

Abstract
Desmosomes are dynamic junctions between cells that maintain the structural integrity of skin and heart tissues by withstanding shear forces. Mutations in component genes cause life-threatening conditions including arrhythmogenic right ventricular cardiomyopathy, and desmosomal proteins are targeted by pathogenic autoantibodies in skin blistering diseases such as pemphigus. Here, we review a set of newly discovered pathogenic alterations and discuss the structural repercussions of debilitating mutations on desmosomal proteins. The architectures of native desmosomal assemblies have been visualized by cryo-electron microscopy and cryo-electron tomography, and the network of protein domain interactions is becoming apparent. Plakophilin and desmoplakin mutations have been discovered to alter binding interfaces, structures, and stabilities of folded domains that have been resolved by X-ray crystallography and NMR spectroscopy. The flexibility within desmoplakin has been revealed by small-angle X-ray scattering and fluorescence assays, explaining how mechanical stresses are accommodated. These studies have shown that the structural and functional consequences of desmosomal mutations can now begin to be understood at multiple levels of spatial and temporal resolution. This review discusses the recent structural insights and raises the possibility of using modeling for mechanism-based diagnosis of how deleterious mutations alter the integrity of solid tissues.
Abbreviations: ARVC, arrhythmogenic right ventricular cardiomyopathy; DIFC, desmosome–intermediate filament complex; EC, extracellular cadherin; EGFR, epidermal growth factor receptor; IA, intracellular anchor; iPSC, induced pluripotent stem cell; ICS, intracellular cadherin-typical sequence; Lef, lymphoid enhancer factor; NMD, nonsense-mediated RNA decay; PPAR, peroxisome proliferator-activated receptor; PRD, plakin repeat domain; SR, spectrin repeat; SH3, Src homology 3; Tcf, T-cell factor
Keywords: desmosomal cadherin, desmoplakin, desmosome, arrhythmogenic right ventricular cardiomyopathy, plakoglobin
Graphical Abstract

Introduction to Desmosomes
How cells form tissues fundamentally depends on how they connect physically. The four major types of connections are desmosomes, adherens, tight, and gap junctions. Desmosomes are intercellular junctions found in cardiac muscle, epithelia, and some other tissues. They are located at the cell membrane where they act as anchors for intermediate filaments of the cell cytoskeleton. By linking intermediate filaments of adjacent cells, desmosomes form a network of adhesive bonds that radiates throughout a tissue, providing mechanical strength. The desmosome–intermediate filament complex (DIFC) [1] is essential for maintaining the integrity of tissues. When desmosomal adhesion is compromised, as occurs in some genetic and autoimmune diseases, cells lose cohesiveness, often with severe consequences for the tissue as a whole. This is particularly true of those tissues, such as the heart and skin, which are subjected to mechanical stress.
In the DIFC, desmosomal cadherins provide the coupling between adjacent cells. Seven desmosomal cadherins are expressed by human cells, four desmogleins (DSG1–DSG4) and three desmocollins (DSC1–DSC3). Desmosomal cadherins interact with other desmosomal cadherins produced by adjacent cells in the intercellular space. Within the cell, four members of the armadillo family of proteins are found in association with desmogleins and desmocollins. The armadillo proteins plakoglobin (γ-catenin) and plakophilins 1–3 (PKP1–3), together with the plakin protein desmoplakin, act as linkers between desmosomal cadherins at the membrane and intermediate filaments in the DIFC. Desmoplakin interacts with a variety of intermediate filaments including desmin in cardiac muscle, keratin in epithelial tissues, and vimentin in certain specialized tissues such as the arachnoidal tissue of the brain meninges and the dendritic reticulum of lymph node follicles.
Plakoglobin and desmoplakin are obligatory desmosomal components. Of the other proteins, at least one desmoglein, one desmocollin, and one plakophilin are always present and required for normal desmosomal adhesion. However, it should be noted that desmocollins are dispensable for the efficient formation of desmosomes in some experimental situations [2]. Cardiomyocyte desmosomes contain five proteins, namely, DSG2, DSC2, plakoglobin, PKP2, and desmoplakin. The situation is more complicated in other tissues such as the epidermis where all seven desmosomal cadherins and all three plakophilins, as well as plakoglobin and desmoplakin, are expressed. Desmosomes can contain more than one desmocollin [3] and presumably more than one desmoglein and more than one plakophilin, thus yielding a diversity of intercellular networks within the fabric of the epidermis. Further diversity may be present in cardiac muscle as a unique type of junction (the area composita) has been identified in the intercalated disk connecting cardiomyocytes. This novel junction is of unusually high molecular complexity and contains a mixture of proteins that are usually associated with either desmosomes or adherens junctions [4,5].
Diseases Caused by Desmosomal Dysfunction
Due to their central role in maintaining the integrity of stressed solid tissues, the disruption of the assembly or structure of desmosomes leads to debilitating conditions including inherited, infectious, and autoimmune diseases. The molecular consequences include loss of adhesion between cells, disorganization within cells, ineffectual cell communication and regulation, and misguided tissue development. The pathological conditions that result are diverse and include cardiomyopathies, epidermolysis bullosa, epidermal and mucosal blistering, ectodermal dysplasia, palmoplantar keratoderma, keratosis, woolly hair, and alopecia. The disorders reflect the tissue-specific expression of desmosomal variants. For example, mutations in the genes of the cardiomyocyte desmosome lead to arrhythmogenic right ventricular cardiomyopathy (ARVC). The foundation laid by many structural, cellular, and mutational studies provides a basis for predicting the effects of desmosomal mutations, which are found in 1 in 200 people of some populations [6] and may be used for mechanism-based diagnosis.
The role for desmosomal components in cancer progression is emerging but is as of yet poorly understood. Colorectal and breast cancers show decreases in DSC2 and DSC3 levels, respectively [7,8], whereas DSG3 is overexpressed in squamous cell carcinoma and head and neck cancer [9,10]. Altered DSG2 expression is found in melanoma, squamous cell carcinomas and gastric cancers [9,11–13], and alterations in plakophilin expression are associated with lung and prostate cancer [14,15]. Oropharyngeal tumors exhibit reduced expression of desmoplakin [16]. Whereas some studies have clearly shown increases or decreases in desmosomal components in various epithelial cancers [17], others have found no apparent changes in desmosomal protein levels during tumor progression [9]. Few cancer-linked mutations have been found in desmosomal components with the exception of the JUP gene that encodes plakoglobin [18,19]. Although oncogenic roles have been suggested, the weight of evidence infers that desmosomes play tumor-suppressive functions analogous to E-cadherin and underscore the need for detailed genetic studies to define the contributions in cancer model systems.
Architecture of the Intact Desmosome
Desmosomes were first visualized by the Italian pathologist Bizzozero in 1864 as 200- to 500-nm-wide “buttons” linking epithelial cells. One hundred fifty years later, the molecular composition of these junctions is being resolved in detail. Their structural layers are apparent by atomic force microscopy, cryo-electron tomography, and electron microscopy [20–23], enabling detailed molecular models of the overall architecture of the desmosome to be produced (Fig. 1).
Fig. 1.
