

Abstract
CRK (c-Crk) as an adaptor protein is involved in several oncogenic signal transduction pathways, conveying oncogenic signals to its downstream effectors and thereby affecting multiple cellular processes including proliferation, differentiation, and migration. For example, we have observed that CRK expression and phosphorylation influence the invasiveness of non–small cell lung cancer (NSCLC) cells. To intervene in CRK signaling pathway, we examined whether CRK protein domains can be used as therapeutic tools to interrupt CRK signaling, thus influencing the biological behavior of NSCLC cells. For this purpose, Src Homology domains of CRK-I (i.e., SH2 and SH3N domains) were overexpressed in H157, Rh2, and A549 cells. CRK-SH3N domain expression induced epithelial morphology in H157 cells and enhanced epithelial morphology of A549 and Rh2 cells as compared to cells transfected with CRK-SH2 domain or empty vector. In addition, CRK-SH3N domain expression significantly decreased the motility and invasiveness of A549 and H157 cells. Furthermore, CRK-SH3N domain expression disrupted the interaction of CRK-II with DOCK180. In summary, these data provide evidence that the CRK-SH3N domain can be used to influence the malignant phenotype of NSCLC cells and also reduce the metastatic potential of these cells.
Keywords: cell adhesion, cell invasion, c-Crk, lung cancer, signal transduction, protein domains
Introduction
CRK (c-Crk) family of adaptor proteins (i.e., CRK-I, CRK-II, and their paralogue CRKL) are cellular homologues of v-Crk (CT10 avian sarcoma oncogene).1,2 These adaptor proteins are widely expressed and are involved in signal transduction from a variety of receptors, signaling scaffold proteins, and oncoproteins, thereby affecting several cellular functions including proliferation, differentiation, and migration. Examples of cellular stimuli that result in CRK-mediated signal transduction include prominent oncogenes (e.g., BCR-ABL, Tel-Abl, Erythropoietin receptor, EGFR, Met, Insulin receptor substrate, PDGF, and VEGFR).3,4 In addition to CRK’s role as an adaptor protein that transduces the upstream oncogenic signals, overexpression of CRK has been observed in several malignancies including non–small cell lung cancer (NSCLC) and brain and colon cancers.5-7 CRK overexpression can cause cellular transformation as it is well demonstrated in the case of v-Crk and CRK-I.1,6 Therefore, it seems that CRK participates in the malignant process through activation of its downstream effectors by either transducing the upstream oncogenic signals or directly playing the role of an oncoprotein.
With respect to NSCLC, CRK seems to be positioned at the intersection of multiple abnormally regulated signaling pathways. For example, CRK is shown to be downstream of EGFR and MET signaling pathways in NSCLC.8-10 Furthermore, as mentioned above, CRK overexpression at protein and mRNA levels are observed in NSCLC.5,7
CRK does not contain a catalytic domain yet it participates in signaling pathways by establishing protein-protein interactions through its Src Homology domains. v-Crk and its cellular homologues CRK-I and CRK-II contain Src Homology domains (SH2 and SH3; Fig. 1). CRK-I contains 1 SH2 domain and 1 SH3 domain (SH3N) whereas CRK-II contains an additional SH3 domain (SH3C). CRK-I domains SH2 and SH3N are structurally conserved protein structures of close to 100 and 60 amino acids, respectively. The SH2 domain binds to tyrosine phosphorylated proteins whereas the SH3N domain recognizes a consensus sequence X-P-p-X-P where the first and fourth residues are aliphatic amino acids and the second, fifth, and sometimes third residues are proline. Through its SH2 and SH3 domains, CRK physically bridges phosphorylated proteins and forms protein complexes to transduce their respective signals (Fig. 1A). Considering CRK participates in transduction of multiple oncogenic signaling pathways, it seems that CRK signaling is a key element of the malignant transformation; therefore, it is conceivable that interruption of CRK-mediated signals could influence the malignant phenotype. Here, we examined whether interruption of CRK signaling by overexpression of CRK SH2 or SH3 (SH3N) domains might affect the biological behavior of NSCLC cells.
Figure 1.

Schematic view of the CRK and CRK domains in signaling pathways. (A) Multiple oncogenic pathways in NSCLC and other malignancies that involve CRK are demonstrated. Additionally, a number of downstream effectors of CRK are shown. (B) Protein domains of v-crk, CRK-I, and CRK-II including Src Homology domains SH2, SH3N, and SH3C are shown. **Percentage of sequence similarity between CRK-II and CRK-L domains. (C) Schematic view of CRK SH2 and SH3 domains during effective signal transduction. Additionally, the strategy shown is of using CRK-I domain overexpression to saturate the upstream stimuli and downstream CRK effectors, thereby blocking the CRK signaling.
Results
NSCLC cells show epithelial morphology following stable expression of the CRK-SH3N domain
Adheres junctions can be destabilized through CRK-mediated signaling. This phenomenon can be the reflection of an upstream stimulus that involves CRK or as a result of CRK overexpression. For example, activation of the MET receptor tyrosine kinase through its ligand hepatocyte growth factor (HGF) recruited CRK-II, which in turn resulted in disruption of adherens junctions.11 Furthermore, overexpression of Crk-II in Madin-Darby canine kidney (MDCK) cells promoted cell spreading and resulted in loss of adherens junctions. These investigators also noticed that overexpression of Crk-II promoted loss of adherens junctions and cell spreading in human breast cancer epithelial cells in the absence of HGF. In addition to the MET pathway, overexpression of CRK-II has been shown to synergize with epidermal growth factor (EGF)-stimulated pathways to induce cell dispersal in 2-dimensional cultures.12 These results point to the key role of CRK signaling in disruption of the adherens junctions in epithelial cells.
