

Abstract
Tsetse flies (Glossina spp.) are the sole vectors of Trypanosoma brucei—the agent of human (HAT) and animal (AAT) trypanosomiasis. Glossina fuscipes fuscipes (Gff) is the main vector species in Uganda—the only country where the two forms of HAT disease (rhodesiense and gambiense) occur, with gambiense limited to the northwest. Gff populations cluster in three genetically distinct groups in northern, southern, and western Uganda, respectively, with a contact zone present in central Uganda. Understanding the dynamics of this contact zone is epidemiologically important as the merger of the two diseases is a major health concern. We used mitochondrial and microsatellite DNA data from Gff samples in the contact zone to understand its spatial extent and temporal stability. We show that this zone is relatively narrow, extending through central Uganda along major rivers with south to north introgression but displaying no sex-biased dispersal. Lack of obvious vicariant barriers suggests that either environmental conditions or reciprocal competitive exclusion could explain the patterns of genetic differentiation observed. Lack of admixture between northern and southern populations may prevent the sympatry of the two forms of HAT disease, although continued control efforts are needed to prevent the recolonization of tsetse-free regions by neighboring populations.
1. Introduction
Tsetse flies, Glossina spp. (Diptera: Glossinidae), are the sole vectors of the trypanosomes causing human (HAT) and animal (AAT) African trypanosomiasis [1]. Currently, there are no vaccines, and available drugs are expensive, toxic, and logistically difficult to administer [2, 3]. As vector density reduction can effectively reduce HAT transmission [4, 5], in 2001, the African Union established the Pan African Tsetse and Trypanosomiasis Eradication Campaign (PATTEC) for a large-scale control of HAT and AAT [6]. Control methods used in the campaign include sterile insect technique (SIT), odor-baited insecticide-treated traps, live baits, targeted aerial insecticide spraying, and sequential aerosol [7–13].
The efficacy of the methods that target vector population reduction can be improved by understanding the population dynamics of the vector species. Genetic tools can be very helpful in this regard, as they allow for identifying barriers to gene flow, predicting fly movements, and assessing reinvasion risks from neighboring sites, where control is not implemented [14]. Population genetic studies can also define the spatial extent, temporal stability, and size of the population targeted for control and thus help determine the appropriate scale at which control can be effective. This information has become a vital tool in guiding the implementation of tsetse control strategies geared towards suppression and complete elimination of flies [15], since the pattern of spatial genetic structure provides quantitative information on population densities and dispersal rates, which are important parameters for designing an efficient control strategy [16]. For example, the density of traps or targets impregnated with insecticides needed to reduce tsetse densities will depend on the dispersal capacities of the flies [17]. The number of sterile males and the distance between release sites to achieve an SIT campaign will also depend on the abundance and dispersal capacities [18–20].
Population genetic studies of riverine G. palpalis gambiensis in Guinea and Senegal have identified populations that are sufficiently isolated to warrant attempts at complete eradication [15, 21]. Population genetic studies in G. p. gambiensis and G. p. palpalis in Senegal, Burkina Faso, and Equatorial Guinea show patterns of high gene flow characterized by spatial and temporal heterogeneity influenced by landscape fragmentation [15, 22, 23]. These studies demonstrate the importance of gene flow in determining the degree of fine-scale genetic structure, the size of the local genetic neighborhoods within populations [15, 23], and the need to integrate information regarding barriers to gene flow in tsetse elimination schemes [14].
In Uganda, Gff, a riverine subspecies in the palpalis group, is the major HAT transmission vector. Except for a disjunct region in Ethiopia/Sudan, Uganda and western parts of Kenya represent the eastern edge of its range [28], where it occurs in localized vegetation thickets along water bodies, which offer tsetse seasonal refugia and access to host species in their search for food and relief from heat. Gff densities are strongly influenced by ecological and climatic features, since temperature and precipitation may change the vegetation landscape and thus density and size of tsetse populations [29–31].
Ecological data suggest that Gff has great capacity for dispersal and recolonization of suitable habitats [32]. Such rapid dispersal within the habitat would cause genes to spread rapidly leading to genetic homogenization of tsetse flies across the geographic landscape. This is not what genetic studies from our group have revealed. The genetic screening of about 37 Gff populations across Uganda showed that this taxon is structured into three major genetic population clusters, with a southern and northern cluster separated by Lake Kyoga [24] and a third one present in western Uganda [25]. Although gene flow can occur between these genetic clusters with a few migrants detected over a radius of about 100 km, genetic mixing is quite frequent between the northern and southern population genetic clusters along a contact zone along Lake Kyoga in the areas of Bunghazi in eastern, Masindi in western, and Junda in central Uganda. Our studies have also suggested female-biased dispersal into the contact zone from sites within the southern cluster [25]. Genetic studies also demonstrated the temporal stability of Gff populations in Uganda, including those from the contact zone [20, 26, 33].
The patterns of genetic differentiation between Gff populations might also be impacted by the symbiotic bacteria they carry. All tsetse species harbor a vertically transmitted mutualistic symbiont, Wigglesworthia glossinidia, which is necessary for host physiology. As expected, the genetic structure of Wigglesworthia reflected the Gff host mtDNA patterns of genetic differentiation [34]. On the other hand this congruence between host mtDNA and parasite patterns of genetic diversity was not found in another maternally inherited bacteria, the parasitic Wolbachia [34–36]. This symbiont has been shown to manipulate host reproduction in Glossina morsitans morsitans, causing cytoplasmic incompatibility (CI) in the laboratory; crosses between Wolbachia infected females and uninfected males result in embryonic lethality, while the reciprocal cross are fertile [37]. Our studies on Wolbachia in Gff in Uganda has shown that infection prevalence and density in different populations vary, and that individual flies can carry more than one Wolbachia strain [34]. Thus, Wolbachia mediated incompatibilities between populations can contribute to the genetic disjunction we observe in Gff as a result of CI mediated effects.
Due to lack of obvious vicariant barriers, the contact zone was speculated to result from secondary contact of flies following allopatric divergence and expansion [25]. However, the limited sampling of the region did not allow for the determination of its precise geographic extent and dynamics. The low vagility of Gff together with its tendency to cluster in discrete habitats (thickets of vegetation along river bodies) and its strong association with density-independent factors suggest that local adaptation to environmental parameters may also contribute to the maintenance of population divergence. This implies that habitat availability will largely control densities of populations and their connectivity.
The Gff populations in northwestern Uganda transmit only the gambiense form of HAT caused by Trypanosoma brucei gambiense, while Gff populations in the southern genetic clusters transmit the rhodesiense form of HAT caused by T. b. rhodesiense. In recent years, the rhodesiense disease has been expanding its range from the historical loci in the southeast into new foci in central Uganda north of Lake Kyoga [35]. In fact, the two disease belts are separated only by a disease free belt of less than <100 km just north of Lake Kyoga [37]. Since the pathology, diagnosis, and treatment vary between the two forms of disease, a potential merger of the two disease belts would cause major public health crises [2]. Thus, understanding the vector dynamics in this contact zone will provide insights on the potential risk of the sympatry of these two currently allopatric HAT forms, which were never in contact before. If the flies can acquire and transmit both forms of the human parasite, this could result in unknown epidemiological outcomes since the parasites can undergo recombination in the tsetse salivary glands [20, 38–42].
In the current study, we comprehensively sampled the area where the genetically differentiated populations of Gff are in contact in central Uganda and used multilocus genetic data to examine population structure and dynamics and to evaluate if environmental differences might be involved in maintaining the genetic difference between the two genetic clusters north and south of Lake Kyoga. We discuss our data on the stability of the different genetic populations around Lake Kyoga in the context of the potential merger of the two disease belts and the ongoing vector control programs.
