

Abstract
Bacteria require explicit control over their proteomes in order to compete and survive in dynamic environments. The Lyme disease spirochete Borrelia burgdorferi undergoes substantial protein profile changes during its cycling between vector ticks and vertebrate hosts. In an effort to understand regulation of these transitions, we recently isolated and functionally characterized the borrelial nucleic acid-binding protein BpuR, a PUR domain-containing protein. We now report that this regulatory protein governs its own synthesis through direct interactions with bpuR mRNA. In vitro and in vivo techniques indicate that BpuR binds with high affinity and specificity to the 5′ region of its message, thereby inhibiting translation. This negative feedback could permit the bacteria to fine-tune cellular BpuR concentrations. These data add to the understanding of this newly described class of prokaryotic DNA- and RNA-binding regulatory proteins.
INTRODUCTION
Global regulatory factors that bind nucleic acids act on diverse targets to modulate bacterial physiology and pathogenesis. Due to the inherent biochemical features of nucleic acid-binding proteins, self-regulation is a common theme (1–7). This mode of action affords precise feedback regulation, creating rheostat-like control over the protein's regulon. It could be surmised that this feature is a critical component of bacterial physiology to ensure that cellular concentrations do not surpass a critical threshold, which might result in deleterious effects (5, 8–11).
In addition to transcription initiation, accumulating evidence indicates that, like their multicellular counterparts, bacteria regulate their proteomes at the posttranscriptional level. Strategies that they use to do so are diverse and include antisense RNAs, secondary structures that respond to small molecules (riboswitches), and mRNA-binding proteins that can stabilize or promote transcript degradation (12–16). Known functions of bacterial RNA-binding proteins include roles in virulence, cellular physiology, DNA replication, and molecular trafficking (17–29). As knowledge of the diverse targets of RNA-binding proteins continues to expand, the need to understand how these factors are regulated becomes more pressing.
We recently biochemically characterized Borrelia burgdorferi BpuR, a novel type of prokaryotic nucleic acid-binding protein (19). BpuR shares significant structural and sequence identity with eukaryotic PUR-domain proteins, which are critical pre- and posttranscriptional regulatory factors (30–36). BpuR is a transcriptional regulatory factor in the Lyme disease spirochete Borrelia burgdorferi and its own production is controlled by the bacterium (19). We now demonstrate that BpuR can bind with high affinity to its own mRNA and inhibit its own translation.
MATERIALS AND METHODS
Bacterial strains and culture conditions.
B. burgdorferi was cultured in Barbour-Stoenner-Kelly II medium (37, 38). When appropriate, kanamycin, gentamicin, or both were added to cultures of transformed bacteria at final concentrations of 200 μg/ml and 70 μg/ml, respectively. For transformation studies, a clonal derivative of B. burgdorferi type strain B31, known as B31e2, was used. RNA immunoprecipitation and primer extension experiments were performed using the infectious clonal strain B31 MI-16 (39).
5′ RACE to determine the bpuR transcriptional start site.
B. burgdorferi B31 MI-16 was grown at either 23 or 34°C to mid-exponential growth phase (approximately 5 × 107 cells/ml). Cells were harvested, and RNA was isolated as previously described (40). Purified RNA was treated with DNase to remove contaminating DNA and reverse transcribed into cDNA following the manufacturer's recommended procedures (Roche). Primer extension was performed on each RNA preparation using 5′ rapid amplification of cDNA ends (RACE; Invitrogen, Carlsbad, CA). Controls consisted of reaction mixtures that lacked reverse transcriptase, hexameric oligonucleotide primers, or template DNA. The resulting fragments were cloned into pCR2.1 (Invitrogen). Twelve clones were selected at random, and the inserts were sequenced. The sequences of the products were aligned, compared to the B. burgdorferi B31 genome sequence, and aligned with the sequences of other Lyme disease-causing spirochetes using the Geneious program.
In vivo production of BpuR or EbfC from inducible promoter constructs.
We previously described plasmid constructs in which either bpuR or ebfC is under the transcriptional control of the inducible Post promoter system (19, 41–43). Each plasmid was individually introduced into B. burgdorferi B31e2. To evaluate the dose-dependent response to inducer, anhydrotetracycline (ATc) was added to early-exponential-phase cultures (approximately 105 bacteria/ml) of each transformed strain at a final concentration of 0, 0.2, 0.4, or 0.8 μg/ml. After cultivation to a final density of approximately 107 bacteria/ml, bacteria were harvested by centrifugation, washed, and lysed and proteins were separated by SDS-polyacrylamide gel electrophoresis (PAGE). Total proteins were detected by Coomassie brilliant blue staining to assess them for equal loading.
Immunoblot analyses.
EbfC was identified by immunoblotting using monospecific antiserum (41). The constitutively expressed FlaB protein served as a loading control (44, 45). Antiserum directed against BpuR was produced commercially in New Zealand White rabbits by NeoPeptide (Cambridge, MA). A polypeptide corresponding to the BpuR sequence VESKRSPSGDFERH was used for vaccination. Antibodies were affinity purified from serum using the vaccinogen polypeptide.
Lysates from transformed strains were boiled, separated by SDS-PAGE, and transferred to nitrocellulose membranes. Membranes were blocked with Sea Block blocking buffer (Thermo Fisher, Hudson, NH). Primary antibody detection was accomplished by secondary donkey anti-rabbit (EbfC and BpuR) or goat anti-mouse (FlaB) IgG conjugated to horseradish peroxidase and detected by SuperSignal West Pico chemiluminescent substrate (Thermo Fisher, Hudson, NH). Band intensities were normalized to the band intensity of FlaB by densitometric analysis and graphed using GraphPad Prism (version 5.0) software (46).
Recombinant proteins.
Recombinant BpuR was expressed and purified as previously described (19). Purified proteins were dialyzed against a buffer compatible with electrophoretic mobility shift assay (EMSA) (1 nM dithiothreitol, 25 mM Tris [pH 7.5], 5 mM NaCl, 0.01% [vol/vol] Tween 20, 10% glycerol, 0.1% [vol/vol] phenylmethanesulfonyl fluoride) or compatible with in vitro transcription/translation reactions (25 mM Tris [pH 7.5], 5 mM NaCl, 1% [vol/vol] glycerol) (19, 41, 42). Protein concentrations were determined by Bradford analysis (Bio-Rad, Hercules, CA). Protein preparation purity was determined by SDS-PAGE and staining with Coomassie brilliant blue. Aliquots of purified protein were stored at −80°C.
EMSA.
Nucleic acids used as probes or PCR primers are described in Table 1. For double-stranded DNA (dsDNA) probes, one oligonucleotide primer was 5′ end labeled with biotin and annealed as previously described (Integrated DNA Technologies [IDT], Coralville, IA) (47). Single-stranded RNA probes labeled with biotin at the 5′ end were synthesized chemically by IDT. All probe concentrations were determined spectrophotometrically.
Table 1.
