

Abstract
The alloimmune response can be divided into specific junctures where critical decisions between tolerance and immunity are made which define the outcome of the transplant. At these “decision nodes” various cytokines direct alloresponsive T cells to develop either a proinflammatory response aimed at graft destruction or an immunoregulatory response facilitating graft acceptance. This review will focus on the role of these cytokines in influencing the progression of an alloimmune response leading ultimately to either allograft survival or rejection.
Introduction
Self-tolerance refers to the normal homeostatic balance in which the immune system exists without any pathologic response to self-antigens. Similarly, acquired transplant tolerance refers to the absence of a pathologic immune response to the allograft without the need for chronic, nonspecific immunosuppression. In other words, the host’s immune system should be fully competent to respond adequately to further antigenic challenge without targeting the transplanted organ or tissue. Although the attainment of true transplantation tolerance (with acceptable treatment toxicity) in the clinical setting remains elusive, it can often be induced in a variety of rodent models. In these settings, intense investigation of the mechanisms involved has led to a greater understanding of the requirements for tolerance and the obstacles to be overcome if long-term allograft acceptance is to be achieved in the future.
As with other immune responses, cytokines play an integral role in alloimmunity, and the pattern of cytokine expression is central to the mechanisms which regulate the development of immune tolerance to transplanted tissues. This review will provide an overview of current concepts of alloimmunity and allotolerance with an emphasis on the part played by cytokines in these processes and discuss strategies which have been employed to exploit this knowledge as a means of inducing transplantation tolerance.
Overview of Allorecognition and Graft Rejection
Allograft rejection is primarily driven by the ability of host T cells to recognize polymorphisms encoded within the MHC as well as multiple minor histocompatibility loci. While all components of the innate and adaptive immune systems participate in graft rejection, models in which T cell- or CD4 T cell-deficient mice indefinitely accept allografts demonstrate the paramount importance of T lymphocytes and particularly CD4 T cells in this process (Rosenberg and Singer, 1992). Host T cells can be activated in response to an allogeneic stimulus by one of two separate mechanisms (Batchelor and Lechler, 1982). Direct allorecognition defines the response of host T cells recognizing intact donor MHC molecules present on the surface of donor-derived antigen-presenting cells (APCs). In the indirect pathway of allorecognition, host T cells respond to processed donor-derived peptides bound to syngeneic MHC molecules and presented in a self-restricted manner.
Studies over the past twenty years suggest separate but complementary roles for these two pathways of allo-recognition in rejection and tolerance. This does not appear to be the result of differences in the quality of the T cell response, or the cytokines elicited, but rather is based on differences in cell quantity and locale. For example, donor-derived APCs, primarily dendritic cells, are present in grafted tissues as passenger leucocytes that migrate posttransplant to host lymphoid tissues where they directly stimulate host T cells. Dendritic cells are extremely powerful activators of naive T cells, and this, coupled with the very large frequency of cells exhibiting direct alloreactivity, has led to the concept that the direct pathway of allorecognition is dominant during acute rejection (Game and Lechler, 2002; Suchin et al., 2001; Womer et al., 2001).
As migratory dendritic cells are relatively short-lived after transplantation, the role of direct alloreactivity may be temporally limited, allowing the indirect pathway to predominate in the later stages of the allograft response. Evidence in favor of this includes the findings of large numbers of indirectly reactive T cells (by limiting dilution analysis) in patients undergoing chronic rejection, but not in controls with good graft function, and the requirement for indirect allorecognition to provide B cell help for alloantibody production, a key feature of chronic rejection (Hornick et al., 2000; Pettigrew et al., 1998). Nonetheless, direct alloreactivity may not necessarily be limited to the early phases of the allograft response. Donor endothelial cells, expressing many of the same costimulatory and adhesion molecules found on dendritic cells, are able to directly activate recipient CD8 T cells (Kreisel et al., 2002). As donor endothelial cells persist for the life of the graft, they may provide a constant source of stimulation for directly alloreactive T cells. It is also important to keep in mind that the indirect pathway of allorecognition may be utilized by regulatory T cells which are required to maintain tolerance in a number of different experimental transplant models (reviewed in Cobbold and Waldmann, 1998; further discussion below).
Overview of Peripheral Tolerance
One of the most successful experimental strategies to induce allograft acceptance is to create a state of mixed hematopoietic chimerism (Sykes, 2001). This strategy exploits the natural homeostatic mechanisms used to create self-tolerance during T cell development in the thymus. Mixed chimerism and other strategies of central (i.e., thymic) tolerance appear to act almost exclusively via deletional mechanisms, with scant if any evidence for anergy or regulation (Nikolic and Sykes, 1997). As a result, the requirement for cytokines, as they may exist, in this process has not been investigated in depth. Thus, for the purposes of this review, we will restrict our discussion to the role of cytokines in the regulation of peripheral tolerance, i.e., strategies primarily targeted at mature T cells in secondary lymphoid organs.
It is conceptually useful to divide peripheral transplantation tolerance into two distinct but not mutually exclusive temporal phases (Li et al., 2001b). The induction phase encompasses events which occur immediately posttransplant when a large number of alloresponsive T cells are stimulated by graft antigens presented by either donor or host APCs. The development of such a robust immune response will eventually lead to acute graft rejection unless mechanisms are set in place which hold the inflammatory process in check. Thus, the induction phase of tolerance typically is characterized by depletion or ignorance of alloreactive T cells with an implicit role for a number of different cytokines.
Once this initial acute immune response has been restricted, the threat of rejection persists unless the tolerant state is maintained. The maintenance phase of allograft tolerance is an active process of T cell-mediated regulation, which suppresses the potentially injurious graft-reactive T cells. While there has long been a general acceptance of the requirement of active immunoregulatory mechanisms for the maintenance of transplantation tolerance, it is only recently that such mechanisms have been identified and characterized more closely (Wood and Sakaguchi, 2003).
Role of Cytokines in the Response to Allografts
From the above considerations, it is clear that rejection and tolerance can be considered alternative outcomes of the allograft response, i.e., host encounter with allo-antigens. Once activated, CD4+ T cells primarily direct the progression of the response by secreting cytokines which activate, expand, and/or recruit other effector cells such as macrophages and CD8+ T cells, B cells, and NKT T cells. The primary consideration is defining key junctures at which alternate outcomes are favored and determining the parameters that dictate the decision ultimately made by the T cell. Here, we consider five such decision nodes and will examine the role of cytokines in these fate determinations. They are APC maturation, T helper cell expansion, T cell survival, T helper cell differentiation, and regulatory T cell homeostasis. The first four events largely take place during the induction phase of tolerance while the latter is likely of primary importance during the maintenance phase.
APC Maturation
Dendritic cells (DCs) are found in most peripheral tissues (Banchereau and Steinman, 1998) and, as noted above, migrate immediately posttransplant to regional lymph nodes where the host response to the allograft is then initiated. While the requirements for DC development and homeostasis in vivo have not been clearly defined, in vitro systems indicate an important role for cytokines, including IL-4 and granulocyte-macrophage colony-stimulating factor (GM-CSF) in particular, in promoting the development of these cells from bone marrow- and peripheral blood-derived precursors. Type-1 interferons, which are secreted from a variety of different cell types in response to inflammatory damage, can also lead to DC development in vitro. Infection or tissue damage (such as takes place in the context of transplantation) promote the maturation of DCs. In contrast to their immature counterparts, mature DCs can efficiently induce T cell expansion and differentiation, which has led to the emergence of a model whereby infection or inflammatory tissue damage helps provide the danger signals required for the development of an immune response. The importance of DC maturation in the context of transplantation is illustrated by studies showing that immunization of recipients with immature DCs from the same donor strain prolongs subsequent murine heart allograft survival in a donor-specific fashion (Lutz et al., 2000; O’Connell et al., 2002).