To interrupt the CRK-mediated loss of cell adhesions, we decided to disrupt CRK signaling by overexpressing CRK protein domains (Fig. 1B). In this fashion, CRK protein domains would saturate the CRK effectors, thereby interrupting protein complex formation by wild type CRK, which in turn would result in the disruption of CRK signals. For this purpose, we stably overexpressed CRK-I Src Homology domains (SH2) and (SH3N) in NSCLC cell lines. Following selection of the transfected cells in the presence of a selecting agent (G418), the growing colonies that were transfected with the CRK-SH3N domain demonstrated a distinct epithelial morphology (Fig. 2). H157 (squamous cell carcinoma) NSCLC cells that typically have a mesenchymal phenotype established an epithelial formation following stable expression of the CRK-SH3N domain. In the case of A549 (adenocarcinoma) and Rh2 cells (squamous cell carcinoma) that routinely represent an epithelial morphology, stable expression of the CRK-SH3N domain resulted in the formation of colonies with smooth borders without scattering cells around the colony. The colonies of A549 and Rh2 cells transfected with the CRK-SH3N domain stood out compared to colonies from cells transfected with empty vector or SH2 domain (Fig. 2). Further views demonstrating multiple colonies from each cell line are demonstrated in Supplementary Figure S1A-C. The CRK SH2 and SH3N domains were flag tagged, and their expression level in each cell line is demonstrated in Figure 8. These findings reveal that disruption of CRK signaling affects the morphology and adhesiveness of multiple NSCLC cells lines and that the CRK-SH3N domain can function as an effective tool for the inhibition of CRK-mediated loss of cell adhesions.
Figure 2.
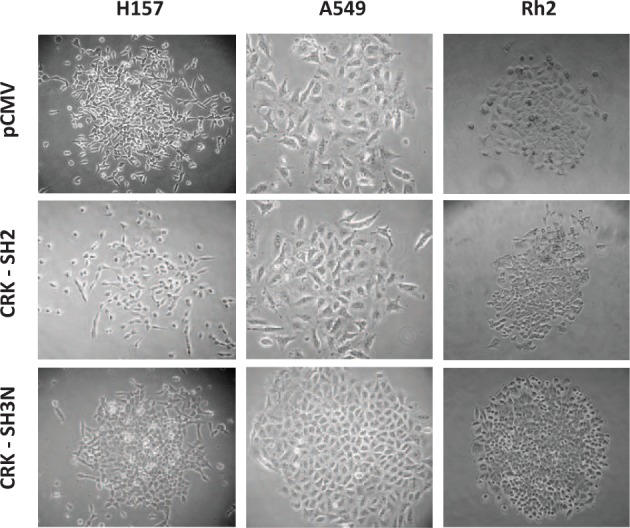
CRK SH3N domain expression induces morphological changes in NSCLC cells. CRK-I Src Homology domains (SH2 and SH3N) were stably expressed in 3 NSCLC cell lines H157, A549, and Rh2. The selected colonies in all cells lines demonstrate epithelial morphology on expression of the CRK-SH3N domain as compared to cells transfected with pCMV (empty vector) or the CRK-SH2 domain.
Figure 8.
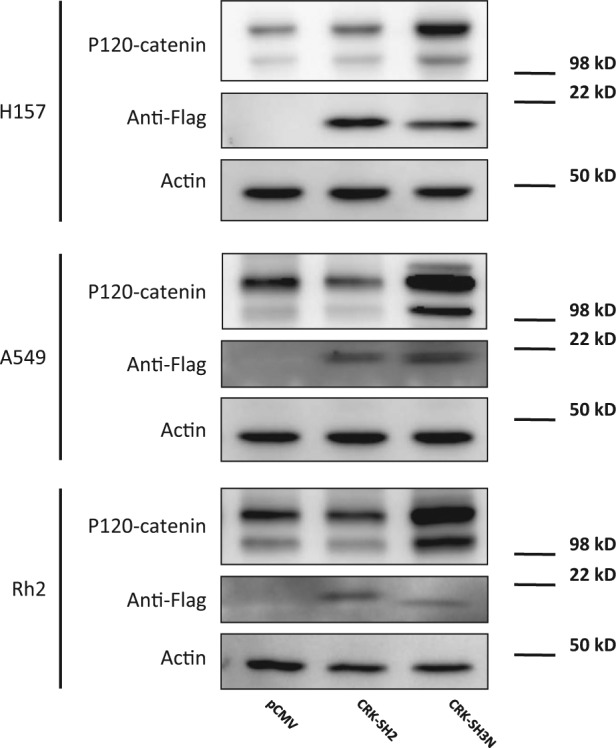
p120-catenin protein level is enhanced following CRK-SH3N domain expression. Western blots demonstrating the expression levels of CRK-SH2 and CRK-SH3N domains in H157, A549, and Rh2 NSCLC cells. Cells were stably transfected with plasmid constructs expressing empty vector, CRK-SH2 domain, or CRK-SH3N domain fused with a flag tag. Additionally, the relative p120-catenin expression level in each group is demonstrated.
