

Abstract
Attempts to prevent HIV infection through pre-exposure prophylaxis (PrEP) include topical application of anti-HIV drugs to the mucosal sites of infection; however, a potential role for local drug metabolizing enzymes in modulating the exposure of the mucosal tissues to these drugs has yet to be explored. Here we present the first report that enzymes belonging to the cytochrome P450 (CYP) and UDP-glucuronosyltransferase (UGT) families of drug metabolizing enzymes are expressed and active in vaginal and colorectal tissue using biopsies collected from healthy volunteers. In doing so, we discovered that dapivirine and maraviroc, a non-nucleoside reverse transcriptase inhibitor and an entry inhibitor currently in development as microbicides for HIV PrEP, are differentially metabolized in colorectal tissue and vaginal tissue. Taken together, these data should help to guide the optimization of small molecules being developed for HIV PrEP.
Keywords: HIV drug metabolism, CYP, colorectal, vaginal
1. Introduction
Pre-exposure prophylaxis (PrEP) is a promising new strategy to prevent the spread of human immunodeficiency virus (HIV) by using antiretroviral (ARV) therapy to interrupt the viral life cycle before infection is established in newly exposed individuals [1]; however, oral ARV drugs may select for drug resistant viral strains in patients who become infected while on the prophylactic regimen. Topically applied microbicides may overcome this disadvantage by preventing the establishment of infection within the mucosal tissues without resulting in significant systemic exposure [2]. A pharmacokinetics study of tenofovir, which is one of the two drugs that comprise the therapy Truvada®, currently the only antiretroviral therapy FDA approved for HIV PrEP, corroborates this by demonstrating lower systemic and higher mucosal concentrations of tenofovir and its active metabolite when dosed vaginally versus orally [3].
Dapivirine is a non-nucleoside reverse transcriptase inhibitor (NNRTI) currently undergoing phase III clinical trials for use in HIV PrEP [4]; however, due to its poor oral bioavailability and remarkable inhibitory activity against both cell-associated and cell-free virions [2], dapivirine is in development solely as a topical microbicide. Maraviroc is an entry inhibitor co-formulated into a vaginal ring with dapivirine that is currently undergoing phase I clinical trials [5]. The factors that might modulate dapivirine and maraviroc exposure, including a role for metabolism of dapivirine in mucosal tissues, are unknown. A small subset of CYP isozymes are responsible for the phase I metabolism of 70–80% of all clinically used drugs, including maraviroc and other antiretrovirals used to treat HIV [6]. The CYP superfamily of enzymes are heme-containing monooxygenases that increase the hydrophilicity of their substrates, facilitating their clearance [7]. Further, CYP-dependent metabolism plays a major role in drug-drug interactions and can result in the formation of metabolites that exhibit toxicity. Phase II drug metabolism that is mediated by the UGT family of enzymes transfers a glucuronic acid moiety onto the substrate, further facilitating their elimination.
CYP enzymes are primarily expressed in the liver, which is the major site of oral xenobiotic metabolism; however, it is unknown whether drugs applied topically to vaginal and colorectal tissues, such as topical microbicides used in HIV PrEP, are metabolized locally. Topically applied drugs are unlikely to encounter the liver, and as such local metabolism would likely be a primary mediator of the pharmacokinetics of these drugs. Further, it is unclear whether a drug would be metabolized similarly if it were administered orally versus topically. In the present study, we demonstrate for the first time that CYP isozymes are expressed and active in vagina and colon. In addition, our findings indicate that metabolism of dapivirine and maraviroc differs between colorectal and vaginal tissues, potentially resulting in differential drug exposure. Collectively, these data provide novel insight that can be leveraged in interpreting HIV PrEP clinical pharmacology data and in developing drugs that will be administered topically to vaginal and/or colorectal tissue including antiretrovirals to be used in HIV PrEP.
2. Materials and Methods
2.1 Materials
Dapivirine, dapivirine-d11, and maraviroc were obtained from Toronto Research Chemical (Toronto, Ontario, Canada). Furafylline, sulfaphenazole, (+)-N3-benzylnirvanol, quinidine, and ketoconazole were obtained from Sigma-Aldrich (St. Louis, MO) and 2-phenyl-2-(1-piperidenyl) propane (PPP) purchased from Santa Cruz Biotechnology, Inc (Santa Cruz, CA). Optima liquid chromatography/mass spectrometry grade water, acetonitrile, and formic acid were obtained from Thermo Fisher Scientific (Pittsburg, PA).
2.2 Metabolism Assays
All experiments were performed in Fisherbrand Siliconized Low-Retention Microcentrifuge Tubes (Thermo Fisher Scientific). Dapivirine (10 μM) was incubated with human liver microsomes (50 donor pool, Xenotech, LLC, Lenexa, KS), cDNA-expressed individual human CYPs (Supersomes™, BD Biosciences, San Jose, CA, CYP -1A2, -2A6, -2B6, -2C8, -2C9, -2C19, -2D6, -3A4, -3A5) or UGTs (Supersomes™, BD Biosciences, UGT1A1, -1A3, -1A4, -1A6, -1A7, -1A8, -1A9, -1A10, -2B4, -2B7, -2B15, -2B17). Final concentrations were as follows: human liver microsomes at 1 mg/mL, CYPs at 10 pmol/mL, and UGTs at 0.2 mg/mL. Human liver microsomes or CYPs were combined with dapivirine in 100 mM potassium phosphate buffer, pH 7.4, and incubated for 5 min in a 37 °C water bath. Reactions were initiated by the addition of a NADPH-regenerating system (BD Biosciences) and allowed to proceed for 30 min at 37 °C. For inhibition studies, small molecule inhibitors were prepared as 500x solutions in DMSO and preincubated with the human liver microsomes in potassium phosphate buffer with the NADPH-regenerating system for 5 min at 37 °C. Inhibitors used were: furafylline (20 μM) for CYP1A2, 2-phenyl-2-(1-piperidenyl) propane (30 μM) for CYP2B6, sulfaphenazole (20 μM) for CYP2C9, (+)-N3-benzylnirvanol (10 μM) for CYP2C19, quinidine (1 μM) for CYP2D6, and ketoconazole (1 μM) for CYP3A4/5. After the addition of dapivirine, the reactions were allowed to proceed for 30 min at 37 °C. For reactions containing UGTs, the enzymes were combined with dapivirine in UGT reaction mix (Tris buffer, pH 7.5, alamethicin, and MgCL2; BD Biosciences) and incubated for 5 min in a 37 °C water bath before initiating the reaction with 2 mM UDPGA. Reactions were allowed to proceed for 60 min at 37 °C and the total reaction volumes were 100 μL. After incubations, reactions were quenched with 100 μL acetonitrile, incubated for 10 min at 4 °C, and the proteins removed by centrifugation at 3000g for 10 min at 4 °C. The supernatant was dried at 65 °C under vacuum pressure and the resulting residue reconstituted in 100 μL methanol for uHPLC-MS/MS analysis.