EMSA probes and oligonucleotide primers used in this studya
| Oligonucleotide name | Modification | Sequence (5′→3′) | Target/purpose |
|---|---|---|---|
| BioRNAbpuRp-F (RNA) | 5′ biotin | CUUAAAUGUAGUCAAGUACAAAAACUUGUGUGGAGGAAAUUGAUGGGAGAGAGAGGGGAAGUAUACUCU | bpuR TSS to nt +30/EMSA |
| BioRNAbpuRORF (RNA) | 5′ biotin | AAACUAUUUACAGAGUCUGAGAGAACUUAUUUUUUUAAUGUCAAGGAAAAUAGAAAAGGAGAUUAUUUU | bpuR nt +31 to +100/EMSA |
| BioRNA61-F (RNA) | 5′ biotin | AAUGGAGAGAUUUUGGGGGAGUUGUUUAAAAUUACAUUUGCGUUUUGUUAAAAUG | erp operator/EMSA |
| bpuRp-12 | None | GCTAGCTAAAAATAACATTAC | bpuR NCD/EMSA-dsDNA |
| BiobpuRp-13 | 5′ biotin | GTAATGTTATTTTTAGCTCGA | bpuR NCD/EMSA-dsDNA |
| bpuRp-14 | None | CCACACAAGTTTTTGTACTTGAC | bpuR NCD/EMSA-dsDNA |
| Bioerp 129-F | 5′ biotin | GAGACGGGGAGTTGTTAAATT | erp operator/EMSA-dsDNA |
| A69-R | None | GTAACAGCTGAATGTAAC | erp operator/EMSA-dsDNA |
| GFP-5 | None | GTGACAAGTGTTGGCCATGGAAC | gfp mRNA |
| GFP-6 | None | CACTGGAGTTGTCCCAATTCTTGTTG | gfp mRNA |
| BpuR-1 | None | GGAGAGAGAGGGGAACTATAC | bpuR RIP-PCR |
| BpuR-2 | None | GCCTTGCAAAGGAGCCAACG | bpuR RIP-PCR |
| Fla-3 | None | GGGTCTCAAGCGTCTTGG | flaB RIP-PCR |
| Fla-4 | None | GAACCGGTGCAGCCTGAG | flaB RIP-PCR |
All nucleic acids were DNA, except as noted. NCD, noncoding DNA region; ORF, open reading frame; TSS, transcription start site.
EMSAs were performed essentially as previously described (48, 49). Protein-nucleic acid combinations were subjected to electrophoresis using 10% nondenaturing polyacrylamide gels (Invitrogen). Following transfer to Biodyne nylon membranes (Thermo Pierce) and UV cross-linking (Stratalinker 1800; Stratagene, San Diego, CA), biotin-labeled DNAs were visualized using nucleic acid detection kits (Thermo Pierce) and autoradiography.
For RNA-binding assays, all equipment was treated with diethyl pyrocarbonate prior to use, and RiboGuard RNase inhibitor (Epicenter, Madison WI) was added to each reaction mixture to a final concentration of 1 U/ml.
RNA immunoprecipitation (RIP).
Culture conditions, cross-linking, and soluble fraction preparation have been previously described, with the exception that cross-linking time was reduced to 4 min (41, 42). We modified the immunoprecipitation (IP) buffer to contain 5 U/ml of RiboGuard. To shear the bacterial RNA, lysates were sonicated using a Branson 102C sonicator (Branson Ultrasonics, Danbury, CT) with 10 pulses of 15 s each at 15% amplitude.
Anti-BpuR or anti-IgG control (Santa Cruz) antibodies or phosphate-buffered saline (PBS) alone (bead control) was incubated with equal amounts of cleared lysate overnight at 4°C. Protein G resin particles were added to each IP or IP control reaction for 2 h at 4°C in the presence of RiboGuard. Target antigen-bead complexes were washed 5 times as previously described with the addition of 2 U/ml of RiboGuard (41, 42). The formaldehyde cross-link was reversed in RNase-free TE (Tris-EDTA) at 75°C for 10 min, RNA was purified and DNase treated, and cDNA synthesis was performed as described previously (50).
Oligonucleotides specific for the bpuR or flaB open reading frame (ORF) were used in separate PCRs, with BpuR IP eluates, the bead control, or the IP control serving as the template cDNA (Table 1). Amplicons were separated by agarose gel electrophoresis, stained with ethidium bromide, and imaged.
bpuR::gfp transcriptional/translational fusions and flow cytometry.
The plasmid containing a promoterless green fluorescent protein (GFP) gene (pBLS590) is described elsewhere and served as a background fluorescence control (51). In addition, this construct served as the backbone to splice the noncoding region of bpuR DNA into a site immediately 5′ of gfp, generating pGJ1. Derivative pBLJ370 was produced by site-directed mutagenesis of pGJ1 to add the first 30 bp of bpuR in frame with the beginning of gfp.
These three constructs were individually introduced into B. burgdorferi. In addition, B. burgdorferi strains that contained pBLJ307, which constitutively expresses high levels of bpuR, plus either pGJ1 or pBLJ370, were produced. pBLJ307 confers resistance to gentamicin, whereas pGJ1 and pBLJ370 confer resistance to kanamycin. Each strain was cultured at 34°C to mid-exponential growth phase. Bacteria were independently harvested, washed in PBS, and resuspended in PBS at approximately 106 cells/ml. The mean GFP fluorescence per bacterial cell was analyzed using a FACSCalibur flow cytometer (BD Biosciences, San Jose, CA), with excitation at 488 nm and detection at 530 nm. Each experiment involved measuring a minimum of 50,000 individual bacteria. The results reported represent a mean of three independent experiments.
In vitro coupled transcription and translation.
A linear DNA fragment was produced by PCR from template pGJ1 or pBLJ370 using oligonucleotide primers M13 Forward (5′-GTAAAACGACGGCCAG-3′) and M13 Reverse (5′-CAGGAAACAGCTATGAC-3′). Coupled in vitro transcription and translation reactions, using Escherichia coli S30 extracts (Promega) were performed as previously described, with slight modifications (19, 41). Each reaction mixture was identical, with the exception that the final BpuR concentration was 0, 2, 4, or 8 nM. In addition, 1 U of RNase inhibitor was added to all reaction mixtures to prevent mRNA decay, which could influence the total amount of RNA present.
Quantitative reverse transcription-PCR (qRT-PCR) and enzyme-linked immunosorbent assay (ELISA) were used to evaluate the influence of BpuR on gfp transcript and protein levels, respectively. Briefly, after completion, the reaction mixtures were separated into equal volumes and gfp transcript or GFP protein levels were evaluated as described previously (19, 41). For transcript production, total RNA was extracted using an Epicenter MasterPure RNA extraction kit following the manufacturer's recommended procedure. Contaminating DNA was removed by DNase treatment, and RNA was reverse transcribed into cDNA as previously described (50). Quantitative PCRs (qPCRs) were performed as previously described (42, 52). ELISAs were performed using standard methods, as previously described (41, 53). GFP detection utilized anti-GFP–horseradish peroxidase conjugate (MACSmolecular; Miltenyi Biotec, Auburn, CA).
