; Applied Biosystems) and analyzed with Vector NTI Advance 11 (Invitrogen). Amino acid sequence alignments were conducted using BLAST and Clustal W2.
Construction of P. multocida TargeTron mutants.
For inactivation of genes in P. multocida strain P1059, we used the TargeTron method (Sigma-Aldrich) of mutagenesis as described previously (20), but with the following modifications. To assist in the selection of P. multocida transformants containing an integrated intron, the plasmid pAL692 (20) was modified to introduce a kanamycin resistance gene, aph3, into the intron. Briefly, the aph3 gene was amplified from pAL99 (Table 1) by using the primers BAP6796 and BAP6797 and then cloned into MluI-digested pAL692, generating the plasmid pAL953 (Table 1). Retargeting of the intron within pAL953 was performed per the instructions of the TargeTron user's manual, using oligonucleotides designed by the TargeTron design site (Sigma-Aldrich) (see Table S1 in the supplemental material). Each plasmid containing an intron correctly targeted to the target gene (pAL1003, pAL1004, and pAL1006) (Table 1) was used to transform the P. multocida serovar 3 type strain (P1059) by electroporation; transformants containing the plasmid and/or integrated intron were selected on solid agar with kanamycin. Mutants were then cured of replicating plasmid by overnight growth in nutrient broth without antibiotic selection, and transformants containing the intron were identified by patching onto HI agar containing spectinomycin or kanamycin. Kanamycin-resistant, spectinomycin-sensitive transformants were then screened for the correct intron insertion by colony PCR using one oligonucleotide located within the target gene and one located within the intron (EBS universal). To confirm integration into the target gene and to determine if there were any additional intron insertions elsewhere in the genome, direct sequencing from genomic DNA was performed using the intron-specific EBS universal primer (Table 1) together with DNA isolated from each TargeTron mutant. Mutants that gave unambiguous sequencing data identical to the target gene sequence were selected for further study.
trans-Complementation of mutants.
For complementation of the kanamycin-resistant TargeTron mutants, the P. multocida expression plasmid pAL99 was modified to remove the kanamycin resistance gene, aph3, and replace it with the aadA or tetM gene, conferring spectinomycin (pAL99S) or tetracycline (pAL99T) resistance, respectively (Table 1). The plasmid pAL99 was amplified using two outward-firing primers (BAP5358 and BAP5359 [see Table S1 in the supplemental material]) flanking aph3, digested with EcoRV, and then ligated to an EcoRV-digested PCR-amplified fragment containing either the aadA gene (amplified from pUA826 by using primers BAP5360 and BAP5361) or the tetM gene (amplified from Tn916 [in strain AL523] by using primers BAP7134 and BAP7135), generating pAL99S or pAL99T, respectively.
For complementation of each of the P. multocida mutants, PCR amplifications were performed using P1059 genomic DNA and the appropriate pair of oligonucleotides flanking the target gene (see Table S1 in the supplemental material). Each product was digested with the appropriate restriction enzyme and then ligated to appropriately digested pAL99, pAL99S, or pAL99T (Table 1), such that transcription of each gene would be driven by the constitutive P. multocida tpiA promoter. Each plasmid was then introduced into the corresponding P. multocida mutant, generating the complemented strain (Table 1). As a control, the appropriate empty vector was introduced separately into each mutant (Table 1).
Isolation and purification of lipopolysaccharide.
LPS was isolated and purified as described previously (16). O-deacylated LPS (LPS-OH), core oligosaccharide (OS), and completely deacylated LPS were all isolated and purified from LPS as described previously (16).
Analytical methods, MS, and NMR spectroscopy.
Sugars were determined as their alditol acetate derivatives, and linkage analysis was performed following methylation analysis by gas-liquid chromatography–mass spectrometry (GLC-MS) and electrospray mass spectrometry (ES-MS) as described previously. To determine exact locations and linkage patterns of residues, nuclear magnetic resonance (NMR) techniques were performed as described previously (16). The assignment of 1H resonances was achieved using correlation spectroscopy (COSY), total correlation spectroscopy (TOCSY), and nuclear Overhauser effect spectroscopy (NOESY). The assignment of 13C resonances was achieved using 13C-1H heteronuclear single-quantum coherence (HSQC) and 13C-1H HSQC-TOCSY NMR. To identify the locations of PEtn substitutions, 31P-1H heteronuclear multiple-quantum coherence (HMQC) experiments were performed (16).
Nucleotide sequence accession numbers.
Sequence data for the P1059 and P1662 outer core biosynthesis loci have been deposited in GenBank under accession numbers KF314825 and KF314826, respectively.
RESULTS
Analyses of LPS outer core biosynthesis loci.
The region of the genome containing the LPS outer core biosynthesis locus in the serovar 3 and 4 type strains (P1059 and P1662, respectively) was sequenced, and the sequences were found to be 99% identical to each other and also to the equivalent locus in Pm70 (Fig. 1A) (18). However, the sequence of the locus in the serovar 4 type strain (P1662) contained a nonsense mutation within natC due to a C291A nucleotide substitution. In keeping with our function-based nomenclature of P. multocida LPS glycosyltransferases (19), we have named the orthologues of the Pm70 glycosyltransferase genes in the serovar 3 and 4 type strains natC (pm1138), gatG (pm1139), natB (pm1140), gatF (pm1141), gctC (losA), and hptE (pm1144) (Fig. 1A). The LPS outer core biosynthesis locus common to all three strains (Pm70, P1059, and P1662) was named L3 (Fig. 1A).
Fig 1.

(A) Schematic representation of the genetic organization of the P. multocida L3 LPS outer core biosynthesis locus, which is common to serovars 3 and 4. Genes involved in LPS outer core assembly are shown in white. Conserved genes unrelated to LPS biosynthesis are shown in gray. Original gene annotation numbers for Pm70 are shown below the diagram. (B) Schematic representation of the LPS structure expressed by the P. multocida serovar 3 and 4 type strains (P1059 and P1662, respectively). Only inner core glycoform A is shown. The P. multocida glycosyltransferase genes predicted to be required for assembly of the outer core are shown below each linkage on the structure expressed by P1059. The rare addition of GalNAc II onto the P1059 LPS is shown with a dotted line and in gray. The specific number and position of phosphoethanolamine (PEtn) residues attached to each LPS structure are strain dependent; nonstoichiometric additions of PEtn are shown with dotted lines. Regions of the L3 LPS that are identical to the oligosaccharide components of the vertebrate glycosphingolipids, Forssman, P, and Pk, are shown boxed in the Pm70 structure. GalNAc, N-acetyl-galactosamine; Gal, galactose; Glc, glucose; Hep, heptose; PEtn, phosphoethanolamine; Kdo, 3-deoxy-d-manno-octulosonic acid; P, phosphate.