2.3 Ultra high performance liquid chromatography mass spectrometry (uHPLC-MS)
A uHPLC-MS assay was developed for the quantification and identification of dapivirine metabolites, using a Dionex Ultimate 3000 uHPLC system coupled to a TSQ Vantage Triple Stage Quadrupole mass spectrometer (Thermo Fisher Scientific). Compounds were separated using a Polaris 5 C18-A column (5 μm, 100 ×2.0 mm, Agilent Technologies, Santa Clara, CA) at a flow rate of 0.4 mL/min. Solvent A was 0.1% formic acid in H2O and solvent B was 0.1% formic acid in acetonitrile. The gradient used is as follows: from 0% B to 50% B from 0.0–9.0 min, 50% B to 100% B from 9.0 min to 9.3 min, held at 100% B until 12.3 min, from 100% B to 0% B from 12.3 to 13.3 min, and held at 0% B until 16.3 min. In MS/MS mode, metabolites were detected in positive ion mode as m/z = 346.4 (monohydroxy dapivirine), 362.4 (dihydroxy dapivirine), 506.4 (glucuronidated dapivirine), and 522.4 (glucuronidated monohydroxy dapivirine). In selected reaction monitoring mode, fragment ions were detected in positive ion mode using the following transitions (Q1→Q3): dapivirine (m/z 330.4→158.4); M1 (m/z 346.4→316.0); M2 (m/z 346.4→185.0); M3 (m/z 346.4→174.0); M4 (m/z 346.4→119.0); M5 (m/z 362.4→171.0); M6 (m/z 362.4→332.0); M7 (m/z 506.4→330.4); M8 (m/z 522.4→31.06); M9 (m/z 522.4→346.4); M10 (m/z 522.4→312.0); and M11 (m/z 522.4→346.6). The assay used to detect maraviroc metabolites used the same uHPLC and mass spectrometer system as the dapivirine assay and a uHPLC BEH C8 column (1.7 μm, 2.1 ×100 mm, Waters, Milford, MA). Solvent A was 0.1% formic acid, 5% acetonitrile, and 95% H2O and solvent B was 0.1% formic acid, 95% acetonitrile, and 5% H2O. The gradient started at 0% B and increased to 10% B over 1.1 min, 14.3% B at 10.0 min, 25% B at 15.0 min, held at 25% B for 1 min, then brought down to 0% B at 16.1 min and held at 0% B for 0.9 min. Transitions for selected reaction monitoring were identical to those used in a previously reported assay [8].
2.4 Tissue Biopsy Culture
Healthy subjects were recruited for tissue donation after providing written informed consent for participation in a protocol approved by the institutional review board of Johns Hopkins Medical Institutions. Each colorectal tissue donor provided 30 biopsies (approximately 25 mg each) and each vaginal donor provided 5 biopsies (approximately 25 mg each). Donor information is presented as sex and age: colorectal donors were female 49 years old, male 24 years old, and female 28 years old (denoted as colon 1, colon 2 and colon 3, respectively) while vaginal donors were female 27 years old, female 27 years old, and female 28 years old (vagina 1, vagina 2 and vagina 3, respectively). The third colon and third vagina samples were obtained from the same individual. Upon collection, biopsies were placed into RPMI 1640 medium (Corning Life Sciences, Tewksbury, MA) supplemented with 10% heat inactivated fetal bovine serum, penicillin-streptomycin, and L-glutamine and stored on ice for transport. The medium was replaced with fresh medium containing the appropriate treatment: methanol (vehicle control, 0.1%), dapivirine (10 μM), or maraviroc (10 μM). After incubation for 6, 12, or 24 hours at 37 °C in a 5% CO2 humidified environment, culture medium was collected, dried at 65 °C under vacuum, and the residue reconstituted in 100 μL methanol for uHPLC-MS/MS analysis.
2.5 RNA Isolation, Endpoint PCR, and qRT-PCR Analysis
Immediately after culture medium was removed and tissue biopsies were immersed in TRIzol® (Life Technologies, Grand Island, NY) and RNA isolated following the manufacturer’s instructions. RNA was quantitated spectrophotometrically, and 2 μg of each used to synthesize cDNA using a Maxima First-Strand cDNA Synthesis Kit (Thermo Fisher Scientific) for use in endpoint reverse transcriptase PCR or qRT-PCR. For qRT-PCR, a standard curve was generated using glyceraldehyde-3-phosphate dehydrogenase (GAPDH) PCR products cloned into pJET1.2/blunt cloning vectors (Thermo Fisher Scientific). Primers used are presented in Table 1. Endpoint reverse transcriptase PCR was run using PCR Master Mix (2X) (Thermo Fisher Scientific), with the following thermal cycling conditions: 95 °C for 1 min, followed by 30 cycles of 95 °C for 30 s, 60 °C for 30 s, and 72 °C for 1 min, and a final extension step of 72 °C for 5 min. Products were analyzed via 1% agarose gel electrophoresis and visualized with SYBR® Safe DNA Gel Stain (Life Technologies). qRT-PCR was run using Maxima SYBR Green qPCR Master Mix (Thermo Fisher Scientific), with the following thermal cycling conditions: 95 °C for 10 min, followed by 40 cycles of 95 °C for 15 s, 60 °C for 30 s, and 72 °C for 30 s. GAPDH was used for normalization of mRNA levels.
Table 1.
Primers used for qRT-PCR for CYPs.