Statistical analyses.
Statistical significance between samples was determined by Student's t test, assuming unequal variance. Protein level differences were evaluated densitometrically using ImageJ software and normalized against the levels for the loading controls. Each experiment was performed at least twice, unless otherwise described in the text.
RESULTS
Defining the bpuR transcriptional unit.
Extending our observations that B. burgdorferi controls cellular levels of the BpuR protein (19), we sought to determine the basis of this regulation. The bpuR gene is flanked on either side by divergently transcribed genes, indicating that bpuR forms a monocistronic operon (54) (Fig. 1A). Mapping by 5′ RACE detected a single transcriptional start site 42 bp upstream of the first coding methionine (Fig. 1B). Identical results were obtained from analyses of RNA purified from bacteria cultured under conditions that yielded either high or low levels of BpuR production, suggesting that bpuR is transcribed from only a single promoter (19, 46).
Fig 1.

Mapping the bpuR transcriptional start site. (A) Schematic of the bpuR operon and neighboring loci in Lyme disease spirochetes. 5′ RACE analyses located the bpuR transcriptional start site 42 nt upstream of the first translational start codon (+1). Diamonds, locations of primers used for RIP-PCR. (B) Nucleotide sequence alignment of the bpuR operons indicated conservation in Lyme disease-causing spirochetes. Green arrow, site of the transcription initiation nucleotide; shaded boxes, predicted −10 and −35 regions; solid vertical black lines, bpuR ribosome-binding site (RBS); blue line, DNA sequence used for the bpuR operon fusion pBLJ370; red line, the corresponding DNA of pBLJ370 used for RNA; black line, the bpuR mRNA required for negative autoregulation shown at the level of DNA.
BpuR binds bpuR mRNA.
The first clue about BpuR autoregulation came from studies of B. burgdorferi strains that contain a construct that should enhance cellular concentrations of BpuR. In order to evaluate the effects of BpuR levels on B. burgdorferi physiology, the bpuR ORF was placed under the control of the inducible Post promoter (19, 43). This TetR-repressible promoter permits the experimenter to precisely regulate transcription by titrating the inducer molecule anhydrotetracycline (ATc) into culture medium (43). In all previously tested Post-regulated chimeras, increasing concentrations of ATc induced protein in a dose-dependent manner (41–43, 47). For example, B. burgdorferi containing a Post::ebfC transcriptional fusion produced steadily increasing concentrations of EbfC protein as ATc was titrated into the culture medium (Fig. 2) (41, 42). However, equivalent titration of ATc into cultures of B. burgdorferi carrying the Post::bpuR construct initially increased BpuR levels approximately 1.8-fold, but BpuR levels did not increase further, regardless of how much ATc was added (Fig. 2). Noting that this construct lacks the native bpuR promoter, the observed regulation of BpuR protein production must be an intrinsic property of BpuR and/or the bpuR ORF.
Fig 2.
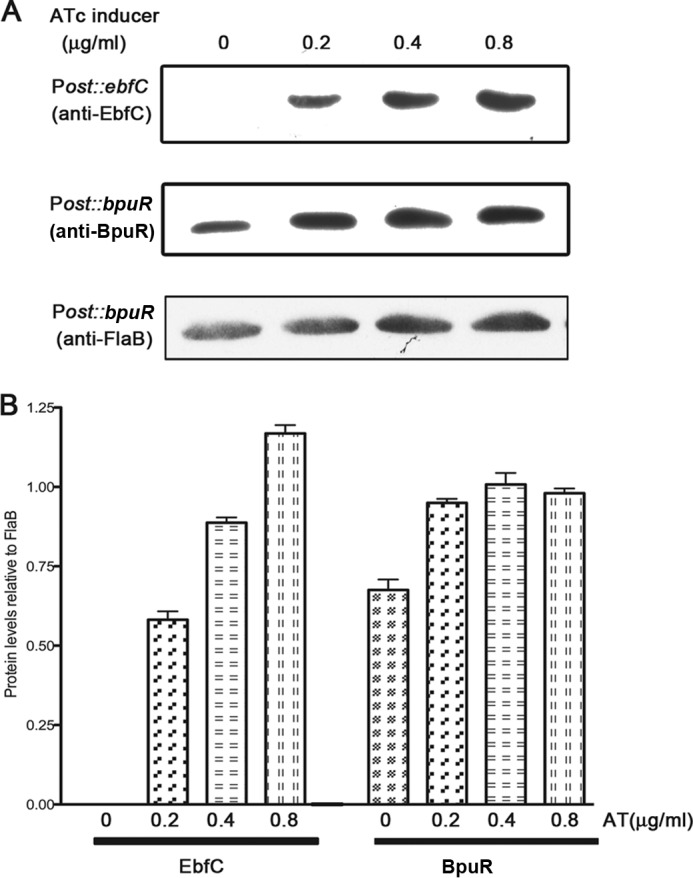
Autogenous regulation of BpuR occurs independently of the bpuR promoter. (A) B. burgdorferi was independently transformed with plasmids carrying Post::ebfC or Post::bpuR. The levels of EbfC and BpuR were assessed by immunoblotting following induction with 0, 0.2, 0.4, or 0.8 μg/ml ATc. Levels of FlaB (flagellin), which served as a loading control, were determined by immunoblotting. Illustrated are FlaB immunoblotting results for the bacteria carrying Post::bpuR. (B) Densitometric analysis of Post::ebfC and Post::bpuR immunoblotting results normalized to the FlaB data for each strain. Note that the anti-BpuR antibody detected BpuR produced by the bacterium's native bpuR locus (lane with no ATc added).
BpuR binds RNA with a 10-fold higher affinity than dsDNA (19). The protein exhibits sequence specificity with both substrates, preferentially interacting with guanine-rich sequences (Fig. 3; see Fig. 5) (19). The initial nucleotides of bpuR mRNA include an extensive stretch of purines (Fig. 1). Centering on the translational start site, 18 of 34 (53%) nucleotides (nt) of bpuR mRNA are guanine, an oddity in this bacterium that overall contains less than 30% G+C (54).
Fig 3.
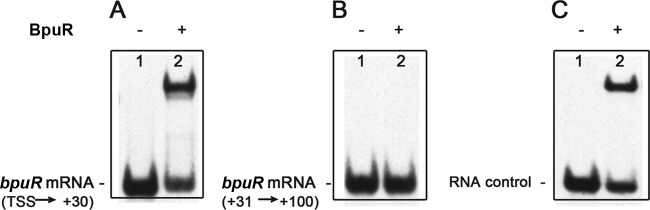
BpuR binds specifically to bpuR mRNA in vitro. EMSAs were performed with recombinant BpuR and a labeled bpuR RNA transcript. (A) Labeled RNA (2 nM) corresponding to the bpuR transcriptional start site to position +30 of the bpuR ORF; (B) labeled RNA (2 nM) corresponding to positions +31 to +100 of the bpuR ORF; (C) labeled erp operator sequence (2 nM), as RNA, serving as a positive control (19). Each probe was incubated either with 10 nM BpuR protein (lanes +) or without added BpuR (lanes −).