2. Materials and Methods
2.1. Tsetse Collection and Study Area
Tsetse flies were collected using biconical traps [43] during field expeditions between 2009 and 2012 from 49 sites. Sampling details are summarized in Figure 1, Table 1 and Supplementary Material. Collections in each locality were carried out for 3 days with an average of 6 traps per site. Traps were located within a radius of 5 km2. Each fly was stored individually in 90% ethanol. Localities generally reflect the riverine/woodland habitat preferred by Gff.
Figure 1.

Localities where Glossina f. fuscipes samples were collected. The inset map shows the geographical position of Uganda in Africa. The dark grey shows the current species range. Lakes are indicated by name and with a light shade of grey. The three major rivers (Nile, Kafu, and Mpologoma) are also indicated by name. Sampling sites are identified by abbreviations and expanded in Table 1 and Supplementary Table S1 (see Supplementary Material available online at http://dx.doi.org/10.1155/2013/614721). Empty circle corresponds to areas previously sampled in Abila et al. [24], Beadell et al. [25], and Echodu et al. [26]. Circles indicate new sampling sites (black circles: new sites with tsetse; grey circles: sites with no tsetse) and triangles indicate sites examined in previous studies.
Table 1.
Sample sizes and genetic diversity statistics for the mitochondrial CO1 and 15 microsatellite loci in 23 populations of G. f. fuscipes, on samples analyzed for this study (a) and from previous studies (Abila et al., 2008 [24]; Beadell et al., 2010 [25]; and Echodu et al., 2011 [26]).
| Population | Code | Date of sampling | Microsatellites | Mitochondrial DNA | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| N | A R | H O | H E | F IS | N | No. haplotypes |
Hd | π | |||
| Kafu | KF-0 | Feb 2010 | 30 | 4.500 | 0.501 | 0.526 | 0.065 | 16 | 2 | 0.233 | 0.00345 |
| KF-10 | Jul 2011 | 4 | 3.438 | 0.509 | 0.525 | 0.197 | 2 | 1 | 0.000 | 0.0000 | |
| Karuma | KR-0 | Feb 2010 | 40 | 7.667 | 0.571 | 0.652 | 0.137 | 12 | 3 | 0.439 | 0.00641 |
| KR-10 | Jul 2011 | 20 | 6.056 | 0.604 | 0.639 | 0.08 | 8 | 3 | 0.714 | 0.00664 | |
| Putiputi | PT | Oct 2009 | 26 | 4.000 | 0.525 | 0.564 | 0.038 | 22 | 2 | 0.247 | 0.00104 |
| Tororo | TR | Oct 2009 | 2 | 1.944 | 0.472 | 0.347 | −0.03 | — | — | — | — |
| Iganga | IGG | Feb 2011 | 65 | 6.000 | 0.535 | 0.556 | 0.033 | 10 | 1 | 0 | 0 |
| Kisoko | KIS | Feb 2011 | 8 | 3.555 | 0.527 | 0.542 | −0.06 | — | — | — | — |
| Mayuge | MGG | Feb 2011 | 59 | 5.600 | 0.545 | 0.557 | 0.027 | 15 | 4 | 0.371 | 0.00334 |
| Namutumba | NAM | 16 | 5.267 | 0.553 | 0.610 | 0.09 | 31 | 4 | 0.574 | 0.00517 | |
| Nambogo | NB | Jun 2011 | 17 | 4.333 | 0.592 | 0.584 | −0.03 | 14 | 2 | 0.143 | 0.00272 |
| Sangalo | SA | Dec 2011 | 15 | 4.000 | 0.450 | 0.496 | 0.087 | 15 | 3 | 0.257 | 0.0056 |
| Tuba | TB | Jun 2011 | 28 | 5.267 | 0.579 | 0.583 | 0.005 | 15 | 2 | 0.248 | 0.00105 |
| Aminakwach | AM | Jul 2011 | 30 | 4.733 | 0.537 | 0.555 | 0.068 | 16 | 2 | 0.125 | 0.00056 |
| Bukedea | BKD | May 2011 | 25 | 4.429 | 0.495 | 0.541 | 0.098 | 14 | 3 | 0.473 | 0.00170 |
| Oculoi | OC | Jul 2011 | 20 | 4.786 | 0.639 | 0.612 | −0.015 | 16 | 3 | 0.592 | 0.0036 |
| Kitgum | KT | Jan 2012 | 17 | 5.333 | 0.55201 | 0.62790 | 0.126 | 10 | 3 | 0.600 | 0.00184 |
| Budaka | BK* | 80 | 4.778 | 0.509 | 0.527 | 0.041 | 11 | 1 | 0.000 | 0.0000 | |
| Bunghazi | BN-0* | Mar 2008 | 32 | 4.2 | 0.529 | 0.578 | 0.079 | 15 | 3 | 0.648 | 0.00538 |
| BN-08+ | Mar 2009 | 40 | 3.9 | 0.568 | 0.609 | 0.068 | |||||
| BN-12+ | Oct 2009 | 65 | 4.1 | 0.549 | 0.574 | 0.116 | 16 | 4 | 0.692 | 0.00466 | |
| BN-24 | Jun 2011 | 18 | 5.348 | 0.631 | 0.631 | 0.06 | 13 | 2 | 0.385 | 0.00244 | |
| Busiime | BU-0∗+ | Mar 2008 | 39 | 3.5 | 0.476 | 0.485 | 0.046 | 17 | 1 | 0.000 | 0.0000 |
| BU-8+ | Mar 2009 | 40 | 3.4 | 0.464 | 0.477 | 0.025 | |||||
| BU-12+ | Oct 2009 | 40 | 3.4 | 0.464 | 0.489 | 0.037 | 19 | 1 | 0.0000 | 0.0000 | |
| Junda | JN-0∗+ | Mar 2008 | 40 | 3.2 | 0.479 | 0.489 | 0.023 | 19 | 3 | 0.444 | 0.00731 |
| JN-13+ | Jan 2010 | 18 | 3.1 | 0.460 | 0.485 | 0.065 | 18 | 1 | 0.000 | 0.00000 | |
| JN-24 | Jul 2011 | 3 | 3.091 | 0.407 | 0.333 | −0.023 | 1 | ||||
| Mukongoro | MK-0∗+ | Mar 2008 | 40 | 2.9 | 0.487 | 0.460 | −0.041 | 21 | 2 | 0.495 | 0.00093 |
| MK-08+ | Mar 2009 | 24 | 3.0 | 0.455 | 0.431 | −0.043 | |||||
| MK-12+ | Nov 2009 | 22 | 3.1 | 0.418 | 0.445 | 0.068 | 21 | 2 | 0.467 | 0.00088 | |
| Masindi | MS-0∗+ | Mar 2008 | 40 | 3.7 | 0.568 | 0.547 | −0.025 | 18 | 2 | 0.467 | 0.001886 |
| MS-13+ | Jan 2010 | 17 | 4.4 | 0.562 | 0.597 | 0.055 | 17 | 2 | 0.471 | 0.00964 | |
| MS-25 | Jul 2011 | 30 | 4.722 | 0.542 | 0.5533 | 0.039 | 15 | 2 | 0.419 | 0.00620 | |
| Okame | OK-0∗+ | Mar 2008 | 39 | 3.3 | 0.452 | 0.507 | 0.081 | 17 | 3 | 0.471 | 0.0094 |
| OK-8+ | Mar 2009 | 40 | 3.4 | 0.563 | 0.546 | 0.008 | — | — | — | — | |
| OK-12+ | Oct 2009 | 39 | 3.4 | 0.547 | 0.552 | 0.036 | 18 | 2 | 0.294 | 0.00055 | |
| Otuboi | OT-0∗+ | Jul 2008 | 53 | 4.0 | 0.508 | 0.535 | 0.085 | 20 | 3 | 0.426 | 0.0122 |
| OT-11+ | Nov 2009 | 40 | 3.7 | 0.514 | 0.540 | 0.076 | 20 | 4 | 0.537 | 0.00131 | |
N: number of individuals analyzed, A R: allelic richness, H O: observed heterozygosity, H E: expected heterozygosity, F IS: Fisher's inbreeding coefficient; Hd: haplotype diversity, π: nucleotide diversity. Dashes indicate sites from which mtDNA was not collected, *indicates samples used in Beadell et al. (2010) [25], and +indicates samples used in Echodu et al. 2011 [26].