Of note, CRK SH2 has been reported to induce apoptosis in other species.13 Therefore, to assess any alteration in the level of apoptosis as a result of CRK domain expression, we measured cleaved caspase-3 expression as described in the Material and Methods section. We did not observe any significant change in the percentage of cells expressing cleaved caspase-3 among the groups following transient transfection of CRK-I domains in H157 cells at 48 hours (Suppl. Fig. 2).
CRK-SH3N domain diminishes the motility of NSCLC cells
CRK phosphorylation by various stimuli (i.e., upstream kinases) affects cell motility.14-16 For example, phosphorylation of Crk on Y221 by Abl, which acts as a molecular switch to dissociate p130Cas/Crk/DOCK180 complex, resulted in blocking the motility of pancreatic cancer cells.15 To examine whether CRK protein domains can be used to interrupt CRK signaling, thereby influencing the motility of NSCLC cells, we stably expressed CRK SH2 or SH3N domains in A549, Rh2, and H157 NSCLC cell lines and subjected the pool of stably selected cells to a standard wound healing assay (Fig. 3). H157 cells expressing the CRK-SH3N domain demonstrated a significant reduction of cell motility as compared to cells transfected with empty vector or the SH2 domain. This effect was less pronounced in A549 cells at 48 hours. These findings also point to a role for the utility of CRK-SH3N domain expression in interruption of CRK-mediated signals, thus affecting the behavior of NSCLC cells.
Figure 3.

CRK-SH3N domain expression reduces the motility of NSCLC cells. (A) Standard would healing assay in H157 and A549 NSCLC cells stably expressing pCMV (empty vector), CRK-SH2 domain, or CRK-SH3N domain. (B) Quantification of the wound area among cells expressing pCMV, CRK-SH2 domain, or CRK-SH3N domain at 48 hours. Experiments were done in triplicate. Error bars represent ± standard deviation. Comparison of the wound surface area means between the groups was performed by a 2-sided Student’s t test. NS = nonsignificant (P > 0.05); ***P < 0.0001.
CRK-SH3N domain diminishes cell invasiveness of NSCLC cell lines
As shown earlier, interruption of CRK signaling by overexpression of the CRK-SH3N domain results in an epithelial morphology and reduced cell motility of NSCLC cells. To examine whether the CRK-SH3N domain can affect the invasiveness of NSCLC cells, we subjected the selected pool of A549 and H157 cells that stably express CRK SH2 or SH3N to a Matrigel cell invasion assay (Fig. 4). We observed a significant decrease in the invasiveness of A549 and H157 cells expressing the CRK-SH3N domain as compared to the cells transfected with empty vector or the CRK-SH2 domain. These findings also establish the effectiveness of CRK-SH3N domain overex0pression as a tool for influencing the biological behavior of NSCLC cells. To assess the effect of CRK protein domain expression on cell growth, the growth pattern of each cell line was examined. We did not observe any meaningful difference between the growth patterns among the groups (Suppl. Fig. 3).
Figure 4.

CRK-SH3N domain expression diminishes invasiveness of NSCLC cells. (A) Matrigel invasion assays in H157 and A549 NSCLC cells stably expressing pCMV (empty vector), CRK-SH2 domain, or CRK-SH3N domains at 24 hours following seeding of 1 × 105 cells in the upper chambers of the invasion assay. (B) Quantification of invaded cells through the Matrigel membrane following 24 hours of incubation. Experiments were done in triplicate. Five separate areas of each membrane were counted. Error bars represent ± standard deviation. Comparison of the invaded cells means between the groups was performed by a 2-sided Student’s t test. NS = nonsignificant (P > 0.05); **P < 0.001; ***P < 0.0001.
CRK-SH3N domain expression disrupts the interaction between CRK and DOCK180
As mentioned earlier, CRK participates in signal transduction pathways by physically bridging proteins and forming protein complexes. Here we attempt to disrupt CRK-containing protein complexes by overexpression of CRK protein domains, thereby disrupting the corresponding signaling pathway. CRK is known to interact with several downstream effectors including DOCK180, C3G, and SOS1. To determine which CRK downstream effector is expelled from its complex with CRK on overexpression of CRK-I domains, we performed co-immunoprecipitation experiments. Specifically, we sought to examine whether stable expression of the CRK-SH3N domain would disrupt co-immunoprecipitation of DOCK180, C3G, and SOS1 with CRK. Cell lysates from A549 cells that were stably transfected with the CRK-SH3N domain or empty vector were incubated with anti-CRK-II antibody or IgG control. To avoid the pull down of overexpressed CRK-I domains (i.e., SH2 and SH3N), an anti-CRK-II antibody was selected for this experiment. Subsequently, the presence of DOCK180, C3G, or SOS1 in the immunoprecipitate was examined by western blotting (Fig. 5). We observed that expression of the CRK-SH3N domain did not significantly alter the co-immunoprecipitation of C3G and SOS1 along with CRK-II; however, CRK-SH3N expression significantly diminished co-immunoprecipitation of CRK-II with DOCK180.
Figure 5.