Fig 5.
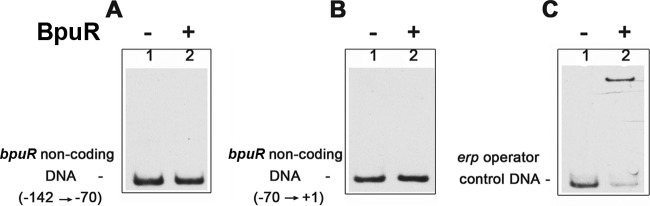
BpuR does not bind bpuR promoter DNA. EMSAs of recombinant purified BpuR and bpuR 5′ noncoding DNA. (A) Labeled dsDNA (1 nM) corresponding to positions −142 to −71 relative to the bpuR translational start site; (B) labeled dsDNA (1 nM) corresponding to positions −70 to +1 relative to the bpuR translational start site; (C) labeled erp operator dsDNA (1 nM) serving as a positive control (19). Each probe was incubated either with 60 nM BpuR protein (lanes +) or without added BpuR (lanes −).
To test whether BpuR can bind its own mRNA, EMSAs were performed using labeled RNAs derived from the transcriptional start site through the first 30 nt of the bpuR ORF, or nt 31 to 100 of the bpuR ORF. In addition, an RNA molecule consisting of the high-affinity BpuR-binding sequence in the erp operator served as a positive control (19). BpuR bound to the RNA that consisted of the bpuR ribosome-binding site and the first 30 nucleotides of the open reading frame (Fig. 3A). The affinity for this labeled RNA was similar to that displayed for the control probe, which has a dissociation constant (Kd) of 13 nM (Fig. 3A and C) (19). In contrast, BpuR did not bind to the RNA sequence derived from the downstream region of its own open reading frame (Fig. 3B). Since each RNA probe was approximately the same size (∼70 nt), those differences cannot be attributed to a probe length bias (55).
Extending our in vitro analysis, we next directly tested whether BpuR binds its native mRNA in live B. burgdorferi, using RNA immunoprecipitation (RIP) (56–58). Much like chromatin immunoprecipitation (ChIP), RIP utilizes cross-linking of proteins to nucleic acids in live bacteria, followed by immunoprecipitation of complexes that contain a specific protein. However, RNA rather than DNA is purified from the immunoprecipitate. The RNA was reverse transcribed and then subjected to PCR with oligonucleotide primers specific for bpuR mRNA.
BpuR-specific RIP yielded a single amplicon of the same size as a genomic DNA control (Fig. 4, top, lanes 3 and 4, respectively). RIP control reactions included a mock immunoprecipitation using an irrelevant IgG affixed to protein G beads or protein G beads alone. Those reactions failed to produce PCR products (Fig. 4, top, lanes 1 and 2). Since molecular crowding and absolute transcript levels can cause transient, nonspecific interactions between proteins and nucleic acids, the flaB mRNA was tested as a control to confirm RIP specificity (59, 60). flaB expression is constitutive, and cellular mRNA levels are abundant relative to the levels of other mRNAs in the borrelial cell, making it a suitable control locus for RIP evaluation (44, 45). Control or BpuR IP PCR of immunoprecipitated mRNA did not yield a flaB amplicon (Fig. 4, bottom). Thus, these RIP assays may be concluded to be specific, demonstrating that BpuR binds to its own mRNA in live B. burgdorferi.
Fig 4.

RIP demonstration that BpuR binds bpuR mRNA in vivo. Live B. burgdorferi bacteria were cross-linked, fixing BpuR to mRNAs within the cell. IP was conducted with BpuR-specific antibodies. BpuR-bound mRNAs were eluted, DNase treated, and reverse transcribed. PCR was performed using oligonucleotides specific for the bpuR or flaB ORF (top and bottom panels, respectively). Lane 1, control reactions of protein A beads alone; lane 2, control reactions of protein A beads plus nonspecific IgG; lane 3, RIP reactions of protein A beads plus BpuR-specific antibodies; lane 4, PCR of purified B. burgdorferi genomic DNA, serving as a control.
BpuR does not bind bpuR promoter DNA.
Many nucleic acid-binding proteins control their own production by interacting with DNA adjacent to their own promoter (5, 61, 62). Noting that BpuR can bind double-stranded DNA (19), we could not disregard that possibility for BpuR. Moreover, eukaryotic PUR-domain proteins may govern their own transcription through direct interactions with noncoding DNA (35). To that end, EMSAs were performed with recombinant BpuR protein and two dsDNA probes that span the bpuR 5′ noncoding region. BpuR did not bind to either of these probes (Fig. 5A and B). Since both dsDNA probes were the same length as the bpuR RNA probes described above, we could confidently discount probe length as a variable influencing BpuR-nucleic acid interactions (55). Furthermore, controls with a labeled erp operator dsDNA probe confirmed that BpuR was active and bound to that high-affinity dsDNA sequence (Fig. 5C) (19). While it is possible that BpuR might bind dsDNA outside the bpuR 5′ noncoding region, additional studies described below demonstrated that BpuR does not detectably affect transcription from its own promoter.
BpuR is a negative autoregulator.
To test the effect of BpuR binding to mRNA in live borreliae, two GFP reporter constructs were created: pGJ1, which consists of the bpuR promoter driving transcription of gfp, and pBLJ370, which consists of the bpuR promoter and the first 30 bp of bpuR fused in frame to gfp (Fig. 6A). B. burgdorferi was transformed with pGJ1 or pBLJ370 to produce strains BJ26 and BJ27, respectively. Each of those plasmids was additionally transformed into a B. burgdorferi strain that carries a second plasmid, pBLJ307, which caused maximal, constitutive expression of BpuR. Thus, strain BJ30 carries pGJ1 and pBLJ307 and strain BJ31 carries pBLJ370 and pBLJ307. This strategy provided a means for comparison between increased BpuR production and the potential effect on both the transcription and translation of the bpuR gene. Each construct harbors a different selectable marker, so incubation with both kanamycin and gentamicin ensured maintenance of both plasmids.
Fig 6.