2.2. Genetic Data Collection
DNA was extracted from tsetse legs using PrepGEM Insect DNA extraction kit (ZYGEM 79, New Zealand) or DNeasy kits (Qiagen, USA). PCR was used to amplify a 530 base pair (bp) fragment of the mitochondrial gene cytochrome C oxidase II (COII) from a subset of flies from each locality following Beadell et al. [25]. Individual flies were genotyped at 18 microsatellite loci (Supplementary Material) using previously described protocols [25, 44, 45] with the exception of C07 and GmL11, where 0.5 units of Taq Gold polymerase (Life Technologies, USA) were used. PCR products were multiplexed in groups of two or three loci and then genotyped on an ABI 3730xL sequencer (Life technologies, USA). Alleles were scored using Genemarker v2.4.0 (SoftGenetics, USA) with manual editing of the automatically scored peaks.
2.3. Statistical Analyses of Genetic Data
We tested microsatellite loci for within site deviations from Hardy-Weinberg equilibrium (HWE) and linkage disequilibrium (LD) using Genepop v4.0 [46]. Markov chain parameters were set at 10,000 dememorizations, 1000 batches, and 10,000 iterations per batch. Locus and locality specific estimates of microsatellite allele frequencies were generated using GenAlex v6.4 [47]. We used the Fstat v2.9.3.2 [48] to calculate site specific inbreeding coefficients (Fis) and Arlequin v3.5 [49] to calculate allelic richness (A) and observed (H O) and expected (H E) heterozygosity of populations. DnaSP v5.0 [50] was used to calculate mtDNA haplotype (Hd) and nucleotide (π) diversity.
For all sites from which we had temporal samplings, we characterized the proportion of the variance attributable to differences in sampling dates using the analysis of molecular variance (AMOVA) as implemented in Arlequin v3.5 [49] on both microsatellite and mtDNA datasets. For the microsatellite dataset we calculated pairwise F ST using Genepop v4.0 [46] and generated locus and population specific estimates of microsatellite allele frequencies using GenAlex v6.4 [47] for the different temporal samplings. Temporal samples that were not genetically distinct were pooled in subsequent analyses.
Overall genetic differentiation among localities was assessed by estimating pairwise F ST values [51], and significance was determined using Fisher's G-based exact test for genotypic differentiation using Genepop v4.0 [46]. For both microsatellite and mtDNA data, we used an analysis of molecular variance (AMOVA), in Arlequin v3.1 [49], to analyze the partitioning of genetic variance within and between sampling localities. Relationships among haplotype lineages were inferred by constructing a parsimony network using TCS v1.21 [52].
Population structure was inferred from microsatellite data using the Bayesian clustering method implemented in STRUCTURE 2.3 [53]. Three independent runs for each K = 1–15 were carried out. For all runs, an admixture model and independent allele frequencies were used with a burn-in value of 250,000 steps followed by 1,000,000 iterations. The optimal value of K was determined using STRUCTURE HARVESTER v0.6 [54] to calculate ad hoc statistic “ΔK” [55].
To determine whether the patterns of genetic structure were a result of sampling related individuals, we estimated relatedness and examined relationships for all individual pairs using the program ML-Relate [27]. Each study site was run individually to determine if sites differed significantly in the proportion of related individuals detected. We compared results for individuals from which we had both mtDNA and microsatellite data to identify mismatches between data types ((10) of the Appendix).
To identify first generation migrants, we used the Bayesian approaches implemented in STRUCTURE and GeneClass 2.0 [53, 56]. Previously obtained STRUCTURE results were used to assign individuals to each of the K populations. Samples were placed into the respective population based upon the highest percentage of membership (q) using a threshold value of q ≥ 0.90 [57]. We used the “detect migrants” function of GeneClass to calculate the likelihood of finding an individual in the locality in which it was sampled (L h), the greatest likelihood among all sampled localities (L max), and their ratio (L h/L max). Because migrants from unsampled populations can be misclassified as residents, we selected the Rannala and Mountain [58] criterion with the resampling method of Paetkau et al. [57] to determine the critical value of the test statistics, L h and L h/L max, using 1,000 simulated individuals and the default 0.01 Type I error (α).
We used Fstat v2.9.3.2 [48] to test whether the observed population structure could be attributed to differences in dispersal between sexes and used four statistics: differentiation among populations (F ST), mean assignment indices (mAIc), the variance in assignment indices (vAIc), and mean pairwise relatedness (mPr) [48, 59, 60].
3. Results
3.1. Sampling
We visited 49 sites spanning the known Gff distribution but could trap flies in only 23 sites despite similar environmental conditions and equivalent collection efforts. We collected 2918 tsetse flies with an average of 127 per locality (Figure 1 and Table S1). The absence of flies or the extremely low (n < 2) capture rates occurred at sites near cattle corridors, where farmers apply synthetic pyrethroid acaricides for tick and tsetse control on cattle weekly, and in districts subjected to control efforts for tsetse flies using insecticide treated traps [9]. In addition, low capture rates of flies in a trap do not necessarily reflect the abundance of tsetse in an area, as trap efficiencies for Gff are particularly low [60].
3.2. Genetic Diversity
In all analyses for both microsatellite and mtDNA markers, we included published data from seven sites [25, 26]. The final data set for the microsatellite loci analyses included 23 sites and 1221 flies averaging 53 flies/site. For the mtDNA analyses, we screened a subset of these samples (244 flies from 19 sites, averaging 13 flies/site).
Of the 18 microsatellite loci, GpC29 and C5 did not adhere to HWE expectations (Supplementary Material) and were excluded from further analysis. Pgp17 was also excluded because of scoring inaccuracies likely due to the large range of allele sizes (70 bp to >200 bp). We detected no significant linkage among the 15 remaining loci. Overall, all sites showed moderate to high levels of genetic variability; A R ranged from 2.900 to 7.677, H O ranged from 0.487 to 0.604, H E ranged from 0.46 to 0.652, and FIS ranged from −0.015 to 0.126 (Table 1). H E and H O microsatellite diversities were similar, indicating random mating within sites.
The mtDNA dataset consisted of 489 COII sequences (530 bp), including 244 new and 245 published sequences [25, 26], and resulted in a total of 57 haplotypes, including 15 new ones (Figure 2(a), Supplementary material). The number of haplotypes at each site ranged from 1 to 4, haplotype diversity (Hd) ranged from 0 to 0.714, and nucleotide diversity (π) ranged from 0 to 0.00664 (Table 1). In 12% of the samples (26 individuals) cluster assignment for mtDNA and microsatellite data was incongruent ((10) of the Appendix).
Figure 2.