A number of microbial products such as LPS and bacterial DNA as well as necrotic cell products such as heat shock proteins have been reported to provide such an activating stimulus for DCs. These diverse stimuli are recognized by members of the Toll-like receptor family (TLRs) expressed on the surface of DCs. They promote DC maturation through activation of a variety of signaling cascades involving adaptor proteins such as MyD88 and TRAF family members leading to NF-κB-mediated transcription (Dunne and O’Neill, 2003; Kobayashi et al., 2003). These signaling events in turn lead to the upregulation of T cell costimulatory molecules including CD40, CD80, and CD86 as well as the secretion of cytokines such as IL-12 and TNFα (Akira et al., 2001). The importance of this sequence of events in the initiation of an alloresponse has been highlighted by a recent report demonstrating the critical role of MyD88 expression in acute allograft rejection. In this report, minor antigen-mismatched allografts were not rejected when both donor and recipient mice were MyD88 deficient. Prolonged graft survival was associated with a decreased number of phenotypically mature DCs found in draining lymph nodes as well as a reduced capacity for priming of alloreactive T cells (Goldstein et al., 2003). It also has been reported that in vitro manipulation of DCs with low doses of GM-CSF or with IL-10 renders the cells resistant to maturation and can facilitate prolonged allograft survival (Lutz et al., 2000; Mitra et al., 1995).
Several mechanisms have been proposed by which immature DCs can promote tolerance (Figure 1) (Steinman et al., 2003). First, immature DCs constitutively express only very low (or absent) levels of T cell costimulatory molecules such as CD80/86 and CD40 as well as a relatively low level of MHC complexes on the cell surface. Moreover, immature DCs do not secrete significant levels of proinflammatory cytokines such as IL-12 and TNFα. Lack of appropriate costimulation may induce T cell anergy or apoptosis, clearly favoring tolerance. However, the relevance of this observation to in vivo tolerance may be questioned by the fact that, as opposed to naive T cells, activation of memory T cells is relatively independent of costimulation (Harris and Ronchese, 1999) and otherwise unmanipulated inbred mice contain substantial numbers of memory T cells (Lee et al., 1996), which can exhibit crossreactivity with alloantigens (Adams et al., 2003).
Figure 1. Dendritic Cell Maturation Determines the Fate of Responding CD4+ T Cells.
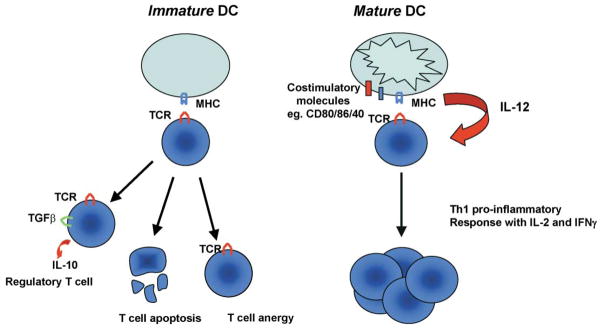
Immature DCs can induce a number of responses from CD4+ T cells which are conducive to tolerance. These include anergy, apoptosis, or differentiation into a regulatory T cell producing immunosuppressive cytokines such as IL-10. Mature DCs, on the other hand, express elevated levels of costimulatory molecules and secrete proinflammatory cytokines such as IL-12. This allows mature DCs to efficiently prime CD4+ cells to differentiate into Th1 type effector cells secreting IL-2 and IFN-γ.
Second, and perhaps more intriguing, is the reported induction of a regulatory T cell phenotype by repeated stimulation with immature allogeneic DCs (Jonuleit et al., 2000). These induced regulatory T cells (Tr1 cells) appear to be a distinct population of cells from those Tregs which arise naturally in the thymus (e.g., CD4+CD25+ T cells, further discussion below). Similar regulatory populations have been described in models of autoimmune disease (Th3 cells), and their suppressive effect has been associated with a number of cytokines, particularly IL-10 and TGF-β (Cobbold and Waldmann, 1998; Josien et al., 1998; Roncarolo and Levings, 2000). Inducible Tregs are likely to mediate the occurrence of antigen-specific hyporesponsiveness in human T lymphocytes in vivo after injection of antigen-loaded immature DCs (Dhodapkar et al., 2001).
Although the extent of maturation of DCs undoubtedly plays an important role in determining the decision between tolerance and immunity, there also exists considerable evidence indicating that specific DC subsets may suppress T cell responses independent of their maturation state. CD8+ DCs can in certain models induce tolerance of CTLs upon crosspresentation of exogenous antigen. The tolerizing potential of this DC subset appears to be related to their ability to promote deletion of the activated CD8+ T cells (Heath and Carbone, 2001). There is also accumulating evidence for the existence of a discrete population of DCs expressing the enzyme idoleamine 2,3-dioxygenase (IDO) which constitutes the rate-limiting step in the catabolism of tryptophan. Several reports have now documented the role of IDO in tolerance to allogeneic fetuses. While it was originally postulated that IDO acted by depriving T cells of a requisite growth factor (tryptophan) (Mellor and Munn, 1999), more recent studies indicate that it is the tryptophan metabolites themselves which exert a cytotoxic effect on T cells (Frumento et al., 2002; Terness et al., 2002). Stimulation of mature IDO expressing DCs with IFN-γ results in downregulation of this enzyme, an effect which is inhibited by the presence of IL-10 (Munn et al., 2002).
T Helper Cell Expansion
An obvious role for cytokines in allogeneic responses, and indeed their first identified role for T cells, is to promote T cell proliferation. Limiting the expansion of effector T cells is an integral part to any approach toward promoting tolerance, but until recently, it has been unclear which cytokines, if any, might be specifically and, if possible, nonredundantly important for T cell expansion in vivo. The cytokines which can act as T cell growth factors are primarily those whose multimeric receptor complexes contain the common γ (γc) chain, e.g., IL-2, IL-4, IL-7, IL-15, and IL-21. IL-2 was the first such cytokine identified. Its discovery and the production of antibodies against the α chain of the IL-2R (CD25) prompted studies which demonstrated that anti-CD25 mAbs could induce transplantation tolerance (Kupiec-Weglinski et al., 1987; Reed et al., 1989). Initially, it was assumed that this was mediated by blockade of clonal expansion of pathogenic alloreactive T cells. However, a variety of lines of evidence subsequently indicated a far more complex situation. First, calcineurin inhibitors such as cyclosporine and tacrolimus are potent inhibitors of IL-2 production, yet they fail to induce tolerance in mice and humans. Second, IL-2-deficient, as well as IL-2/IL-4 doubly deficient mice readily reject heart and islet allografts (Li et al., 1998a). Indeed, IL-2-deficient animals are in fact difficult to tolerize (see below). Third, it is clear that IL-2 is not the only, or perhaps even the most, important in vivo growth factor for activated T cells. While the use of IL-2 by T cells may predominate in vitro where T cells themselves are the source of cytokines, in vivo T cell proliferation appears to be much more dependent on IL-15, which is produced by diverse cell types such as APCs and endothelial cells (Li et al., 2001a).
So how does anti-CD25 work? It is important to remember that in fact there is a hierarchy of tolerance susceptibility in murine models: liver > kidney > heart > islet ≫ skin. Anti-CD25 by itself works only in more permissive models. A recent study may provide some insight into this. Here, it was found that anti-CD25 lysed activated CD25+ effector T cells, while also targeting CD25+ Tregs (Zheng et al., 2003). In contrast, use of an agonistic IL-2/Fc fusion protein selectively targeted CD25+ effector T cells while sparing CD25+ Tregs and promoting allograft survival in a very stringent model. This approach exploits the ability of IL-2 to induce activation-induced cell death (AICD) in effector T cells while promoting the survival of CD25+ Tregs.
T Cell Survival
In the last several years, a very complex story has emerged regarding the ability of cytokines to alternately promote T cell survival and death and the role these functions play in the net effect of cytokines on transplantation tolerance (Figure 2). Particular attention has focused on IL-2, and much of this evidence stems from investigations in mice, which are deficient in expression of IL-2 or the α chain of the IL-2R. Such mice have a severe lymphoproliferative disorder caused by an apparent failure to clear activated T cells from the periphery through AICD (Sadlack et al., 1995; Willerford et al., 1995). T cells from IL-2−/− mice were observed to be insensitive to Fas-mediated apoptosis in vitro leading to the hypothesis that a major function of IL-2R signaling in vivo is to prime activated T cells for Fas-induced apoptosis (Refaeli et al., 1998). At present, it does not appear that any of the other γ chain cytokines can also act as a feedback regulator of clonal expansion similar to IL-2. Interestingly, the other major cytokine associated with an inflammatory immune response, IFN-γ, also has an important role in regulating T cell expansion, as revealed by studies in IFN-γ-deficient mice, which display enhanced proliferation in response to allogeneic stimulation in vitro when compared to their wild-type counterparts (Konieczny et al., 1998).