| Primer | Accession Number | Role | Primers (5′→3′) | Coordinates |
|---|---|---|---|---|
| GAPDH | NM_002046.3 | Forward | CTTCTTTTGCGTCGCCAGCCGA | 62–83 |
| Reverse | CACGACGTACTCAGCGCCAGC | 390–370 | ||
| CYP1A1 | NM_000499.3 | Forward | GTACCTCAGCCACCTCCAAGAT | 88–109 |
| Reverse | GCTCCTGCAACGTGCTTATC | 632–651 | ||
| CYP1A2 | NM_000761.3 | Forward | CATCCCCCACAGCACAACAAGGG | 1219–1241 |
| Reverse | CAGTTGATGGAGAAGCGCAGCC | 1593–1614 | ||
| CYP2B6 | NM_000767.4 | Forward | GTCGACCCATTCTTCCGGGGAT | 317–338 |
| Reverse | GAAGAGCTCAAACAGCTGGCCGAA | 644–667 | ||
| CYP2C19 | NM_000769.1 | Forward | TGGAGAAGGAAAAGCAAAACCAAC | 812–835 |
| Reverse | GGTTGTGCCCTTGGGAATGAGG | 1137–1158 | ||
| CYP2E1 | NM_000773.3 | Forward | ACCCAAGGCCAGCCTTTCGAC | 514–534 |
| Reverse | TCCACGAGCAGGCAGTCGGT | 829–848 | ||
| CYP3A4 | NM_017460.5 | Forward | GGAAAAGTGTGGGGCTTTTATGATGG | 307–333 |
| Reverse | GCCTGTCTCTGCTTCCCGCC | 589–608 | ||
| CYP3A5 | NM_000777.3 | Forward | TTCACCATGACCCAAAGTACTGGAC | 1304–1321 |
| Reverse | TAACTCATTCTCCACTTAGGGTTCC | 1591–1615 | ||
| PXR | NM_022002.2 | Forward | CAAGCGGAAGAAAAGTGAACG | 540–560 |
| Reverse | CTGGTCCTCGATGGGCAAGTC | 961–981 |
2.6 Protein Isolation and Immunoblotting Analysis
Biopsies were washed once in 500 μL phosphate buffered saline (PBS) and homogenized on ice in cell lysis buffer (Cell Signaling Technology, Danvers, MA) containing Halt Protease + Phosphatase Inhibitor Cocktail (Thermo Fisher Scientific) and 1 mM phenylmethanesulfonylfluoride (Sigma-Aldrich) using disposable pellet mixer pestles attached to a pestle motor (VWR International, Radnor, PA). Samples were centrifuged for 10 min at 3000 g, 4 °C and the resulting supernatant was stored at −20 °C. Protein concentrations were determined using a Pierce® BCA Protein Assay Kit (Thermo Scientific), following the manufacturer’s instructions. For immunoblots, 10 μg of each sample were loaded onto 10% or 12% Mini-PROTEAN® TGX™ Precast Gels (Bio-Rad, Hercules, CA) for separation via SDS-polyacrylamide gel electrophoresis. Proteins were transferred onto 0.2 μm pore nitrocellulose membranes (Life Technologies) and blotted with commercially available antibodies. Antibodies for CYP -2B6, -2C9, -2C19, -3A4, and -3A5 were obtained from BD Biosciences, anti-CYP2D6 was obtained from Xenotech. Anti-β-actin was used for normalization and was obtained from Cell Signaling. Proteins were visualized using SuperSignal West Dura Chemiluminescent Substrate (Thermo Fisher Scientific) according to the manufacturer’s protocol and imaged with a Carestream 4000R system. Blots were stripped with ReBlot Plus Strong Antibody Stripping Solution (Millipore, Billerica, MA), then blotted with different antibodies following confirmation of antibody stripping.
2.7 In situ Extract Preparation
For analysis of in situ metabolite concentrations the tissue biopsies were washed with PBS 3 times prior to homogenizing in 500 μL ethyl acetate. The solution was dried at 65 °C under vacuum followed by reconstitution in 100 μL methanol for uHPLC-MS/MS analysis.
2.8 Statistical Analyses
All data presented are means ± S. E. from three independent experiments. Two-tailed unpaired t tests were performed to compare datasets, and p ≤ 0.05 was considered significant. Significance was denoted as follows: *, p ≤ 0.05; **, p ≤ 0.01; ***, p ≤ 0.001.
3. Results
3.1 Cytochrome P450 Expression in Mucosal Tissues that are Sites of HIV Infection
The ability of the colorectal and vaginal mucosa to biotransform xenobiotics has not been clearly defined; thus the expression of CYP isozymes found to metabolize HIV antiretroviral drugs was explored in these tissues. Vaginal and colorectal mRNA expression of CYPs that play a prominent role in drug metabolism was examined through quantitative reverse transcriptase PCR (qRT-PCR). Expression of CYP1A1, -1A2, -2B6, -2C19, -2E1, -3A4, and -3A5 mRNA was detected in all samples (Figure 1A). Comparing the expression levels of these mRNAs in colorectal tissue to those of the vaginal tissue samples revealed that CYP3A5 mRNA levels were4- (p-value = 0.04) fold higher in colorectal tissues. Expression was also probed on the protein level via immunoblotting using cell lysates isolated from the vaginal and colorectal biopsies (Figure 1B). Prior to screening, we verified that all of the antibodies were able to detect the cDNA expressed CYP isozymes that they were designed to target (data not shown). While CYP2B6, -2C19, -3A4, and -3A5 were readily detected in both tissue types, CYP1A2, -2A6, -2C9, and -2D6 were not. These results were consistent when comparing samples “colon 3 (C3)” and “vagina 3 (V3)” that were collected from the same individual. Additionally, protein expression of CYP2B6, -2C19, and -3A4 was markedly higher in vaginal tissue than in colorectal.
Fig. 1.
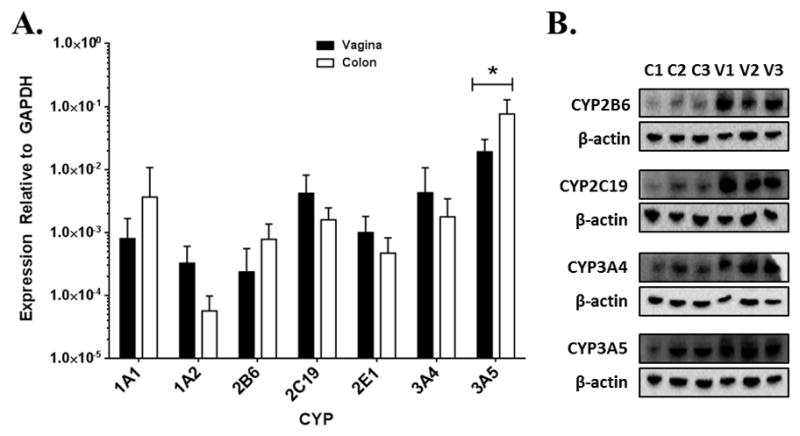
Expression of CYP mRNA and protein in colorectal and vaginal tissues. A. Quantification of mRNA expression levels of CYPs (n=6) in colorectal and vaginal tissue. B. Immunoblots of CYP isozymes in protein lysates isolated from vaginal and colorectal tissue biopsies. C=colon donor, V=vagina donor. Colon 3 and vagina 3 are from a single individual. * = p ≤ 0.05.