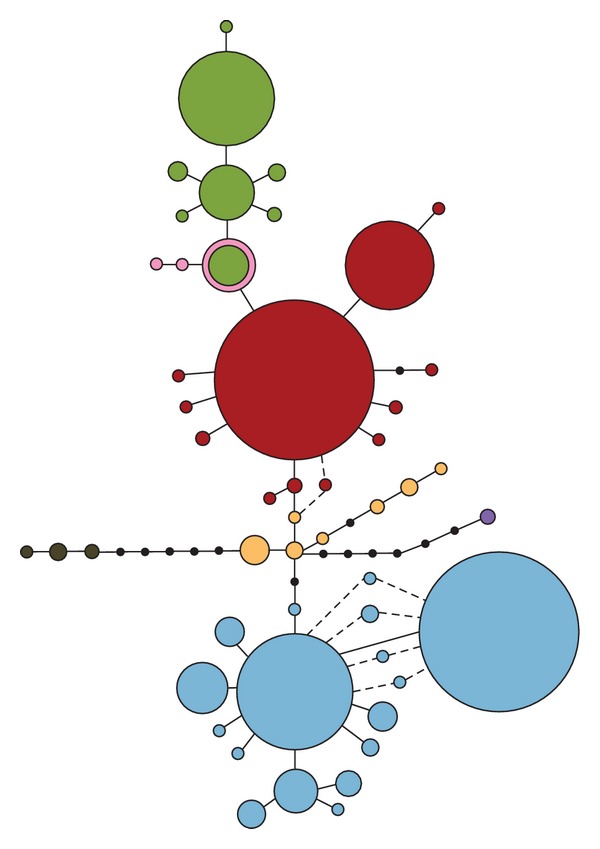
Parsimony network of mitochondrial haplotypes recovered from G. f. fuscipes and their distribution in Uganda, Kenya, and Sudan. Haplotypes are represented by circles. Their size is proportional to their frequency. Haplotypes are shaded to represent genetic populations—Green is western, red is northern, blue is southern, and yellow is intermediate. The purple haplotype represents a divergent haplotype discovered in MGG, pink haplotypes are from a disjunct population of G. f. fuscipes in Sudan, and brown haplotypes are from a population of G. f. quanzensis from the Democratic Republic of the Congo.
3.3. Temporal Variation in Genetic Diversity
We tested for differences in genetic diversity between samples collected in 2008 and 2011 at 4 sites ((5) of the Appendix). AMOVA using both microsatellite allele frequencies and mtDNA haplotype frequencies suggested that differences between temporally-spaced samples were not significant; however, differences among localities were significant (Supplementary Material). The variation explained by site was greater for mtDNA than for microsatellite data. Pairwise F ST values between samples were also low (Table S6). MtDNA haplotype frequencies were not different in three of the four sites between the two time points for each site (MS, KF, or KR, Figure S1), although some change was observed in BN, likely due to the loss of the two least common haplotypes in the 2011 samples.
3.4. mtDNA Network Analyses
The phylogenetic relationships among the 57 mtDNA haplotypes are shown in Figure 2 and confirm the existence of the two major northern (N) and southern (S) haplogroups [25] with most of the new haplotypes included in one of the two haplogroups. The increased sampling density allowed for the recovery of intermediate haplotypes between the two major haplogroups, and also of more distantly related haplotypes, separated by a maximum of 10 substitutions from the most common haplotypes from each haplogroup. The haplotype geographic distribution is shown in Figure 3; both N and S haplogroups were found in 6 sites (KF, KR, MS, JN, NAM, and BN), 3 of which (BN, JN, and MS) had been previously identified as having both haplogroups.
Figure 3.

Geographic distribution of mitochondrial sequence diversity. Pie charts on maps indicate the frequency with which mtDNA haplotypes (identified in Figure 2) occurred in each sampling site. Background shading indicates the predicted range of G. f. fuscipes in the region and in Africa (inset). Haplotypes are shaded to represent genetic populations—Green is western, red is northern, blue is southern, and yellow is intermediate. The purple haplotype represents a divergent haplotype discovered in MGG, pink haplotypes are from a disjunct population of G. f. fuscipes in Sudan, and brown haplotypes are from a population of G. f. quanzensis from the Democratic Republic of the Congo. The dark grey shows the current species range.
3.5. Measures of Genetic Differentiation and Structure
Bayesian clustering analyses performed in STRUCTURE identified 3 genetic clusters including multiple sampling sites—northern (KT, AM, OC, OT, MK, BKD, PT, BK, and BN), southern (NAM, IGG, MGG, KIS, TB, BU, NB, OK, and SA), and western (KF, MS, and KR). The analysis also showed admixture across genetic clusters with flies from Junda (JN) showing significant ancestry with flies from both southern and western genetic clusters (Figure 4).
Figure 4.
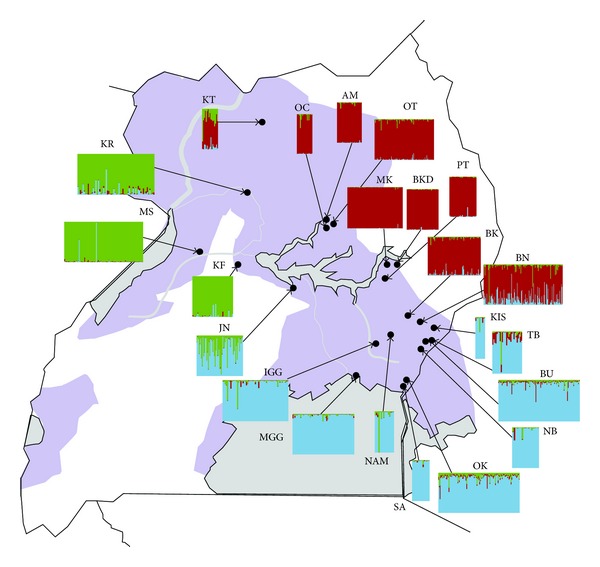
Geographic distribution of the three genetic clusters detected using microsatellite data based on STRUCTURE results. Each rectangle depicts the genetic assignment of each individual from that sampling site. Each individual within each rectangles is represented by a vertical bar, with assignment to the three genetic clusters shown as the proportion of each color making up each bar. Colors represent genetic populations—Green is western, red is northern, and blue is southern. The dark grey shows the current species range.
F ST values between sites based on microsatellites ranged from 0.06 to 0. (Supplementary Material). F ST values between clusters detected by STRUCTURE ranged from 0.14 to 0.21 (Supplementary Material). AMOVA results using microsatellite allele frequencies (Table 2) showed that differences both within (79.3%, P ≥ 0.01) and between (18.03%, P ≥ 0.01) sites were significant, though most of the variation was between individuals within sites.
Table 2.
Results of AMOVA analyses on 15 microsatellite loci and mtDNA.
| D.f | Sum of squares | Variance component | % Variation | P value | |
|---|---|---|---|---|---|
| Microsatellites | |||||
| Among sites | 21 | 2215.937 | 0.99981 | 18.03 | 0.0000 |
| Within sites | 1118 | 5250.224 | 0.15046 | 2.71 | 0.0000 |
| mtDNA | |||||
| Among sites | 428.246 | 0.94483 | 64.01 | 0.05 | |
| Within sites | 239.629 | 0.53133 | 35.99 | 0.05 |
D.f: degrees of freedom.