Structural analyses of P. multocida LPS derived from the serovar 3 and 4 type strains (P1059 and P1662).
Sugar analysis of the purified LPS isolated from the serovar 4 type strain (P1662) revealed glucose (Glc), galactose (Gal), and l-glycero-d-manno-heptose (ld-Hep), in an approximate ratio of 2:1:3. A small amount of N-acetylglucosamine (GlcNAc) was also identified, as is often observed for sugar analyses of LPS and presumably derives from lipid A. In order to separate and detect closely related glycoforms, capillary electrophoresis coupled with electrospray ionization mass spectrometry (CE-MS) was used to analyze the O-deacylated LPS (LPS-OH) (Table 2). This analysis revealed major triply charged ions at m/z 1,025.4 and 1,066.2, corresponding to a composition of 5Hex, 4Hep, Kdo-P, 2PEtn, and O-deacylated lipid A (lipid A-OH) for the smaller molecule and an additional PEtn moiety for the larger molecule. Glycoforms with one or no PEtn residues were also observed. Similarly, MS analysis of the nonfractionated core OS suggested a composition of 5Hex, 4Hep, 2PEtn, and Kdo as the major glycoform. Interestingly, there was no evidence of any glycoforms containing two Kdo residues (inner core glycoform B). Methylation analysis of the fractionated core OS (fraction 15) allowed determination of the linkage pattern of the molecule, which revealed the presence of terminal Glc, terminal Gal, 4-substituted Glc, and terminal ld-Hep in approximately equimolar amounts, with 6-substituted Glc, 2-substituted ld-Hep, 4,6-disubstituted ld-Hep, and 3,4,6-trisubstituted ld-Hep identified in smaller amounts.
Table 2.
Negative-ion CE-ES-MS data and proposed compositions of O-deacylated LPS (LPS-OH) and core OS from the Heddleston serovar 3 and 4 type strains (P1059 and P1662), and proposed compositions of LPS-OH from the P1059 TargeTron mutants
| Study and serovar (description) | Observed ions (m/z) |
Molecular mass (Da)a |
Relative intensity | Proposed composition | ||||
|---|---|---|---|---|---|---|---|---|
| (M − H)− | (M − 4H)4− | (M − 3H)3− | (M − 2H)2− | Observed | Calculated | |||
| Serovar study | ||||||||
| Serovar 3 (P1059) | 1,106.5 | 3,322 | 3,320.0 | 0.2 | HexNAc, 6Hex, 4Hep, Kdo-P, PEtn, lipid A-OH | |||
| LPS-OH | 1,147.3 | 1,721.2 | 3,444 | 3,443.0 | 1.0 | HexNAc, 6Hex, 4Hep, Kdo-P, 2PEtn, lipid A-OH | ||
| 889.8 | 1,188.2 | 3,568 | 3,566.0 | 0.8 | HexNAc, 6Hex, 4Hep, Kdo-P, 3PEtn, lipid A-OH | |||
| Serovar 4 (P1662) | ||||||||
| LPS-OH | 707.4 | 943.5 | 2,833.6 | 2,831.6 | 0.2 | 5Hex, 4Hep, Kdo-P, lipid A-OH | ||
| 738.0 | 984.3 | 1,477.2 | 2,956.0 | 2,954.7 | 0.5 | 5Hex, 4Hep, Kdo-P, PEtn, lipid A-OH | ||
| 768.6 | 1,025.4 | 1,538.7 | 3,079.0 | 3,077.7 | 1.0 | 5Hex, 4Hep, Kdo-P, 2PEtn, lipid A-OH | ||
| 799.5 | 1,066.2 | 3,202.2 | 3,200.7 | 0.5 | 5Hex, 4Hep, Kdo-P, 3PEtn, lipid A-OH | |||
| Core OS | 899.1 | 1,800.2 | 1,799.5 | 0.2 | 5Hex, 4Hep, aKdob | |||
| 960.3 | 1,922.6 | 1,922.6 | 0.8 | 5Hex, 4Hep, PEtn, aKdob | ||||
| 1,022.1 | 2,046.2 | 2,045.7 | 1.0 | 5Hex, 4Hep, 2PEtn, aKdob | ||||
| GctC study | ||||||||
| AL539 | ||||||||
| LPS-OH (gatA polar mutant) | 709.7 | 1,064.9 | 2,131.9 | 2,130.9 | 0.2 | Hex, 3Hep, 2Kdo, lipid A-OH | ||
| 717.0 | 1,075.9 | 2,153.9 | 2,152.9 | 1.0 | 2Hex, 3Hep, Kdo-P, lipid A-OH | |||
| LPS-OH | 758.0 | 1,137.4 | 2,276.9 | 2,276.0 | 0.6 | 2Hex, 3Hep, Kdo-P, PEtn, lipid A-OH | ||