3.2 Maraviroc Metabolism by Colorectal and Vaginal Tissues
In order to probe the CYP activity in the mucosal tissues, colorectal and vaginal biopsies were obtained from healthy human donors for maraviroc treatment, followed by metabolite detection using uHPLC-MS/MS. We have previously demonstrated that maraviroc is a substrate of CYP3A4/5 [8], identifying 6 monooxygenated metabolites (M1–M6), 4 dioxygenated metabolites (M7–M10), and 2 glucuronidated metabolites (M11, M12) in human liver microsomes, plasma, and urine. Maraviroc is also being tested for use as a topical microbicide for HIV PrEP. After 24 hours of maraviroc treatment, colon tissue from all three donors produced a monooxygenated metabolite that was detectable in both the culture medium and in situ (Figure 2A and 2B). In contrast, of the two vaginal tissue donors that were treated with maraviroc, one produced this same monooxygenated metabolite (as measured in both culture medium and in situ) while the other did not (Figure 2C–2F).
Fig. 2.

Representative chromatograms depicting metabolism of maraviroc by colon and vagina tissue biopsies. Biopsies were incubated with maraviroc (10 μM) for 24 hours at 37 °C. After incubation, the medium was collected and biopsies were homogenized in ethyl acetate. Medium and homogenates were analyzed via uHPLC-MS/MS in selected reaction monitoring mode. A. Colon 3 biopsy medium. B. Colon 3 biopsy in situ. C. Vagina 2 biopsy medium. D. Vagina 2 biopsy in situ. E. Vagina 3 biopsy medium. F. Vagina 3 biopsy in situ.
3.3 Identification of Dapivirine Metabolites
Since dapivirine is under development as a topical microbicide for HIV PrEP we sought to determine whether dapivirine is also metabolized in the mucosal tissues to which it would be applied; however, since the metabolism of dapivirine has yet to be reported, we began our studies by using human liver microsomes to identify potential metabolites of dapivirine and to develop our uHPLC-MS methods for qualitatively detecting dapivirine and dapivirine products. Dapivirine (m/z = 330.4) was incubated with human liver microsomes and the phase I- and phase II-dependent metabolites of dapivirine were analyzed using uHPLC-MS/MS in product ion mode. In this manner, four monooxygenated (m/z = 346.4), two dioxygenated (m/z = 362.4), and five glucuronidated (m/z = 506.4 and m/z = 522.4) metabolites were identified. The monooxygenated metabolites were designated M1–M4, dioxygenated products M5–M6 and glucuronides M7–M11. No additional analytes corresponding to metabolites were detectable above background. Figure 3 shows the chromatograms for these metabolites that were obtained using uHPLC-MS/MS performed in selected reaction monitoring mode. Transitions for each metabolite were identified from the corresponding MS/MS fragmentation spectra and are detailed under Materials and Methods. Since NADPH is required for CYP activity and UDPGA (uridine diphosphate glucuronic acid) is required for UGT activity, it was possible to confirm which biotransformations were catalyzed by each enzyme class in the human liver microsomal system. When the reaction contained only NADPH, M1 was the most abundant metabolite and only products M1–M6 were detected. Reactions containing dapivirine in the presence of only UDPGA produced metabolite M7 as the sole detectable metabolite while formation of M8–M11 required incubation with both NADPH and UDPGA. In reactions that were performed in the absence of either NADPH or UDPGA no oxygenated or glucuronidated products, respectively, were detected. These data demonstrate that metabolites M1–M6 are CYP-dependent, M7 is UGT-dependent, and that the formations of M8–M11 are catalyzed by both CYPs and UGTs acting in concert. Of the glucuronides, M7 and M11 are the most abundant metabolites.
Fig. 3.

Extracted ion chromatograms showing the transitions used to monitor dapivirine metabolite formation. Dapivirine (10 μM) was incubated with human liver microsomes (1 mg/mL) for 30 min at 37 °C in the presence of a NADPH-regenerating system. Fragmentation spectra were used to derive the following transitions (Q1→Q3): M1 (m/z 346.4→316.0); M2 (m/z 346.4→185.0); M3 (m/z 346.4→174.0); M4 (m/z 346.4→119.0); M5 (m/z 362.4→171.0); M6 (m/z 362.4→332.0); M7 (m/z 506.4→330.4); M8 (m/z 522.4→316.0); M9 (m/z 522.4→346.4); M10 (m/z 522.4→312.0); and M11 (m/z 522.4→346.6).
In order to elucidate the chemical structures of each metabolite, the MS/MS spectra were analyzed. Potential structures and proposed origins of fragment ions for the mono- and di-oxygenated metabolites are presented in Figure 4. The fragments 316.2, 173.1, and 144.2 m/z found in the spectrum of metabolite M1 are proposed to originate from the loss of C2H6, C9H7N3O, and C12H14N2O, respectively. Fragments of 299.2, 185.1, and 156.1 m/z in MS/MS spectrum collected for product M2 are due to the loss of C2H7O, C10H11NO, and C10H12N3O, respectively. For M3, we propose that fragments 186.2, 174.1, 161.1, and 135.1 m/z derive from the loss of C10H12N2, C12H14N, C12H13N2, and C11H7N4O, respectively. The spectrum of M4 has fragments 146.0, 135.8, and 119.2 m/z, corresponding to losses of C10H8N4O, C12H10N4, and C14H15N2O, respectively. Metabolite M5 has ions of 285.0, 171.1, and 156.0 m/z, proposed to result from losses of C3H9O2, C8H7N4O2, and C9H10N4O2, respectively. Lastly, the fragmentation spectrum of M6 shows ions of 332.1, 328.2, 156.1, 144.0, and 131.8 m/z, which are proposed to originate from the loss of H2O2, C2H6, C9H10N4O2, C11H12N3O2, and C12H14N4O. Quality spectra were difficult to obtain for M7–M11, as for each of these products the glucuronic acid dissociated from the parent ion in the mass spectrometer; however, the remainder of the molecule did not fragment to detectable ions. Since M7 is only dependent on UDPGA, it must be the result of the direct glucuronidation of dapivirine. It is known that O- and N-glucuronidation are the most common glucuronidation reactions [9], thus the glucuronic acid moiety of M7 is most likely located on one of the secondary amines or a nitrogen atom within the pyrimidine ring. The other four glucuronides result from glucuronidation of monooxygenated metabolites M1–M4, although it remains to be determined whether the glucuronide conjugation to these metabolites occurs on the oxygen inserted by the CYP or on a nitrogen atom already present in dapivirine.
Fig. 4.

Fragmentation analysis of MS/MS spectra for M1–M6. Human liver microsomes (1 mg/mL) were incubated with 10 μM dapivirine for 30 min at 37 °C in the presence of a NADPH-regenerating system. Metabolites were detected and fragmented using uHPLC-MS/MS in product ion mode. From left to right, then top to bottom are M1, M2, M3, M4, M5, and M6.