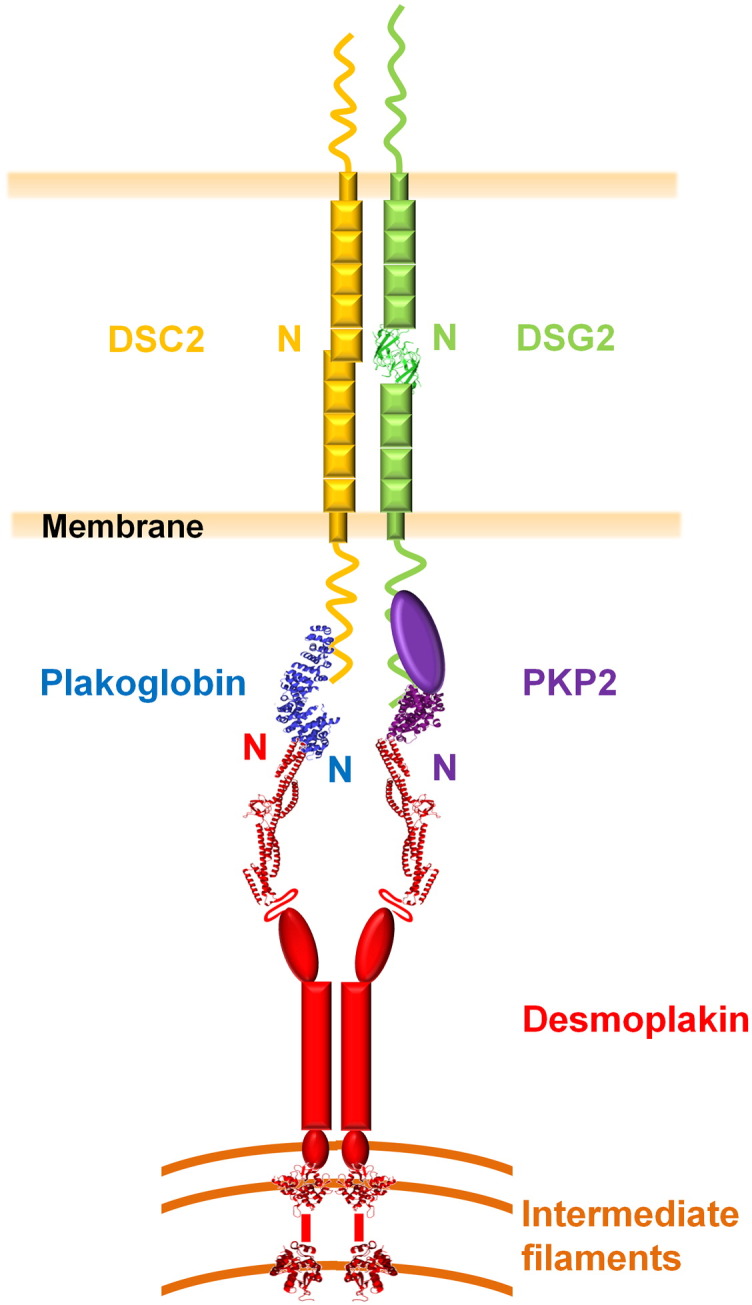
Architecture of the cardiac desmosome. The approximate locations of the core proteins are shown, including the structures of DSG2's EC1 domain (green ribbon) and the arm repeat domains of plakoglobin (blue) and PKP2 (purple). Also shown are crystal structures of the first four SRs of the desmoplakin plakin domain (SR3–6) and PRDs B and C. The unstructured DSC2 and DSG2 C-termini are shown as wavy lines, as is the protease-sensitive hinge between the long and short arms of the desmoplakin plakin domain [24]. The N-termini of the proteins are labeled and their respective binding sites are juxtaposed. Both homophilic and heterophilic interactions between desmosomal cadherins may take place in the extracellular space, but for simplicity, only homophilic interactions are shown.
The center of the junction is composed of the extracellular regions of cadherin family members, and a dense midline of their interleaved N-termini runs through this. Just inside the plasma membrane is the outer dense plaque, which contains plakoglobin and plakophilin. The intracellular domains of the desmogleins and desmocollins contribute to the outer dense plaque, as do plakoglobin and plakophilin. Beyond this lies a translucent zone and a further inner dense plaque that is composed largely of desmoplakin. Together, these proteins provide a highly organized supermolecular assembly that mediates stable yet adaptable mechanical coupling between points of cell–cell adhesion and the cytoskeleton.
The zipper-like organization of desmosomal molecules that are arrayed perpendicular to the membrane is becoming apparent [25,26]. This array shows a periodic repeat pattern of 5.3 nm, suggesting regular packing of straight cadherin extracellular domains. Inside the cell, this is reflected by a periodic spacing of around 6.6 nm for the sets of protein chains that link to intermediate filaments [21]. The determinants of the regular spacing between the elongated protein complexes that line the desmosome are unclear but presumably stem from multimerization surfaces inside and outside the cell. Desmosomal protein structures can be accommodated within the density map of desmosomes, indicating the meshwork of possible orientations within the intact junction. This model infers that extracellular organization is dictated by plakoglobin packing, with subsequent addition of desmoplakin and plakophilin then reinforcing the overall scaffold [27].
Desmoglein Structure and Alterations
In common with classical cadherins, desmogleins and desmocollins are expressed with N-terminal leader and pro-peptides that are cleaved during maturation. The pro-peptides of the desmogleins (~ 26 residues) are shorter than those of the desmocollins (~ 108 residues), which are of a similar length to that of E-cadherin. The mature proteins span the membrane once with the extreme N-termini of their extracellular domains interacting in the intercellular space and their C-terminal tails embedded in the cytoplasmic plaque. They share a similar architecture, with approximately 30% sequence identity being maintained across the four desmogleins and three desmocollins that are differentially expressed in solid tissues [28]. These specialized cadherins contain five extracellular cadherin domains (EC1–EC5) that are rigidified by Ca2 + binding sites in their linkers [29], followed by a transmembrane helix and disordered C-terminal region.
The solution structure of the EC1 domain of human DSG2 has been determined by Yokoyama et al. (unpublished results) and contains the expected β sandwich fold and a signature β helix [30,31] (Fig. 2). Cadherin extracellular domains mediate two types of protein interactions, as revealed by the crystal structures of C-, E-, and N-cadherin extracellular domains [32–34]. Trans-interactions between cells are mediated by reciprocal exchange of an N-terminal strand and a conserved Trp at the extreme N-terminus of EC1 (“Trp2”) in the mature classical or desmosomal cadherins [35] while unique cis-interactions involve contacts between EC1 and the EC2–EC3 linker within the three classical cadherins. The two interaction modes occur at almost perpendicular angles in these molecules and are weak individually, with affinities of 20–100 μM and over 1 mM, respectively [36]. Together, this forms a zipper-like array of contacts to stably join cells within an adherens junction. Extrapolation from the individual sites and affinities allows the assembly of the extracellular adhesive lattice to be modeled [37]. The resulting assembly shows how classical cadherins could accumulate in contact zones, followed by the synergistic trans-dimerization and lateral cis-association to allow formation of stable junctions with a regular molecular spacing that extends into the cell. This is consistent with the related desmosomal architecture resolved by cryo-electron tomography [21] in which β1 strands are also swapped at the adhesive interface. However, the details of the packing of desmosomal protomers appears to differ. The spacings between adjacent cell membranes reported for desmosomes are 320–350 Å [21,38], which is larger than those for adherens junctions (150–300 Å) [38–41], despite the fact that both are composed of cadherin ectodomains of similar lengths. Exactly how desmosomal cadherin packing in desmosomes differs from classical cadherin packing in adherens junctions remains to be seen. Since both extracellular domains are similar in length, this difference involves distinct angles from which they exit the plasma membrane, with cryo-electron microscopy maps showing a more perpendicular orientation in desmosomes with only the EC1 domains bridging the junction [21].
Fig. 2.

Desmosomal cadherin structures. The DSG2 EC1 domain solution structure [Protein Data Bank (PDB) code: 2YQG] is shown as a ribbon, as is a homology-derived model of DSC2's EC1 (based on the crystal structure of the corresponding region of the mouse N-cadherin extracellular domain) (PDB code: 3Q2W). Point mutations that are known to be pathogenic are shown as copper (buried residues) or green (surface-exposed residues). Alignment of the EC1 sequences of DSG2, DSC2, and mouse N-cadherin indicates that mutated residues are all highly conserved, supporting their critical functional or structural roles.
The mechanism by which desmosomal cadherins interact in the intercellular space is not yet clear. Strong cell–cell adhesion may occur via both homophilic and heterophilic interactions between desmogleins and desmocollins that cross the divide between cells [35,42]. It seems likely that upon proteolytic activation, the desmosomal cadherins interact via their EC1 domains in a Ca2 +- and “Trp2”-dependent manner [35,43], reminiscent of the proposed C-cadherin cis-multimerization event [32,34]. The loops and linkers within successive cadherin domains typically coordinate three Ca2 + to form a rigidified extracellular domain [29,44], although a dramatically kinked, calcium-free linker in DN-cadherin has also been resolved [45]. The specific contributions of the interdomain contacts and linkers to homophilic or heterophilic recognition of desmosomal cadherins is unclear. However, it is likely to vary from the heterodimeric structure of tandem EC domains of two atypical cadherins, which reveals a particularly extensive trans-interaction responsible for linking hair cells to mediate mechanotransduction in the inner ear [46].