| 773.5 | 1,160.9 | 2,323.7 | 2,323.1 | 0.1 | Hex, 4Hep, 2Kdo, lipid A-OH | |||
| 780.5 | 1,171.5 | 2,344.8 | 2,345.1 | 0.2 | 2Hex, 4Hep, Kdo-P, lipid A-OH | |||
| 822.1 | 1,233.2 | 2,469.2 | 2,468.2 | 0.1 | 2Hex, 4Hep, Kdo-P, PEtn, lipid A-OH | |||
| AL806 LPS-OH (AL539 + gctC, hptE) | 691.8 | 923.2 | 2,772.1 | 2,770.5 | 0.5 | 3Hex, 4Hep, 2Kdo, PEtn, lipid A-OH | ||
| 930.4 | 2,794.2 | 2,792.4 | 0.3 | 4Hex, 4Hep, Kdo-P, PEtn, lipid A-OH | ||||
| 728.5 | 971.4 | 2,917.6 | 2,915.5 | 1.0 | 4Hex, 4Hep, Kdo-P, 2PEtn, lipid A-OH | |||
| GatF study | ||||||||
| AL2116 LPS-OH (gatF mutant) | 697.7 | 930.7 | 1,395.5 | 2,794.0 | 2,792.4 | 0.3 | 4Hex, 4Hep, Kdo-P, PEtn, lipid A-OH | |
| 728.3 | 971.2 | 1,457.2 | 2,916.6 | 2,915.5 | 1.0 | 4Hex, 4Hep, Kdo-P, 2PEtn, lipid A-OH | ||
| AL2297 LPS-OH (gatF mutant + gatF) | 1,174.0 | 3,525.0 | 3,523.2 | 0.4 | 2HexNAc, 6Hex, 4Hep, Kdo-P, PEtn, lipid A-OH | |||
| 1,215.0 | 3,648.0 | 3,646.3 | 1.0 | 2HexNAc, 6Hex, 4Hep, Kdo-P, 2PEtn, lipid A-OH | ||||
| GatG study | ||||||||
| AL2155 LPS-OH (gatG mutant) | 768.9 | 1,025.3 | 3,079.2 | 3,077.7 | 1.0 | 5Hex, 4Hep, Kdo-P, 2PEtn, lipid A-OH | ||
| 799.6 | 1,066.2 | 3,202.0 | 3,200.8 | 1.0 | 5Hex, 4Hep, Kdo-P, 3PEtn, lipid A-OH | |||
| AL2192 LPS-OH (gatG mutant + gatG) | 1,106.5 | 3,322.5 | 3,320.0 | 1.0 | HexNAc, 6Hex, 4Hep, Kdo-P, lipid A-OH | |||
| 1,147.5 | 3,445.5 | 3,443.1 | 0.9 | HexNAc, 6Hex, 4Hep, Kdo-P, lipid A-OH | ||||
| 1,174.0 | 3,525.0 | 3,523.2 | 0.3 | 2HexNAc, 6Hex, 4Hep, Kdo-P, PEtn, lipid A-OH | ||||
| 1,215.0 | 3,648.0 | 3,646.3 | 0.4 | 2HexNAc, 6Hex, 4Hep, Kdo-P, 2PEtn, lipid A-OH | ||||
| NatB study | ||||||||
| AL2117 LPS-OH (natB mutant) | 768.7 | 1,025.4 | 3,079.0 | 3,077.7 | 0.8 | 5Hex, 4Hep, Kdo-P, 2PEtn, lipid A-OH | ||
| 809.4 | 1,079.5 | 3,241.5 | 3,239.9 | 1.0 | 6Hex, 4Hep, Kdo-P, 2PEtn, lipid A-OH | |||
| AL2299 LPS-OH (natB mutant + natB) | 1,174.0 | 3,525.0 | 3,523.2 | 0.4 | 2HexNAc, 6Hex, 4Hep, Kdo-P, PEtn, lipid A-OH | |||
| 1,215.0 | 3,648.0 | 3,646.3 | 1.0 | 2HexNAc, 6Hex, 4Hep, Kdo-P, 2PEtn, lipid A-OH | ||||
Average mass units were used for calculation of molecular mass based on the proposed composition, as follows: lipid A-OH, 952.00; Hex, 162.15; Hep, 192.17; Kdo, 220.18; anhydro Kdo (aKdo), 202.18; PEtn, 123.05; P, 79.95.
aKdo, hydrolysis products of Kdo formed during core hydrolysis.
In order to elucidate the exact locations and linkage patterns of the carbohydrate residues, NMR studies were performed on the core oligosaccharide fraction that gave the most resolved and homogeneous spectrum (fraction 14). The inner core oligosaccharide 1H and 13C resonances were assigned with reference to the published data for the structurally related oligosaccharides from P. multocida strains belonging to serovars 1, 2, 3, and 5 and revealed that the serovar 4 strain elaborated the previously identified conserved inner core structure (Hep I to IV and Glc I and II) (Table 3) (12, 15–17).
Table 3.
1H- and 13C-NMR chemical shifts for the core OS from the Pasteurella multocida serovar 4 type strain, P1662a
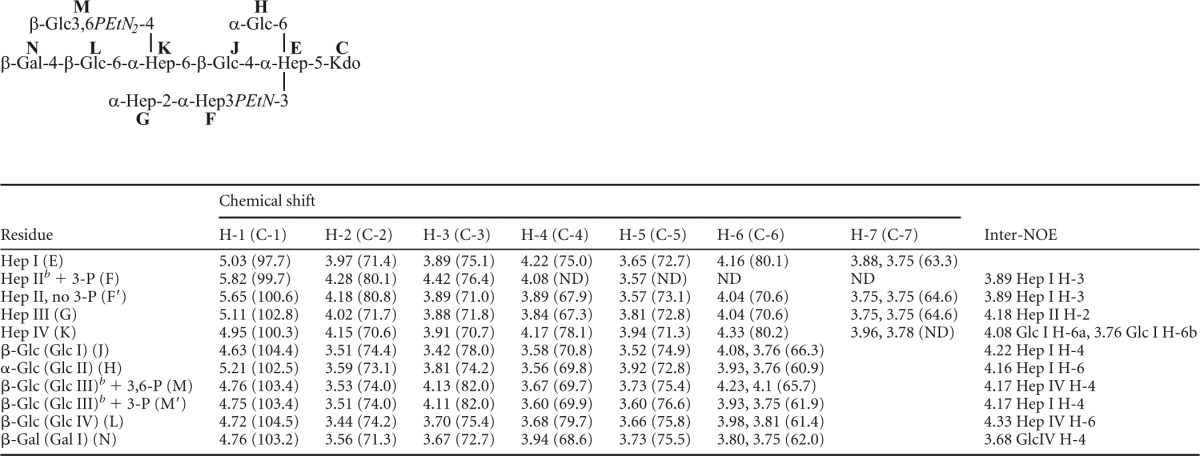
Chemical shifts were recorded at 25°C and referenced to internal acetone at 2.225 ppm (1H)/31.07 ppm (13C).
Signals for ethanolamine of phosphoethanolamine at F-3, M-3, and M-6 were observed at 4.19/63.1 and 3.31/41.3.