In order to further probe which sites of dapivirine were oxygenated by CYP enzymes metabolism assays were carried out using deuterated dapivirine, with 11 deuteriums replacing the 11 hydrogens on the mesitylene ring. Due to the isotope effects, oxygen insertions occurring at the deuterated locations proceed at a slower rate, resulting in decreased abundance of the oxygenated metabolites as compared to parallel assays performed using the non-isotopically labeled dapivirine. As can be seen in Figure 5, the relative abundances of M1 (p-value = 0.003), M2 (p-value = 0.014), and M5 (p-value = 0.018) were decreased when the deuteriums are present. Taken together, the fragmentation analyses and dapivirine-d11 studies indicate that M1 and M2 each result from monooxygenation of one of the three methyl groups of the mesitylene ring while M3 appears to be oxygenated on one of the four carbons of the benzene ring, M4 is oxygenated at the nitrogen connecting the mesitylene to the pyrimidine, M5 is oxygenated on two of the mesitylene carbons, and M6 is oxygenated on one mesitylene carbon and a secondary amine.
Fig. 5.
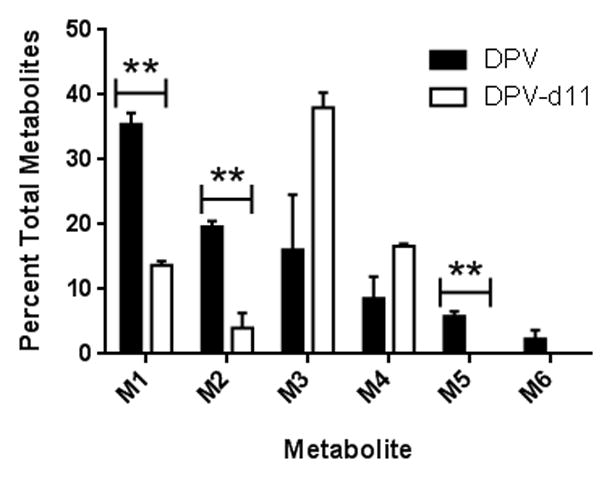
Relative contributions to total metabolite profile when dapivirine or dapivirine-d11 are incubated with human liver microsomes (1 mg/mL) for 60 min at 37 °C in the presence of an NADPH-regenerating system and UDPGA. All metabolites were detected in product ion mode. The experiment was carried out in triplicate. ** = p ≤ 0.01.
3.4 Enzymes Responsible for Dapivirine Metabolism
In order to determine which CYP isozymes were responsible for producing the observed dapivirine metabolites, metabolism assays were carried out using individual cDNA-expressed CYP1A2, -2A6, -2B6, -2C8, -2C9, -2C19, -2D6, -3A4, and -3A5 (Figure 6A). M1 was primarily produced by CYP1A2, -2B6, -2D6, -3A4, and -3A5 and to a lesser extent by CYP2C8 and -2C19. M2 had a similar profile, although CYP2A6 and -2D6 were minor contributors to the formation of this metabolite. M3 was primarily produced by CYP3A4 and -3A5, with minor production by CYP1A2, -2C8, -2C19, and -2D6. Formation of M4 was primarily catalyzed by CYP1A2 with all of other CYP isozymes tested being minor contributors. M5 was produced solely by CYP3A5 and M6 was most abundant in the incubations containing CYP3A4 with CYP2D6 and -3A5 as lesser contributors.
Fig. 6.

Formation of dapivirine metabolites by CYP isozymes. A. Ability of individual cDNA-expressed CYPs to produce dapivirine metabolites. CYP isozymes (10 pmol/mL) were incubated individually with dapivirine (10 μM) for 30 min at 37 °C in the presence of an NADPH-regenerating system. B. Inhibition of individual CYPs in order to examine dapivirine metabolite production when multiple CYP isozymes are present. Human liver microsomes (1 mg/mL) were preincubated with small molecule CYP inhibitors in the presence of a NADPH-regenerating system for 5 min at 37 °C prior to the addition of dapivirine (10 μM), then incubated for 30 min at 37°C. Dapivirine metabolites were detected via uHPLC-MS/MS in selected reaction monitoring mode. The experiments were performed in triplicate. * = p ≤ 0.05; ** = p ≤ 0.01; *** = p ≤ 0.001.
The relative contributions of each CYP to overall formation of these metabolites in vitro was explored by performing human liver microsome metabolism assays in the presence and absence of small molecule inhibitors of the individual CYP isozymes (Figure 6B). There were no statistically significant differences in the formation of any of the metabolites when furafylline, which inhibits CYP1A2, nor sulfaphenazole, which inhibits CYP2C9, nor when quinidine, which inhibits CYP2D6, were present. When a CYP2B6 inhibitor, 2-phenyl-2-(1-piperidenyl), was present, the production of M2, M3, and M4 was decreased. An inhibitor for CYP2C19, benzylnirvanol, resulted in decreased production of M3 and M6. The greatest decreases in M2–M6 production were observed when ketoconazole, an inhibitor of CYP3A4/5, was present. In addition, M1 production was also decreased in the incubations containing ketoconazole.
Relative contributions of UGT isozymes to the formation of glucuronides M7–M11 were also probed (Figure 7). Dapivirine was incubated with human liver microsomes in the presence of a NADPH-regenerating system, so that all CYP-mediated metabolites were produced. The reaction product was then incubated with individual cDNA-expressed UGT1A1, -1A3, -1A4, -1A6, -1A7, -1A8, -1A9, -1A10, -2B4, -2B7, -2B15, and -2B17. M7 was primarily produced by UGT1A4 and -2B7, with UGT1A1 as a minor producer. UGT1A1 was the major contributor to M8 formation and UGT1A3, -1A4, -2B7, and -2B15 were minor contributors. M9 was most abundant in the incubations containing UGT1A7, -1A9, and -2B7 while UGT1A6, -1A8, -1A10, and -2B4 were minor producers. M10 formation was primarily catalyzed by UGT2B7 and to a lesser extent by UGT1A3 and -1A4. Lastly, UGT1A1 was the major contributor to M11 production and UGT1A3, -1A7, -1A8, -1A9, -2B4, and -2B7 were minor contributors.
Fig. 7.

Ability of individual UGTs to produce dapivirine metabolites. For the UGT incubations, dapivirine (10 μM) was incubated with human liver microsomes (2 mg/mL) for 30 min at 37 °C in the presence of NADPH-regenerating system, the reactions were quenched and dried under vacuum followed by resuspension of the residue in methanol for use as a substrate for UGTs. UGT (0.2 mg/mL) incubations were for 60 min at 37 °C in the presence of UDPGA. Dapivirine metabolites were detected using uHPLC-MS/MS in selected reaction monitoring mode. Experiments were carried out in triplicate.