Of the 148 variants of the human DSG2 gene in the ARVC database [47], 51 are likely to have pathogenic effects including three novel mutations (P157L, S194L, and F833I) which are reported here. Note that here and in the following discussion of ARVC mutations in the DSG2 and DSC2 genes, residue numbers refer to the immature proteins, before removal of N-terminal leader and pro-peptides. Most DSG2 mutations map to the extracellular domain, emphasizing its central role within the desmosome. Here, we focus on clinically identified ARVC mutations that alter protein sequence, rather than those without clear phenotypic consequences, or deletions or premature stops which would abrogate surface expression altogether. Defects in processing of the DSG2 protein contribute to ARVC progression, as exemplified by M1I, R46W, R46Q, and R49H mutations, which are only found in the immature, non-adhesive protein [48–50]. The functional integrity of the EC1 domain that mediates trans- and cis-interactions could be compromised structurally by I73V and Y87C alterations, as well as exposed G100R and R146H substitutions and the D154E mutant that alters a calcium coordinating residue [49,51–54]. The EC2 domain that may also mediate lateral cis-interactions is predicted to be compromised by D187G, P205L, or E230G pathogenic mutations [52,54,55] based on homology modeling. The N266S substitution alters the second calcium coordination site responsible for rigidification and cis-interactions of the extracellular domain. Mice that express the corresponding DSG2 mutation show separated junctions and widened intercellular spaces, suggesting defective adhesion, and developed macroscopic lesions and ARVC features including sudden death, spontaneous ventricular arrhythmias, and cardiac dysfunction [52,56,57]. The more dramatic deletion of the first two EC domains of DSG2 also causes an ARVC-like phenotype in mice. Their hearts develop differentiated scar tissue in place of myofibril-rich cardiomyocytes, which are seen to be disconnected by transmission electron microscopy [58]. Their sarcomers are disrupted, and abnormal mitochondria and autophagic vacuoles develop, suggesting removal of dead cardiomyocytes and tissue remodeling. The EC3 domain has the largest number of mutations that are associated with ARVC, including K294E, D297G, F321I, N326V, N330D, E331K, and T335A [48,50,53,54,59,60]. Two ARVC-linked nonsense mutations, Q461X and D419X [51,53], induce premature stops and would have dramatic consequences including nonsense-mediated RNA decay (NMD).
Just inside the plasma membrane of the desmosome are the membrane proximal regions of desmoglein and desmocollin proteins, which contain an intracellular anchor (IA) region and an intracellular cadherin-typical sequence (ICS). ARVC-linked mutations in the DSG2 IA region include short deletions due to frameshifts as well as a G638R substitution next to the transmembrane domain, which could conceivably promote binding to negatively charged lipids in the plasma membrane [61]. The ICS region is required for DSG1 binding to plakoglobin through a 1:1 complex, with key hydrophobic residues involved having been mapped by alanine scanning mutagenisis [62,63]. Missense mutations in the plakoglobin binding site including G812C, G812S, and C813R are known to be pathogenic [50,64]. Surprisingly, these mutant DSG2 proteins display localizations, stabilities, and plakoglobin binding similar to the wild-type form and differ instead in their abilities to be posttranslationally modified and bind PKP2. The ARVC database includes a L831F point mutation [47] and F833I, which is reported here, both of which map to the DSG2 ICS region. These point mutations are likely to affect plakoglobin protein interactions, although determination of their specific effects requires resolution of the complexed structures.
The desmoglein-specific cytoplasmic region contains a repeat unit domain, which is intrinsically disordered and monomeric but contains inducible structure [65]. This region can form multimers with a globular head and a thin tail, as seen by electron microscopy [66]. The DSG2 protein's desmoglein-specific cytoplasmic region forms stable complexes between tails that block internalization and promotes strong cell–cell adhesion. An ARVC-linked frameshift deletion (V977fsX1006) that disrupts this interaction leads to rapid DSG2 endocytosis in cardiac muscle cells [67], and pathogenic mutations linked to ARVC include P925S and Y1047R substitutions in the second and fifth repeat unit domain motifs, respectively [55,68,69]. This is consistent with a crucial role for this region in mediating desmosomal protein interactions, although the specific partners are yet to be identified.
The formation of desmosomes involves several stages. The component proteins are transported to the plasma membrane in separate compartments. These assemble into less adhesive desmosomes that mature into hyperadhesive desmosomes that cannot be dissociated by calcium depletion [70,71]. This transition from calcium-dependent to -independent junctions involves protein kinase C-α activity [71]. Moreover, specific phosphorylation sites have been identified experimentally at Ser680, Ser703, Ser968, Tyr1013, and Ser1118 residues of DSG2 [72–74]. Reversion to a calcium-dependent junction occurs upon wound formation, allowing cells to move as the tissue heals [75], and similar processes may occur in mitotically active basal cells and during tumor invasion.
Epidermal desmosomes found in the outer and inner layers of the skin contain primarily DSG1 and DSG3, respectively. Targeting of DSG1 by autoantibodies leads to blistering in the outer layers of the skin in the disease pemphigus foliaceus. By contrast, in pemphigus vulgaris, targeting of DSG3 results in blistering deep within mucous membranes, and targeting of both DSG3 and DSG1 leads to mucocutaneous blistering with blisters appearing just above the basal cell layer of the skin [76]. Pathogenic antibodies isolated from mouse models recognize the first cadherin domain of DSG3 that directly mediates trans-adhesion between cells [77,78], and those isolated from human patients also bind here [79]. The antibodies are conformationally specific, binding preferentially to the mature protein, which has been proteolytically cleaved to initiate cadherin strand dimer formation [80]. The sites in the first and second cadherin domains of DSG3 that may mediate cis-adhesion are targeted by serum and pathogenic antibodies of pemphigus patients [81]. Epitope mapping reveals that the EC1 and EC2 domains of desmogleins are most commonly recognized by pathogenic antibodies from both pemphigus foliaceus [82] and pemphigus vulgaris sera [83]. An endemic form of pemphigus foliaceus (fogo selvagem) evolves from autoantibodies that recognize an epitope in desmoglein's EC5 domain and progresses when anti-EC1:EC2 antibodies develop [84]. The mechanism by which these pathogenic antibodies result in a loss of intercellular adhesion and blister formation remains unclear. Pathogenic antibodies may block the assembly of desmosome adhesion complexes by occluding the sites that directly mediate trans and cis contacts. Alternatively, binding of autoantibodies to desmogleins may trigger endocytosis of desmogleins followed by desmosome disassembly [85].
Desmocollin mutations linked to ARVC
Both desmocollins and desmogleins are required for normal desmosomal adhesion. Homophilic interactions between desmocollins [86] and between desmogleins [87,88] have been reported in solution. However, it appears that desmocollins and desmogleins can also engage in heterophilic interactions in solution [86] and both homophilic and heterophilic interactions in cultured cells [2,35,43]. The ARVC-associated mutations that could alter processing of DSC2 include E2K, C32X, E102K, and R132C substitutions in the signal and pro-sequences [48,54,61,69,89,90]. The EC1 domain of DSC2 exhibits potentially destabilising ARVC-linked mutations including G150A, R203C, R203H, G220R, and I231T [64,68,69,89]. ARVC-linked pathogenic mutations in the EC2 domain of DSC2 include T275M, P292S, T340A, I342V, I345T, and D350Y, with the latter mutation removing a Ca2 + coordinating group that rigidifies the linkage to EC3 [52,54,64,89]. The E102K and I345T mutant DSC2 proteins delocalize from the plasma membrane to the cytosol [90], inferring trafficking defects, while the R203C and T275M mutants show impaired proteolytic processing [64]. A pair of NMD mutations, R375X and Q554S, are found in the EC3 and EC4 domains, respectively, as well as deletions due to frameshift mutations [54]. The EC5 domain contains a single pathogenic mutation, I603T [91], as well as frameshifts and deletions that would cause NMD. Like desmogleins, desmocollins have cytoplasmic IA and ICS domains. The ICS domain is truncated in desmocollin “b” proteins, which are shorter than desmocollin “a” proteins and arise as a result of alternative splicing. The “b” proteins have a number of amino acids (11 in DSC2b) at the extreme C-terminus of the protein that are not found in the “a” proteins. DSC2b retains the ability to bind PKP2 but is unable to bind plakoglobin [92]. A pathogenic ARVC mutation at S824L is found in both DSC2a and DSC2b isoforms whereas another at G863R is found in DSC2a only, along with a pathogenic nonsense mutation in PKP2. The latter resides next to a potential phosphorylation site at Ser864 [69,91,93]. Hence, the frequency and types of mutations roughly mirrors those found in DSG2, consistent with their similar roles and orientations in cardiac desmosomes.