Figure 2. Role of Cytokines at Major Decision Nodes between Allograft Tolerance and Rejection.

Cytokines play a pivotal role in the development of both transplant tolerance and rejection by regulating critical points during the generation of an allospecific immune response. Proinflammatory cytokines such as IL-12 and IL-15 appear to exclusively promote graft rejection through directing T effector differentiation or promoting expansion, survival, and memory development, respectively. Conversely, immunosuppressive cytokines such as IL-10 and TGFβ can promote tolerance through mediating regulatory T cell function. IL-2 can either be tolerogenic (priming for AICD and regulating Treg homeostasis) or promote rejection (effector T cell expansion and survival). The pleiotropic effects of IL-2 appear to be temporally and contextually dependent.
However, while IL-2 has the ability to prime T cells to ultimately undergo AICD, its initial role in the immune response is actually to promote T cell survival, primarily by upregulation of the antiapoptotic gene Bcl-2 (Mueller et al., 1996; Van Parijs et al., 1999). Expression of Bcl-2 and the related gene Bcl-xL, which is upregulated through CD28 costimulation (Boise et al., 1995), enables activated cycling T cells to survive in cytokine-depleted environments. Thus, the net effects of IL-2 are both time and context dependent. Early in the immune response, and in the face of limiting cytokines, IL-2 promotes T cell growth and survival. Later in the immune response, and in the face of continued antigenic stimulation, IL-2 promotes cell death.
In addition to IL-2, other cytokines such as IL-15, IL-4, and IL-7 have been reported to play a role in activated T cell survival in vivo and as such can directly regulate the clonal size of alloresponsive T cells (Vella et al., 1998). The overlapping effects of these cytokines are thought to be linked to the shared expression of a common γ chain by their respective receptors. Redundancy of function between these related cytokines may help explain why strategies which target expression of a single family member have proved largely unsuccessful in inducing allospecific tolerance (Li et al., 1998a; Steiger et al., 1995). In support of this hypothesis, recent studies have demonstrated that blockade of γ chain signaling induces the long term survival of islet allografts while inducing T cell apoptosis (Li et al., 2000). Similarly, administration of a humanized monoclonal antibody, which blocks signaling through the IL-2Rβ chain, prolongs renal allograft survival in cynomolgus monkeys (Tinubu et al., 1994). As the IL-2R β chain is shared by the receptors for both IL-2 and IL-15, this agent inhibits both the discrete and overlapping effects of these cytokines on T cell-mediated immune responses (Waldmann et al., 2001). These observations indicate that it may be more efficient to target multiple cytokines over individual cytokine pathways as a means of inducing tolerance.
The question of T cell survival may be particularly important for transplantation tolerance. Recently, we and others have demonstrated the necessity of a reduction in alloresponsive T cell clone size through deletion as a prerequisite for achieving tolerance (Li et al., 1999; Wells et al., 1999; Dai et al., 1998). In these studies, we found that mice whose T cells were defective in undergoing either AICD (IL-2-deficient mice) or in undergoing apoptosis in response to cytokine withdrawal (Bcl-xL transgenic recipients) resisted tolerance induction to MHC mismatched allografts by costimulatory blockade. Furthermore, calcineurin inhibition, which blocks the induction of apoptosis following T cell activation, antagonized the ability of costimulatory blockade to induce tolerance. Conversely, rapamycin, which potentiates AICD induced through costimulatory blockade, enhanced the tolerizing potential of this approach in highly resistant models such as skin. The importance of cell death in the induction of tolerance further explains why both IL-2 and IFN-γ, proinflammatory cytokines which might be considered a barrier to tolerance induction, can in fact, through their ability to promote T cell apoptosis, also facilitate rather than impede allograft acceptance. These observations suggest that these so-called “proinflammatory” cytokines play an additional, if not predominantly limiting, role in the context of an in vivo alloimmune reaction (Figure 2).
During the course of a normal T cell immune response, initiation is followed by clonal expansion and a subsequent large-scale apoptotic cascade, which memory cells escape. This is likely due to an inherent survival advantage of memory T cells over their effector counterparts, and as a consequence memory cells may evade tolerizing strategies, which induce the deletion of allore-active T cells during the induction phase. While the requirements for the generation of CD4+ T cell memory remain largely undefined, it is increasingly evident that IL-15 plays a central role in the generation and survival of CD8+ memory cells (Figure 2) (Schluns and Lefrancois, 2003). In contrast, IL-2 has been reported to have little or no effect on CD8+ memory cell homeostasis, indicating that this function is specific to IL-15 and not shared by related cytokines (Dai et al., 2000). An important feature of memory T cells is their decreased requirement for costimulation to induce optimal responses. In fact, costimulatory blockade has been demonstrated to be ineffective in preventing allograft rejection in certain models and this resistance may be mediated by CD8+ memory T cells (Valujskikh et al., 2002; Zhai et al., 2002) Other studies have demonstrated that antagonism of IL-15 signaling and/or lysis of IL-15R+ cells can promote allospecific tolerance in models which are resistant to the effects of costimulatory blockade (Ferrari-Lacraz et al., 2001; Smith et al., 2000). However, it has not been conclusively proven that all of these effects are mediated specifically through memory cells, as primary CD8+ T cells also have a reduced requirement for costimulation when compared to their CD4+ counterparts (Newell et al., 1999).
T Helper Cell Differentiation
Th2 responses are often observed in the context of long-term allograft acceptance and/or tolerance. This initially led to the proposal that Th2 immune deviation was causally linked to transplant tolerance; however, many subsequent investigations indicate that the situation is somewhat more complex. For example, in most circumstances, deliberate skewing of alloimmunity toward Th2 responses either through antagonism of IL-12 signaling or in vivo administration of stable Th2 cytokines still results in rejection of MHC mismatched grafts (Li et al., 1998b; Piccotti et al., 1996; Zheng et al., 1995). However, redirecting a Th1 response toward a Th2 response is by itself sufficient to prevent graft rejection across selected minor histocompatibility barriers (Li et al., 1998b). We have interpreted this in the context of the size of the alloreactive T cell pool, being much larger in across MHC barriers than across minor histocompatibility differences (Kishimoto et al., 2002). Consistent with an important role for pool size, while costimulatory blockade cannot induce long-term MHC mismatched allograft survival in models where T cell apoptosis is impaired (see above), CTLA4Ig is very effective at inducing long-term allograft survival across mH barriers in these same models (IL-2- or IFN-γ-deficient recipients or Bcl-xL-transgenic recipients). Thus, the relative ease of creating a tolerant state, and the requirements to do so, may differ depending on quantitative issues (e.g., the number of alloreactive cells). Nonetheless, other reports indicate that Th2 alloreactive clones can be effective mediators of graft rejection (Barbara et al., 2000) and murine strain specific differences in the Th1 versus Th2 dominance of allograft rejection have been reported, with either type of response culminating in graft destruction (Wang et al., 2003). While these studies make it clear that Th2 responses can be directed at allograft rejection, the ultimate role of Th2 cytokines in graft rejection versus tolerance remains unresolved. Any such role is undoubtedly related to a number of parameters including the size of the alloreactive T cell repertoire, the extent and types of antigenic disparities between donor and recipient, and the tissue being transplanted, as well as the background genes of the recipient which influence the nature of the allograft response.
There is, however, one well-described system in which immune deviation can induce allograft tolerance and whose mechanism is understood at a cellular level. The term anterior chamber-associated immune deviation (ACAID) was coined to describe a naturally occurring phenomenon which facilitates the transplantation of MHC-mismatched corneas with the absence of a proinflammatory T cell response (Streilein et al., 2002). ACAID occurs as a result of a number of factors, which combine to promote the development of a regulatory response by APCs normally resident within the anterior chamber of the eye. APCs at this site are phenotypically modulated by the high levels of constitutive expression of TGF-β in the intraocular microenvironment, leading to the downregulation of T cell costimulatory factors such as CD40 and a failure to secrete IL-12. After antigen capture these APCs are mobilized to leave this environment and migrate to the spleen. Here the APCs continue to secrete immunosuppressive cytokines, in particular, TGF-β and IFNα/β, and upon initial interaction with NK T cells attract antigen-specific CD4+ and CD8+ T cells. The net effect of such phenotypic alterations in antigen presentation is the development of an immunoregulatory T cell response in place of a Th1-type response. Immunoregulatory T cells are induced to produce cytokines such as TGFβ and IL-10 and disseminate from the spleen to contribute to the maintenance of tolerance against the antigen in question.