The outer core residues of the oligosaccharide extension beyond Hep IV in strain P1662 were characterized by COSY, TOCSY, and NOESY experiments (Fig. 2). In addition to the residues of the conserved inner core structure, a glucose residue (Glc III) was identified, based on a characteristic spin system in TOCSY experiments. Two spin systems were actually identified for this residue, with proton resonances at H-1 (4.76 ppm), H-2 (3.53 ppm), H-3 (4.13 ppm), H-4 (3.67 ppm), H-5 (3.73 ppm) and H-6ab (4.23, 4.10 ppm) in one system and at H-1 (4.75 ppm), H-2 (3.51 ppm), H-3 (4.11 ppm), H-4 (3.60 ppm), H-5 (3.60 ppm), and H-6ab (3.93, 3.75 ppm) in the other system. Both exhibited internuclear Overhauser effect (inter-NOE) connectivities from the anomeric protons to a resonance at 4.17 ppm (Fig. 2). This resonance was assigned as the proton at the 4-position of the Hep IV residue by virtue of 13C-1H HSQC and 13C-1H HSQC-TOCSY experiments (data not shown). The identification of two spin systems became apparent when 31P-1H HMQC experiments were performed in conjunction with the homonuclear NMR data, which revealed the presence of two PEtn residues linked to both the H-3 (4.13 ppm) and H-6ab (4.23, 4.10 ppm) resonances in one spin system and to only the H-3 resonance (4.11 ppm) in the other system (Fig. 2). A further glucose residue (Glc IV) was identified, based on a characteristic spin system in TOCSY experiments, with H-1 (4.72 ppm), H-2 (3.44 ppm), H-3 (3.70 ppm), H-4 (3.68 ppm), H-5 (3.66 ppm), and H-6ab (3.98, 3.81 ppm) resonances being identified. An inter-NOE connectivity from the anomeric proton of Glc IV to a resonance at 4.33 ppm was identified (Fig. 2). This resonance was assigned as the proton at the 6-position of the Hep IV residue by virtue of 13C-1H HSQC and 13C-1H HSQC-TOCSY experiments (data not shown). This assignment, coupled with the data for the Glc III residue, is consistent with the methylation analysis data which identified a 4,6-substituted ld-Hep residue. A terminal galactose residue (Gal I) was identified based on a characteristic spin system in TOCSY experiments, with H-1 (4.46 ppm), H-2 (3.56 ppm), H-3 (3.67 ppm), and H-4 (3.94 ppm) resonances being identified. An inter-NOE connectivity from the anomeric proton of Gal I to a resonance at 3.68 ppm was identified (Fig. 2). This resonance was assigned as the proton at the 4-position of the Glc IV residue by virtue of 13C-1H HSQC and 13C-1H HSQC-TOCSY experiments (data not shown). This assignment is also consistent with the methylation analysis data, which identified a 4-linked Glc residue and a terminal Gal residue. The identification of two PEtn residues substituting the 3- and 6-positions of Glc III, along with evidence for a PEtn residue at the 3-position of Hep II in some glycoforms, combined with the identification of only 2 PEtn residues in total in the core OS, led us to examine closely the 31P-1H heteronuclear and homonuclear NMR data (Fig. 2). It became clear that the PEtn at the 6-position of the Glc III residue was present nonstoichiometrically; however, the PEtn residue at the 3-position was found to always be present. Furthermore, the PEtn residue was identified at the 3-position of the Hep II residue (in the inner core) approximately 20% of the time. However, the simultaneous expression of all three PEtn residues in the core OS was not identified, perhaps suggesting a mutual exclusivity. Taken together, these data show that the serovar 4 type strain (P1662) produces LPS that is a truncated derivative of the Pm70 LPS (17), with the exception of one or two additional PEtn residues on Glc III of the P1662 outer core (Fig. 1B).
Fig 2.
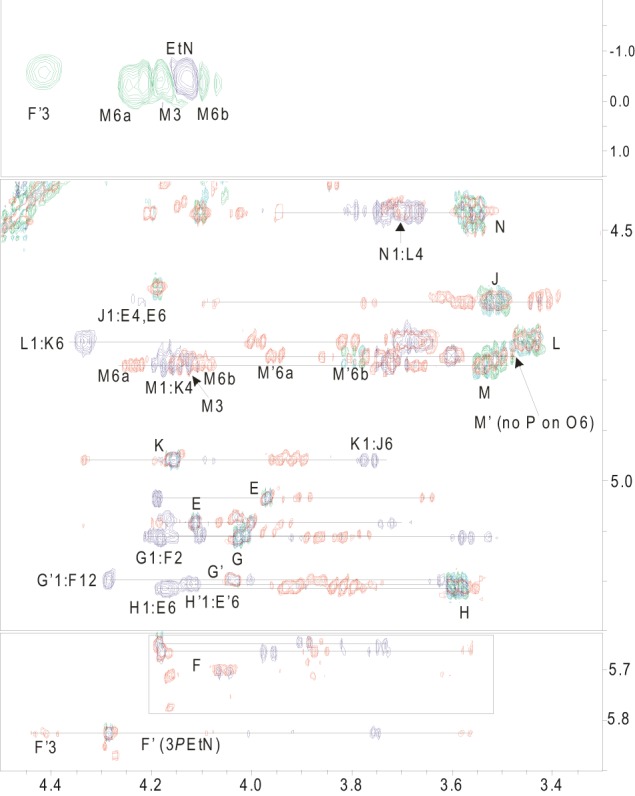
NMR analyses of core OS fraction 14 from P. multocida Heddleston serovar 4 strain P1662. Regions of overlapped COSY (green), TOCSY (red), and NOESY (purple) spectra (bottom two panels) and the 1H-31P HMQC (green and purple) spectrum (top panel) of the core OS are shown. Residues are labeled as indicated in Table 3.
As the full structure of the LPS core oligosaccharide from the serovar 3 strain Pm70 has been determined previously (17), only CE-MS analysis of LPS-OH was conducted on the serovar 3 type strain (P1059). Analysis of the P1059 LPS revealed that all LPS glycoforms lacked the terminal HexNAc; CE-MS analysis of LPS-OH revealed a major triply charged ion at m/z 1,147.3, corresponding to a composition of HexNAc, 6Hex, 4Hep, 2PEtn, Kdo, and lipid A-OH (Table 2). To confirm this finding, we undertook CE-MS analysis of LPS-OH isolated from two independent freeze-dried cultures of strain P1059 that had been sourced from different collections and found that the LPS composition consisted mainly of HexNAc, 6Hex, 4Hep, 2PEtn, Kdo, and lipid A-OH, but trace amounts of the 2HexNAc, 6Hex, 4Hep, 2PEtn, Kdo, and lipid A-OH glycoform were detected in both samples (data not shown). A trace amount of the inner core glycoform B (HexNAc, 5Hex, 4Hep, 2Kdo, PEtn, and lipid A-OH) was also detected in one of the samples (data not shown).