Armadillo protein structure, interactions, and mutations in ARVC
Plakoglobin is closely related to β-catenin, an important component of adherens junctions and the Wnt/β-catenin signaling pathway, which interacts with the cytoplasmic domains of classical cadherins such as E-cadherin. Both plakoglobin and β-catenin have a central “armadillo” domain, consisting of 12 non-identical 42-amino-acid “arm” repeats, flanked by N- and C-terminal “head” and “tail” regions. The head and tail domains are sensitive to proteolysis and may be flexible. The structures of plakoglobin and β-catenin are similar; both are elongated molecules with closely packed arm repeats, most of which consist of three α-helices [94,95]. Their superhelical structures offer a positively charged groove where numerous β-catenin binding partners engage. For example, E-cadherin's cytoplasmic tail binds to this β-catenin site via an extended conformation [94]. E-cadherin shows almost identical interactions with plakoglobin [95], explaining why plakoglobin is interchangeable with β-catenin in adherens junctions. Both plakoglobin and β-catenin bind strongly to E-cadherin, whereas plakoglobin interacts with DSG1 much more strongly than does β-catenin, which is absent from desmosomes [95].
Alignment of desmoglein or desmocollin sequences with those of E-cadherin shows that most of the observed β-catenin/E-cadherin interactions are likely to be conserved in plakoglobin/desmosomal cadherin complexes [94]. Desmosomal cadherin binding sites on plakoglobin overlap, with arm repeats 1–3 being important for binding both desmoglein and desmocollin. Residues Ile127, Ile165, and Leu231 within these repeats are absolutely required for binding of either desmogleins or desmocollins [62]. While repeats 1–3 may be sufficient for desmoglein binding, desmocollin association requires both ends of the armadillo repeat domain [96]. As well as interacting with desmosomal cadherins, plakoglobin interacts with PKP2 and PKP3, but not PKP1, by immunoprecipitation [97,98]. The arm domain of plakoglobin is responsible for the interaction with PKP2 [98] and it also interacts with desmoplakin [99]. Tyrosine phosphorylation of plakoglobin is important in regulating its interaction with desmoplakin. In particular, the epidermal growth factor receptor (EGFR) phosphorylates Tyr693, Tyr724, and Tyr729 in the C-terminal tail of plakoglobin, so abolishing its interaction with desmoplakin [100]. Conversely, Src phosphorylates residue Tyr643 in plakoglobin, increasing its interaction with desmoplakin, but decreasing its interaction with E-cadherin and another adherens junction protein, α-catenin [101].
A homozygous 2-bp deletion in the gene encoding plakoglobin was the first genetic mutation in a desmosomal gene to be associated with ARVC [102]. The mutation results in a frameshift and premature termination of the plakoglobin protein (G680fsX690), causing the cardiocutaneous syndrome Naxos disease. As well as heart problems, patients exhibit the skin disorder palmoplantar keratoderma and have woolly hair. Recently, a missense mutation in the central armadillo repeat domain of plakoglobin (R256H) was reported, causing ARVC with palmoplantar keratoderma and alopecia [103]. Mutations in plakoglobin can result exclusively in skin disease. Thus, a homozygous nonsense mutation in the JUP gene (Q539X), which causes premature termination in the 10th arm repeat and complete loss of plakoglobin expression in the skin, has been described in a single patient [104]. The patient suffered from extreme skin fragility and massive fluid loss causing early death, but showed no signs of heart dysfunction [104]. Conversely, mutations that result in ARVC alone, without cutaneous manifestations, have been documented. One such mutation leads to the insertion of a Ser residue into the protease-sensitive head domain (S39_K40insS). The mutation results in a marked decrease in the amount of plakoglobin at the intercalated disk [105], a common feature of ARVC (see below). Similarly, a pathogenic missense mutation in the protease-sensitive head domain (T19I) is known to cause ARVC without skin disease [59]. Mutations in the central armadillo repeat domain (Fig. 3), including deletion of an Ile residue from the first arm repeat (I131del) [54], and a variety of missense mutations throughout the central domain (V159L, S225L, V407I, and V603L) [69,106], also cause ARVC without skin disease, while a homozygous E301G mutation was also detected in two patients with Naxos disease. It is not clear how mutations in plakoglobin cause ARVC. A reduction in ligand binding, leading to a decrease in desmosomal adhesion, is one possibility, as is loss of plakoglobin from the intercalated disk, leading to changes in intracellular signaling (see below).
Fig. 3.

Plakoglobin structure and locations of ARVC mutations. Crystal structure of the plakoglobin arm domain (PDB code: 3IFQ) (left) and electrostatic potential of surface-exposed residues (right). In the ribbon diagram, point mutations that are known to be pathogenic are shown as either copper (buried) or green (surface exposed). Although plakoglobin exhibits comparatively fewer pathogenic mutations than other desmosomal proteins, they are balanced between exposed and buried positions across the domain, with consequences on protein stability and binding properties that require further examination. In the electrostatic potential map, blue and red represent positively and negatively charged regions, respectively. The surface shows the charged E-cadherin binding groove, which is also proposed to bind to desmoglein and desmocollin [95]. Amino acids proposed to mediate desmoglein and desmocollin binding are highlighted in purple [95].
All three plakophilins contain a central armadillo repeat domain containing nine arm repeats and unstructured N- and C-terminal head and tail domains. Two PKP1 and two PKP2 isoforms are known. PKP1a and PKP1b are identical with the exception of a 21-amino-acid insert between arm repeats 3 and 4 and PKP2b is larger than PKP2a because of the insertion of 44 amino acids between arm repeats 2 and 3 [107,108]. The crystal structure of the plakophilin 1a central domain [109] is similar to that of plakoglobin [95], β-catenin [94], N458Y and p120 catenin [110]. The nine arm repeats, each consisting of three α-helices, pack together to form a superhelical structure [109]. Overall, the domain is sickle shaped, as a result of a long flexible insert between arm repeats 5 and 6 that introduces the bend into the structure. As well as interacting with plakoglobin, plakophilins interact with desmosomal cadherins, although some differences in affinity between different plakophilins and desmogleins may exist [97,98,111]. The unstructured plakophilin head domain appears to be primarily responsible for these interactions. All three plakophilins interact with desmoplakin [97,98,111].
Mutations leading to human disease have been described in both PKP1 and PKP2, but not PKP3. Mutations in PKP1 result in ectodermal dysplasia/skin fragility syndrome. In the first case reported, the disease was caused by two recessively inherited stop codons within DNA encoding the first and third arm repeats and was characterized by skin fragility, resulting in erosions, fissures, and keratoderma. The patient also exhibited defective hair growth, nail dystrophy, and a reduced ability to sweat [112]. Mutations in PKP2 are the most common cause of ARVC [59]. A total of 131 pathogenic mutations that occur throughout the PKP2 gene are listed in the ARVC database [47]. Four pathogenic missense mutations (Q59L, K112N, E137K, and P238L) are located in the unstructured head domain and may affect binding of other desmosomal proteins. Thus, the Q59L mutation disrupts interaction with desmoplakin in in vitro binding assays [113]. A large number of pathogenic missense mutations have been documented in the PKP2 central armadillo repeat domain (Fig. 4). To determine the effect of mutations in this domain, Kirchner et al. expressed wild-type PKP2, as well as three proteins containing single pathogenic missense mutations (C796R, S615F, and K654Q), a truncated protein containing a pathogenic frameshift mutation (C693fsX741) and two proteins containing amino acid changes of unknown significance (I531S and V587I) in cultured cardiac-derived HL-1 cells. Expression levels of the proteins containing pathogenic mutations were reduced, whereas those of the I531S and V587I proteins were not significantly changed when compared to the wild-type protein. Furthermore, the pathogenic mutant proteins exhibited a cytoplasmic localization, whereas the other two were localized at the membrane as expected. The most likely explanation for the results is that the pathogenic mutations adversely affect protein stability, with the unstable proteins being targeted for degradation, perhaps by calpain proteases [114]. One further pathogenic missense mutation, L847P, is found in the last arm repeat of PKP2 but its functional effects are not known. In addition to its role in intercellular adhesion, PKP2 interacts with protein kinase C-α [116], β-catenin [98], and RNA polymerase III [117], and may have additional roles in intercellular signaling and gene transcription. Whether these potential roles are affected by ARVC mutations remains to be determined.
Fig. 4.