Regulatory T Cell Homeostasis and Function
While deletional events during the induction phase of transplantation tolerance prevent early rejection and facilitate graft acceptance, an active process of immune regulation is required to maintain host tolerance to transplanted tissues over a prolonged time period.
Initial observations which established that tolerance to alloantigens is transferable to a naive animal by T cells provided evidence of the existence of immunoregulatory mechanisms within the host’s T cell repertoire (Qin et al., 1993). The existence of distinct T cell subsets, which function to restrict the expansion of effector T cells is now widely accepted, and the identification and mechanism of action of such regulatory T cell (Treg) subsets remain areas of intense investigation. While the majority of Treg subsets defined thus far develop within the CD4+ population, regulatory activity is by no means exclusively restricted to CD4+ T cells. Indeed, regulatory activity has also been demonstrated in CD8+ as well as in TCR+CD4−CD8− T cells after transplantation (Colovai et al., 2000; Zhang et al., 2001). Tregs have been reported to have a variety of actions on immune effector cells, most notably the inhibition of proliferation and cytokine production. However, the precise mechanisms of immunosuppression by Treg subsets have yet to be defined, and it is becoming increasingly clear that various cytokines play an integral role in the development, homeostasis, and function of these cells.
Most recent studies on Tregs have focused on a naturally occurring subset of CD4+ T cells which constitutively express the IL-2R α chain (CD25). While CD4+CD25+ Tregs were originally characterized as being important in the maintenance of self-tolerance (Takahashi et al., 1998), they are also recognized to play a role in many models of allograft tolerance. The suppressive effect of these cells appears to be cell contact dependent, and there is evidence to suggest that Treg-mediated production of IL-10 and cell surface expression of TGFβ may play an important role in vivo. Inhibitory antibodies against both IL-10 and its cognate receptor have been shown to inhibit the suppressive effects of Treg cells in skin allograft models (Hara et al., 2001; Kingsley et al., 2002). However, a defined role for IL-10 in transplantation tolerance remains controversial, perhaps reflecting both the immunostimulatory and immunosuppressive properties of this cytokine (Figure 4). Accordingly, while some studies reveal the beneficial effects of IL-10 signaling in enhancing graft survival many others suggest neutral or even detrimental effects (Moore et al., 2001). Interestingly, a homolog of IL-10 expressed in many viral genomes appears to retain the immunosuppressive properties of this cytokine without the capacity for immunostimulation observed with endogenous IL-10. Indeed, studies involving ectopic expression of viral IL-10 within the allograft have had considerably more success in prolonging graft survival than models which manipulate endogenous IL-10 (Bromberg, 1995; De-Bruyne et al., 1998; Wang et al., 1999). The observation that Treg cells express a cell surface bound isoform of TGFβ has led to the hypothesis that it may be the mechanism through which cell contact-mediated suppression occurs. Indeed, a role for TGFβ in mediating suppression by Tregs has been described in a number of allograft models (Jonuleit et al., 2002; Josien et al., 1998). However, it is noteworthy that functional CD4+CD25+ T regs are present in mice with deficiencies in both IL-10 and TGFβ, perhaps indicating that these cytokines alone are not solely responsible for suppression (Piccirillo et al., 2002; Suri-Payer and Cantor, 2001). It should also be noted that most reported data would also be consistent with a model in which cytokines such as IL-10 and TGFβ are not necessarily direct products of the Tregs, but may be induced by Tregs in other target populations.
Figure 4. Cytokines Regulate the Polarity of the Immune Response toward the Transplant.

Many studies demonstrate that IL-10 plays a role in the induction of T cell tolerance due to its immunosuppressive function; however, other reports indicate that it may also act in an immunostimulatory fashion to promote rejection. On the other hand, it is believed that IL-15 promotes the development of a solely proinflammatory T cell response leading to allograft rejection. IL-2 can induce both tolerizing and proinflammatory effects, depending on the phase and context of the response, demonstrating its critical role in determining the fate of the allograft.
Along those lines, an interesting feature of regulatory T cells in the context of allograft transplantation is an apparent ability to promote the development of an im-munosuppressive phenotype in otherwise nonregulatory T cells. Furthermore, an allounresponsive state can be generated without subjecting the host to the entire range of donor graft antigens. Preexposure to a single antigen expressed within the graft can be sufficient (Madsen et al., 1988). This linked suppression appears to act via inducing new T cells which enter the host repertoire after transplantation to develop a regulatory rather than effector phenotype (Figure 3). The exact mechanism by which linked suppression occurs has not been defined; however, it has been reported that increased Th2 type cytokines (IL-4 and IL-10) are present at the graft site in models where tolerance is achieved through such mechanisms (Onodera et al., 1997). IL-10 and TGFβ have also been implicated in the suppressive function of these induced Treg subsets (Dieckmann et al., 2002; Jonuleit et al., 2002).
Figure 3. The Phenomenon of Linked Suppression.

Linked suppression refers the induction of tolerance to a third party alloantigen B by regulatory T cells recognizing alloantigen A when both antigens are processed and presented by the same antigen-presenting cell. This effect is mediated at least in part by the secretion of cytokines such as TGFβ and IL-10 by the regulatory T cells which cause the effector cell recognizing alloantigen B to become hyporesponsive and perhaps develop a regulatory phenotype (infectious tolerance).
In addition to the ability of cytokines to mediate suppressive functions of regulatory T cells, there is mounting evidence that cytokines, in particular IL-2, play a critical role in the development and homeostasis of Treg cells (Figure 2). CD4+CD25+ T cells are not detectable in IL-2−/− mice, which suffer from a severe lymphoproliferative disorder and autoimmune disease. Significantly, the generation of IL-2−/− mice on a TCR-transgenic background blunts the progression of autoimmune disease, presumably because of the lack of autoreactive T cells generated naturally in the thymus (Wolf et al., 2001). Malek et al. have demonstrated the requirement of IL-2R signaling through the β chain of the receptor for the development of CD4+CD25+ Tregs in vivo (Malek et al., 2002), and it has also been reported that IL-2 contributes to the survival of Tregs in the periphery (Almeida et al., 2002; Murakami et al., 2002). A characteristic feature of CD4+CD25+ Tregs is their hypoproliferative response to IL-2 stimulation in vitro, despite constitutive expression of all three chains of the IL-2R. Proliferation of Tregs can be induced in vitro upon stimulation through either the TCR or TNFR superfamily member GITR when either stimulus is used in combination with IL-2 (McHugh et al., 2002). While recent studies have highlighted the requirement for TCR-derived signals to induce Treg expansion in vivo (Walker et al., 2003), it is highly likely that such a stimulus alone is not sufficient to drive Treg expansion and that a secondary cytokine signal is necessary. Whether IL-2 contributes to cell cycle progression or simply functions to maintain Treg viability thus contributing to efficient expansion, driven by another cytokine (e.g., IL-15), remains unclear.
Cytokines as Potential Therapeutic Targets
Arguably, the greatest single barrier to the induction of allospecific tolerance is the extraordinarily large number of responding T cells with direct allospecificity. Mounting evidence indicates that in order for tolerance to be induced an initial wave of deletion of effector T cells is required. This creates a more level playing field on which regulatory T cells can suppress the now reduced number of alloreactive cells available and primed for graft destruction.
At present, the most effective single therapeutic agents which prolong graft survival are calcineurin inhibitors, such as cyclosporine A and tacrolimus. The primary effect of these agents is to broadly block T cell cytokine gene transcription by inhibiting activation of NFAT transcription factors. However, cytokines both promote and terminate immune responses. This is well demonstrated by the findings that calcineurin inhibitors abrogate tolerance induction by costimulatory blockade, in association with inhibition of T cell apoptosis (Li et al., 1999).