BpuR transcriptional and translational fusions. (A) Schematic of the plasmids transformed into B. burgdorferi. pBLJ307 carries a gentamicin resistance cassette and the bpuR locus driven by the constitutively expressed flgB promoter. pGJ1 possesses the bpuR promoter and 5′ noncoding DNA fused to gfp. pBLJ370 additionally contains the first 30 bp of the bpuR ORF fused to gfp. Both pGJ1 and pBLJ370 carry kanamycin resistance markers. Nucleotide sequences displayed inside vertical lines show the similarities (gray, lowercased, and italicized nucleotides) and differences (uppercased and black nucleotides) between pGJ1 and pBLJ370. The same ribosome-binding site (underlined nucleotides) and start codon (bold, lowercased nucleotides) were present in each construct. For continuity, the 2nd and 3rd codons of the GFP ORF are shown (broken lines). (B) Mean GFP expression assessed by flow cytometry from three independent experiments (arbitrary units). All values were normalized against the values obtained from a strain that carried a promoterless gfp construct (51). *, statistically significant difference between BJ26 and BJ27; **, statistically significant difference between BJ27 and BJ31. (C) Immunoblot analyses of each B. burgdorferi strain to assess cellular levels of BpuR and the constitutively expressed FlaB protein (loading control).
There were no significant differences between GFP expression by BJ26 and BJ30, which carry the wild-type gfp gene and do not and do express high levels of BpuR, respectively (Fig. 6B and C). However, addition of the first 30 bp of bpuR to gfp reduced GFP levels by 50% in bacteria that produced wild-type levels of BpuR (strain BJ27) and by 75% in bacteria that expressed higher levels of BpuR (strain BJ31) (Fig. 6B and C).
Considering that constitutive BpuR expression might alter reporter plasmid copy numbers, which could explain the reduction in GFP production, qPCR was performed on strains BJ27 and BJ31. Plasmid amplicons from each strain were determined quantitatively, and their amounts were normalized to the relative amount of the B. burgdorferi main linear chromosome present. The relative ratio of the amount of pBLJ370 in strain BJ27 to that in strain BJ31 was 1.095, indicating that elevated BpuR levels did not impact reporter plasmid copy number.
As a further, independent approach to determining the effects of BpuR on its own translation, we used an in vitro cell-free transcription/translation system (41). Two different linear templates were generated: the bpuR promoter fused to gfp (pGJ1 template) and the bpuR promoter fused to the first 30 nt of the bpuR ORF in frame with gfp (pBLJ370 template) (Fig. 6A). Purified BpuR was titrated into E. coli S30 transcription/translation reaction mixtures containing each template, and then qRT-PCR and ELISA were performed to determine the relative levels of mRNA and protein produced, respectively. The addition of purified BpuR did not have any significant effects on transcription from either template DNA (Fig. 7A). BpuR did not significantly affect the synthesis of GFP from the pGJ1 bpuR promoter fusion. In contrast, addition of 8 mM BpuR significantly inhibited translation from the pBLJ370 bpuR::gfp chimera (Fig. 7B). Collectively, these findings indicate that BpuR inhibits its own translation but does not significantly affect its transcription.
Fig 7.

In vitro transcription and translation reactions indicating that BpuR inhibits its own translation. Note that the use of two different templates for these analyses means that absolute numbers cannot be compared between templates. (A) Mean qRT-PCR results for the gfp transcript produced from DNA templates consisting of either the bpuR promoter fused to gfp (unshaded bars) or the bpuR promoter plus the first 30 bp of the bpuR ORF fusion in frame to gfp (shaded bars). Reactions were conducted either without added BpuR protein or in the presence of 2, 4, or 8 nM recombinant BpuR. None of the differences in results without and with BpuR were statistically significant. (B) Mean ELISA values of GFP production levels from the same reactions. Values were normalized to those for a reaction mixture containing no template DNA. *, statistically significant difference (P < 0.05).
DISCUSSION
In vitro and in vivo studies indicated that BpuR binds bpuR mRNA and, consequently, inhibits its own translation. The BpuR binding site includes a region proximal to its ribosome-binding site. Thus, BpuR could effectively compete with 30S binding to nascent mRNA, since the calculated Kd for ribosome binding is higher than that of BpuR (19, 63). Physical boundaries for the translational machinery on mRNA extend from positions −20 to +15 relative to the translational start site, which constitutes a considerable portion of BpuR's high-affinity binding site on bpuR mRNA (64, 65). Alternatively, since ribosome occupancy has been proposed to include the first 12 nt from the charged tRNA P site, BpuR may act as a roadblock to prevent productive translation following ribosome loading (15, 66). It is also possible that bpuR transcription and translation are not directly coupled but are instead slightly delayed, creating a larger window for newly synthesized BpuR to adhere to the bpuR message and exert its inhibitory effect (67, 68). Any or all of these mechanisms of translational inhibition could be at play, and further molecular and atomic analyses will be required to determine the kinetics and limits of BpuR-mRNA binding in vivo.
Bacterial Hfq proteins also exhibit self-limiting, posttranscriptional autoregulation (14, 69). However, the mechanisms differ from what appears to occur with BpuR, with Hfq binding to the 5′ untranslated leader of hfq mRNA. BpuR's posttranscriptional autoregulation is reminiscent of the effect of E. coli Hfq on ompA mRNA, where binding adjacent to the ribosome-binding site reduces OmpA translation (15, 70). In that BpuR controls its own translation, it is possible that BpuR may similarly be a posttranscriptional regulator of other B. burgdorferi genes.
Functional similarities between BpuR and Hfq are not limited to autoregulation. BpuR was isolated due to its affinity for erp operator dsDNA (19, 71), and E. coli Hfq has been reported to interact with dsDNA (72–74). Hfq hexamers have two distinct RNA-binding faces, one of which interacts with purine-rich regions of RNA (75). PUR-domain proteins are widespread throughout the Spirochetes and Bacteroidetes phyla, but recognizable homologues are absent from most other bacterial genomes. In contrast, Hfq is almost ubiquitous throughout the Eubacteria and yet absent from the Bacteroidetes (76). An intriguing possibility is that bacterial PUR-domain proteins may be functionally equivalent to Hfq. B. burgdorferi has the most complex known prokaryotic genome and encodes both an Hfq and BpuR, which may together coordinate the different genetic elements (23, 54, 77).
In summary, B. burgdorferi BpuR binds to its own mRNA and inhibits translation. As demonstrated through the use of inducible transcription constructs, this prevents cellular BpuR concentrations from exceeding a certain level. BpuR is a transcriptional and posttranscriptional regulator, binding to both DNA and RNA (19; this study). This novel, multifunctional protein thus presents itself as a significant model for exploring the burgeoning field of bacterial posttranscriptional regulation.
ACKNOWLEDGMENTS
The National Institutes of Health funded this work (grant R01-AI044254), and an exploratory grant to Brian Stevenson was received from the University of Kentucky College of Medicine.
We thank Martha Peterson for her advice and experimental suggestions on the RIP studies and Will Arnold, Cody Creekmore, Gabriela Farina Camara, Michael Fried, Jason Johnston, and Brett Spear for their insightful comments during manuscript preparation.