3.5 Dapivirine Metabolism by Vaginal and Colorectal Tissues
Following the identification of the metabolites that result from dapivirine metabolism and the demonstration that mucosal tissues produce maraviroc metabolites, dapivirine was similarly used as a probe for enzymatic activity in mucosal tissues (Figure 8). The biopsies were submerged in culture medium and following 24 hours of dapivirine treatment, monooxygenated metabolites M1 and M4 as well as glucuronides M7 and M11 were consistently detected in medium that was incubated with colorectal biopsies. However, only monooxygenated metabolites M1, M2, and M4 could be detected in the medium used to treat vaginal biopsies. Tissue biopsies were also homogenized in ethyl acetate in order to detect metabolites in situ. M1, M2, M3, M4, M7, and M11 were all detected within colon tissues while only M1, M2, M3, and M4 were present in vagina tissue. These differences were consistent in the matched colon and vagina biopsy samples from the same donor. In addition, no metabolites were identified following incubation with colorectal or vaginal tissue that were not present in the human liver microsome or cDNA-expressed enzyme systems. Taken together, these data are a demonstration of CYP activity in both colon and vagina tissues, whereas UGTs that catalyze the formation of dapivirine and dapivirine metabolite glucuronic acid conjugates may be active in colon but not vagina.
Fig. 8.

Representative chromatograms depicting metabolism of dapivirine by colon and vagina tissue biopsies. Biopsies were incubated with dapivirine (10 μM) for 24 hours at 37 °C. After incubation, the medium was collected and biopsies were homogenized in ethyl acetate. Medium and homogenates were analyzed via uHPLC-MS/MS in selected reaction monitoring mode. A. Colon 3 biopsy medium. B. Colon 3 biopsy in situ. C. Vagina 3 biopsy medium. D. Vagina 3 biopsy in situ.
Since drug-induced changes in CYP expression are a major mechanism of drug-drug interactions, dapivirine treatment dependent CYP mRNA changes were explored in colorectal tissue (Figure 9). Such analysis could not be performed in vaginal tissue due to limited tissue availability as a result of the marked difference in the maximum amount of tissue that could reasonably be collected from the colon versus the vagina. Colon donor 1 (age 49, female) demonstrated a 3-fold decrease of CYP2B6 and a 4-fold decrease in CYP2E1 mRNA levels following 24 hours of dapivirine treatment and no changes in CYP1A1, -1A2, -2C19, -3A4, nor -3A5 mRNA, as compared to the vehicle-treated control tissue that was cultured in parallel with the drug-treated samples. The second colon donor (age 24, male) exhibited a 4-fold, 5-fold, 5-fold and 7-fold increase in CYP1A2, CYP2C19, CYP2E1 and CYP3A4 mRNA levels, respectively, after 24 hours, and no changes in mRNA levels of CYP1A1, -2B6, nor -3A5. The third donor (age 28, female) demonstrated a 2.5-fold increase in CYP3A4 mRNA levels with no other changes. It was previously reported that etravirine modulates CYP3A4 expression in a pregnane x receptor (PXR)-dependent manner [10]. With this in mind, PXR mRNA expression in the colon tissues was measured and showed positive correlation with the observed changes in CYP3A4 mRNA, indicating that there may be a relationship between PXR levels and dapivirine-treatment dependent CYP3A4 mRNA expression changes in colon tissue (Figure 9D).
Fig. 9.

Dapivirine-treatment mediated changes in colon CYP mRNA levels and the relationship to basal PXR expression. Colon biopsies (A–C) were incubated with dapivirine (10 μM) for 24 hours at 37 °C. After incubation, RNA was isolated and used in qRT-PCR to quantify CYP and PXR mRNA levels (D).
4. Discussion
Through an examination of dapivirine and maraviroc metabolism in colon and vagina tissues, the present study demonstrates, for the first time, CYP activity in both colon and vagina and UGT activity in colon. While dapivirine metabolism was consistent across donors, and maraviroc was metabolized in all colon donors, maraviroc was only metabolized in one of the two vagina biopsies treated. This hints at inter-individual differences in antiretroviral drug metabolism and disposition which will have to be explored further. While the relatively small number of samples employed in the current study is a potential limitation, these data lay the groundwork for future studies of drug metabolism in the vaginal and colorectal mucosa. Analyses of larger study populations will be required to gain a comprehensive understanding of tissue-specific biotransformation in these compartments.
While the liver is the primary organ responsible for xenobiotic metabolism, when drugs are dosed orally, mucosal tissues may be important to the local metabolism of topical drugs used as microbicides. Previous studies have examined colorectal CYP mRNA expression [11, 12]. These studies found that multiple members of the CYP2C, CYP2E, and CYP3A families are expressed on the mRNA and protein level in human colon biopsy tissue, and that this expression may vary in different parts of the colon. There have also been studies that detect protein or mRNA expression of CYP1A, CYP2B, CYP2C, and CYP3A in cervical tissue [13–15]. However, enzymatic activity of CYPs and other metabolizing enzymes in the colon and vagina has not yet been reported. Using qRT-PCR, the present study demonstrates differences in CYP mRNA expression levels between colon and vagina tissues, finding higher CYP1A2 and -2B6 mRNA in vagina and greater expression of CYP3A5 mRNA in colon. Immunoblots also revealed that protein expression of CYP2B6, -2C19, and -3A4 is higher in the vagina than in the colon. Interestingly, in colon samples, dapivirine stimulated increases in CYP3A4 mRNA levels that appeared to be positively correlated with basal PXR mRNA expression. PXR is a nuclear receptor that contributes to the transcriptional regulation of CYP3A4 [6] and we have previously demonstrated that etravirine, which is structurally similar to dapivirine, upregulates CYP3A4 mRNA expression in a PXR-dependent manner [10]. As such, PXR may play a role in mediating the dapivirine-dependent increases in CYP3A4 mRNA levels in colorectal tissue that were observed here.