3.6. Relatedness, Dispersal and Migration
Measures of relatedness based on microsatellite variation using both likelihood and Bayesian methods showed that the majority (85.8%) of the individuals are unrelated (Table 3). Migrant detection using GeneClass and STRUCTURE yielded similar results (Table 4 and Supplementary Material). STRUCTURE results indicated that although there was significant genetic differentiation between clusters, there was also gene flow between them, as 69 migrants (33 males and 36 females, Supplementary Material) were identified. While most migrants moved between sites within the genetic clusters, a small number moved between them. Within the northern cluster our analyses identified 5 migrants: one female with southern ancestry, one male with ancestry in the mixed southern/western locality (JN), and two females and one male with ancestry to the western cluster. Within the southern cluster we detected 3 migrants: two males with ancestry in the mixed southern/western locality (JN) and one male with ancestry to the western cluster. In the western cluster we found 2 migrants: one male individual with southern ancestry and one male individual with ancestry in the mixed southern/western locality (JN). In this site we inferred 7 migrants: six females and one male all with southern ancestry.
Table 3.
Percent of pairwise comparisons of individuals that fell into each relatedness category (e.g., unrelated, half-sibling, or full sibling) as calculated in ML-Relate (Kalinowski, 2011 [27]). Comparisons were made among all individuals (All) and among individuals within each sampling site.
| All | BU | SA | OK | KIS | NB | TB | NAM | MGG | IGG | BN | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Unrelated | 85.8 | 82.5 | 94.2 | 83 | 92.8 | 85.3 | 89.9 | 92.5 | 85.3 | 84.7 | 83.9 |
| Half-sibling | 12 | 15.2 | 4.8 | 14.8 | 3.6 | 11.8 | 9 | 7.5 | 12.5 | 13.6 | 15 |
| Full sibling | 1 | 0.9 | 0 | 1.2 | 0 | 0.7 | 0.8 | 0 | 1.3 | 1 | 0.7 |
| Parent-offspring | 1.2 | 1.4 | 1 | 1 | 3.6 | 2.2 | 0.3 | 0 | 0.9 | 0.7 | 0.4 |
|
| |||||||||||
| BK | PT | BKD | MK | OT | OC | AM | MS | KF | KR | JN | |
|
| |||||||||||
| Unrelated | 82.3 | 82.8 | 84.3 | 77.6 | 84.1 | 92.1 | 84.8 | 82.1 | 84.3 | 89.2 | 83.2 |
| Half-sibling | 15.8 | 13.9 | 11.7 | 17.3 | 14.2 | 4.2 | 12.2 | 15.4 | 14.1 | 9.9 | 14.5 |
| Full sibling | 1 | 1.8 | 2 | 2.1 | 0.9 | 1.6 | 1.8 | 1.1 | 1.1 | 0.8 | 1.3 |
| Parent-offspring | 0.9 | 1.5 | 2 | 3 | 0.8 | 2.1 | 1.2 | 1.4 | 0.5 | 0.1 | 1 |
Table 4.
Summary of first generation migrants by sampling locality. The first four columns report the sampling site, its symbol, and microsatellite and mtDNA haplogroups assignment. The last four columns list the total number of migrants detected in each site and how many of these migrants come from a site from the northern, the southern, or the western clusters.
| Locality | Microsatellite cluster | mtDNA cluster | Total migrants | Northern | Southern | Western |
|---|---|---|---|---|---|---|
| BU | S | S | 2 | — | 2 | — |
| SA | S | S | 2 | — | 2 | — |
| OK | S | S | 3 | — | 3 | — |
| NB | S | S | 4 | — | 4 | — |
| TB | S | S | 3 | — | 2 | 1 |
| NAM | S | Mixed | 6 | — | 4 | 2 |
| MGG | S | S | 4 | — | 4 | — |
| IGG | S | S | 4 | — | 4 | — |
| BN | N | Mixed | 8 | 5 | 1 | 2 |
| BK | N | N | 2 | 2 | — | — |
| PT | N | N | 1 | 1 | — | — |
| BKD | N | N | 0 | — | — | — |
| MK | N | N | 3 | 3 | — | — |
| OT | N | N | 3 | 3 | — | — |
| OC | N | N | 2 | 2 | — | — |
| AM | N | N | 2 | 2 | — | — |
| KT | N | W | 3 | 1 | 1 | 1 |
| MS | W | W | 5 | — | 3 | 2 |
| KF | W | W | 3 | — | — | 3 |
| KR | W | W | 2 | — | 1 | 1 |
| JN | Mixed | Mixed | 4 | — | 4 | — |
| TR | S | S | 3 | — | 3 | — |
The four statistical methods implemented did not detect significant differences in sex-biased dispersal in both one-sided and two-sided tests (Table 5).
Table 5.
Sex-biased dispersal test results. Four different statistics were used: F ST, mean assignment indices (mAIc), the variance in assignment indices (vAIc), and mean pairwise relatedness (mPr). Results for both two- and one-sided tests are reported. Sex and samples sizes are reported in the first column. P values for each statistics are reported in the last row.
| F ST | mAIc | vAIc | mPr | |
|---|---|---|---|---|
| Two-sided | ||||
| F (540) | 0.1912 | 0.02368 | 22.5165 | 0.3106 |
| M (581) | 0.1915 | 0.02201 | 16.36478 | 0.3112 |
| P value | 0.9775 | 0.8642 | 0.8552 | 0.9503 |
| One-sided | ||||
| F (540) | 0.1913 | 0.02368 | 22.5165 | 0.3106 |
| M (581) | 0.1915 | 0.02201 | 16.36478 | 0.3112 |
| P value | 0.774 | 0.4392 | 0.5695 | 0.7847 |
4. Discussion
In this study, we undertook an expanded temporal and spatial analysis of population genetic structures of Gff flies collected in central Uganda where multiple genotypes from genetically distinct population clusters were shown to exist in a zone of contact. Our analyses included multiple sampling sites and identified three main genetic clusters with northern, southern, and western distributions in concordance with previous studies [25, 26]. We find a narrow contact zone between northern and southern genetic clusters with low levels of migration between clusters.
Increased sampling in the Lake Kyoga region revealed a much higher spatial resolution of the contact between these three clusters and the associated admixture zone. However, our additional mtDNA sampling recovered intermediate haplotypes between the northern and southern haplogroups, shown in yellow in Figure 2, reducing the division between these two populations groups and suggesting that their separation was more recent than previously thought [25]. Additionally, mtDNA analysis revealed many more sites at which northern and southern haplotypes occur sympatrically (Figure 3) than previously detected, providing greater spatial resolution of the contact zone. The contact zone between the three clusters appears to be relatively narrow, extending through central Uganda to the western areas in Karuma and Kafu. All admixed sampling localities are along the path of major rivers; the Mpologoma River, the Kafu River, and the Nile River that drain into and out of Lake Kyoga (Figure 1). The narrow extent of this contact zone, in addition to the apparent south to north direction of mitochondrial introgression between the northern and southern population clusters, supports the role of contiguous riverine habitat facilitating the dispersal of tsetse flies along watercourses [14, 25]. Despite relatively short distances between sites within different genetic clusters (e.g., 10.5 km between NB and OK), we detected significant genetic differentiation between them (Supplementary Material), which is concordant with previous studies [25, 26, 61].
There was a substantial mismatch in the assignment of mitochondrial haploytpes and ancestry based on microsatellite data. This mismatch could be explained by sex bias in dispersal, as has been detected in other population studies of Gff [24, 25], but we did not detect any significant bias in this study. It is also possible that Wolbachia-induced mating incompatibility is driving the differentiation between the population structure inferred from mitochondrial and nuclear DNA. Wolbachia mediated effects on host fitness and host population genetic can drive patterns of mtDNA variation regardless of the nuclear DNA background [36, 61, 62]. A finer scale analysis of Wolbachia infection prevalence and strain types present in flies in the contact zone is necessary to further clarify the role of Wolbachia mediated reproductive effects.