Footnotes
Published ahead of print 23 August 2013
REFERENCES
- 1.Dersch P, Schmidt K, Bremer E. 1993. Synthesis of the Escherichia coli K-12 nucleoid-associated DNA-binding protein H-NS is subjected to growth-phase control and autoregulation. Mol. Microbiol. 8:875–889 [DOI] [PubMed] [Google Scholar]
- 2.Heroven AK, Nagel G, Tran HJ, Parr S, Dersch P. 2004. RovA is autoregulated and antagonizes H-NS-mediated silencing of invasin and rovA expression in Yersinia pseudotuberculosis. Mol. Microbiol. 53:8718–8788 [DOI] [PubMed] [Google Scholar]
- 3.Ninnemann O, Koch C, Kahmann R. 1992. The E. coli fis promoter is subject to stringent control and autoregulation. EMBO J. 11:1075–1083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brinkman AB, Ettema TJ, de Vos WM, van der Oost J. 2003. The Lrp family of transcriptional regulators. Mol. Microbiol. 48:287–294 [DOI] [PubMed] [Google Scholar]
- 5.Soncini FC, Véscovi EG, Groisman EA. 1995. Transcriptional autoregulation of the Salmonella typhimurium phoPQ operon. J. Bacteriol. 177:4364–4371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhang Y, Gao H, Wang L, Xiao X, Tan Y, Guo Z, Zhou D, Yang R. 2011. Molecular characterization of transcriptional regulation of rovA by PhoP and RovA in Yersinia pestis. PLoS One 6:e25484. 10.1371/journal.pone.0025484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Santangelo MP, Blanco FC, Bianco MV, Klepp LI, Zabal O, Cataldi AA, Bigi F. 2008. Study of the role of Mce3R on the transcription of mce genes of Mycobacterium tuberculosis. BMC Microbiol. 8:38. 10.1186/1471-2180-8-38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.García-Calderón CB, García-Quintanilla M, Casadesús J, Ramos-Morales F. 2005. Virulence attenuation in Salmonella enterica rcsC mutants with constitutive activation of the Rcs system. Microbiology 151:579–588 [DOI] [PubMed] [Google Scholar]
- 9.Humphreys S, Rowley G, Stevenson A, Anjum MF, Woodward MJ, Gilbert S, Kormanec J, Roberts M. 2004. Role of the two-component regulator CpxAR in the virulence of Salmonella enterica serotype Typhimurium. Infect. Immun. 72:4654–4661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lemon KP, Freitag NE, Kolter R. 2010. The virulence regulator PrfA promotes biofilm formation by Listeria monocytogenes. J. Bacteriol. 192:3969–3976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mouslim C, Delgado M, Groisman EA. 2004. Activation of the RcsC/YojN/RcsB phosphorelay system attenuates Salmonella virulence. Mol. Microbiol. 54:386–395 [DOI] [PubMed] [Google Scholar]
- 12.Lee ER, Baker JL, Weinberg Z, Sudarsan N, Breaker RR. 2010. An allosteric self-splicing ribozyme triggered by a bacterial second messenger. Science 329:845–848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sudarsan N, Hammond MC, Block KF, Welz R, Barrick JE, Roth A, Breaker RR. 2006. Tandem riboswitch architectures exhibit complex gene control functions. Science 314:300–304 [DOI] [PubMed] [Google Scholar]
- 14.Vecerek B, Moll I, Bläsi U. 2005. Translational autocontrol of the Escherichia coli hfq RNA chaperone gene. RNA 11:976–984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vytvytska O, Moll I, Kaberdin VR, von Gabain A, Bläsi U. 2000. Hfq (HF1) stimulates ompA mRNA decay by interfering with ribosome binding. Genes Dev. 14:1109–1118 [PMC free article] [PubMed] [Google Scholar]
- 16.Nomura M, Gourse R, Baughman G. 1984. Regulation of the synthesis of ribosomes and ribosomal components. Annu. Rev. Biochem. 53:75–117 [DOI] [PubMed] [Google Scholar]
- 17.Baker CS, Eory LA, Yakhnin H, Mercante J, Romeo T, Babitzke P. 2007. CsrA inhibits translation initiation of Escherichia coli hfq by binding to a single site overlapping the Shine-Dalgarno sequence. J. Bacteriol. 189:5472–5481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Franze de Fernandez MT, Hayward WS. 1972. Bacterial proteins required for replication of phage Q ribonucleic acid. Purification and properties of host factor I, a ribonucleic acid-binding protein. J. Biol. Chem. 247:824–831 [PubMed] [Google Scholar]
- 19.Jutras BL, Chenail AM, Carroll DW, Miller MC, Zhu H, Bowman A, Stevenson B. 2013. Bpur, the Lyme disease spirochete's PUR-domain protein: identification as a transcriptional modulator and characterization of nucleic acid interactions. J. Biol. Chem. 288:26220–26234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kaminski PA, Desnoues N, Elmerich C. 1994. The expression of nifA in Azorhizobium caulinodans requires a gene product homologous to Escherichia coli HF-I, an RNA-binding protein involved in the replication of phage Q beta RNA. Proc. Natl. Acad. Sci. U. S. A. 91:4663–4667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Karna SLR, Sanjuan E, Esteve-Gassent MD, Miller CL, Maruskova M, Seshu J. 2011. CsrA modulates levels of lipoproteins and key regulators of gene expression critical for pathogenic mechanisms of Borrelia burgdorferi. Infect. Immun. 79:732–744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lapouge K, Schubert M, Allain FH, Haas D. 2008. Gac/Rsm signal transduction pathway of gamma-proteobacteria: from RNA recognition to regulation of social behaviour. Mol. Microbiol. 67:241–253 [DOI] [PubMed] [Google Scholar]
- 23.Lybecker MC, Abel CA, Feig AL, Samuels DS. 2010. Identification and function of the RNA chaperone Hfq in the Lyme disease spirochete Borrelia burgdorferi. Mol. Microbiol. 78:622–635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Robertson GT, Roop RMJ. 1999. The Brucella abortus host factor I (HF-I) protein contributes to stress resistance during stationary phase and is a major determinant of virulence in mice. Mol. Microbiol. 34:690–700 [DOI] [PubMed] [Google Scholar]
- 25.Sanjuan E, Esteve-Gassent MD, Maruskova M, Seshu J. 2009. Overexpression of CsrA (BB0184) alters the morphology and antigen profiles of Borrelia burgdorferi. Infect. Immun. 77:5149–5162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sharma CM, Papenfort K, Pernitzsch SR, Mollenkopf HJ, Hinton JC, Vogel J. 2011. Pervasive post-transcriptional control of genes involved in amino acid metabolism by the Hfq-dependent GcvB small RNA. Mol. Microbiol. 81:1144–1165 [DOI] [PubMed] [Google Scholar]