Plakophilin 2 structure and locations of ARVC mutations. A sequence homology-based model of the entire PKP2 arm domain is shown based on a fragment of the PKP2 arm domain (PDB code: 3TT9) [114] and the PKP1 arm repeat domain (PDB code 1XM9) [109] as calculated by the PHYRE server [115]. Point mutations that are known to be pathogenic are shown as either copper (buried) or green (surface exposed). PKP2 exhibits the most numerous and diverse pathogenic mutations found in any desmosomal protein. This includes a mutation “hotspot” involving residues N613 and S615 as well as the newly identified L611R and L614P mutations that are linked to ARVC and are clustered within the hydrophobic core of the PKP2 arm domain.
Desmoplakin Structure and Function
Desmoplakin is an obligatory component of the desmosomal complex. Like other plakin family members, it has a tripartite structure and includes a globular N-terminal plakin domain, a central coiled-coil domain and a C-terminal tail domain. The latter consists of three plakin repeat domains (PRDs) designated A, B, and C, with a conserved linker joining PRDs B and C. A short glycine–serine–arginine (GSR)-rich element is found at the extreme C-terminal end of the protein. The plakin domain, together with a short region at the N-terminal end of the protein, which is predicted to be unstructured, is responsible for interacting with plakoglobin and plakophilin. The coiled-coil rod domain facilitates homodimerization and the C-terminal tail interacts with intermediate filament proteins [118,119]. Two isoforms created by alternative splicing are known: DPI (~ 332 kDa) and DPII (~ 260 kDa). They are identical except that DPII has a shorter rod domain. DPI and DPII are expressed in comparable amounts in most tissues except the heart, where DPII is detected only at low levels.
The desmoplakin plakin domain consists of six spectrin repeats (SRs) (SR3–8) and an Src homology 3 (SH3) domain [120]. The structure of a desmoplakin plakin construct spanning the first four SRs reveals a high level of structural homology to typical SR structures [121]. The four repeats form a rigid elongated structure with the SH3 domain located within a loop between two helices of the third SR. The SH3 domain extensively interacts with the preceding SR in a fashion that rigidifies that part of the plakin domain. The interdigitation between the SR4 and SH3 domains suggests that the SH3 domain is unlikely to interact with ligands [121]. Small-angle X-ray scattering studies of the entire desmoplakin plakin domain with all six SRs have shown that it is not a rigid rod but consists of two arms of four and two SRs, respectively, which are able to rotate about a protease-sensitive hinge. The desmoplakin plakin domain appears to alternate between extended “I”- and jack-knifed “U”-shaped conformers, while the plakin domains of its envoplakin and periplakin relatives predominantly form “L” shapes [24,122]. The recently elucidated plakin domain flexibility may be important in desmosome assembly, allowing the domain to “fish” for ligands, and the hinge could extend when desmosomes are subjected to mechanical force, so limiting SR unfolding and preventing damage to the desmosome.
Desmoplakin's central coiled-coil region forms a dimeric rod 130 nm in length [123,124], suggesting that it spans much of the desmosomal plaque. The crystal structures of desmoplakin PRDs B and C domains have been solved, and consist of 4.5 repeats of a 38-amino-acid plakin repeat motif [125]. The motif itself consists of a β-hairpin followed by two antiparallel α-helices (Fig. 5). A negatively charged residue at the fourth position of the first β-strand of the β-hairpin contacts a positively charged residue at position 19 of the first helix to “fix” the β-hairpin into place. Multiple hydrophobic contacts are used to stabilize the two antiparallel α-helices [125]. Each PRD forms a compact globular structure as a result of further hydrophobic contacts between the motifs. A positively charged groove is located on the surfaces of PRDs B and C between each of their paired plakin repeat motifs. Intermediate filaments possess many negatively charged patches and may form electrostatic interactions within the complementary groove. It appears that binding of desmoplakin to intermediate filaments is predominantly dependent on its PRDs. However, the linker domain between PRDs B and C could contribute to efficient binding, although by itself it only interacts weakly with intermediate filaments [126,127]. Interestingly, phosphorylation of Ser2849 within the GSR element at the extreme C-terminal end of desmoplakin is thought to regulate its interaction with the cytoskeleton [128].
Fig. 5.

Desmoplakin structures and locations of ARVC mutations. Crystal structures of desmoplakin (a) SRs 3-6 (PDB code: 3R6N), (b) PRD-B (PDB code: 1LM7), and (c) PRD-C (PDB code: 1LM5) are depicted as ribbon diagrams. Point mutations that are known to be pathogenic are shown as either copper (buried) or green (surface exposed). For PRDs B and C, electrostatic potential maps are also shown with blue and red representing positively and negatively charged groups, respectively. Both PRDs B and C possess a conserved basic groove, which it is speculated may represent intermediate filament binding sites [125]. Amino acid residues that are proposed to interact with vimentin [125] are indicated in purple. Also depicted are the newly identified and as of yet unpublished mutations of R222L and H618P.
Mutations in desmoplakin result in diseases that affect the heart, skin, and hair. Autosomal dominant mutations in DSP, resulting in a null allele and haploinsufficiency, cause striate palmoplantar keratoderma, which is characterized by thickened areas of skin, particularly on the palms and soles [129,130]. Carvajal syndrome is characterized by cardiomyopathy, palmoplantar keratoderma, and woolly hair. It is inherited in an autosomal recessive fashion and is similar in many ways to Naxos syndrome (caused by mutations in JUP; see above), although in Carvajal syndrome, it is primarily the left ventricle that is affected [131]. Carvajal syndrome can occur as a result of a homozygous one-base deletion leading to premature termination and a truncated protein lacking the PRD-C and GSR domains (S2542fsX2560). Skin fragility/woolly hair syndrome is characterized by palmoplantar keratoderma and woolly hair, but with no apparent cardiac involvement [132]. The syndrome has been described in two unrelated individuals. Both were compound heterozygotes, each with one nonsense mutation resulting in haploinsufficiency, and each with a missense mutation (either N287K in the second SR of the plakin domain, or R2366C in PRD-B). Compound heterozygosity for two DSP nonsense mutations, leading to the production of two truncated polypeptides lacking all three PRDs, can result in lethal acantholytic epidermolysis bullosa [133]. In lethal acantholytic epidermolysis bullosa, loss of intermediate filament binding leads to severe skin blistering, catastrophic fluid loss, and early neonatal death. In one case, a homozygous mutation (R1267X) that occurs in an exon encoding part of the DPI-specific rod region has been reported [134]. This mutation results in loss of expression of DPI while that of DPII is retained. The patient exhibited palmoplantar keratoderma, woolly hair, and early-onset cardiomyopathy but survived until early childhood (4 years), suggesting that DPII can compensate, at least to some extent, for the absence of DPI.
Structural Implications of Desmoplakin Mutations in ARVC
Mutations throughout the desmoplakin protein are linked to ARVC. Two missense mutations (V30M and Q90R) are located within the unstructured N-terminal region of the protein. One of these mutations adversely affects binding of plakoglobin to desmoplakin, although binding of PKP2 is unaffected [135]. Of the 25 pathogenic missense mutations listed in the ARVC database [47], 14 are located in the plakin domain, 6 are in the rod domain, and 5 are in the tail domain. Six of the mutations in the plakin domain are located within the SH3 domain [121]. Pathogenic mutations at conserved SH3 positions including S299R, N375I, I445V, and S507F are clustered near the SR4 binding site and are predicted to disrupt core stability and interdomain contact or both. Another mutation, K470E, occupies an exposed loop position and does not cause significant destabilization of the domain [121,122]. Other pathogenic mutations scattered along the length of the SR3–6 rod include R222L, D230N, N287K, N375I, E422K, S442F, I445V, N458Y, Y494F, S507F, S597L, and H618P (Fig. 5). Pathogenic R808H/C mutations within the fifth SR reduce the stability of a construct consisting of the last two SRs of the plakin domain, although the overall folded structure is maintained [122]. Thus, most of the pathogenic mutations in desmoplakin's plakin domain appear to perturb its structural stability and surface properties and hence could have severe consequences for desmosome function.