The proposed scheme for CYP-mediated metabolism of dapivirine is presented in Figure 10. Both colon and vagina tissues were able to metabolize dapivirine, producing metabolites that were determined to be CYP-mediated using human liver microsomes. This is a definitive demonstration of CYP activity in colorectal and vaginal tissues. Metabolites M1–M4 were produced by colon and vagina tissue biopsies, and the CYP isozymes that we determined were responsible for the formation of these dapivirine metabolites were all found to be expressed in these same biopsies using immunoblotting. Dapivirine oxygenations occurred on the mesitylene and phenyl rings but not the pyridine ring. This may be due to constraints on substrate positioning within the active site of the CYP isozymes, which can be further examined through the use of molecular docking. Incubations of dapivirine with individual cDNA-expressed CYPs or human liver microsomes with small molecule inhibitors of individual CYPs established which CYP isozymes produce the observed metabolites of dapivirine. There is strong evidence for major contributions from CYP3A4 and -3A5, since human liver microsomes pre-incubated with the CYP3A inhibitor ketoconazole exhibited significantly lower production of all oxygenated metabolites with the exception of M1. Production of M1 was shown to be catalyzed by cDNA expressed CYP1A2, -2B6, -2D6, -3A4, and -3A5. Thus, even in the presence of chemical inhibition of CYP3A4/5 activity in human liver microsomes this metabolite may be formed by other microsomal CYP isozymes. It was also noted that CYP1A2 appeared to be able to produce metabolite M4 in our cDNA-expressed CYP assays; however, incubation of human liver microsomes with the CYP1A2 inhibitor furafylline did not result in a significant decrease in M4 levels. These data indicate that, while CYP1A2 may have the ability to form M4, this enzyme may not be a major contributor to the formation of this metabolite in a system where all hepatic microsomal CYP isozymes are present. Interestingly, while CYP-dependent metabolites of dapivirine were detected in the culture medium collected following incubation with both vaginal and colorectal tissue as well as in situ in both tissues the UGT-dependent metabolites of this compound were only produced by colorectal tissue. This difference in metabolism may lead to differential rates of clearance between the two tissues.
Fig. 10.
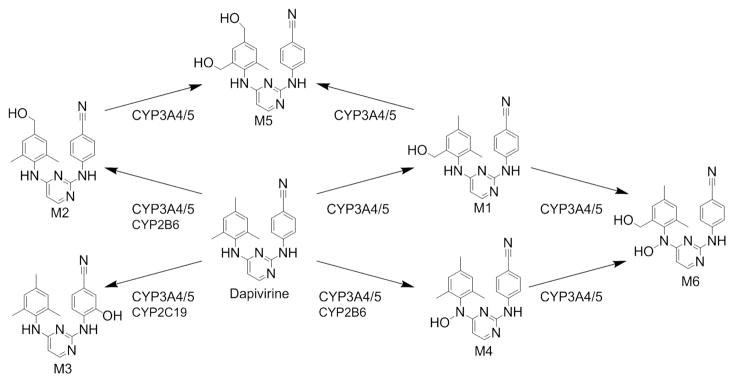
Proposed CYP-mediated metabolism of dapivirine.
Further, our data indicate that metabolism of dapivirine and maraviroc by vaginal and colorectal tissue may vary from hepatic metabolism of these drugs. The ideal comparison to examine differences in hepatic and mucosal metabolism would be to compare human liver microsomes to microsomes isolated from the colorectal and vaginal mucosa. However, due to constraints on the amount of tissue that can be collected from living donors, neither colorectal nor vaginal microsomes were able to be isolated in large enough quantities to facilitate metabolism assays. Additionally, it is not feasible to collect liver tissue from living donors to be used as a direct comparison to the mucosal biopsies we have studied. A potential difference in metabolism of a drug dosed orally versus topically is particularly evident in the case of maraviroc since in the present study we observed a marked difference in metabolite formation by colorectal and vaginal tissues as compared to the metabolite profiles that we have previously described in plasma and urine of human subjects following oral administration of maraviroc [8]. As such, both tissue and hepatic biotransformation may need to be examined in working to gain a complete understanding of the clearance of antiretrovirals and other drugs being developed for both oral and topical administration. Since dapivirine is being developed for topical use only, metabolism by the tissue upon which dapivirine would be administered is likely to be the most relevant in understanding the biotransformation of this compound.
In order to facilitate the success of HIV PrEP while concurrently avoiding selection for resistant strains of virus it is crucial that the factors that regulate the exposure to individual antiretrovirals be understood. While dapivirine is highly potent with an IC50 in the nanomolar range for both cell-free and cell-associated virus [16, 17], it has been shown that suboptimal concentrations of dapivirine can still lead to resistance [18]. Patient non-adherence is the most widely cited factor for lower than intended concentrations of drugs, but drug metabolism must also be taken into account. If the metabolites are not pharmacologically active against HIV, individuals with high metabolic activity may experience decreased efficacy and select for resistant strains of virus. For instance, CYP3A5 which we have demonstrated here to be a major contributor to dapivirine metabolism, is highly polymorphic [19] and as such CYP3A5 genotype could potentially influence the vaginal and colorectal tissue exposure to dapivirine. Biotransformation of drugs may also produce toxic metabolites, as is the case with the 8-hydroxy metabolite of efavirenz, another NNRTI [20]. Toxic metabolites formed locally by the vaginal and colorectal tissues have the potential to stimulate inflammation in the mucosa, whether from mucosal cell death or pathogenic bacteria displacing the commensal species, which may initiate the recruitment of immune cells and make establishment of HIV infection more likely [21]. While studies have already shown that dapivirine is not toxic to commensal vaginal Lactobacillus [22] this testing has not been performed for the dapivirine metabolites. To this end, now that the metabolites of dapivirine have been identified, future studies should include an analysis of the toxicity and pharmacologic activity of these compounds. Such studies will help to understand how inter-individual differences in metabolizing enzyme activity may contribute to differences in toxicity and efficacy across populations. Because these dapivirine metabolites were previously unreported, reference standards were unavailable for development of a quantitative assay. The current qualitative assay serves the desired purpose of metabolite identification and preliminary characterization, and could be developed into a quantitative assay which would be required for studying the in vivo pharmacokinetics of dapivirine metabolites.
In summary, the studies described here examine the metabolism in vaginal and colorectal tissues of dapivirine and maraviroc, a NNRTI and an entry inhibitor, respectively, being developed for use as topical microbicides for HIV PrEP. CYP activity was demonstrated for the first time in vaginal and colorectal mucosal tissues and potential differences in CYP and UGT expression and activity between these two tissues were revealed. We anticipate that these data can be leveraged in order to inform the development of antiretroviral drugs as topical microbicides for HIV PrEP as well as a broader spectrum of drugs that may be developed for administration to vaginal and colorectal tissues for other indications.
Acknowledgments
Our thanks go to the Drug Development Unit and Clinical Pharmacology Analytical Laboratory at the Johns Hopkins University School of Medicine for acquiring the human tissue specimens for this study. This work was supported by the HIV Prevention Trials Network (NIH grant U01 AI068613) and NIH grant R01 GM103853 awarded to N.N.B.
Footnotes
5.2 Author Contributions
E. E. T., C. W. H., and N. N. B. designed the experiments and performed analyses, E. E.T. carried out the experiments, and E. E. T., C. W. H., and N. N. B. contributed to manuscript preparation.
5.3 Conflict of Interest
The authors hereby declare no conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Elaine E. To, Email: eto1@jhmi.edu.
Craig W. Hendrix, Email: chendrix@jhmi.edu.