It is likely that tolerance-opposing effects of calcineurin inhibitors are multifactorial. For example, by blocking the production of IL-2, these agents may prevent T cell priming for AICD which might otherwise occur during the natural evolution of an immune response or as a deliberate effect by other pharmacological or biological agents. The inability to tolerize IL-2-deficient mice by costimulatory blockade is consistent with this concept. In addition, it is likely that agents which target costimulatory molecules, such as CTLA4Ig or anti-CD154, are effective in inducing peripheral tolerance by targeting activated T cells. Inhibition of costimulatory pathways decreases secretion of prosurvival cytokines rendering activated T cells susceptible to apoptosis due to cytokine withdrawal. By preventing the early stages of T cell activation, calcineurin inhibitors may prevent the dependency on cytokines that T cells develop upon cell cycle entry. As a result, costimulatory blockade does not result in T cell apoptosis in the context of calcineurin inhibition (Li et al., 1999).
An additional major problem with the use of nonspecific immunosuppressive agents is the diverging effects of such regimens on different T cell subsets such as Tregs. As IL-2 is critical for Treg development and survival in the periphery, any therapy which inhibits IL-2 production is likely to have a detrimental effect on the maintenance of a regulatory T cell pool. Thus, while calcineurin inhibition may be effective in preventing the progression of an alloreactive T cell response during the acute phase after allograft transplantation, it may also prevent the equally important maintenance of allograft tolerance through its negative effect on Treg populations (Figure 4).
A more rewarding approach may be to target cytokine signaling distal to receptor engagement, when selected effects of intracellular cytokine signaling may be specifically blocked while allowing those pathways which are important to tolerance induction and maintenance to remain intact. Rapamycin is one such agent which blocks IL-2-mediated proliferation without preventing the induction of susceptibility to AICD of activated effector T cells. As a result, when administered in combination with strategies to induce costimulatory blockade, rapamycin is permissive (if not additive) for promoting cell death and inducing peripheral tolerance (Li et al., 1999). The effects of selective inhibition of IL-2-dependent signaling with rapamycin on Treg cells are largely undefined. However, it is possible that they may differ from the effects observed in activated effector T cells given that CD4+CD25+ T cells have somewhat distinct responses to IL-2R stimulation.
The redundancy of cytokine functions, where any of several cytokines can promote T cell growth and survival, as well as the pleiotropic nature of specific cytokines, which can have both time- and context-dependent enhancing and terminating effects on immune responses, has made it very difficult to target specific individual cytokines or their receptors to promote transplantation tolerance (Table 1). Our own belief is that the key to generating and sustaining tolerance is reducing the overall number of alloreactive T cells while allowing for the development of immunoregulatory mechanisms, some of which are likely to involve Th2 cytokines. In this context, the emergence of Th2 responses may be viewed as permissive for tolerance. A recent report demonstrating tolerance induction in a very stringent model through the combined blockade of IL-15 signals (critical for T cell proliferation in vivo) and the use of an agonistic IL-2/Fc fusion protein to promote the selective lysis of nonregulatory CD25+ T cells is just one example of how this may be achieved (Zheng et al., 2003). Clearly, a greater understanding of the specific roles these cytokines play in the context of allograft transplantation may allow other specific targeting for therapeutic strategies in the future.
Table 1.
Roles of Major Cytokines in the Development of Transplant Tolerance or Rejection
| Cytokine | Tolerance | Rejection | Reference |
|---|---|---|---|
| Il-2 | + | + | Wells et al., 1999; Malek et al., 2002; Refaeli et al., 1998 |
| IFN-γ | + | + | Konieczny et al., 1998; Akira et al., 2001 |
| IL-10 | + | + | Moore et al., 2001; Bromberg, 1995 |
| TGFβ | + | − | Jonuleit et al., 2002 |
| IL-15 | − | + | Ferrari-Lacraz et al., 2001 |
| IL-12 | − | + | Banchereau and Steinman, 1998 |
| IL-4 | + | + | Onodera et al., 1997; Vella et al., 1998 |
The role of cytokines in transplantation tolerance and rejection is based on the reported influence on T cell-mediated responses as well as investigation of their respective roles in models of transplantion.
Acknowledgments
The authors would like to thank Scott Adler and Andrew Wells for critical reading of this manuscript. L.A.T. is supported by N.I.H. grants AI-37691 and AI-41521.
References
- Adams AB, Williams MA, Jones TR, Shirasugi N, Durham MM, Kaech SM, Wherry EJ, Onami T, Lanier JG, Kokko KE, et al. Heterologous immunity provides a potent barrier to transplantation tolerance. J Clin Invest. 2003;111:1887–1895. doi: 10.1172/JCI17477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akira S, Takeda K, Kaisho T. Toll-like receptors: critical proteins linking innate and acquired immunity. Nat Immunol. 2001;2:675–680. doi: 10.1038/90609. [DOI] [PubMed] [Google Scholar]
- Almeida AR, Legrand N, Papiernik M, Freitas AA. Homeostasis of peripheral CD4+ T cells: IL-2R alpha and IL-2 shape a population of regulatory cells that controls CD4+ T cell numbers. J Immunol. 2002;169:4850–4860. doi: 10.4049/jimmunol.169.9.4850. [DOI] [PubMed] [Google Scholar]
- Ardavin C. Origin, precursors and differentiation of mouse dendritic cells. Nat Rev Immunol. 2003;3:582–590. doi: 10.1038/nri1127. [DOI] [PubMed] [Google Scholar]
- Banchereau J, Steinman RM. Dendritic cells and the control of immunity. Nature. 1998;392:245–252. doi: 10.1038/32588. [DOI] [PubMed] [Google Scholar]
- Barbara JA, Turvey SE, Kingsley CI, Spriewald BM, Hara M, Witzke O, Morris PJ, Wood KJ. Islet allograft rejection can be mediated by CD4+, alloantigen experienced, direct pathway T cells of TH1 and TH2 cytokine phenotype. Transplantation. 2000;70:1641–1649. doi: 10.1097/00007890-200012150-00017. [DOI] [PubMed] [Google Scholar]
- Batchelor JR, Lechler RI. Why MHC incompatible grafts induce strong primary alloimmunity. Transplant Proc. 1982;14:535–537. [PubMed] [Google Scholar]
- Boise LH, Minn AJ, Noel PJ, June CH, Accavitti MA, Lindsten T, Thompson CB. CD28 costimulation can promote T cell survival by enhancing the expression of Bcl-XL. Immunity. 1995;3:87–98. doi: 10.1016/1074-7613(95)90161-2. [DOI] [PubMed] [Google Scholar]
- Bromberg JS. IL-10 immunosuppression in transplantation. Curr Opin Immunol. 1995;7:639–643. doi: 10.1016/0952-7915(95)80070-0. [DOI] [PubMed] [Google Scholar]
- Cobbold S, Waldmann H. Infectious tolerance. Curr Opin Immunol. 1998;10:518–524. doi: 10.1016/s0952-7915(98)80217-3. [DOI] [PubMed] [Google Scholar]