The specific effects of the six pathogenic ARVC mutations of the desmoplakin rod domain are unknown but could conceivably affect its coiled-coil structure, surface properties, and multimeric state. In the C-terminal region, there are three mutations—G2056R, G2375R, and R2639Q—that are found within PRDs A, B, and C, respectively. The mutations in PRDs B and C are not in the proposed filament binding grooves [125]. The sharp bend formed by the conserved Gly2375 could not be accommodated by replacement with an Arg residue. A positively charged residue is generally found at the position of Arg2639 and contacts a negatively charged amino acid (Asp2624) to “lock” a β-hairpin and stabilize the fold in a way that could be lost with the Gln substitution. Thus, either ARVC-linked mutation would be predicted to directly destabilize the PRD fold and could conceivably induce aggregation of the full-length desmoplakin protein. Two other mutations are found in the desmoplakin C-terminal tail. One (R2541K) is found in the conserved linker, and another (T2595I) is found in a 30-amino-acid region that is likely to be flexible and that follows the linker and precedes PRD-C. The effects of these mutations are unknown.
Intermediate Filament Structures and ARVC Mutations in Desmin
Desmoplakin interacts with three types of intermediate filament protein: desmin, keratin, and vimentin. Of these, only desmin is expressed in cardiac muscle. It is also expressed in skeletal and smooth muscle cells, and mutations in the DES gene are associated with a broad range of myopathies, including myofibrillar myopathies and/or dilated cardiomyopathy [136]. Desmin consists of a central 308-amino-acid α-helical rod domain that is flanked by non-α-helical head and tail domains. The central rod domain shows a seven-residue periodicity with the first and fourth positions for the most part occupied by apolar, hydrophobic amino acids. The heptad repeat arrangement allows for the formation of a homodimer, the elementary unit of desmin filaments. The α-helical rod domain is interrupted by three 8- to 16-amino-acid “linkers” (L1, L12, and L2) that interrupt the heptad periodicity and subdivide the domain into four separate segments (1A, 1B, 2A, and 2B). During formation of desmin intermediate filaments, homodimerization of central rod domains occurs to form a parallel coiled-coil dimer. Dimers then associate in a half-staggered antiparallel manner to form tetramers that associate laterally to form “unit-length filaments”. The latter then anneal longitudinally to ultimately yield long compacted intermediate filaments [137].
A number of mutations in the desmin gene have been associated with ARVC Ref. [138]. In the majority of cases, these are associated with muscle pathologies. One mutation (N116S) has been reported in a patient with ARVC and terminal heart failure, and without signs of clinical myopathy (although subclinical skeletal muscle disease was reported) [139]. The mutation is located in segment 1A of the rod domain and disrupts desmin filament formation and results in desmin immunoreactive protein aggregates (aggresomes) in cardiac and skeletal muscle. In a recent study, 91 ARVC index patients were screened for mutations in the desmin gene. Only two potential pathogenic missense mutations were found, suggesting that the frequency of DES mutations in ARVC in the absence of skeletal muscle involvement is low [138].
Potential Mechanisms for Fibrofatty Replacement in ARVC
ARVC is characterized by gradual loss of cardiomyocytes and their replacement by fibrous and fatty tissue. A number of potential mechanisms could explain the appearance of this fibrofatty tissue. Cardiomyocyte death, inflammation, and regional fibrosis could occur as a result of reductions in desmosomal adhesion. However, the appearance of adipocytes in the hearts of ARVC patients may be the result of changes in intracellular signal transduction pathways, in particular the Wnt/β-catenin pathway, in response to release of plakoglobin from defective desmosomal junctions. In the canonical Wnt/β-catenin pathway, cytoplasmic β-catenin is degraded in the absence of a Wnt signal. In the presence of Wnt, β-catenin is translocated to the nucleus where it interacts with transcription factors of the T-cell factor (Tcf)/lymphoid enhancer factor (Lef) family and drives transcription of β-catenin-responsive genes [140]. Plakoglobin interacts with many of the same proteins as β-catenin, including Tcf/Lef transcription factors. There is some evidence that suggests that plakoglobin may not be as effective in activating transcription of Tcf/Lef-dependent transcription as β-catenin itself [141] and that, in the heart, plakoglobin interferes with β-catenin transcriptional activity by competing for binding to Tcf/Lef [142]. This is important because loss of plakoglobin from the intercalated disk appears to occur in the majority of ARVC cases [143,144]. Wnt/β-catenin signaling enhances myogenesis and inhibits adipogenic transcription factors such as CCAAT/enhancer-binding protein α and peroxisome proliferator-activated receptor (PPAR) γ [145,146] and it is not difficult to envisage a mechanism whereby suppression of Wnt/β-catenin signaling by plakoglobin allows activation of adipogenic genes, thereby accounting for the characteristic fibrofatty replacement in the right ventricle of ARVC patients.
There is some experimental evidence that supports such a tissue replacement mechanism. Cardiac-specific deletion of one DSP allele in transgenic mice leads to nuclear localization of plakoglobin, reduced Wnt/β-catenin signaling, and increased numbers of adipocytes and fibrosis in the myocardium. Furthermore, cardiac dysfunction and ventricular arrhythmias are observed in the animals, recapitulating the phenotype of ARVC in patients [147]. A similar phenotype is seen in mice that are engineered to overexpress plakoglobin [148], suggesting that release of plakoglobin from the membrane and subsequent nuclear localization could be important in the etiology of ARVC. Interestingly, cardiac-specific knockout of plakoglobin leads to loss of cardiomyocytes, inflammation, fibrous tissue replacement, and cardiac dysfunction, but no increase in adipocytes [142,149]. An increase in Wnt/β-catenin signaling was reported in one such mouse model, so it may be that nuclear localization of plakoglobin, as well as inhibition of Wnt/β-catenin signaling, is required for adipogenesis in ARVC [142]. In a recent report [150], induced pluripotent stem cells (iPSCs) were generated from fibroblasts from a patient with ARVC (as a result of a homozygous frameshift mutation in PKP2). When the iPSCs were differentiated to beating embryoid bodies (using a defined cardiogenic medium), the mutant iPSC cardiomyocytes showed abnormal nuclear localization of plakoglobin and reduced Wnt/β-cateninin signaling. However, this was insufficient to induce pathological features of ARVC such as lipogenesis and apoptosis. Activation of the adipogenic transcription factor PPARγ alone did not induce ARVC pathologies, but when it was co-activated with PPARα, pronounced lipogenesis and apoptosis were observed. Adult cardiomyocytes produce most energy by fatty acid oxidation (regulated by PPARα), whereas embryonic cardiomyocytes utilize glycolysis so it may be that adult-like energy utilization is required for the development of ARVC [150].
The cellular origin of the excess adipocytes in ARVC has been something of a mystery. Cardiac progenitor cells isolated from mice overexpressing plakoglobin show enhanced adipogenesis and increased levels of adipogenic factors such as CCAAT/enhancer-binding protein α. Significantly, pharmacological activation of Wnt/β-catenin signaling in these cells prevented adipogenesis, and cardiac progenitor cells isolated from plakoglobin null mice were resistant to adipogenesis [148]. The right ventricle and outflow tract are derived from progenitor cells of the second heart field, and recently, genetic fate mapping following deletion of desmoplakin in different sets of cardiac progenitor cells has established these cells as the source of adipocytes in ARVC [148]. This then may explain why ARVC is primarily a disease of the right ventricle.
Future Directions
Structural analysis has now yielded models of parts of each protein within the desmosome and, together with binding studies, suggests how the domains could assemble into the supramolecular machines that mediate cell adhesion. The effects of pathogenic mutations and antibodies include incomplete assembly, loss of stability, and altered sites of interaction and articulation, allowing more accurate molecular phenotyping and diagnostics. This has yielded many new questions that remain unanswered. For example, most of the linkers, intrinsically disordered, coiled-coil and transmembrane regions remain unresolved yet mediate important molecular interactions. Although the entire desmosomal machine has been visualized at low resolution, it has yet to be modeled, either in mutant, compressed, or extended states. The unique mechanisms of each desmosomal protein isoform and their mixed assemblies remain unknown. How desmosomal cadherins mediate cis and trans coupling in the extracellular space and parallel dimerization and regular spacing inside the cell is unclear and may differ significantly from the classical cadherins in terms of architectural details. The desmosome assembly and internalization process is particularly opaque, yet is perhaps most critical for understanding how mutants fail to assemble correctly and are degraded. Finally understanding the compensation mechanisms whereby tissues tolerate apparently deleterious mutations yet manage to adapt to preserve their integrity remain a challenge, yet also presents opportunities for intervention.