Namandjé N. Bumpus, Email: nbumpus1@jhmi.edu.
References
- 1.Baeten J, Celum C. Systemic and topical drugs for the prevention of HIV infection: antiretroviral pre-exposure prophylaxis. Annu Rev Med. 2013;64:219–32. doi: 10.1146/annurev-med-050911-163701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Adams JL, Kashuba AD. Formulation, pharmacokinetics and pharmacodynamics of topical microbicides. Best Pract Res Clin Obstet Gynaecol. 2012;26:451–62. doi: 10.1016/j.bpobgyn.2012.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hendrix CW, Chen BA, Guddera V, Hoesley C, Justman J, Nakabiito C, et al. MTN-001: randomized pharmacokinetic cross-over study comparing tenofovir vaginal gel and oral tablets in vaginal tissue and other compartments. PLoS One. 2013;8:e55013. doi: 10.1371/journal.pone.0055013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lewi P, Heeres J, Arien K, Venkatraj M, Joossens J, Van der Veken P, et al. Reverse transcriptase inhibitors as microbicides. Curr HIV Res. 2012;10:27–35. doi: 10.2174/157016212799304643. [DOI] [PubMed] [Google Scholar]
- 5.Fetherston SM, Boyd P, McCoy CF, McBride MC, Edwards KL, Ampofo S, et al. A silicone elastomer vaginal ring for HIV prevention containing two microbicides with different mechanisms of action. Eur J Pharm Sci. 2012;48:406–15. doi: 10.1016/j.ejps.2012.12.002. [DOI] [PubMed] [Google Scholar]
- 6.Zanger UM, Schwab M. Cytochrome P450 enzymes in drug metabolism: Regulation of gene expression, enzyme activities, and impact of genetic variation. Pharmacol Ther. 2013;138:103–41. doi: 10.1016/j.pharmthera.2012.12.007. [DOI] [PubMed] [Google Scholar]
- 7.Guengerich FP. Common and uncommon cytochrome P450 reactions related to metabolism and chemical toxicity. Chem Res Toxicol. 2001;14:611–50. doi: 10.1021/tx0002583. [DOI] [PubMed] [Google Scholar]
- 8.Lu Y, Hendrix CW, Bumpus NN. Cytochrome P450 3A5 plays a prominent role in the oxidative metabolism of the anti-human immunodeficiency virus drug maraviroc. Drug Metab Dispos. 2012;40:2221–30. doi: 10.1124/dmd.112.048298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kaivosaari S, Finel M, Koskinen M. N-glucuronidation of drugs and other xenobiotics by human and animal UDP-glucuronosyltransferases. Xenobiotica. 2011;41:652–69. doi: 10.3109/00498254.2011.563327. [DOI] [PubMed] [Google Scholar]
- 10.Yanakakis LJ, Bumpus NN. Biotransformation of the antiretroviral drug etravirine: metabolite identification, reaction phenotyping, and characterization of autoinduction of cytochrome P450-dependent metabolism. Drug Metab Dispos. 2012;40:803–14. doi: 10.1124/dmd.111.044404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Thorn M, Finnstrom N, Lundgren S, Rane A, Loof L. Cytochromes P450 and MDR1 mRNA expression along the human gastrointestinal tract. Br J Clin Pharmacol. 2005;60:54–60. doi: 10.1111/j.1365-2125.2005.02389.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bergheim I, Bode C, Parlesak A. Distribution of cytochrome P450 2C, 2E1, 3A4, and 3A5 in human colon mucosa. BMC Clin Pharmacol. 2005;5:4. doi: 10.1186/1472-6904-5-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Patel KR, Astley S, Adams DJ, Lacey CJ, Ali SW, Wells M. Expression of cytochrome P450 enzymes in the cervix. An immunohistochemical study. Int J Gynecol Cancer. 1993;3:159–63. doi: 10.1046/j.1525-1438.1993.03030159.x. [DOI] [PubMed] [Google Scholar]
- 14.Farin FM, Bigler LG, Oda D, McDougall JK, Omiecinski CJ. Expression of cytochrome P450 and microsomal exposide hydrolase in cervical and oral epithelial cells immortalized by human papillomavirus type 16 E6/E7 genes. Carcinogenesis. 1995;16:1670. doi: 10.1093/carcin/16.7.1670. [DOI] [PubMed] [Google Scholar]
- 15.Yokose T, Doy M, Taniguchi T, Shimada T, Kakiki M, Horie T, et al. Immunohistochemical study of cytochrome P450 2C and 3A in human non-neoplastic and neoplastic tissues. Virchows Arch. 1999;434:401–11. doi: 10.1007/s004280050359. [DOI] [PubMed] [Google Scholar]
- 16.Van Herrewege Y, Michiels J, Van Roey J, Fransen K, Kestens L, Balzarini J, et al. In vitro evaluation of nonnucleoside reverse transcriptase inhibitors UC-781 and TMC120-R147681 as human immunodeficiency virus microbicides. Antimicrob Agents Chemother. 2004;48:337–9. doi: 10.1128/AAC.48.1.337-339.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fletcher P, Harman S, Azijn H, Armanasco N, Manlow P, Perumal D, et al. Inhibition of human immunodeficiency virus type 1 infection by the candidate microbicide dapivirine, a nonnucleoside reverse transcriptase inhibitor. Antimicrobial agents and chemotherapy. 2009;53:487–95. doi: 10.1128/AAC.01156-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schader SM, Oliveira M, Ibanescu RI, Moisi D, Colby-Germinario SP, Wainberg MA. In vitro resistance profile of the candidate HIV-1 microbicide drug dapivirine. Antimicrobial agents and chemotherapy. 2012;56:751–6. doi: 10.1128/AAC.05821-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Guengerich FP. Cytochrome P450s and other enzymes in drug metabolism and toxicity. AAPS J. 2006;8:E101–11. doi: 10.1208/aapsj080112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bumpus NN. Efavirenz and 8-hydroxyefavirenz induce cell death via a JNK- and BimEL-dependent mechanism in primary human hepatocytes. Toxicol Appl Pharmacol. 2011;257:227–34. doi: 10.1016/j.taap.2011.09.008. [DOI] [PubMed] [Google Scholar]
- 21.Eade CR, Cole AL, Diaz C, Rohan LC, Parniak MA, Marx P, et al. The anti-HIV microbicide candidate RC-101 inhibits pathogenic vaginal bacteria without harming endogenous flora or mucosa. Am J Reprod Immunol. 2013;69:150–8. doi: 10.1111/aji.12036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Moncla BJ, Pryke K, Rohan LC, Yang H. Testing of viscous anti-HIV microbicides using Lactobacillus. J Microbiol Methods. 2012;88:292–6. doi: 10.1016/j.mimet.2011.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]