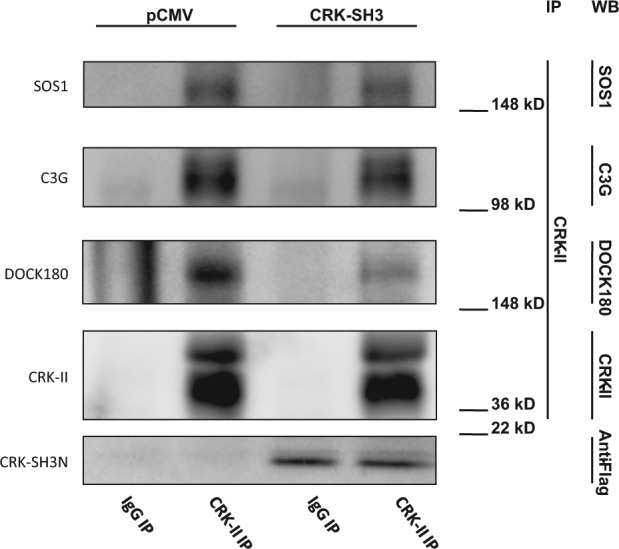
CRK-SH3N expression dissociates CRK interaction with DOCK180. Co-immunoprecipitation experiments demonstrating CRK interaction with its downstream effectors on expression of the CRK-SH3N domain in A549 cells. Lysates from cells that stably express the CRK-SH3N domain or empty vector were incubated with an anti-CRK-II antibody. Anti-CRK-II was chosen for this experiment to avoid pulling down the CRK-SH3N expressed domain. Cells expressing the CRK-SH3N domain demonstrate a significant loss of DOCK180 co-immunoprecipitation with CRK-II.
CRK-SH3N expression alters PAK1 phosphorylation on serine 144 in the Pak1 kinase inhibitory domain
Since we observed that the CRK-SH3N domain expression diminishes the motility and invasiveness of NSCLC cells, we sought to investigate the molecular mechanism behind this biological phenotype. Cell motility is a complex biological process that is regulated by several biochemical events including activation of GTPases. For example, activation of Transforming Protein RhoA (RHOA), Ras-related C3 Botulinum Toxin Substrate 1 (RAC1), and Cell Division Control Protein 42 Homolog (CDC42) GTPases are known to regulate cell motility. These GTPases mediate their pro-motility effects through interaction with their downstream effectors. RHOA interacts with several effectors including ROCK while RAC1 and CDC42 interact with PAK1 as one of their downstream effectors. The activity level of these GTPases can be measured by examining the interaction of the GTPase with its downstream effector. For example, RAC1/CDC42 activities are indirectly measured by examining the RAC1/CDC42 interaction with PAK1 as activated RAC1/CDC42 binds to and alters PAK1 dimerization.17-19 PAK1 can be phosphorylated in several residues including S21, S57, S144, S149, S199, S204, and T423, among others. PAK1 serine 144 residue is located in the PAK1 kinase inhibitory domain, and its phosphorylation is reportedly mediated by RAC1.20 Therefore, we assessed the activity level of RAC1 in our model by measuring phospho-PAK1 Ser144 in A549 cells. Interestingly, CRK-SH3N expressing A549 cells demonstrated an increased phospho-PAK1 Ser144 compared to cells expressing empty vector. Of note, increased phospho-PAK1 Ser144 is closely correlated with increased E-cadherin and p120-catenin expression in a panel of NSCLC cells (Suppl. Fig. 4). These data suggest that expression of the CRK-SH3N domain most likely influences the motile machinery of the cells by affecting RAC1 and its downstream effector PAK1.
p120-catenin promoter activity and protein expression are affected by CRK protein domain expression
CRK affects several downstream effectors including JNK1, SP1, and guanine exchange factors DOCK180 and SOS1. Our previous work demonstrated the involvement of CRK in transcriptional downregulation of p120-catenin (CTNND1) in NSCLC cells21 (Fig. 1). We observed that CRK transduced its repressive signal for p120-catenin (CTNND1) downregulation via interaction with transcription factor SP1. CRK directly interacted with SP1, which was shown in a co-immunoprecipitation assay. Furthermore, overexpression of CRK-II diminished p120-catenin (CTNND1) promoter activity and p120-catenin protein level in a SP1 dependent manner. To examine the downstream biochemical effects as a result of CRK signaling interruption by CRK-I protein domain expression, we measured p120-catenin (CTNND1) promoter activity in NSCLC cells. For this purpose, H157, A549, and Rh2 cells stably expressing CRK SH2 or SH3N domains were transfected with a promoter reporter construct harboring p120-catenin (CTNND1) promoter region as well as Renilla luciferase expressing control vector (pRL-SV40) (Fig. 7). Forty-eight hours after transfections, cells were subjected to a dual luciferase reporter assay. H157, A549, and Rh2 cells expressing CRK-SH3N domains showed significantly enhanced p120-catenin (CTNND1) promoter activity compared to their counterpart cells expressing empty vector or CRK-SH2 domain. We also measured the protein expression level of p120-catenin in the 3 mentioned NSCLC cell lines that stably express CRK-I domains. All 3 cell lines (i.e., H157, A549, and Rh2) harboring the CRK-SH3N domain demonstrated enhanced p120-catenin protein levels (Fig. 8). These findings further demonstrate that overexpression of the CRK-SH3N domain influences CRK’s downstream targets.
Figure 7.