Characterization of the glycosyltransferases involved in biosynthesis of the L3 LPS outer core.
The functions of two glycosyltransferases (HptE and NatC) encoded within the L3 LPS outer core biosynthesis locus were determined previously: HptE is required for the addition of the first outer core sugar, Hep IV (19), and NatC (annotated as PM1138 in strain Pm70) is an N-acetyl-galactosyltransferase required for the addition of the last outer core sugar, GalNAc II (22) (Fig. 1). Previous bioinformatic analysis of the equivalent region in strain Pm70 indicated that pm1141 (glycosyltransferase family 25) and pm1139 (glycosyltransferase family 8) likely encode 1,4-galactosyltransferases that add Gal I and Gal II, respectively, and pm1140 (glycosyltransferase family A) encodes an N-acetyl-galactosyltransferase that adds GalNAc I (17).
To confirm the predicted functions of these enzymes, the pm1141, pm1139, and pm1140 orthologues (gatF, gatG, and natB, respectively) were separately inactivated in the serovar 3 type strain (P1059), and the effect of each mutation on LPS structure was determined (Table 2). To confirm that any LPS structural changes were due specifically to inactivation of the mutated glycosyltransferase, each mutant was complemented with a functional copy of the appropriate transferase provided in trans. Structural analysis of the mutants revealed that the gatF mutant (AL2116) was unable to assemble LPS beyond Glc IV and that the gatG mutant (AL2155) was unable to assemble LPS beyond Gal I (Fig. 1; Table 2), confirming bioinformatic predictions that GatF is the galactosyltransferase which adds Gal I to the 4th position of the Glc IV and GatG is a galactosyltransferase which adds Gal II to the 4th position of Gal I (Fig. 1). Similarly, analysis of the LPS produced by the P1059 natB mutant (AL2117) confirmed that NatB is the 1,3-N-acetyl-galactosyltransferase responsible for the addition of GalNAc I to Gal II (Fig. 1; Table 2). Importantly, in each case, complementation experiments showed that mutants provided with a functional copy of the appropriate transferase in trans produced full-length LPS (Table 2), while mutants transformed with vector only did not (data not shown).
To determine the role of gctC in LPS outer core assembly, we used an LPS mutant, AL539, constructed in the serovar 1 strain VP161, which expresses only the highly conserved inner core LPS, including Glc I, which is the acceptor molecule for the HptE heptosyltransferase (19). This strain was transformed with the expression plasmid pAL446 containing a functional copy of the L3 glycosyltransferase genes hptE and gctC (PCR amplified from Pm70) (Table 1). MS (Table 2) and NMR structural analyses (data not shown) of the LPS produced by the complemented strain (AL806) revealed that it elaborated an LPS outer core of two terminal Glc residues linked to the 4- and 6-positions of Hep IV, confirming the role of the L3 HptE as the 1,6-heptosyltransferase and identifying that GtcC is a bifunctional glucosyltransferase that adds glucose to both the 4- and 6-positions of d-Hep IV.
Analysis of genetics underlying P1662 LPS truncation.
The serovar 4 type strain, P1662, produced a truncated LPS structure terminating at Gal I, suggesting that the 1,4-galactosyltransferase, GatG, was nonfunctional in this strain. Analysis of the P1662 gatG sequence indicated that the translated product contained eight amino acid changes (R85K, V240A, A253D, N259K, R272K, E277K, V279M, and T284N) compared to the functional GatG glycosyltransferases expressed by Pm70 and P1059. The Neisseria meningitidis galactosyltransferase LgtC is 39% identical (57% conserved) to GatG, and the crystal structure of LgtC has been determined in complex with both its donor and acceptor molecules (23). Alignment of LgtC with other family 8 glycosyltransferases, including GatG (PM1139), identified a number of highly conserved amino acids critical for interaction with the donor molecule, the acceptor molecule, or the divalent cation Mn2+ (23) (Fig. 3). Only one of the eight GatG substitutions identified in strain P1662 (R85K) was in a highly conserved position (R86 in LgtC), and this residue is critical for the interaction of LgtC with the galactose portion of the donor. Therefore, we suggest that the R85K mutation is responsible for the loss of GatG function in P1662.
Fig 3.

Amino acid sequence alignment of GatG (PM1139) from Pasteurella multocida strain Pm70 (Swiss-Prot accession no. Q9CLS0) aligned with the galactosyltransferase LgtC from Neisseria meningitidis (Swiss-Prot accession no. P96945). Amino acids that are highly conserved within LgtC-related glycosyltransferases are highlighted in gray (23). LgtC amino acids involved in binding to Mn2+ are marked with squares. Amino acids in LgtC known to interact with the UDP or galactose region of the nucleotide sugar UDP-galactose are marked with open or filled circles, respectively, while those involved in binding to the acceptor molecule are marked with triangles (23).
Structural and genetic analyses of LPS biosynthesis in P. multocida field isolates belonging to genotype L3.
To determine the diversity of LPS structures produced by P. multocida field isolates belonging to LPS genotype L3, we identified 23 P. multocida isolates from Australian poultry farms that contained L3 LPS outer core biosynthesis loci. Proposed compositions determined from CE-MS analysis of LPS-OH purified from each of the isolates (see Table S2 in the supplemental material) indicated that a number of different LPS structures were produced, ranging from inner core-only structures (terminating at Glc I) to full-length structures terminating at GalNAc II (Fig. 4; Table 4). Interestingly, 14 of the isolates simultaneously expressed multiple LPS glycoforms containing outer core sugar extensions ranging from 3 to 7 residues (see Table S2), and none of the field isolates analyzed elaborated an LPS structure terminating with Hep IV (Fig. 4; see Table S2).
Fig 4.

Schematic representation showing the range of LPS outer core structures expressed by the 23 Australian field isolates containing the L3 outer core biosynthesis locus. Only the longest LPS outer core structure is shown for strains expressing multiple LPS glycoforms. The specific number and position of phosphoethanolamine (PEtn) residues attached to each LPS structure are strain dependent and are not shown. GalNAc, N-acetyl-galactosamine; Gal, galactose; Glc, glucose; Hep, heptose.
Table 4.