Migration of flies between sites from different genetic clusters is extremely low, particularly in relation to north-south migration. We detected only a single migrant from a southern locality to a northern locality (BN to NAM) and no migrants in the other direction. Despite this, it does appear that mitochondrial introgression is occurring in a south to north direction (Figure 2(b)), especially along the path of major rivers. Conversely, admixture between both northern and southern population clusters with the western one was slightly higher, particularly in the mixed JN locality.
The contrast between the occurrence of intracluster migration and the low level of interacluster migration could be caused by two mechanisms—restriction of movement between clusters due to vicariant barriers or through reciprocal competitive exclusion of flies originating from adjacent clusters following local adaptation. As the contact zone lies around Lake Kyoga and major rivers and flies can cross riverine barriers separating sites within clusters, there is a lack of obvious physical barriers to gene flow that would explain the observed genetic differentiation among the genetic clusters seen in this study.
Competitive exclusion could also be driving the observed pattern, and experimental mating studies could test this hypothesis. This is a compelling biological hypothesis and one that has significant implications for vector control, thus representing an important direction for future study. Environmental barriers could also be important in maintaining the genetic distinctiveness between clusters, and we have preliminary data that show a substantial gradient in climatic conditions between Gff sites, north and south of Lake Kyoga, suggesting congruence between the genetic differentiation of Gff and the differentiation of its environment which we are exploring further.
4.1. Implications for Gff Vector Control and Future Directions
The observation of high gene flow amongst localities within the three genetic clusters indicates that control efforts, undertaken solely at local scales, are unlikely to produce long-lasting results due to reinvasion from adjacent areas and that, in the absence of continued control, areas presently free of tsetse, such as the ones in south central Uganda where control is currently enforced and that yielded low tsetse captures, are likely to become recolonized, especially given the habitat suitability of many of these regions.
Whilst the low genetic admixture between northern and southern populations of Gff could suggest that the two regions they occupy could be managed as separate entities, this interpretation bears caution, as elimination of one population could result in rapid population expansion of the other. Experimental control with strict monitoring at sites in the contact zone could assist in understanding how the dynamics between these two populations change in response to control measures. In addition, this study highlights the importance of ongoing monitoring of Gff in Uganda to provide quantitative information on population densities and dispersal rates to inform efficient control strategies into the future.
Future work will be directed at exploring further the underlying causes of the genetic differentiation between the three genetic clusters by performing mating experiments to look at mating compatibility, exploring further the role of Wolbachia mediated bidirectional CI in determining such patterns, and looking at the association between genetic and environmental variables and the impact of climatic change in shaping the distribution of Gff and thus disease risk.
Supplementary Material
Appendix 1: Information on the study site, time of collection and number of flies caught in each site.
Appendix 2: Details of microsatellite markers used in the study.
Appendix 3: Per locus estimates of FIS at 18 microsatellite loci for each sampling locality.
Appendix 4: Mitochondrial haplotype information, including frequencies observed across studied populations. New haplotypes recovered from this study are indicated by ∗.
Appendix 5: Results of an AMOVA testing for temporal genetic structure in four populations of G. fuscipes sampled in 2008 and also in 2011.
Appendix 6: Fst values for temporal samples calculated on microsatellite data. Non-significant values are in bold.
Appendix 7: Microsatellite-based FST values pairwise comparison between sampling localities of G. f. fuscipes in Uganda.
Appendix 8: Microsatellite-based FST values for pairwise comparisons among the three populations detected using Bayesian clustering.
Appendix 9: Details of all first generation migrants detected by Geneclass 2.0, using Lh, Lh/Lmax and STRUCTURE.
Appendix 10: Comparison of mtDNA clade and microsatellite assignment for each individual where both data types were collected.
Figure 1: Microsatellite allele frequencies observed in seven populations of G. f. 52 fuscipessampled at different time points. Numbers after location code indicate the time interval 53 (in generations) since the first sampling.
Authors' contribution
The authors one and two are the first two authors listed in alphabetical order.
Appendix
Information on the study site, time of collection, and number of flies caught in each site.
Details of microsatellite markers used in the study.
Per locus estimates of F IS at 18 microsatellite loci for each sampling locality.
Mitochondrial haplotype information, including frequencies observed across studied populations. New haplotypes recovered from this study are indicated by Richard Echodu and Mark Sistrom.
Results of an AMOVA testing for temporal, genetic structure in four populations of G. fuscipes sampled in 2008 and also in 2011.
F ST values for temporal samples calculated on microsatellite data. Nonsignificant values are in bold.
Microsatellite-based F ST values pairwise comparison between sampling localities of G. f. fuscipes in Uganda.
Microsatellite-based F ST values for pairwise comparisons among the three populations detected using Bayesian clustering.
Details of all first generation migrants detected by GeneClass 2.0, using L h, L h/L max, and STRUCTURE.
Comparison of mtDNA clade and microsatellite assignment for each individual where both data types were collected.
References
- 1.Itard J, Cuisance D, Tacher G. Europe et Régions Chaudes. Editions Tec et Doc and Editions Médicales Internationales. Vol. 2. Paris, France: Lavoisier; 2003. Trypanosomoses: historique repartition géographique. Principales maladies infectieuses et parasitaires du bétail; pp. 1607–1615. [Google Scholar]
- 2.Jannin J, Cattand P. Treatment and control of human African trypanosomiasis. Current Opinion in Infectious Diseases. 2004;17(6):565–570. doi: 10.1097/00001432-200412000-00009. [DOI] [PubMed] [Google Scholar]
- 3.Simarro PP, Franco J, Diarra A, Postigo JA, Jannin J. Update on field use of the available drugs for the chemotherapy of human African trypanosomiasis. Parasitology. 2012;139(7):842–846. doi: 10.1017/S0031182012000169. [DOI] [PubMed] [Google Scholar]
- 4.Rogers DJ, Randolph SE. Population ecology of tsetse. Annual Review of Entomology. 1985;30(1):197–216. doi: 10.1146/annurev.en.30.010185.001213. [DOI] [PubMed] [Google Scholar]
- 5.Laveissière C, Penchenier L. Manuel de Lutte Contre La MaLadie du Sommeil, IRD. Paris, France: Institut de Recherche pour le Développement, Collections Didactiques; 2005. [Google Scholar]
- 6.Kabayo JP. Aiming to eliminate tsetse from Africa. Trends in Parasitology. 2002;18(11):473–475. doi: 10.1016/s1471-4922(02)02371-1. [DOI] [PubMed] [Google Scholar]
- 7.Vreysen MJB, Saleh KM, Ali MY, et al. Glossina austeni (diptera: glossinidae) eradicated on the Island of Unguja, Zanzibar, using the sterile insect technique. Journal of Economic Entomology. 2000;93(1):123–135. doi: 10.1603/0022-0493-93.1.123. [DOI] [PubMed] [Google Scholar]
- 8.Brightwell R, Dransfield RD, Stevenson P, Williams B. Changes over twelve years in populations of Glossina pallidipes and Glossina longipennis (diptera: Glossinidae) subject to varying trapping pressure at Nguruman, south-west Kenya. Bulletin of Entomological Research. 1997;87(4):349–370. [Google Scholar]
- 9.Thompson JW, Mitchell M, Rees RB, Shereni W, Schoenfeld AH, Wilson A. Studies on the efficacy of deltamethrin applied to cattle for the control of tsetse flies (Glossina spp.) in southern Africa. Tropical Animal Health and Production. 1991;23(4):221–226. doi: 10.1007/BF02357104. [DOI] [PubMed] [Google Scholar]
- 10.Leak SGA. Tsetse Biology and Ecology: Their Role in the Epidemiology and Control of Trypanosomosis. Wallingford, UK: CABI Publishing; 1999. [Google Scholar]
- 11.Allsopp R. Options for vector control against trypanosomiasis in Africa. Trends in Parasitology. 2001;17(1):15–19. doi: 10.1016/s1471-4922(00)01828-6. [DOI] [PubMed] [Google Scholar]
- 12.Vale GA, Hargrove JW, Cockbill GF, Phelps RJ. Field trials of baits to control populations of Glossina morsitans morsitans Westwood and G. pallidipes Austen (Diptera: Glossinidae) Bulletin of Entomological Research. 1986;76(2):179–193. [Google Scholar]
- 13.Catley A, Leyland T. Community participation and the delivery of veterinary services in Africa. Preventive Veterinary Medicine. 2001;49(1-2):95–113. doi: 10.1016/s0167-5877(01)00171-4. [DOI] [PubMed] [Google Scholar]
- 14.Bouyer J, Balenghien T, Ravel S, et al. Population sizes and dispersal pattern of tsetse flies: rolling on the river? Molecular Ecology. 2009;18(13):2787–2797. doi: 10.1111/j.1365-294X.2009.04233.x. [DOI] [PubMed] [Google Scholar]
- 15.Solano P, Kaba D, Ravel S, et al. Population genetics as a tool to select tsetse control strategies: suppression or eradication of Glossina palpalis gambiensis in the niayes of senegal. PLoS Neglected Tropical Diseases. 2010;4(5, article e692) doi: 10.1371/journal.pntd.0000692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vreysen M, Robinson AS, Hendrichs J. Area-Wide Control of Insect Pests, From Research to Field Implementation. Dordrecht, The Netherlands: Springer; 2007. [Google Scholar]
- 17.Hargrove JW. Tsetse eradication: sufficiency, necessity and desirability. DFID Animal Health Programme, 2003.