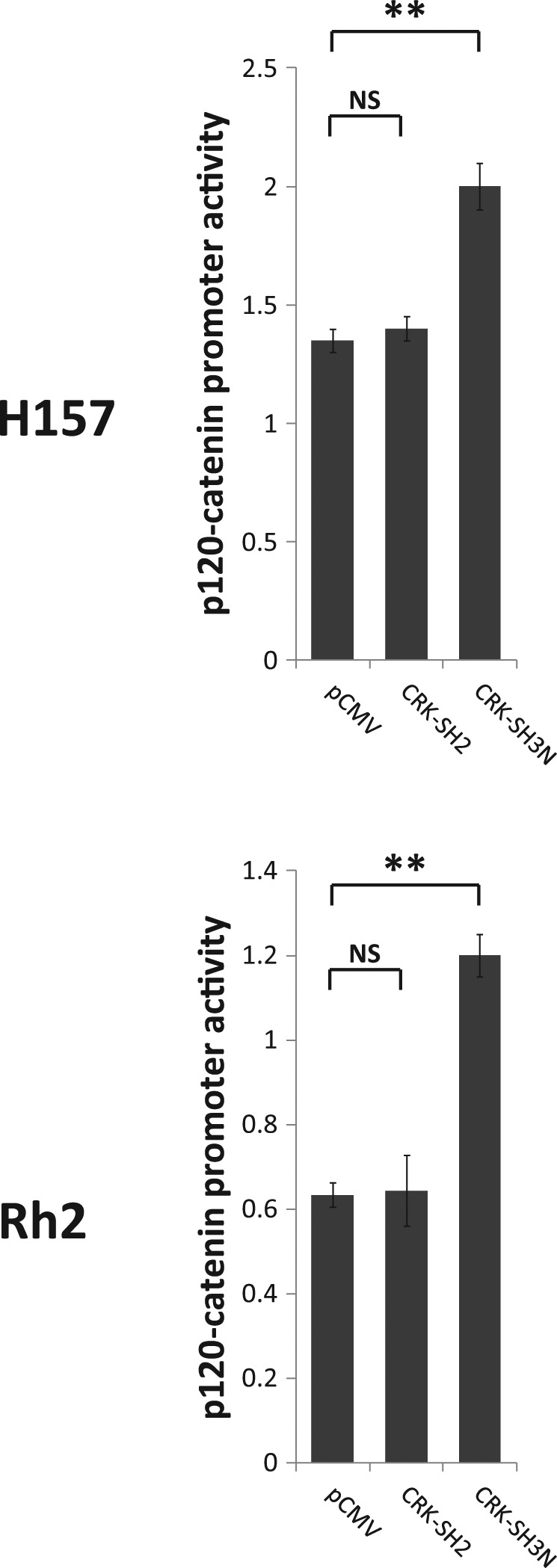
p120-catenin (CTNND1) promoter activity increases following CRK-SH3N expression. Dual luciferase reporter assays in A549 and H157 NSCLC cells stably expressing pCMV (empty vector), CRK-SH2 domain, and CRK-SH3N domain. Experiments are performed in triplicate. Error bars represent ± standard deviation. Comparison between the promoter activity of p120-catenin (CTNND1) mean between the groups was performed by a 2-sided Student’s t test. NS = nonsignificant (P > 0.05); **P < 0.001.
Figure 6.
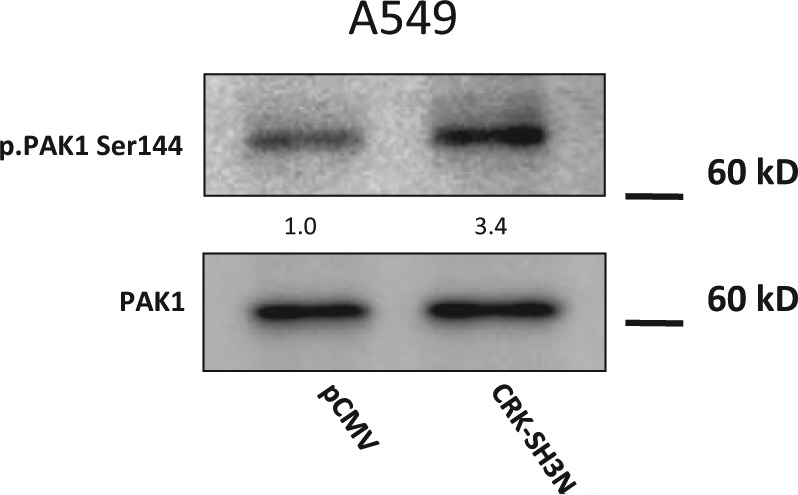
CRK-SH3N domain expression enhances PAK1 phosphorylation in the PAK1 kinase inhibitory domain. Western blot demonstrating PAK1 and phospho-PAK1 Ser144 in A549 cells expressing the CRK-SH3N domain or empty vector. Quantification of the phospho-PAK1 Ser144 signal in cells relative to the signal of cell expressing empty vector is shown.
Discussion
CRK participates in malignant transformation through (a) transducing the upstream oncogenic signals as an adaptor protein and (b) by playing the role of an oncoprotein (c-Crk), as discovered by Mayer, Hamaguchi and Hanafusa.1,2 CRK signaling influences several cellular processes including proliferation, differentiation, and migration, which are among the key dysregulated processes in malignant phenotype. Therefore, it seems that CRK is positioned in a way that alterations in its signal transduction can affect several aspects of the tumorigenesis.
Multiple oncogenic pathways transduce their respective signal through adaptor protein CRK (Fig. 1A). This fact highlights the role of CRK as a prominent component of the signaling pathways, which is shared among several oncogenic signals in different tumor types. For example, BCR-ABL fusion protein famously forms complexes with CRK-L22; however, both CRK-II and CRK-L are known to bind to BCR-ABL and both have been used to evaluate BCR-ABL activity in CML in order to monitor drug resistance.23,24 In the case of NSCLC, 2 major known oncogenic signals (i.e., MET and EGFR) are transduced through CRK. These observations suggests that interruption of signals that are transduced through CRK could block multiple oncogenic pathways in NSCLC, and furthermore, blocking CRK family signals can potentially have implications for treatment of several tumor types.