- Colovai AI, Liu Z, Ciubotariu R, Lederman S, Cortesini R, Suciu-Foca N. Induction of xenoreactive CD4+ T-cell anergy by suppressor CD8+CD28− T cells. Transplantation. 2000;69:1304–1310. doi: 10.1097/00007890-200004150-00016. [DOI] [PubMed] [Google Scholar]
- Dai Z, Konieczny BT, Baddoura FK, Lakkis FG. Impaired alloantigen-mediated T cell apoptosis and failure to induce long-term allograft survival in IL-2-deficient mice. J Immunol. 1998;161:1659–1663. [PubMed] [Google Scholar]
- Dai Z, Konieczny BT, Lakkis FG. The dual role of IL-2 in the generation and maintenance of CD8+ memory T cells. J Immunol. 2000;165:3031–3036. doi: 10.4049/jimmunol.165.6.3031. [DOI] [PubMed] [Google Scholar]
- DeBruyne LA, Li K, Chan SY, Qin L, Bishop DK, Bromberg JS. Lipid-mediated gene transfer of viral IL-10 prolongs vascularized cardiac allograft survival by inhibiting donor-specific cellular and humoral immune responses. Gene Ther. 1998;5:1079–1087. doi: 10.1038/sj.gt.3300694. [DOI] [PubMed] [Google Scholar]
- Dhodapkar MV, Steinman RM, Krasovsky J, Munz C, Bhardwaj N. Antigen-specific inhibition of effector T cell function in humans after injection of immature dendritic cells. J Exp Med. 2001;193:233–238. doi: 10.1084/jem.193.2.233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dieckmann D, Bruett CH, Ploettner H, Lutz MB, Schuler G. Human CD4(+)CD25(+) regulatory, contact-dependent T cells induce interleukin 10-producing, contact-independent type 1-like regulatory T cells. J Exp Med. 2002;196:247–253. doi: 10.1084/jem.20020642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunne A, O’Neill LA. The interleukin-1 receptor/Toll-like receptor superfamily: signal transduction during inflammation and host defense. Science’s STKE. 2003 doi: 10.1126/stke.2003.171.re3. http://stke.sciencemag.org/cgi/content/full/OC_sigtrans;2003/171/re3. [DOI] [PubMed]
- Ferrari-Lacraz S, Zheng XX, Kim YS, Li Y, Maslinski W, Li XC, Strom TB. An antagonist IL-15/Fc protein prevents costimulation blockade-resistant rejection. J Immunol. 2001;167:3478–3485. doi: 10.4049/jimmunol.167.6.3478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frumento G, Rotondo R, Tonetti M, Damonte G, Benatti U, Ferrara GB. Tryptophan-derived catabolites are responsible for inhibition of T and natural killer cell proliferation induced by indoleamine 2,3-dioxygenase. J Exp Med. 2002;196:459–468. doi: 10.1084/jem.20020121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Game DS, Lechler RI. Pathways of allorecognition: implications for transplantation tolerance. Transpl Immunol. 2002;10:101–108. doi: 10.1016/s0966-3274(02)00055-2. [DOI] [PubMed] [Google Scholar]
- Goldstein DR, Tesar BM, Akira S, Lakkis FG. Critical role of the Toll-like receptor signal adaptor protein MyD88 in acute allograft rejection. J Clin Invest. 2003;111:1571–1578. doi: 10.1172/JCI17573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hara M, Kingsley CI, Niimi M, Read S, Turvey SE, Bushell AR, Morris PJ, Powrie F, Wood KJ. IL-10 is required for regulatory T cells to mediate tolerance to alloantigens in vivo. J Immunol. 2001;166:3789–3796. doi: 10.4049/jimmunol.166.6.3789. [DOI] [PubMed] [Google Scholar]
- Harris NL, Ronchese F. The role of B7 costimulation in T-cell immunity. Immunol Cell Biol. 1999;77:304–311. doi: 10.1046/j.1440-1711.1999.00835.x. [DOI] [PubMed] [Google Scholar]
- Heath WR, Carbone FR. Cross-presentation in viral immunity and self-tolerance. Nat Rev Immunol. 2001;1:126–134. doi: 10.1038/35100512. [DOI] [PubMed] [Google Scholar]
- Hornick PI, Mason PD, Baker RJ, Hernandez-Fuentes M, Frasca L, Lombardi G, Taylor K, Weng L, Rose ML, Yacoub MH, et al. Significant frequencies of T cells with indirect anti-donor specificity in heart graft recipients with chronic rejection. Circulation. 2000;101:2405–2410. doi: 10.1161/01.cir.101.20.2405. [DOI] [PubMed] [Google Scholar]
- Jonuleit H, Schmitt E, Schuler G, Knop J, Enk AH. Induction of interleukin 10-producing, nonproliferating CD4(+) T cells with regulatory properties by repetitive stimulation with allogeneic immature human dendritic cells. J Exp Med. 2000;192:1213–1222. doi: 10.1084/jem.192.9.1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonuleit H, Schmitt E, Kakirman H, Stassen M, Knop J, Enk AH. Infectious tolerance: human CD25(+) regulatory T cells convey suppressor activity to conventional CD4(+) T helper cells. J Exp Med. 2002;196:255–260. doi: 10.1084/jem.20020394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Josien R, Douillard P, Guillot C, Muschen M, Anegon I, Chetritt J, Menoret S, Vignes C, Soulillou JP, Cuturi MC. A critical role for transforming growth factor-beta in donor transfusion-induced allograft tolerance. J Clin Invest. 1998;102:1920–1926. doi: 10.1172/JCI4221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kingsley CI, Karim M, Bushell AR, Wood KJ. CD25+CD4+ regulatory T cells prevent graft rejection: CTLA-4- and IL-10-dependent immunoregulation of alloresponses. J Immunol. 2002;168:1080–1086. doi: 10.4049/jimmunol.168.3.1080. [DOI] [PubMed] [Google Scholar]
- Kishimoto K, Sandner S, Imitola J, Sho M, Li Y, Langmuir PB, Rothstein DM, Strom TB, Turka LA, Sayegh MH. Th1 cytokines, programmed cell death, and alloreactive T cell clone size in transplant tolerance. J Clin Invest. 2002;109:1471–1479. doi: 10.1172/JCI14947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi T, Walsh PT, Walsh MC, Speirs KM, Chiffoleau E, King CG, Hancock WW, Caamano JH, Hunter CA, Scott P, et al. TRAF6 is a critical factor for dendritic cell maturation and development. Immunity. 2003;19:353–363. doi: 10.1016/s1074-7613(03)00230-9. [DOI] [PubMed] [Google Scholar]
- Konieczny BT, Dai Z, Elwood ET, Saleem S, Linsley PS, Baddoura FK, Larsen CP, Pearson TC, Lakkis FG. IFN-gamma is critical for long-term allograft survival induced by blocking the CD28 and CD40 ligand T cell costimulation pathways. J Immunol. 1998;160:2059–2064. [PubMed] [Google Scholar]
- Kreisel D, Krupnick AS, Gelman AE, Engels FH, Popma SH, Krasinskas AM, Balsara KR, Szeto WY, Turka LA, Rosengard BR. Non-hematopoietic allograft cells directly activate CD8+ T cells and trigger acute rejection: an alternative mechanism of allorecognition. Nat Med. 2002;8:233–239. doi: 10.1038/nm0302-233. [DOI] [PubMed] [Google Scholar]
- Kupiec-Weglinski JW, Padberg W, Uhteg LC, Towpik E, Lord RH, Ma L, Diamantstein T, Strom TB, Tilney NL. Anti-interleukin-2 receptor (IL-2R) antibody against rejection of organ grafts. Transplant Proc. 1987;19:591–593. [PubMed] [Google Scholar]
- Lee WT, Cole-Calkins J, Street NE. Memory T cell development in the absence of specific antigen priming. J Immunol. 1996;157:5300–5307. [PubMed] [Google Scholar]
- Li XC, Roy-Chaudhury P, Hancock WW, Manfro R, Zand MS, Li Y, Zheng XX, Nickerson PW, Steiger J, Malek TR, Strom TB. IL-2 and IL-4 double knockout mice reject islet allografts: a role for novel T cell growth factors in allograft rejection. J Immunol. 1998a;161:890–896. [PubMed] [Google Scholar]