Detected mutations in LPS outer core biosynthesis loci of P. multocida field isolates
| Longest LPS outer core structure detecteda | Predicted gene disrupted | Field isolate | Detected mutation(s)b |
|---|---|---|---|
| No detectable outer core | hptE | PM1075 | Q229* |
| PM1120 | Q194* | ||
| PM1258 | Single nucleotide deletion at position 61 | ||
| Hep·[Glc]·Glc | gatF | PM3 | None detected |
| PM8 | None detected | ||
| PM18 | A205T | ||
| PM146 | S41P, V138M | ||
| Hep·[Glc]·Glc·Gal | gatG | PM1098 | C128Y |
| PM1205 | Insertion of 42 nucleotides at position 353 | ||
| PM1268 | I50V, K98R, A253D, D254N, N259K, R272K, E277K, V279M, T284N, A288D, F297L | ||
| PM1320 | I50V, K98R, A253D, D254N, N259K, R272K, E277K, V279M, T284N, A288D, F297L | ||
| PM1439 | 14 nucleotides deleted, positions 496–509 | ||
| Hep·[Glc]·Glc·Gal·Gal | natB | PM1369 | None detected |
| PM1474 | None detected | ||
| Hep·[Glc]·Glc·Gal·Gal·GalNAc | natC | PM1 | Nucleotide changes/deletions in promoter region |
| PM48 | S97* | ||
| PM1099 | I132N, I206V | ||
| PM1103 | Deletion/rearrangement between nucleotides 186 and 276 | ||
| PM1441 | K3R, F4L, N7H, G33K, E49K, N62D, F78L, N80S, N105D, E115G, V120I, Y168D, K173N, Q176R, R193Q, I194V, Q207K, Q281K, I231V, E236K, E284D, K285Q, E271A, I292M, K296T, L312I, N396K, S397R | ||
| PM1434 | S97* | ||
| Hep·[Glc]·Glc·Gal·Gal·GalNAc·GalNAc | None | PM72 | All genes functional |
| PM1153 | All genes functional | ||
| PM1470 | All genes functional |
[Glc], residue is attached to the main chain residues via a 1,4 linkage to Hep.
Compared to nucleotide and amino acid sequences of the equivalent functional orthologues in Pm70 and/or P1059. *, stop codon. GatG residue changes shown in bold are in close proximity to amino acids predicted to interact with the donor or acceptor molecule or the divalent cation Mn2+.
To determine if the truncated LPS structures observed in the field isolates were the result of mutations within one of the glycosyltransferase genes in the L3 LPS outer core biosynthesis locus, the glycosyltransferase gene predicted to be responsible for the arrested assembly in each case was amplified by PCR, and the nucleotide sequence was determined using the same oligonucleotides used for complementation experiments (Table 1). The translated amino acid sequences were then compared with those of the functional orthologs in P1059 and Pm70. In eight of the field isolates, there were clear mutations resulting in an immediate stop codon or a reading frameshift leading to a stop codon (PM48, PM1075, PM1120, PM1103, PM1205, PM1258, PM1434, and PM1439) (Table 4). In five field isolates (PM1, PM3, PM8, PM1369, and PM1474), there were no changes detected in the gene examined, although one isolate (PM1) had nucleotide changes and deletions in the predicted promoter region. However, seven of the field isolates contained one or more missense mutations in the genes encoding the predicted inactivated transferase; two strains (PM18 and PM146) contained mutations within gatF, two had changes in natC (PM1099 and PM1441), and three (PM1268, PM1320, and PM1098) contained mutations in gatG (Table 4).
As noted previously, the structure of N. meningitidis LgtC (a GatG ortholog) is known, and alignments of GatG from PM1098, PM1268, and PM1320 (each containing missense mutations) with LgtC revealed that there were amino acid changes located in close proximity to residues that are known to be critical in LgtC. These residues included those required for LgtC to interact with the UDP portion of the sugar donor (I104, G247, and K250), the divalent cation Mn2+ (D103, D105, and H244), or the acceptor molecule (D130 and F132) (23) (Fig. 3). Therefore, it is highly likely that the missense mutations in GatG abrogate transferase function in these field isolates. There are no data available on the structure of GatF and NatC homologs in other bacteria, so it is not possible to predict if the missense mutations identified in gatF and natC were responsible for the altered LPS production observed in those strains.
DISCUSSION
The strains belonging to the L3 LPS genotype include the first genome-sequenced P. multocida strain, Pm70, and the Heddleston serovar 3 and 4 type strains (P1059 and P1662, respectively). Using LPS structural analysis of directed mutants followed by trans-complementation, the role of each of the uncharacterized transferases in the L3 locus (NatB, GatG, GatF, and GctC) was defined, and previous functional predictions were confirmed (17). Interestingly, P1059 elaborates only trace quantities of LPS glycoforms containing GalNAc II. However, when the P1059 gatF, gatG, and natB mutants were complemented with the appropriate wild-type genes, all produced significant levels of the full-length glycoform containing the terminal GalNAc II (Table 2). This finding strongly suggests that the efficiency of addition of GalNAc II by NatC is dependent on the rate of production of the appropriate LPS acceptor molecule (extended to GalNAc I), which in turn is dependent on the efficiency of the transferases involved in the earlier steps of LPS assembly.
LPS structural analysis of the strains belonging to LPS genotype L3, which includes the serovar 3 and 4 type strains, Pm70, and the 23 field isolates analyzed here, revealed that all produced LPS containing the inner core glycoform A (containing Kdo-P), which is the most common LPS inner core structure produced by P. multocida. However, the inner core glycoform B (containing Kdo-Kdo), previously reported by us to be expressed in significant quantities in the serovar 1 strains VP161 and X73 (15, 16), was not detected in strain P1662 or in any of the L3 Australian field isolates, although glycoform B was detected in trace amounts in strain P1059 and was previously found in LPS isolated from Pm70 (17). Although all strains share the same LPS outer core biosynthesis locus, L3, the length of the LPS outer core structure varied considerably; Pm70 produced significant quantities of the fully extended LPS structure, whereas nearly all LPS glycoforms in strain P1059 lacked the terminal GalNAc II, while the longest of the LPS glycoforms produced by P1662 terminated at Gal I. While some of this variation is due to clear mutations inactivating genes within the L3 locus, some field isolates produced a fully extended LPS glycoform as well as up to four shorter, but related, outer core structures. This intrastrain variation in LPS structure as the result of truncations in the outer core region has previously been reported for Haemophilus influenzae (24) but has not been observed in strains belonging to any of the other P. multocida serovars examined to date. There is no genetic evidence of any phase variation in P. multocida, but it is possible that the expression of multiple glycoforms from the same locus could be controlled posttranscriptionally via an unknown mechanism. Alternatively, MsbA, the ABC transporter responsible for “flipping” LPS across the inner membrane (25), may transport an unusually large number of LPS glycoforms before the outer core region of the LPS has fully extended.