CRK does not contain a catalytic domain; however, CRK participates in the signal transduction pathways by physically bridging phosphorylated proteins. Therefore, interruption of CRK-mediated signals seems to be possible by disruption of protein-protein interactions where CRK participates in the protein complex. CRK interacts with protein complexes through its SH2 and SH3 domains in a way that SH2 binds to the phosphotyrosine-containing proteins (incoming pathway) and the SH3 domain interacts with the CRK effectors (output pathway)3 (Fig. 1C). Presented data here demonstrate that expression of the CRK-SH3N domain can effectively interrupt the CRK-mediated signals in a way that results in alterations in the morphology, cell motility, and invasiveness of several NSCLC cell lines with different histological backgrounds. This effect of expressed CRK-SH3N domain appears to be the result of saturating the binding sites for the CRK effectors, thereby preventing the transduction of the oncogenic signals. Although we could detect that our CRK-SH3N expressed domain mainly disrupts the interaction of CRK-II with DOCK180, disruption of other CRK paralogues with their effectors on expression of the CRK-SH3N domain is possible.
The mechanism through which CRK-SH3N domain expression influences cell motility could be multifaceted. Data presented here demonstrate that PAK1 phosphorylation in its kinase inhibitory domain (i.e., Serine 144) is altered in CRK-SH3N expressing cells. In addition, our data demonstrate that PAK1 phosphorylation on serine 144 is closely correlated with higher E-cadherin/p120-catenin in our panel of NSCLC cells (Suppl. Fig. 4), which is concordant with the pattern of PAK1 serine 144 phosphorylation in CRK-SH3N expressing cells. Compared to empty vector, CRK-SH3N expressing cells demonstrate higher phospho-PAK1 Ser144, higher p120-catenin, and also show less invasive behavior. Considering PAK1 is a known effector of RAC1/CDC42,19 it is likely that the activity of RAC1/CDC42 is affected in CRK-SH3N expressing cells.
SH2 and SH3 domains are found in many proteins that participate in the intracellular signaling pathways of the metazoa. There are more than 120 SH225 and more than 500 SH326 domains identified in the human genome. Therefore, overexpression of CRK Src Homology domains seems to have a small likelihood of interfering with signaling pathways that involve other proteins with different SH2 or SH3 domains.
The CRK-SH3N domain is composed of only 61 amino acids. However, even though this is a relatively small size protein, delivery of this protein domain for therapeutic purposes poses a challenge. Of note, transcellular delivery of large macromolecules by using cell penetrating peptides has proven to be effective.27 Considering the significant observed effects as a result of CRK-SH3N domain expression, it seems that CRK domains can serve well as a template for designing peptide-based therapeutics for interruption of pathological CRK signaling, especially in NSCLC.
In summary, the presented data support the utility of CRK-SH3N domain expression for influencing the biological behavior of NSCLC cells through interruption of CRK-mediated signals.
Material and Methods
Cell cultures
A549, H157, and Rh2 cells were routinely cultured in RPMI supplemented with antibiotics and 10% heat-inactivated fetal bovine serum (FBS) (Omega Scientific, Tarzana, CA).
Measurement of p120ctn promoter activity by dual luciferase assay
p120-catenin promoter construct28 was transfected into NSCLC cell lines by Lipofectamine 2000 (Invitrogen, Grand Island, NY). Twenty-four hours after transfection, cells were washed with phosphate-buffered saline (PBS) and lysed using a Branson Sonifier in 1× passive lysis buffer (Promega, Fitchburg, WI) at room temperature. Reporter gene expression was assessed by using the Dual-Luciferase Reporter Assay System kit (Promega) according to the manufacturer’s instructions in a TD-20/20 Luminometer (Turner Biosystems, Sunnyvale, CA). We normalized for transient transfection efficiency (i.e., firefly luciferase activity) by cotransfection of a Renilla luciferase expressing control vector (pRL-SV40). All experiments were performed in triplicate and were reported as means ± standard deviation, and each experiment was performed at least twice.
Western blots
NSCLC cell lines were seeded in 10 cm Petri dishes at 5 × 105 cells per dish, which resulted in 30% to 40% confluency 24 hours after plating. Cells were harvested at 24 hours by adding trypsin, pelleted, and lysed in 100 µL of lysis buffer (NaCl 15 mM; EDTA 0.5 mM; Tris 10 mM) using a Branson Sonifier. Cell debris was collected by centrifugation at 4°C, and protein concentration was measured by the BCA method. Protein was resolved by SDS-PAGE and was transferred to a nitrocellulose membrane. The membrane was blocked with TBS with 5% nonfat powdered milk.
Membranes were immunoblotted with the following primary antibodies: PAK1 (Sigma-Aldrich, Catalogue number SAB4300427), phospho-PAK1 Ser144 (Sigma-Aldrich, Catalogue number p7871), Ecadherin (BD Biosciences, Catalogue number 61081), p120 catenin (BD Biosciences, Catalogue number 610133), and Anti Flag (Sigma, Catalogue number F3165).
Horse radish peroxidase conjugated secondary antibodies were used for detection of bands by chemiluminescence (ECL western blotting detection reagents; Amersham Biosciences, Piscataway, NJ).