Although no therapy is yet available to prevent the progression of ARVC, possible strategies are emerging. The correlation of low plakoglobin expression levels with disease manifestation in individuals with desmosomal gene mutations suggests that plakoglobin's loss from cell junctions is crucial for ARVC causation. When stressed by endurance exercise, plakoglobin deficiency could lead to critical damage to the heart tissue. Consequently, a combination of nitrates and diuretics that reduce preload and right atrial pressure is undergoing trials to reduce the stress inflicted on desmosomes of such individuals [151]. Another therapeutic approach comes from the observation that inhibition of EGFR signaling strengthens adhesion between squamous carcinoma cells [152]. EGFR inhibition leads to increased levels (1.7- to 2.0-fold) of DSC2 and DSG2 protein within the cells, accumulation of both desmosomal cadherins and desmoplakin at cell borders, and recruitment of intermediate filaments to desmosomes. Inhibition of the EGFR kinase blocks tyrosine phosphorylation of DSG2 and plakoglobin and it is likely that the increases in adhesive strength are as a result of increased desmosome assembly. Treatment with EGFR inhibitors can also be used to increase adhesive strength in p63-deficient keratinocytes, which show reduced expression of desmosomal cadherins and desmoplakin [153]. It is tempting to speculate that similar treatments could be developed to increase desmosome assembly in ARVC mutant cardiomyocytes, and so decrease the progression of the disease.
Acknowledgements
This research was funded by the Medical Research Council (C.A.), the Biotechnology and Biological Sciences Research Council, and the Wellcome Trust (M.C. and M.O.).
Edited by M. Sternberg
Footnotes
This is an open-access article distributed under the terms of the Creative Commons Attribution–Noncommercial–No Derivative Works License, which permits noncommercial use, distribution, and reproduction in any medium, provided the original author and source are credited.
References
- 1.Garrod D., Chidgey M. Desmosome structure, composition and function. Biochim Biophys Acta. 2008;1778:572–587. doi: 10.1016/j.bbamem.2007.07.014. [DOI] [PubMed] [Google Scholar]
- 2.Koeser J., Troyanovsky S.M., Grund C., Franke W.W. De novo formation of desmosomes in cultured cells upon transfection of genes encoding specific desmosomal components. Exp Cell Res. 2003;285:114–130. doi: 10.1016/s0014-4827(03)00016-8. [DOI] [PubMed] [Google Scholar]
- 3.North A.J., Chidgey M.A., Clarke J.P., Bardsley W.G., Garrod D.R. Distinct desmocollin isoforms occur in the same desmosomes and show reciprocally graded distributions in bovine nasal epidermis. Proc Natl Acad Sci U S A. 1996;93:7701–7705. doi: 10.1073/pnas.93.15.7701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Franke W.W., Borrmann C.M., Grund C., Pieperhoff S. The area composita of adhering junctions connecting heart muscle cells of vertebrates. I. Molecular definition in intercalated disks of cardiomyocytes by immunoelectron microscopy of desmosomal proteins. Eur J Cell Biol. 2006;85:69–82. doi: 10.1016/j.ejcb.2005.11.003. [DOI] [PubMed] [Google Scholar]
- 5.Borrmann C.M., Grund C., Kuhn C., Hofmann I., Pieperhoff S., Franke W.W. The area composita of adhering junctions connecting heart muscle cells of vertebrates. II. Colocalizations of desmosomal and fascia adhaerens molecules in the intercalated disk. Eur J Cell Biol. 2006;85:469–485. doi: 10.1016/j.ejcb.2006.02.009. [DOI] [PubMed] [Google Scholar]
- 6.Lahtinen A.M., Lehtonen E., Marjamaa A., Kaartinen M., Heliö T., Porthan K. Population-prevalent desmosomal mutations predisposing to arrhythmogenic right ventricular cardiomyopathy. Heart Rhythm. 2011;8:1214–1221. doi: 10.1016/j.hrthm.2011.03.015. [DOI] [PubMed] [Google Scholar]
- 7.Khan K., Hardy R., Haq A., Ogunbiyi O., Morton D., Chidgey M. Desmocollin switching in colorectal cancer. Br J Cancer. 2006;95:1367–1370. doi: 10.1038/sj.bjc.6603453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Oshiro M.M., Kim C.J., Wozniak R.J., Junk D.J., Munoz-Rodriguez J.L., Burr J.A. Epigenetic silencing of DSC3 is a common event in human breast cancer. Breast Cancer Res. 2005;7:R669–R680. doi: 10.1186/bcr1273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kurzen H., Münzing I., Hartschuh W. Expression of desmosomal proteins in squamous cell carcinomas of the skin. J Cutan Pathol. 2003;30:621–630. doi: 10.1034/j.1600-0560.2003.00122.x. [DOI] [PubMed] [Google Scholar]
- 10.Chen Y.J., Chang J.T., Lee L., Wang H.M., Liao C.T., Chiu C.C. DSG3 is overexpressed in head neck cancer and is a potential molecular target for inhibition of oncogenesis. Oncogene. 2007;26:467–476. doi: 10.1038/sj.onc.1209802. [DOI] [PubMed] [Google Scholar]
- 11.Biedermann K., Vogelsang H., Becker I., Plaschke S., Siewert J.R., Höfler H. Desmoglein 2 is expressed abnormally rather than mutated in familial and sporadic gastric cancer. J Pathol. 2005;207:199–206. doi: 10.1002/path.1821. [DOI] [PubMed] [Google Scholar]
- 12.Brennan D., Mahoney M.G. Increased expression of Dsg2 in malignant skin carcinomas: a tissue-microarray based study. Cell Adh Migr. 2009;3:148–154. doi: 10.4161/cam.3.2.7539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yashiro M., Nishioka N., Hirakawa K. Decreased expression of the adhesion molecule desmoglein-2 is associated with diffuse-type gastric carcinoma. Eur J Cancer. 2006;42:2397–2403. doi: 10.1016/j.ejca.2006.03.024. [DOI] [PubMed] [Google Scholar]
- 14.Breuninger S., Reidenbach S., Georg Sauer C., Ströbel P., Pfitzenmaier J., Trojan L. Desmosomal plakophilins in the prostate and prostatic adenocarcinomas: implications for diagnosis and tumor progression. Am J Pathol. 2010;176:2509–2519. doi: 10.2353/ajpath.2010.090737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Furukawa C., Daigo Y., Ishikawa N., Kato T., Ito T., Tsuchiya E. Plakophilin 3 oncogene as prognostic marker and therapeutic target for lung cancer. Cancer Res. 2005;65:7102–7110. doi: 10.1158/0008-5472.CAN-04-1877. [DOI] [PubMed] [Google Scholar]
- 16.Papagerakis S., Shabana A.-H., Pollock B.H., Papagerakis P., Depondt J., Berdal A. Altered desmoplakin expression at transcriptional and protein levels provides prognostic information in human oropharyngeal cancer. Hum Pathol. 2009;40:1320–1329. doi: 10.1016/j.humpath.2009.02.002. [DOI] [PubMed] [Google Scholar]
- 17.Dusek R.L., Attardi L.D. Desmosomes: new perpetrators in tumour suppression. Nat Rev Cancer. 2011;11:317–323. doi: 10.1038/nrc3051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shiina H., Breault J.E., Basset W.W., Enokida H., Urakami S., Li L.-C. Functional loss of the γ-catenin gene through epigenetic and genetic pathways in human prostate cancer. Cancer Res. 2005;65:2130–2138. doi: 10.1158/0008-5472.CAN-04-3398. [DOI] [PubMed] [Google Scholar]
- 19.Caca K., Kolligs F.T., Ji X., Hayes M., Qian J., Yahanda A. Beta- and gamma-catenin mutations, but not E-cadherin inactivation, underlie T-cell factor/lymphoid enhancer factor transcriptional deregulation in gastric and pancreatic cancer. Cell Growth Differ. 1999;10:369–376. [PubMed] [Google Scholar]
- 20.North A., Bardsley W., Hyam J., Bornslaeger E., Cordingley H., Trinnaman B. Molecular map of the desmosomal plaque. J Cell Sci. 1999;112:4325–4336. doi: 10.1242/jcs.112.23.4325. [DOI] [PubMed] [Google Scholar]