- 27.Sze CW, Li C. 2011. Inactivation of bb0184, which encodes carbon storage regulator A, represses the infectivity of Borrelia burgdorferi. Infect. Immun. 79:1270–1279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sze CW, Morado DR, Liu J, Charon NW, Xu H, Li C. 2011. Carbon storage regulator A (CsrA(Bb)) is a repressor of Borrelia burgdorferi flagellin protein FlaB. Mol. Microbiol. 82:851–864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zambrano N, Guichard PP, Bi Y, Cayrol B, Marco S, Arluison V. 2009. Involvement of Hfq protein in the post-transcriptional regulation of E. coli bacterial cytoskeleton and cell division proteins. Cell Cycle 8:2470–2472 [DOI] [PubMed] [Google Scholar]
- 30.Graebsch A, Roche S, Niessing D. 2009. X-ray structure of Pur-α reveals a Whirly-like fold and an unusual nucleic acid binding surface. Proc. Natl. Acad. Sci. U. S. A. 106:18521–18526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Graebsch A, Roche S, Kostrewa D, Söding J, Niessing D. 2010. Of bits and bugs—on the use of bioinformatics and a bacterial crystal structure to solve a eukaryotic repeat-protein structure. PLoS One 5:e13402. 10.1371/journal.pone.0013402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hokkanen S, Feldmann HM, Ding H, Jung CK, Bojarski L, Renner-Müller I, Schüller U, Kretzschmar H, Wolf E, Herms J. 2012. Lack of Pur-alpha alters postnatal brain development and causes megalencephaly. Hum. Mol. Genet. 21:473–484 [DOI] [PubMed] [Google Scholar]
- 33.Gallia GL, Johnson EM, Khalili K. 2000. Pur α: a multifunctional single-stranded DNA and RNA binding protein. Nucleic Acids Res. 28:3197–3205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gallia GL, Darbinian N, Jaffe N, Khalili K. 2001. Single-stranded nucleic acid-binding protein, Pur-α, interacts with RNA homologous to 18S ribosomal RNA and inhibits translation in vitro. J. Cell. Biochem. 83:355–363 [DOI] [PubMed] [Google Scholar]
- 35.Muralidharan V, Sweet T, Nadraga Y, Amini S, Khalili K. 2001. Regulation of Puralpha gene transcription: evidence for autoregulation of Puralpha promoter. J. Cell. Physiol. 186:406–413 [DOI] [PubMed] [Google Scholar]
- 36.White MK, Johnson EM, Khalili K. 2009. Multiple roles for Pur-α in cellular and viral regulation. Cell Cycle 8:414–420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Casjens S, van Vugt R, Tilly K, Rosa PA, Stevenson B. 1997. Homology throughout the multiple 32-kilobase circular plasmids present in Lyme disease spirochetes. J. Bacteriol. 179:217–227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zückert WR. 2007. Laboratory maintenance of Borrelia burgdorferi. Curr. Protoc. Microbiol. Chapter 12C:Unit 12C.1. [DOI] [PubMed] [Google Scholar]
- 39.Miller JC, von Lackum K, Babb K, McAlister JD, Stevenson B. 2003. Temporal analysis of Borrelia burgdorferi Erp protein expression throughout the mammal-tick infectious cycle. Infect. Immun. 71:6943–6952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.von Lackum K, Ollison KM, Bykowski T, Nowalk AJ, Hughes JL, Carroll JA, Zückert WR, Stevenson B. 2007. Regulated synthesis of the Borrelia burgdorferi inner-membrane lipoprotein IpLA7 (P22, P22-A) during the Lyme disease spirochaete's mammal-tick infectious cycle. Microbiology 153:1361–1371 [DOI] [PubMed] [Google Scholar]
- 41.Jutras BL, Verma A, Adams CA, Brissette CA, Burns LH, Whetstine CR, Bowman A, Chenail AM, Zückert WR, Stevenson B. 2012. BpaB and EbfC DNA-binding proteins regulate production of the Lyme disease spirochete's infection-associated Erp surface proteins. J. Bacteriol. 194:778–786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jutras BL, Bowman A, Brissette CA, Adams CA, Verma A, Chenail AM, Stevenson B. 2012. EbfC (YbaB) is a new type of bacterial nucleoid-associated protein and a global regulator of gene expression in the Lyme disease spirochete. J. Bacteriol. 194:3395–3406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Whetstine CR, Slusser JG, Zückert WR. 2009. Development of a single-plasmid-based regulatable gene expression system for Borrelia burgdorferi. Appl. Environ. Microbiol. 75:6553–6558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Barbour AG, Hayes SF, Heiland RA, Schrumpf ME, Tessier SL. 1986. A Borrelia-specific monoclonal antibody binds to a flagellar epitope. Infect. Immun. 52:549–554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bono JL, Tilly K, Stevenson B, Hogan D, Rosa P. 1998. Oligopeptide permease in Borrelia burgdorferi: putative peptide-binding components encoded by both chromosomal and plasmid loci. Microbiology 144:1033–1044 [DOI] [PubMed] [Google Scholar]
- 46.Jutras BL, Chenail AM, Stevenson B. 2013. Changes in bacterial growth rate govern expression of the Borrelia burgdorferi OspC and Erp infection-associated surface proteins. J. Bacteriol. 195:757–764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chenail AM, Jutras BL, Adams CA, Burns LH, Bowman A, Verma A, Stevenson B. 2012. Borrelia burgdorferi cp32 BpaB modulates expression of the prophage NucP nuclease and SsbP single-stranded DNA-binding protein. J. Bacteriol. 194:4570–4578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Burns LH, Adams CA, Riley SP, Jutras BL, Bowman A, Chenail AM, Cooley AE, Haselhorst LA, Moore AM, Babb K, Fried MG, Stevenson B. 2010. BpaB, a novel protein encoded by the Lyme disease spirochete's cp32 prophages, binds to erp operator 2 DNA. Nucleic Acids Res. 38:5443–5455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Riley SP, Bykowski TT, Cooley AE, Burns LH, Babb K, Brissette CA, Bowman A, Rotondi M, Miller MC, DeMoll E, Lim K, Fried MG, Stevenson B. 2009. Borrelia burgdorferi EbfC defines a newly-identified, widespread family of bacterial DNA-binding proteins. Nucleic Acids Res. 37:1973–1983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Miller JC. 2005. Example of real-time quantitative reverse transcription-PCR (Q-RT-PCR) analysis of bacterial gene expression during mammalian infection: Borrelia burgdorferi in mouse tissues. Curr. Protoc. Microbiol. Chapter 1D:Unit 1D.3 [DOI] [PubMed] [Google Scholar]