- Li XC, Zand MS, Li Y, Zheng XX, Strom TB. On histocompatibility barriers, Th1 to Th2 immune deviation, and the nature of the allograft responses. J Immunol. 1998b;161:2241–2247. [PMC free article] [PubMed] [Google Scholar]
- Li Y, Li XC, Zheng XX, Wells AD, Turka LA, Strom TB. Blocking both signal 1 and signal 2 of T-cell activation prevents apoptosis of alloreactive T cells and induction of peripheral allograft tolerance. Nat Med. 1999;5:1298–1302. doi: 10.1038/15256. [DOI] [PubMed] [Google Scholar]
- Li XC, Ima A, Li Y, Zheng XX, Malek TR, Strom TB. Blocking the common gamma-chain of cytokine receptors induces T cell apoptosis and long-term islet allograft survival. J Immunol. 2000;164:1193–1199. doi: 10.4049/jimmunol.164.3.1193. [DOI] [PubMed] [Google Scholar]
- Li XC, Demirci G, Ferrari-Lacraz S, Groves C, Coyle A, Malek TR, Strom TB. IL-15 and IL-2: a matter of life and death for T cells in vivo. Nat Med. 2001a;7:114–118. doi: 10.1038/83253. [DOI] [PubMed] [Google Scholar]
- Li XC, Strom TB, Turka LA, Wells AD. T cell death and transplantation tolerance. Immunity. 2001b;14:407–416. doi: 10.1016/s1074-7613(01)00121-2. [DOI] [PubMed] [Google Scholar]
- Lutz MB, Suri RM, Niimi M, Ogilvie AL, Kukutsch NA, Rossner S, Schuler G, Austyn JM. Immature dendritic cells generated with low doses of GM-CSF in the absence of IL-4 are maturation resistant and prolong allograft survival in vivo. Eur J Immunol. 2000;30:1813–1822. doi: 10.1002/1521-4141(200007)30:7<1813::AID-IMMU1813>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- Madsen JC, Superina RA, Wood KJ, Morris PJ. Immunological unresponsiveness induced by recipient cells transfected with donor MHC genes. Nature. 1988;332:161–164. doi: 10.1038/332161a0. [DOI] [PubMed] [Google Scholar]
- Malek TR, Yu A, Vincek V, Scibelli P, Kong L. CD4 regulatory T cells prevent lethal autoimmunity in IL-2Rβ-deficient mice. Implications for the nonredundant function of IL-2. Immunity. 2002;17:167–178. doi: 10.1016/s1074-7613(02)00367-9. [DOI] [PubMed] [Google Scholar]
- McHugh RS, Whitters MJ, Piccirillo CA, Young DA, Shevach EM, Collins M, Byrne MC. CD4+CD25+ immunoregulatory T cells: gene expression analysis reveals a functional role for the glucocorticoid-induced TNF receptor. Immunity. 2002;16:311–323. doi: 10.1016/s1074-7613(02)00280-7. [DOI] [PubMed] [Google Scholar]
- Mellor AL, Munn DH. Tryptophan catabolism and T-cell tolerance: immunosuppression by starvation? Immunol Today. 1999;20:469–473. doi: 10.1016/s0167-5699(99)01520-0. [DOI] [PubMed] [Google Scholar]
- Mitra RS, Judge TA, Nestle FO, Turka LA, Nickoloff BJ. Psoriatic skin-derived dendritic cell function is inhibited by exogenous IL-10. Differential modulation of B7-1 (CD80) and B7-2 (CD86) expression. J Immunol. 1995;154:2668–2677. [PubMed] [Google Scholar]
- Moore KW, de Waal Malefyt R, Coffman RL, O’Garra A. Interleukin-10 and the interleukin-10 receptor. Annu Rev Immunol. 2001;19:683–765. doi: 10.1146/annurev.immunol.19.1.683. [DOI] [PubMed] [Google Scholar]
- Mueller DL, Seiffert S, Fang W, Behrens TW. Differential regulation of bcl-2 and bcl-x by CD3, CD28, and the IL-2 receptor in cloned CD4+ helper T cells. A model for the long-term survival of memory cells. J Immunol. 1996;156:1764–1771. [PubMed] [Google Scholar]
- Munn DH, Sharma MD, Lee JR, Jhaver KG, Johnson TS, Keskin DB, Marshall B, Chandler P, Antonia SJ, Burgess R, et al. Potential regulatory function of human dendritic cells expressing indoleamine 2,3-dioxygenase. Science. 2002;297:1867–1870. doi: 10.1126/science.1073514. [DOI] [PubMed] [Google Scholar]
- Murakami M, Sakamoto A, Bender J, Kappler J, Marrack P. CD25+CD4+ T cells contribute to the control of memory CD8+ T cells. Proc Natl Acad Sci USA. 2002;99:8832–8837. doi: 10.1073/pnas.132254399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newell KA, He G, Guo Z, Kim O, Szot GL, Rulifson I, Zhou P, Hart J, Thistlethwaite JR, Bluestone JA. Cutting edge: blockade of the CD28/B7 costimulatory pathway inhibits intestinal allograft rejection mediated by CD4+ but not CD8+ T cells. J Immunol. 1999;163:2358–2362. [PubMed] [Google Scholar]
- Nikolic B, Sykes M. Mixed hematopoietic chimerism and transplantation tolerance. Immunol Res. 1997;16:217–228. doi: 10.1007/BF02786391. [DOI] [PubMed] [Google Scholar]
- O’Connell PJ, Li W, Wang Z, Specht SM, Logar AJ, Thomson AW. Immature and mature CD8alpha+ dendritic cells prolong the survival of vascularized heart allografts. J Immunol. 2002;168:143–154. doi: 10.4049/jimmunol.168.1.143. [DOI] [PubMed] [Google Scholar]
- Onodera K, Hancock WW, Graser E, Lehmann M, Sayegh MH, Strom TB, Volk HD, Kupiec-Weglinski JW. Type 2 helper T cell-type cytokines and the development of “infectious” tolerance in rat cardiac allograft recipients. J Immunol. 1997;158:1572–1581. [PubMed] [Google Scholar]
- Pettigrew GJ, Lovegrove E, Bradley JA, Maclean J, Bolton EM. Indirect T cell allorecognition and alloantibody-mediated rejection of MHC class I-disparate heart grafts. J Immunol. 1998;161:1292–1298. [PubMed] [Google Scholar]
- Piccirillo CA, Letterio JJ, Thornton AM, McHugh RS, Mamura M, Mizuhara H, Shevach EM. CD4(+)CD25(+) regulatory T cells can mediate suppressor function in the absence of transforming growth factor beta1 production and responsiveness. J Exp Med. 2002;196:237–246. doi: 10.1084/jem.20020590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piccotti JR, Chan SY, Goodman RE, Magram J, Eichwald EJ, Bishop DK. IL-12 antagonism induces T helper 2 responses, yet exacerbates cardiac allograft rejection. Evidence against a dominant protective role for T helper 2 cytokines in alloimmunity. J Immunol. 1996;157:1951–1957. [PubMed] [Google Scholar]
- Qin S, Cobbold SP, Pope H, Elliott J, Kioussis D, Davies J, Waldmann H. “Infectious” transplantation tolerance. Science. 1993;259:974–977. doi: 10.1126/science.8094901. [DOI] [PubMed] [Google Scholar]
- Reed MH, Shapiro ME, Strom TB, Milford EL, Carpenter CB, Weinberg DS, Reimann KA, Letvin NL, Waldmann TA, Kirkman RL. Prolongation of primate renal allograft survival by anti-Tac, an anti-human IL-2 receptor monoclonal antibody. Transplantation. 1989;47:55–59. doi: 10.1097/00007890-198901000-00013. [DOI] [PubMed] [Google Scholar]
- Refaeli Y, Van Parijs L, London CA, Tschopp J, Abbas AK. Biochemical mechanisms of IL-2-regulated Fas-mediated T cell apoptosis. Immunity. 1998;8:615–623. doi: 10.1016/s1074-7613(00)80566-x. [DOI] [PubMed] [Google Scholar]
- Roncarolo MG, Levings MK. The role of different subsets of T regulatory cells in controlling autoimmunity. Curr Opin Immunol. 2000;12:676–683. doi: 10.1016/s0952-7915(00)00162-x. [DOI] [PubMed] [Google Scholar]
- Rosenberg AS, Singer A. Cellular basis of skin allograft rejection: an in vivo model of immune-mediated tissue destruction. Annu Rev Immunol. 1992;10:333–358. doi: 10.1146/annurev.iy.10.040192.002001. [DOI] [PubMed] [Google Scholar]