- 18.Cuisance D, Politzar H, Merot P, Tamboura I. Les lâchers de mâles irradiés dans la campagne de lutte intégrée contre les glossines dans la zone pastorale de Sidéradougou, Burkina Faso. Revue d’Elevage et de Médecine Vétérinaire des Pays Tropicaux. 1984;37:449–468. [PubMed] [Google Scholar]
- 19.Dyck VA, Hendrichs J, Robinson AS. Sterile Insect Technique Principles and Practice in Area-Wide Integrated Pest Management. Dordrecht, The Netherlands: Springer; 2005. [Google Scholar]
- 20.Aksoy S, Caccone A, Okedi LM, Galvani A. Glossina fuscipes populations provide insights for human African Tryponosomiasis transmission in Uganda. Trends in Parasitology. 2013;29(8):394–406. doi: 10.1016/j.pt.2013.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kagbadouno M, Camara M, Bouyer J, et al. Tsetse elimination: its interest and feasibility in the historical sleeping sickness focus of loos islands, Guinea. Parasite. 2009;16(1):29–35. doi: 10.1051/parasite/2009161029. [DOI] [PubMed] [Google Scholar]
- 22.Bouyer J, Sibert A, Desquesnes M, Cuisance D, De La Rocque S. A diffusion model for Glossina palpalis gambiensis in Burkina Faso. In: Vreysen MJB, Robinson AS, Hendrichs J, editors. Area-Wide Control of Insect Pests. From Research to Field Implementation. Dordrecht, The Netherlands: Springer; pp. 221–228. [Google Scholar]
- 23.Dyer NA, Furtado A, Cano J, et al. Evidence for a discrete evolutionary lineage within Equatorial Guinea suggests that the tsetse fly Glossina palpalis palpalis exists as a species complex. Molecular Ecology. 2009;18(15):3268–3282. doi: 10.1111/j.1365-294X.2009.04265.x. [DOI] [PubMed] [Google Scholar]
- 24.Abila PP, Slotman MA, Parmakelis A, et al. High levels of genetic differentiation between Ugandan Glossina fuscipes fuscipes populations separated by Lake Kyoga. PLoS Neglected Tropical Diseases. 2008;2(5, article e242) doi: 10.1371/journal.pntd.0000242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Beadell JS, Hyseni C, Abila PP, et al. Phylogeography and population structure of Glossina fuscipes fuscipes in Uganda: implications for control of tsetse. PLoS Neglected Tropical Diseases. 2010;4(3, article e636) doi: 10.1371/journal.pntd.0000636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Echodu R, Beadell JS, Okedi LM, Hyseni C, Aksoy S, Caccone A. Temporal stability of Glossina fuscipes fuscipes populations in Uganda. Parasites and Vectors. 2011;4(1, article 19) doi: 10.1186/1756-3305-4-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kalinowski ST. The computer program STRUCTURE does not reliably identify the main genetic clusters within species: simulations and implications for human population structure. Heredity. 2011;106(4):625–632. doi: 10.1038/hdy.2010.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cecchi G, Mattioli RC, Slingenbergh J, De La Rocque S. Land cover and tsetse fly distributions in sub-Saharan Africa. Medical and Veterinary Entomology. 2008;22(4):364–373. doi: 10.1111/j.1365-2915.2008.00747.x. [DOI] [PubMed] [Google Scholar]
- 29.Hargrove JW, Brady J. Activity rhythms of tsetse flies (Glossina spp.) (Diptera: Glossinidae) at low and high temperatures in nature. Bulletin of Entomological Research. 1992;82(3):321–326. [Google Scholar]
- 30.Dyer NA, Ravel S, Choi K-S, et al. Cryptic diversity within the major trypanosomiasis vector Glossina fuscipes revealed by molecular markers. PLoS Neglected Tropical Diseases. 2011;5(8) doi: 10.1371/journal.pntd.0001266.e1266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pollock JN. Training Manual for Tsetse Control Personnel. Vol. 2. Rome, Italy: Food and Agriculture Organization; 1982. (Ecology and Behaviour of Tsetse). [Google Scholar]
- 32.Krafsur ES. Tsetse fly population genetics: an indirect approach to dispersal. Trends in Parasitology. 2003;19(4):162–166. doi: 10.1016/s1471-4922(03)00034-5. [DOI] [PubMed] [Google Scholar]
- 33.Hyseni C, Kato A, Okedi L, Masembe C, Ouma J, Aksoy S, et al. The population structure of Glossina fuscipes fuscipes in the Lake Victoria basin in Uganda: implications for vector control. Parasites and Vectors. 2012;5(1) doi: 10.1186/1756-3305-5-222.e222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Symula RE, Marpuri I, Bjornson RD, et al. Influence of host phylogeographic patterns and incomplete lineage sorting on within-species genetic variability in Wigglesworthia species, obligate symbionts of tsetse flies. Applied and Environmental Microbiology. 2011;77(23):8400–8408. doi: 10.1128/AEM.05688-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Alam U, Hyseni C, Symula R, Brelsfoard C, Wu Y, Kruglov O, et al. Microfauna-host interactions: implications for trypanosome transmission dynamics in Glossina fuscipes fuscipes in Uganda. Applied and Environmental Microbiology. 2012;78(13):4627–4637. doi: 10.1128/AEM.00806-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Alam U, Medlock J, Brelsfoard C, et al. Wolbachia symbiont infections induce strong cytoplasmic incompatibility in the Tsetse fly Glossina morsitans . PLoS Pathogens. 2011;7(12) doi: 10.1371/journal.ppat.1002415.e1002415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rio RVM, Symula RE, Wang J, et al. Insight into the transmission biology and species-specific functional capabilities of tsetse (diptera: glossinidae) obligate symbiont Wigglesworthia . mBio. 2012;3(1) doi: 10.1128/mBio.00240-11.e00240-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fèvre EM, Picozzi K, Fyfe J, et al. A burgeoning epidemic of sleeping sickness in Uganda. The Lancet. 2005;366(9487):745–747. doi: 10.1016/S0140-6736(05)67179-6. [DOI] [PubMed] [Google Scholar]
- 39.Picozzi K, Fèvre EM, Odiit M, et al. Sleeping sickness in Uganda: a thin line between two fatal diseases. British Medical Journal. 2005;331(7527):1238–1241. doi: 10.1136/bmj.331.7527.1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gibson WC. The significance of genetic exchange in trypanosomes. Parasitology Today. 1995;11(12):465–468. [Google Scholar]