- 51.Babb K, McAlister JD, Miller JC, Stevenson B. 2004. Molecular characterization of Borrelia burgdorferi erp promoter/operator elements. J. Bacteriol. 186:2745–2756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bykowski T, Woodman ME, Cooley AE, Brissette CA, Brade V, Wallich R, Kraiczy P, Stevenson B. 2007. Coordinated expression of Borrelia burgdorferi complement regulator-acquiring surface proteins during the Lyme disease spirochete's mammal-tick infection cycle. Infect. Immun. 75:4227–4236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Seling A, Siegel C, Fingerle V, Jutras BL, Brissette CA, Skerka C, Wallich R, Zipfel PF, Stevenson B, Kraiczy P. 2010. Functional characterization of Borrelia spielmanii outer surface proteins that interact with distinct members of the human factor H protein family and with plasminogen. Infect. Immun. 78:39–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Fraser CM, Casjens S, Huang WM, Sutton GG, Clayton R, Lathigra R, White O, Ketchum KA, Dodson R, Hickey EK, Gwinn M, Dougherty B, Tomb J-F, Fleischmann RD, Richardson D, Peterson J, Kerlavage AR, Quackenbush J, Salzberg S, Hanson M, van Vugt R, Palmer N, Adams MD, Gocayne J, Weidmann J, Utterback T, Watthey L, McDonald L, Artiach P, Bowman C, Garland S, Fujii C, Cotton MD, Horst K, Roberts K, Hatch B, Smith HO, Venter JC. 1997. Genomic sequence of a Lyme disease spirochaete, Borrelia burgdorferi. Nature 390:580–586 [DOI] [PubMed] [Google Scholar]
- 55.Jen-Jacobson L, Kurpiewski M, Lesser D, Grable J, Boyer HW, Rosenberg JM, Greene PJ. 1983. Coordinate ion pair formation between EcoRI endonuclease and DNA. J. Biol. Chem. 258:14638–14646 [PubMed] [Google Scholar]
- 56.Baroni TE, Chittur SV, George AD, Tenenbaum SA. 2008. Advances in RIP-chip analysis: RNA-binding protein immunoprecipitation-microarray profiling. Methods Mol. Biol. 419:93–108 [DOI] [PubMed] [Google Scholar]
- 57.Keene JD, Komisarow JM, Friedersdorf MB. 2006. RIP-Chip: the isolation and identification of mRNAs, microRNAs and protein components of ribonucleoprotein complexes from cell extracts. Nat. Protoc. 1:302–307 [DOI] [PubMed] [Google Scholar]
- 58.Sittka A, Lucchini S, Papenfort K, Sharma CM, Rolle K, Binnewies TT, Hinton JC, Vogel J. 2008. Deep sequencing analysis of small noncoding RNA and mRNA targets of the global post-transcriptional regulator, Hfq. PLoS Genet. 4:e1000163. 10.1371/journal.pgen.1000163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Dorman CJ. 2013. Genome architecture and global gene regulation in bacteria: making progress towards a unified model? Nat. Rev. Microbiol. 11:349–355 [DOI] [PubMed] [Google Scholar]
- 60.Mili S, Steitz JA. 2004. Evidence for reassociation of RNA-binding proteins after cell lysis: implications for the interpretation of immunoprecipitation analyses. RNA 10:1692–1694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Knowle D, Lintner RE, Touma YM, Blumenthal RM. 2005. Nature of the promoter activated by C.PvuII, an unusual regulatory protein conserved among restriction-modification systems. J. Bacteriol. 187:488–497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Samarasinghe S, El-Robh MS, Grainger DC, Zhang W, Soultanas P, Busby SJ. 2008. Autoregulation of the Escherichia coli melR promoter: repression involves four molecules of MelR. Nucleic Acids Res. 36:2667–2676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Takahashi S, Akita R, Furusawa H, Shimizu Y, Ueda T, Okahata Y. 2006. Kinetic analysis of ribosome binding process onto mRNA using a quartz-crystal microbalance. Nucleic Acids Symp. Ser. (Oxf.) 50:49–50 [DOI] [PubMed] [Google Scholar]
- 64.Beyer D, Skripkin E, Wadzack J, Nierhaus KH. 1994. How the ribosome moves along the mRNA during protein synthesis. J. Biol. Chem. 269:30713–30717 [PubMed] [Google Scholar]
- 65.Hüttenhofer A, Noller HF. 1994. Footprinting mRNA-ribosome complexes with chemical probes. EMBO J. 13:3892–3901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Steitz JA. 1969. Polypeptide chain initiation: nucleotide sequences of the three ribosomal binding sites in bacteriophage R17 RNA. Nature 224:957–964 [DOI] [PubMed] [Google Scholar]
- 67.Campos M, Jacobs-Wagner C. 2013. Cellular organization of the transfer of genetic information. Curr. Opin. Microbiol. 16:171–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lee EJ, Groisman EA. 2012. Control of a Salmonella virulence locus by an ATP-sensing leader messenger RNA. Nature 486:271–275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sobrero P, Valverde C. 2011. Evidences of autoregulation of hfq expression in Sinorhizobium meliloti strain 2011. Arch. Microbiol. 193:629–639 [DOI] [PubMed] [Google Scholar]
- 70.Vytvytska O, Jakobsen JS, Balcunaite G, Andersen JS, Baccarini M, von Gabain A. 1998. Host factor I, Hfq, binds to Escherichia coli ompA mRNA in a growth rate-dependent fashion and regulates its stability. Proc. Natl. Acad. Sci. U. S. A. 95:14118–14123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Babb K, Bykowski T, Riley SP, Miller MC, DeMoll E, Stevenson B. 2006. Borrelia burgdorferi EbfC, a novel, chromosomally-encoded protein, binds specific DNA sequences adjacent to erp loci on the spirochete's resident cp32 prophages. J. Bacteriol. 188:4331–4339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Azam TA, Ishihama A. 1999. Twelve species of the nucleoid-associated protein from Escherichia coli. Sequence recognition specificity and DNA binding affinity. J. Biol. Chem. 274:33105–33113 [DOI] [PubMed] [Google Scholar]
- 73.Kajitani M, Kato A, Wada A, Inokuchi Y, Ishihama A. 1994. Regulation of the Escherichia coli hfq gene encoding the host factor for phage Q beta. J. Bacteriol. 176:531–534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Updegrove TB, Correia JJ, Galletto R, Bujalowski W, Wartell RM. 2010. E. coli DNA associated with isolated Hfq interacts with Hfq's distal surface and C-terminal domain. Biochim. Biophys. Acta 1799:588–596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Link TM, Valentin-Hansen P, Brennan RG. 2009. Structure of Escherichia coli Hfq bound to polyriboadenylate RNA. Proc. Natl. Acad. Sci. U. S. A. 106:19292–19297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Sobrero P, Valverde C. 2012. The bacterial protein Hfq: much more than a mere RNA-binding factor. Crit. Rev. Microbiol. 38:276–299 [DOI] [PubMed] [Google Scholar]
- 77.Casjens S, Palmer N, van Vugt R, Huang WM, Stevenson B, Rosa P, Lathigra R, Sutton G, Peterson J, Dodson RJ, Haft D, Hickey E, Gwinn M, White O, Fraser C. 2000. A bacterial genome in flux: the twelve linear and nine circular extrachromosomal DNAs of an infectious isolate of the Lyme disease spirochete Borrelia burgdorferi. Mol. Microbiol. 35:490–516 [DOI] [PubMed] [Google Scholar]