- Sadlack B, Lohler J, Schorle H, Klebb G, Haber H, Sickel E, Noelle RJ, Horak I. Generalized autoimmune disease in interleukin-2-deficient mice is triggered by an uncontrolled activation and proliferation of CD4+ T cells. Eur J Immunol. 1995;25:3053–3059. doi: 10.1002/eji.1830251111. [DOI] [PubMed] [Google Scholar]
- Schluns KS, Lefrancois L. Cytokine control of memory T-cell development and survival. Nat Rev Immunol. 2003;3:269–279. doi: 10.1038/nri1052. [DOI] [PubMed] [Google Scholar]
- Smith XG, Bolton EM, Ruchatz H, Wei X, Liew FY, Bradley JA. Selective blockade of IL-15 by soluble IL-15 receptor alpha-chain enhances cardiac allograft survival. J Immunol. 2000;165:3444–3450. doi: 10.4049/jimmunol.165.6.3444. [DOI] [PubMed] [Google Scholar]
- Steiger J, Nickerson PW, Steurer W, Moscovitch-Lopatin M, Strom TB. IL-2 knockout recipient mice reject islet cell allografts. J Immunol. 1995;155:489–498. [PubMed] [Google Scholar]
- Steinman RM, Hawiger D, Nussenzweig MC. Tolero-genic dendritic cells. Annu Rev Immunol. 2003;21:685–711. doi: 10.1146/annurev.immunol.21.120601.141040. [DOI] [PubMed] [Google Scholar]
- Streilein JW, Masli S, Takeuchi M, Kezuka T. The eye’s view of antigen presentation. Hum Immunol. 2002;63:435–443. doi: 10.1016/s0198-8859(02)00393-2. [DOI] [PubMed] [Google Scholar]
- Suchin EJ, Langmuir PB, Palmer E, Sayegh MH, Wells AD, Turka LA. Quantifying the frequency of alloreactive T cells in vivo: new answers to an old question. J Immunol. 2001;166:973–981. doi: 10.4049/jimmunol.166.2.973. [DOI] [PubMed] [Google Scholar]
- Suri-Payer E, Cantor H. Differential cytokine requirements for regulation of autoimmune gastritis and colitis by CD4(+)CD25(+) T cells. J Autoimmun. 2001;16:115–123. doi: 10.1006/jaut.2000.0473. [DOI] [PubMed] [Google Scholar]
- Sykes M. Mixed chimerism and transplant tolerance. Immunity. 2001;14:417–424. doi: 10.1016/s1074-7613(01)00122-4. [DOI] [PubMed] [Google Scholar]
- Takahashi T, Kuniyasu Y, Toda M, Sakaguchi N, Itoh M, Iwata M, Shimizu J, Sakaguchi S. Immunologic self-tolerance maintained by CD25+CD4+ naturally anergic and suppressive T cells: induction of autoimmune disease by breaking their anergic/ suppressive state. Int Immunol. 1998;10:1969–1980. doi: 10.1093/intimm/10.12.1969. [DOI] [PubMed] [Google Scholar]
- Terness P, Bauer TM, Rose L, Dufter C, Watzlik A, Simon H, Opelz G. Inhibition of allogeneic T cell proliferation by indoleamine 2,3-dioxygenase-expressing dendritic cells: mediation of suppression by tryptophan metabolites. J Exp Med. 2002;196:447–457. doi: 10.1084/jem.20020052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tinubu SA, Hakimi J, Kondas JA, Bailon P, Familletti PC, Spence C, Crittenden MD, Parenteau GL, Dirbas FM, Tsudo M, et al. Humanized antibody directed to the IL-2 receptor beta-chain prolongs primate cardiac allograft survival. J Immunol. 1994;153:4330–4338. [PubMed] [Google Scholar]
- Valujskikh A, Pantenburg B, Heeger PS. Primed allo-specific T cells prevent the effects of costimulatory blockade on prolonged cardiac allograft survival in mice. Am J Transplant. 2002;2:501–509. doi: 10.1034/j.1600-6143.2002.20603.x. [DOI] [PubMed] [Google Scholar]
- Van Parijs L, Refaeli Y, Lord JD, Nelson BH, Abbas AK, Baltimore D. Uncoupling IL-2 signals that regulate T cell proliferation, survival, and Fas-mediated activation-induced cell death. Immunity. 1999;11:281–288. doi: 10.1016/s1074-7613(00)80103-x. [DOI] [PubMed] [Google Scholar]
- Vella AT, Dow S, Potter TA, Kappler J, Marrack P. Cytokine-induced survival of activated T cells in vitro and in vivo. Proc Natl Acad Sci USA. 1998;95:3810–3815. doi: 10.1073/pnas.95.7.3810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waldmann TA, Dubois S, Tagaya Y. Contrasting roles of IL-2 and IL-15 in the life and death of lymphocytes: implications for immunotherapy. Immunity. 2001;14:105–110. [PubMed] [Google Scholar]
- Walker LS, Chodos A, Eggena M, Dooms H, Abbas AK. Antigen-dependent proliferation of CD4+ CD25+ regulatory T cells in vivo. J Exp Med. 2003;198:249–258. doi: 10.1084/jem.20030315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang CK, Zuo XJ, Carpenter D, Jordan S, Nicolaidou E, Toyoda M, Czer LS, Wang H, Trento A. Prolongation of cardiac allograft survival with intracoronary viral interleukin-10 gene transfer. Transplant Proc. 1999;31:951–952. doi: 10.1016/s0041-1345(98)01851-x. [DOI] [PubMed] [Google Scholar]
- Wang H, Hosiawa KA, Min W, Yang J, Zhang X, Garcia B, Ichim TE, Zhou D, Lian D, Kelvin DJ, Zhong R. Cytokines regulate the pattern of rejection and susceptibility to cyclosporine therapy in different mouse recipient strains after cardiac allografting. J Immunol. 2003;171:3823–3836. doi: 10.4049/jimmunol.171.7.3823. [DOI] [PubMed] [Google Scholar]
- Wells AD, Li XC, Li Y, Walsh MC, Zheng XX, Wu Z, Nunez G, Tang A, Sayegh M, Hancock WW, et al. Requirement for T-cell apoptosis in the induction of peripheral transplantation tolerance. Nat Med. 1999;5:1303–1307. doi: 10.1038/15260. [DOI] [PubMed] [Google Scholar]
- Willerford DM, Chen J, Ferry JA, Davidson L, Ma A, Alt FW. Interleukin-2 receptor α chain regulates the size and content of the peripheral lymphoid compartment. Immunity. 1995;3:521–530. doi: 10.1016/1074-7613(95)90180-9. [DOI] [PubMed] [Google Scholar]
- Wolf M, Schimpl A, Hunig T. Control of T cell hyperactivation in IL-2-deficient mice by CD4(+)CD25(−) and CD4(+)CD25(+) T cells: evidence for two distinct regulatory mechanisms. Eur J Immunol. 2001;31:1637–1645. doi: 10.1002/1521-4141(200106)31:6<1637::aid-immu1637>3.0.co;2-t. [DOI] [PubMed] [Google Scholar]
- Womer KL, Sayegh MH, Auchincloss H., Jr Involvement of the direct and indirect pathways of allorecognition in tolerance induction. Philos Trans R Soc Lond B Biol Sci. 2001;356:639–647. doi: 10.1098/rstb.2001.0843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood KJ, Sakaguchi S. Regulatory T cells in transplantation tolerance. Nat Rev Immunol. 2003;3:199–210. doi: 10.1038/nri1027. [DOI] [PubMed] [Google Scholar]
- Zhai Y, Meng L, Gao F, Busuttil RW, Kupiec-Weglinski JW. Allograft rejection by primed/memory CD8+ T cells is CD154 blockade resistant: therapeutic implications for sensitized transplant recipients. J Immunol. 2002;169:4667–4673. doi: 10.4049/jimmunol.169.8.4667. [DOI] [PubMed] [Google Scholar]
- Zhang Z, Yang L, Young KJ, Zhang L. Suppression of alloimmune responses in vitro and in vivo by CD3(+)CD8(−)CD4 (−)alphabeta(+) regulatory T cells. Transplant Proc. 2001;33:84–85. doi: 10.1016/s0041-1345(00)01915-1. [DOI] [PubMed] [Google Scholar]
- Zheng XX, Steele AW, Nickerson PW, Steurer W, Steiger J, Strom TB. Administration of noncytolytic IL-10/Fc in murine models of lipopolysaccharide-induced septic shock and allogeneic islet transplantation. J Immunol. 1995;154:5590–5600. [PubMed] [Google Scholar]
- Zheng XX, Sanchez-Fueyo A, Sho M, Domenig C, Sayegh MH, Strom TB. Favorably tipping the balance between cytopathic and regulatory T cells to create transplantation tolerance. Immunity. 2003;19:503–514. doi: 10.1016/s1074-7613(03)00259-0. [DOI] [PubMed] [Google Scholar]