- 41.Peacock L, Ferris V, Sharma R, et al. Identification of the meiotic life cycle stage of Trypanosoma brucei in the tsetse fly. Proceedings of the National Academy of Sciences of the United States of America. 2011;108(9):3671–3676. doi: 10.1073/pnas.1019423108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Peacock L, Ferris V, Bailey M, Gibson W. Intraclonal mating occurs during tsetse transmission of Trypanosoma brucei . Parasites and Vectors. 2009;2(1, article 43) doi: 10.1186/1756-3305-2-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Peacock L, Ferris V, Bailey M, Gibson W. Fly transmission and mating of Trypanosoma brucei brucei strain 427. Molecular and Biochemical Parasitology. 2008;160(2):100–106. doi: 10.1016/j.molbiopara.2008.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Challier A, Laveissiere C. Un nouveau piège pour la capture des glossines (Glossina: Diptera, Muscidae): description et essais sur le terrain. Cahiers ORSTOM, Série Entomologie Médicale Et Parasitologie. 1973;11:251–262. [Google Scholar]
- 45.Agata K, Alasaad S, Almeida-Val VMF, et al. Permanent genetic resources added to molecular ecology resources database 1 December 2010–31 January 2011. Molecular Ecology Resources. 2011;11(3):586–589. doi: 10.1111/j.1755-0998.2011.03004.x. [DOI] [PubMed] [Google Scholar]
- 46.Rousset F. GENEPOP’007: a complete re-implementation of the GENEPOP software for Windows and Linux. Molecular Ecology Resources. 2008;8(1):103–106. doi: 10.1111/j.1471-8286.2007.01931.x. [DOI] [PubMed] [Google Scholar]
- 47.Peakall R, Smouse PE. GENALEX 6: genetic analysis in Excel. Population genetic software for teaching and research. Molecular Ecology Notes. 2006;6(1):288–295. doi: 10.1093/bioinformatics/bts460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Goudet J. FSTAT (Version 1. 2): a computer program to calculate F-statistics. Journal of Heredity. 1995;86(6):485–486. [Google Scholar]
- 49.Excoffier L, Laval G, Schneider S. Arlequin (version 3. 0): an integrated software package for population genetics data analysis. Evolutionary Bioinformatics Online. 2005;1(47) [PMC free article] [PubMed] [Google Scholar]
- 50.Rozas J, Sánchez-DelBarrio JC, Messeguer X, Rozas R. DnaSP, DNA polymorphism analyses by the coalescent and other methods. Bioinformatics. 2003;19(18):2496–2497. doi: 10.1093/bioinformatics/btg359. [DOI] [PubMed] [Google Scholar]
- 51.Weir BS, Cockerham CC. Estimating F-statistics for the analysis of population structure. Evolution. 1984;38(6):1358–1370. doi: 10.1111/j.1558-5646.1984.tb05657.x. [DOI] [PubMed] [Google Scholar]
- 52.Clement M, Posada D, Crandall KA. TCS: a computer program to estimate gene genealogies. Molecular Ecology. 2000;9(10):1657–1659. doi: 10.1046/j.1365-294x.2000.01020.x. [DOI] [PubMed] [Google Scholar]
- 53.Pritchard JK, Stephens M, Donnelly P. Inference of population structure using multilocus genotype data. Genetics. 2000;155(2):945–959. doi: 10.1093/genetics/155.2.945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Earl D, VonHoldt B. STRUCTURE HARVESTER: a website and program for visualizing STRUCTRE output and implementing the Evanno method. Conservation Genetics Resources. 2012;4(2):359–361. [Google Scholar]
- 55.Evanno G, Regnaut S, Goudet J. Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Molecular Ecology. 2005;14(8):2611–2620. doi: 10.1111/j.1365-294X.2005.02553.x. [DOI] [PubMed] [Google Scholar]
- 56.Manel S, Berthier P, Luikart G. Detecting wildlife poaching: identifying the origin of individuals with Bayesian assignment tests and multilocus genotypes. Conservation Biology. 2002;16(3):650–659. [Google Scholar]
- 57.Paetkau D, Slade R, Burden M, Estoup A. Genetic assignment methods for the direct, real-time estimation of migration rate: a simulation-based exploration of accuracy and power. Molecular Ecology. 2004;13(1):55–65. doi: 10.1046/j.1365-294x.2004.02008.x. [DOI] [PubMed] [Google Scholar]
- 58.Rannala B, Mountain JL. Detecting immigration by using multilocus genotypes. Proceedings of the National Academy of Sciences of the United States of America. 1997;94(17):9197–9201. doi: 10.1073/pnas.94.17.9197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Konovalov DA, Heg D. A maximum-likelihood relatedness estimator allowing for negative relatedness values. Molecular Ecology Resources. 2008;8(2):256–263. doi: 10.1111/j.1471-8286.2007.01940.x. [DOI] [PubMed] [Google Scholar]
- 60.Mossman CA, Waser PM. Genetic detection of sex-biased dispersal. Molecular Ecology. 1999;8(6):1063–1067. doi: 10.1046/j.1365-294x.1999.00652.x. [DOI] [PubMed] [Google Scholar]
- 61.Barclay HJ, Hargrove JW. Probability models to facilitate a declaration of pest-free status, with special reference to tsetse (Diptera: Glossinidae) Bulletin of Entomological Research. 2005;95(1):1–11. doi: 10.1079/ber2004331. [DOI] [PubMed] [Google Scholar]
- 62.Symula R, Alam U, Brelsfoard C, Wu Y, Echodu R, Okedi L, et al. Wolbachia association with the tsetse fly, Glossina fuscipes fuscipes, reveals high levels of genetic diversity and complex evolutionary dynamics. BMC Evolutionary Biology. 2013;13(article 1) doi: 10.1186/1471-2148-13-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix 1: Information on the study site, time of collection and number of flies caught in each site.
Appendix 2: Details of microsatellite markers used in the study.
Appendix 3: Per locus estimates of FIS at 18 microsatellite loci for each sampling locality.
Appendix 4: Mitochondrial haplotype information, including frequencies observed across studied populations. New haplotypes recovered from this study are indicated by ∗.
Appendix 5: Results of an AMOVA testing for temporal genetic structure in four populations of G. fuscipes sampled in 2008 and also in 2011.
Appendix 6: Fst values for temporal samples calculated on microsatellite data. Non-significant values are in bold.
Appendix 7: Microsatellite-based FST values pairwise comparison between sampling localities of G. f. fuscipes in Uganda.
Appendix 8: Microsatellite-based FST values for pairwise comparisons among the three populations detected using Bayesian clustering.
Appendix 9: Details of all first generation migrants detected by Geneclass 2.0, using Lh, Lh/Lmax and STRUCTURE.
Appendix 10: Comparison of mtDNA clade and microsatellite assignment for each individual where both data types were collected.
Figure 1: Microsatellite allele frequencies observed in seven populations of G. f. 52 fuscipessampled at different time points. Numbers after location code indicate the time interval 53 (in generations) since the first sampling.