The LPS produced by strains belonging to the L3 LPS genotype contains outer core structures remarkably similar to the oligosaccharide component of P, Pk, and Forssman antigen, found on the surfaces of many cell types in vertebrates (26). In birds, Forssman antigen has been identified in the vascular endothelial tissue, perivascular connective tissue, and hematopoietic organs, including the spleen (27). Forssman antigen is also present on the surfaces of chicken erythrocytes (28). Notably, P and Pk antigen mimicry has been reported for other pathogens that express O-antigen-deficient LPS, including Campylobacter jejuni (29), Haemophilus influenzae (30), and Neisseria spp. (31).
In summary, we have shown that fowl cholera strains containing the LPS outer core biosynthesis locus L3 display a remarkable level of inter- and intrastrain LPS structural heterogeneity. This group includes the serovar 3 and 4 type strains (P1059 and P1662) as well as the first genome-sequenced strain, Pm70. This level of LPS diversity, particularly within a single strain and derived from a single LPS outer core biosynthesis locus, has not been observed in any other P. multocida strains examined to date (37). The role that these multiple LPS glycoforms play in disease progression and immunity is not known, but three of the outer core structures expressed are identical to the carbohydrate structure in the globo series of vertebrate glycosphingolipids, i.e., P, Pk, and Forssman antigen. LPS is considered a major immunogen of Gram-negative bacteria, but as the vertebrate immune system is tolerant of self-antigens, there may be a failure to respond appropriately to strains expressing these antigens. Thus, it is likely that the expression of L3 LPS molecules may aid the in vivo survival/persistence of bacteria through avoidance of the host immune system.
Supplementary Material
ACKNOWLEDGMENTS
We thank Mark Edmunds for PCR analysis of the Australian field isolates, our NRC-IBS colleague Perry Fleming and the Bacterial Culture Facility for large-scale biomass production, and Jacek Stupak for mass spectrometry.
This work was funded in part by the Australian Research Council, Canberra, Australia, and the Poultry Cooperative Research Centre, Armidale, Australia.
Footnotes
Published ahead of print 23 August 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JB.00779-13.
REFERENCES
- 1.Wilkie IW, Harper M, Boyce JD, Adler B. 2012. Pasteurella multocida: diseases and pathogenesis. Curr. Top. Microbiol. Immunol. 361:1–22 [DOI] [PubMed] [Google Scholar]
- 2.Christensen JP, Bisgaard M. 2000. Fowl cholera. Rev. Sci. Tech. 19:626–637 [DOI] [PubMed] [Google Scholar]
- 3.De Alwis MCL. 1992. Haemorrhagic septicaemia—a general review. Br. Vet. J. 148:99–112 [DOI] [PubMed] [Google Scholar]
- 4.Chanter N, Rutter JM. 1989. Pasteurellosis in pigs and the determinants of virulence of toxigenic Pasteurella multocida, p 161–195 In Adlam C, Rutter JM. (ed), Pasteurella and pasteurellosis. Academic Press, London, United Kingdom [Google Scholar]
- 5.Weber DJ, Wolfson JS, Swartz MN, Hooper DC. 1984. Pasteurella multocida infections. Report of 34 cases and review of the literature. Medicine (Baltimore, MD) 63:133–154 [PubMed] [Google Scholar]
- 6.Carter GR. 1952. The type specific capsular antigen of Pasteurella multocida. Can. J. Med. Sci. 30:48–53 [DOI] [PubMed] [Google Scholar]
- 7.Townsend KM, Boyce JD, Chung JY, Frost AJ, Adler B. 2001. Genetic organization of Pasteurella multocida cap loci and development of a multiplex capsular PCR typing system. J. Clin. Microbiol. 39:924–929 (Erratum, 39:2377) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Heddleston KL, Gallagher JE, Rebers PA. 1972. Fowl cholera: gel diffusion precipitin test for serotyping Pasteurella multocida from avian species. Avian Dis. 16:925–936 [PubMed] [Google Scholar]
- 9.Boyce JD, Harper M, Wilkie IW, Adler B. 2010. Pasteurella, p 325–346 In Gyles CL, Prescott JF, Songer JG, Thoen CO. (ed), Pathogenesis of bacterial infections of animals, 4th ed. Blackwell Publishing, Ames, IA [Google Scholar]
- 10.Scott PC, Markham JF, Whithear KG. 1999. Safety and efficacy of two live Pasteurella multocida aro-A mutant vaccines in chickens. Avian Dis. 43:83–88 [PubMed] [Google Scholar]
- 11.Harper M, St Michael F, Vinogradov E, John M, Steen JA, van Dorsten L, Boyce JD, Adler B, Cox AD. 2013. Structure and biosynthetic locus of the lipopolysaccharide outer core produced by Pasteurella multocida serovars 8 and 13 and the identification of a novel phospho-glycero moiety. Glycobiology 23:286–294 [DOI] [PubMed] [Google Scholar]
- 12.St Michael F, Harper M, Parnas H, John M, Stupak J, Vinogradov E, Adler B, Boyce JD, Cox AD. 2009. Structural and genetic basis for the serological differentiation of Pasteurella multocida Heddleston serotypes 2 and 5. J. Bacteriol. 191:6950–6959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Harper M, St Michael F, John M, Vinogradov E, Adler B, Boyce JD, Cox AD. 2011. Pasteurella multocida Heddleston serovars 1 and 14 express different lipopolysaccharide structures but share the same lipopolysaccharide biosynthesis outer core locus. Vet. Microbiol. 150:289–296 [DOI] [PubMed] [Google Scholar]
- 14.Harper M, St Michael F, Vinogradov E, John M, Boyce JD, Adler B, Cox AD. 2012. Characterization of the lipopolysaccharide from Pasteurella multocida Heddleston serovar 9: identification of a proposed bi-functional dTDP-3-acetamido-3,6-dideoxy-alpha-d-glucose biosynthesis enzyme. Glycobiology 22:332–344 [DOI] [PubMed] [Google Scholar]