Immunoprecipitation
We grew A549 cells in 100 cm2 dishes to 90% confluency. Cells were washed with 2 mL PBS and scraped off in 1 mL PBS. Cells were transferred to Eppendorf tubes and spun at 1000 rpm at 4°C for 10 minutes. Then we prepared a cell lysate by resuspending the cells in ice-cold gentle lysis buffer (10 mM Tris–HCl pH 7.5; 10 mM NaCl; 2 mM EDTA; 0.1% Triton-X100; 1 mM PMSF; 2 µg/mL aprotinin; 2 µg/mL leupeptin; approximately 700 µL per 2 × 106 cells). Cells were incubated on ice for 5 minutes before adding NaCl to 150 mM followed by incubating on ice for 10 minutes.
Next, cells were spun again at 14,000 rpm in 4°C for 15 minutes. We split the supernatant into 2 fractions and incubated them with either 4 µg of anti CRK-II antibody (Santa Cruz Biotechnology, Catalogue number sc-289) or 4 µg of control IgG for 4 hours. Subsequently, we added 25 µL of protein G plus/protein A agarose suspension (Calbiochem, Catalogue number IP05) and incubated overnight at 4°C with agitation. We washed the beads 8 times with 1 mL of ice-cold NET (50 mM Tris–HCl pH 7.5; 150 mM NaCl; 0.05% Triton-X100) for 1 minute each time spinning at 1000 rpm at 4°C. Eventually, we eluted the immunoprecipitate by adding SDS directly to beads and proceeded with western blotting with anti DOCK180 (Santa Cruz Biotechnology, Catalogue number sc-6167), anti SOS1 (Santa Cruz Biotechnology, Catalogue number sc-376843), and anti C3G (Santa Cruz Biotechnology, Catalogue number sc-17840).
Wound healing assays and microscopy
A549 and H157 cells were plated in a 6-well plate dish at 1 × 105 cells per well and were grown to confluent stage. By using a sterile P1000 pipette tip, a straight scratch was made along the largest diameter of each well and a baseline photomicrograph was taken from this scratch at 2 different magnifications. A follow-up photomicrograph was taken at 24 hours. Photomicrographs of the cells were obtained by a Nikon Eclipse TS100 inverted microscope equipped with a Cannon A510 digital camera. The digital camera was connected with a Max View Plus adaptor (ScopeTronix) to the inverted microscope. An equal area of the photomicrographs from each condition was imported into the Adobe Photoshop software. The wound area was selected by using the Magic Wand Tool of Adobe Photoshop and the number of pixels in each selected area was determined by the Histogram Tool. Experiments were repeated 3 times and the average wound surface area was compared among the groups.
Cell invasion assay
A Matrigel cell invasion assay was performed by using Matrigel Invasion Chamber (BD Biosciences, Catalogue number 354480) according to the manufacturer’s instructions. A total of 1 × 105 cells were incubated in the upper chambers in RPMI in the absence of serum in triplicate. As the chemoattractant, RPMI with 10% FBS was used in the lower chambers. Cells were incubated for 24 hours in the 37°C incubator. Cells were harvested and stained with Diff-Quik kit according to the manufacturer’s instructions. Photomicrographs were taken by an Olympus BH2 microscope equipped with a Cannon A510 digital camera. The invaded cells in 5 corresponding regions of each Matrigel membrane were counted and the mean of invaded cells were compared among the groups.
Intracellular staining for cleaved caspase-3
Apoptosis was identified by intracellular staining for cleaved caspase-3. H157 cells that stably express pCMV (empty vector), CRK-SH2 domain, or CRK-SH3N domain were fixed, permeabilized, and stained with anti caspase 3-PE antibody by using Active Caspase-3 Apoptosis Kit (BD-Pharmingen, Catalogue number 550914). The expression of active caspase-3 was detected by flow cytometry.
Footnotes
Declaration of Conflicting Interests: The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding: The author(s) received no financial support for the research, authorship, and/or publication of this article.
Supplementary material for this article is available on the Genes & Cancer website at http://ganc.sagepub.com/supplemental.
References
- 1. Mayer BJ, Hamaguchi M, Hanafusa H. A novel viral oncogene with structural similarity to phospholipase C. Nature. 1988;332(6161):272-5 [DOI] [PubMed] [Google Scholar]
- 2. Mayer BJ, Hamaguchi M, Hanafusa H. Characterization of p47gag-crk, a novel oncogene product with sequence similarity to a putative modulatory domain of protein-tyrosine kinases and phospholipase C. Cold Spring Harb Symp Quant Biol. 1988;53(Pt 2):907-14 [DOI] [PubMed] [Google Scholar]
- 3. Birge RB, Kalodimos C, Inagaki F, Tanaka S. Crk and CrkL adaptor proteins: networks for physiological and pathological signaling. Cell Commun Signal. 2009;7:13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Feller SM. Crk family adaptors-signalling complex formation and biological roles. Oncogene. 2001;20(44):6348-71 [DOI] [PubMed] [Google Scholar]
- 5. Miller CT, Chen G, Gharib TG, et al. Increased C-CRK proto-oncogene expression is associated with an aggressive phenotype in lung adenocarcinomas. Oncogene. 2003;22(39):7950-7 [DOI] [PubMed] [Google Scholar]