- 15.St Michael F, Li J, Cox AD. 2005. Structural analysis of the core oligosaccharide from Pasteurella multocida strain X73. Carbohydr. Res. 340:1253–1257 [DOI] [PubMed] [Google Scholar]
- 16.St Michael F, Li J, Vinogradov E, Larocque S, Harper M, Cox AD. 2005. Structural analysis of the lipopolysaccharide of Pasteurella multocida strain VP161: identification of both Kdo-P and Kdo-Kdo species in the lipopolysaccharide. Carbohydr. Res. 340:59–68 [DOI] [PubMed] [Google Scholar]
- 17.St Michael F, Vinogradov E, Li J, Cox AD. 2005. Structural analysis of the lipopolysaccharide from Pasteurella multocida genome strain Pm70 and identification of the putative lipopolysaccharide glycosyltransferases. Glycobiology 15:323–333 [DOI] [PubMed] [Google Scholar]
- 18.May BJ, Zhang Q, Li LL, Paustian ML, Whittam TS, Kapur V. 2001. Complete genomic sequence of Pasteurella multocida, Pm70. Proc. Natl. Acad. Sci. U. S. A. 98:3460–3465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Boyce JD, Harper M, St Michael F, John M, Aubry A, Parnas H, Logan SM, Wilkie IW, Ford M, Cox AD, Adler B. 2009. Identification of novel glycosyltransferases required for assembly of the Pasteurella multocida A:1 lipopolysaccharide and their involvement in virulence. Infect. Immun. 77:1532–1542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Steen JA, Steen JA, Harrison P, Seemann T, Wilkie I, Harper M, Adler B, Boyce JD. 2010. Fis is essential for capsule production in Pasteurella multocida and regulates expression of other important virulence factors. PLoS Pathog. 6:e1000750. 10.1371/journal.ppat.1000750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Murray GL, Ellis KM, Lo M, Adler B. 2008. Leptospira interrogans requires a functional heme oxygenase to scavenge iron from hemoglobin. Microbes Infect. 10:791–797 [DOI] [PubMed] [Google Scholar]
- 22.Houliston RS, Bernatchez S, Karwaski MF, Mandrell RE, Jarrell HC, Wakarchuk WW, Gilbert M. 2009. Complete chemoenzymatic synthesis of the Forssman antigen using novel glycosyltransferases identified in Campylobacter jejuni and Pasteurella multocida. Glycobiology 19:153–159 [DOI] [PubMed] [Google Scholar]
- 23.Persson K, Ly HD, Dieckelmann M, Wakarchuk WW, Withers SG, Strynadka NC. 2001. Crystal structure of the retaining galactosyltransferase LgtC from Neisseria meningitidis in complex with donor and acceptor sugar analogs. Nat. Struct. Biol. 8:166–175 [DOI] [PubMed] [Google Scholar]
- 24.Schweda EK, Richards JC, Hood DW, Moxon ER. 2007. Expression and structural diversity of the lipopolysaccharide of Haemophilus influenzae: implication in virulence. Int. J. Med. Microbiol. 297:297–306 [DOI] [PubMed] [Google Scholar]
- 25.Raetz CR, Whitfield C. 2002. Lipopolysaccharide endotoxins. Annu. Rev. Biochem. 71:635–700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schnaar RL, Suzuki A, Stanley P. 2009. Glycosphingolipids, p 129–141 In Varki A, Cummings RD, Esko JD, Freeze HH, Stanley P, Bertozzi CR, Hart GW, Etzler ME. (ed), Essentials of glycobiology, 2nd ed. CSH Press, New York, NY [Google Scholar]
- 27.Kitamoto N, Ikuta K, Shoji H, Kato S, Naiki M. 1980. Distribution of Forssman antigen in chickens. Biken J. 23:179–192 [PubMed] [Google Scholar]
- 28.Shiraishi T, Uda Y. 1985. Characterization of neutral sphingolipids from chicken erythrocytes. J. Lipid Res. 26:860–866 [PubMed] [Google Scholar]
- 29.Houliston RS, Vinogradov E, Dzieciatkowska M, Li J, St Michael F, Karwaski MF, Brochu D, Jarrell HC, Parker CT, Yuki N, Mandrell RE, Gilbert M. 2011. Lipooligosaccharide of Campylobacter jejuni: similarity with multiple types of mammalian glycans beyond gangliosides. J. Biol. Chem. 286:12361–12370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mandrell RE, McLaughlin R, Aba Kwaik Y, Lesse A, Yamasaki R, Gibson B, Spinola SM, Apicella MA. 1992. Lipooligosaccharides (LOS) of some Haemophilus species mimic human glycosphingolipids, and some LOS are sialylated. Infect. Immun. 60:1322–1328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mandrell RE. 1992. Further antigenic similarities of Neisseria gonorrhoeae lipooligosaccharides and human glycosphingolipids. Infect. Immun. 60:3017–3020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Heddleston KL. 1962. Studies on pasteurellosis. v. Two immunogenic types of Pasteurella multocida associated with fowl cholera. Avian Dis. 6:315–321 [PubMed] [Google Scholar]
- 33.Subaaharan S, Blackall LL, Blackall PJ. 2010. Development of a multi-locus sequence typing scheme for avian isolates of Pasteurella multocida. Vet. Microbiol. 141:354–361 [DOI] [PubMed] [Google Scholar]
- 34.Blackall PJ, Fegan N, Chew GT, Hampson DJ. 1998. Population structure and diversity of avian isolates of Pasteurella multocida from Australia. Microbiology 144:279–289 [DOI] [PubMed] [Google Scholar]
- 35.Harper M, Cox AD, St Michael F, Wilkie IW, Boyce JD, Adler B. 2004. A heptosyltransferase mutant of Pasteurella multocida produces a truncated lipopolysaccharide structure and is attenuated in virulence. Infect. Immun. 72:3436–3443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cardenas M, Fernandez de Henestrosa AR, Campoy S, Perez de Rozas AM, Barbe J, Badiola I, Llagostera M. 2001. Virulence of Pasteurella multocida recA mutants. Vet. Microbiol. 80:53–61 [DOI] [PubMed] [Google Scholar]
- 37.Harper M, Cox AD, Adler B, Boyce JD. 2011. Pasteurella multocida lipopolysaccharide: the long and the short of it. Vet. Microbiol. 153:109–115 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.