- 6. Takino T, Nakada M, Miyamori H, Yamashita J, Yamada KM, Sato H. CrkI adapter protein modulates cell migration and invasion in glioblastoma. Cancer Res. 2003;63(9):2335-7 [PubMed] [Google Scholar]
- 7. Nishihara H, Tanaka S, Tsuda M, et al. Molecular and immunohistochemical analysis of signaling adaptor protein Crk in human cancers. Cancer Lett. 2002;180(1):55-61 [DOI] [PubMed] [Google Scholar]
- 8. Ota S, Kizaka-Kondoh S, Hashimoto Y, et al. Constitutive association of EGF receptor with the CrkII-23 mutant that inhibits transformation of NRK cells by EGF and TGF-beta. Cell Signal. 1998;10(4):283-90 [DOI] [PubMed] [Google Scholar]
- 9. Ishiki M, Sasaoka T, Ishihara H, et al. Evidence for functional roles of Crk-II in insulin and epidermal growth factor signaling in Rat-1 fibroblasts overexpressing insulin receptors. Endocrinology. 1997;138(11):4950-8 [DOI] [PubMed] [Google Scholar]
- 10. Garcia-Guzman M, Dolfi F, Zeh K, Vuori K. Met-induced JNK activation is mediated by the adapter protein Crk and correlates with the Gab1-Crk signaling complex formation. Oncogene. 1999;18(54):7775-86 [DOI] [PubMed] [Google Scholar]
- 11. Lamorte L, Royal I, Naujokas M, Park M. Crk adapter proteins promote an epithelial-mesenchymal-like transition and are required for HGF-mediated cell spreading and breakdown of epithelial adherens junctions. Mol Biol Cell. 2002;13(5):1449-61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lamorte L, Rodrigues S, Naujokas M, Park M. Crk synergizes with epidermal growth factor for epithelial invasion and morphogenesis and is required for the met morphogenic program. J Biol Chem. 2002;277(40):37904-11 [DOI] [PubMed] [Google Scholar]
- 13. Smith JJ, Evans EK, Murakami M, et al. Wee1-regulated apoptosis mediated by the crk adaptor protein in Xenopus egg extracts. J Cell Biol. 2000;151(7):1391-400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Takino T, Tamura M, Miyamori H, et al. Tyrosine phosphorylation of the CrkII adaptor protein modulates cell migration. J Cell Sci. 2003;116(Pt 15):3145-55 [DOI] [PubMed] [Google Scholar]
- 15. Cho SY, Klemke RL. Extracellular-regulated kinase activation and CAS/Crk coupling regulate cell migration and suppress apoptosis during invasion of the extracellular matrix. J Cell Biol. 2000;149(1):223-36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kain KH, Gooch S, Klemke RL. Cytoplasmic c-Abl provides a molecular “Rheostat” controlling carcinoma cell survival and invasion. Oncogene. 2003;22(38):6071-80 [DOI] [PubMed] [Google Scholar]
- 17. Knaus UG, Bokoch GM. The p21Rac/Cdc42-activated kinases (PAKs). Int J Biochem Cell Biol. 1998;30(8):857-62 [DOI] [PubMed] [Google Scholar]
- 18. Manser E, Leung T, Salihuddin H, Zhao ZS, Lim L. A brain serine/threonine protein kinase activated by Cdc42 and Rac1. Nature. 1994;367(6458):40-6 [DOI] [PubMed] [Google Scholar]
- 19. Parrini MC, Lei M, Harrison SC, Mayer BJ. Pak1 kinase homodimers are autoinhibited in trans and dissociated upon activation by Cdc42 and Rac1. Mol Cell. 2002;9(1):73-83 [DOI] [PubMed] [Google Scholar]
- 20. Stockton RA, Schaefer E, Schwartz MA. p21-activated kinase regulates endothelial permeability through modulation of contractility. J Biol Chem. 2004;279(45):46621-30 [DOI] [PubMed] [Google Scholar]
- 21. Mortazavi F, Dubinett S, Rettig M. c-Crk proto-oncogene contributes to transcriptional repression of p120-catenin in non-small cell lung cancer cells. Clin Exp Metastasis. 2011;28(4):391-404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Uemura N, Salgia R, Li JL, Pisick E, Sattler M, Griffin JD. The BCR/ABL oncogene alters interaction of the adapter proteins CRKL and CRK with cellular proteins. Leukemia. 1997;11(3):376-85 [DOI] [PubMed] [Google Scholar]
- 23. Mizutani T, Kondo T, Darmanin S, et al. A novel FRET-based biosensor for the measurement of BCR-ABL activity and its response to drugs in living cells. Clin Cancer Res. 2010;16(15):3964-75 [DOI] [PubMed] [Google Scholar]
- 24. Tunceroglu A, Matsuda M, Birge RB. Real-time fluorescent resonance energy transfer analysis to monitor drug resistance in chronic myelogenous leukemia. Mol Cancer Ther. 2010;9(11):3065-73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Liu BA, Jablonowski K, Raina M, Arce M, Pawson T, Nash PD. The human and mouse complement of SH2 domain proteins-establishing the boundaries of phosphotyrosine signaling. Mol Cell. 2006;22(6):851-68 [DOI] [PubMed] [Google Scholar]
- 26. Harkiolaki M, Lewitzky M, Gilbert RJ, et al. Structural basis for SH3 domain-mediated high-affinity binding between Mona/Gads and SLP-76. EMBO J. 2003;22(11):2571-82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. van den BA, Dowdy SF. Protein transduction domain delivery of therapeutic macromolecules. Curr Opin Biotechnol. 2011;22(6):888-93 [DOI] [PubMed] [Google Scholar]
- 28. Mortazavi F, An J, Dubinett S, Rettig M. p120-catenin is transcriptionally downregulated by FOXC2 in non-small cell lung cancer cells. Mol Cancer Res. 2010;8(5):762-74 [DOI] [PubMed] [Google Scholar]