- 21.Al-Amoudi A., Diez D.C., Betts M.J., Frangakis A.S. The molecular architecture of cadherins in native epidermal desmosomes. Nature. 2007;450:832–837. doi: 10.1038/nature05994. [DOI] [PubMed] [Google Scholar]
- 22.Graham H.K., Hodson N.W., Hoyland J.A., Millward-Sadler S.J., Garrod D., Scothern A. Tissue section AFM: in situ ultrastructural imaging of native biomolecules. Matrix Biol. 2010;29:254–260. doi: 10.1016/j.matbio.2010.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.He W., Cowin P., Stokes D.L. Untangling desmosomal knots with electron tomography. Science. 2003;302:109–113. doi: 10.1126/science.1086957. [DOI] [PubMed] [Google Scholar]
- 24.Al-Jassar C., Bernadó P., Overduin M., Chidgey M. Hinged plakin domains provide specialized degrees of articulation in envoplakin, periplakin and desmoplakin. PLoS One. 2013;8(7):e69767. doi: 10.1371/journal.pone.0069767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Al-Amoudi A., Dubochet J., Norlen L. Nanostructure of the epidermal extracellular space as observed by cryo-electron microscopy of vitreous sections of human skin. J Invest Dermatol. 2005;124:764–777. doi: 10.1111/j.0022-202X.2005.23630.x. [DOI] [PubMed] [Google Scholar]
- 26.Al-Amoudi A., Chang J.-J., Leforestier A., McDowall A., Salamin L.M., Norlen L.P.O. Cryo-electron microscopy of vitreous sections. EMBO J. 2004;23:3583–3588. doi: 10.1038/sj.emboj.7600366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Al-Amoudi A., Castaño-Diez D., Devos D.P., Russell R.B., Johnson G.T., Frangakis A.S. The three-dimensional molecular structure of the desmosomal plaque. Proc Natl Acad Sci U S A. 2011;108:6480–6485. doi: 10.1073/pnas.1019469108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Garrod D.R., Merritt A.J., Nie Z. Desmosomal cadherins. Curr Opin Cell Biol. 2002;14:537–545. doi: 10.1016/s0955-0674(02)00366-6. [DOI] [PubMed] [Google Scholar]
- 29.Nagar B., Overduin M., Ikura M., Rini J.M. Structural basis of calcium-induced E-cadherin rigidification and dimerization. Nature. 1996;380:360–364. doi: 10.1038/380360a0. [DOI] [PubMed] [Google Scholar]
- 30.Overduin M., Harvey T., Bagby S., Tong K., Yau P., Takeichi M. Solution structure of the epithelial cadherin domain responsible for selective cell adhesion. Science. 1995;267:386–389. doi: 10.1126/science.7824937. [DOI] [PubMed] [Google Scholar]
- 31.Shapiro L., Fannon A.M., Kwong P.D., Thompson A., Lehmann M.S., Grubel G. Structural basis of cell–cell adhesion by cadherins. Nature. 1995;374:327–337. doi: 10.1038/374327a0. [DOI] [PubMed] [Google Scholar]
- 32.Boggon T.J., Murray J., Chappuis-Flament S., Wong E., Gumbiner B.M., Shapiro L. C-cadherin ectodomain structure and implications for cell adhesion mechanisms. Science. 2002;296:1308–1313. doi: 10.1126/science.1071559. [DOI] [PubMed] [Google Scholar]
- 33.Harrison O.J., Jin X., Hong S., Bahna F., Ahlsen G., Brasch J. The extracellular architecture of adherens junctions revealed by crystal structures of type I cadherins. Structure. 2011;19:244–256. doi: 10.1016/j.str.2010.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shapiro L., Weis W.I. Structure and biochemistry of cadherins and catenins. Cold Spring Harbor Perspect Biol. 2009;1 doi: 10.1101/cshperspect.a003053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nie Z., Merritt A., Rouhi-Parkouhi M., Tabernero L., Garrod D. Membrane-impermeable cross-linking provides evidence for homophilic, isoform-specific binding of desmosomal cadherins in epithelial cells. J Biol Chem. 2011;286:2143–2154. doi: 10.1074/jbc.M110.192245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Katsamba P., Carroll K., Ahlsen G., Bahna F., Vendome J., Posy S. Linking molecular affinity and cellular specificity in cadherin-mediated adhesion. Proc Natl Acad Sci U S A. 2009;106:11594–11599. doi: 10.1073/pnas.0905349106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wu Y., Jin X., Harrison O., Shapiro L., Honig B.H., Ben-Shaul A. Cooperativity between trans and cis interactions in cadherin-mediated junction formation. Proc Natl Acad Sci U S A. 2010;107:17592–17597. doi: 10.1073/pnas.1011247107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.McNutt N.S., Weinstein R.S. Membrane ultrastructure at mammalian intercellular junctions. Prog Biophys Mol Biol. 1973;26:45–101. doi: 10.1016/0079-6107(73)90017-5. [DOI] [PubMed] [Google Scholar]
- 39.Farquhar M.G., Palade G.E. Junctional complexes in various epithelia. J Cell Biol. 1963;17:375–412. doi: 10.1083/jcb.17.2.375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.McNutt N.S., Weinstein R.S. Carcinoma of the cervix: deficiency of nexus intercellular junctions. Science. 1969;165:597–599. doi: 10.1126/science.165.3893.597. [DOI] [PubMed] [Google Scholar]
- 41.Miyaguchi K. Ultrastructure of the zonula adherens revealed by rapid-freeze deep-etching. J Struct Biol. 2000;132:169–178. doi: 10.1006/jsbi.2000.4244. [DOI] [PubMed] [Google Scholar]
- 42.Getsios S., Amargo E.V., Dusek R.L., Ishii K., Sheu L., Godsel L.M. Coordinated expression of desmoglein 1 and desmocollin 1 regulates intercellular adhesion. Differentiation. 2004;72:419–433. doi: 10.1111/j.1432-0436.2004.07208008.x. [DOI] [PubMed] [Google Scholar]
- 43.Chitaev N.A., Troyanovsky S.M. Direct Ca2 +-dependent heterophilic interaction between desmosomal cadherins, desmoglein and desmocollin, contributes to cell–cell adhesion. J Cell Biol. 1997;138:193–201. doi: 10.1083/jcb.138.1.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mattey D.L., Garrod D.R. Splitting and internalization of the desmosomes of cultured kidney epithelial cells by reduction in calcium concentration. J Cell Sci. 1986;85:113–124. doi: 10.1242/jcs.85.1.113. [DOI] [PubMed] [Google Scholar]
- 45.Jin X., Walker M.A., Felsövályi K., Vendome J., Bahna F., Mannepalli S. Crystal structures of Drosophila N-cadherin ectodomain regions reveal a widely used class of Ca2 +-free interdomain linkers. Proc Natl Acad Sci U S A. 2012;109:E127–E134. doi: 10.1073/pnas.1117538108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sotomayor M., Weihofen W.A., Gaudet R., Corey D.P. Structure of a force-conveying cadherin bond essential for inner-ear mechanotransduction. Nature. 2012;492:128–132. doi: 10.1038/nature11590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.van der Zwaag P.A., Jongbloed J.D., van den Berg M.P., van der Smagt J.J., Jongbloed R., Bikker H. A genetic variants database for arrhythmogenic right ventricular dysplasia/cardiomyopathy. Hum Mutat. 2009;30:1278–1283. doi: 10.1002/humu.21064. [DOI] [PubMed] [Google Scholar]
- 48.Fressart V., Duthoit G., Donal E., Probst V., Deharo J.-C., Chevalier P. Desmosomal gene analysis in arrhythmogenic right ventricular dysplasia/cardiomyopathy: spectrum of mutations and clinical impact in practice. Europace. 2010;12:861–868. doi: 10.1093/europace/euq104. [DOI] [PubMed] [Google Scholar]
- 49.Syrris P., Ward D., Asimaki A., Evans A., Sen-Chowdhry S., Hughes S.E. Desmoglein-2 mutations in arrhythmogenic right ventricular cardiomyopathy: a genotype–phenotype characterization of familial disease. Eur Heart J. 2007;28:581–588. doi: 10.1093/eurheartj/ehl380. [DOI] [PubMed] [Google Scholar]
- 50.Awad M.M., Dalal D., Cho E., Amat-Alarcon N., James C., Tichnell C. DSG2 mutations contribute to arrhythmogenic right ventricular dysplasia/cardiomyopathy. Am J Hum Genet. 2006;79:136–142. doi: 10.1086/504393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Basso C., Czarnowska E., Della Barbera M., Bauce B., Beffagna G., Wlodarska E.K. Ultrastructural evidence of intercalated disc remodelling in arrhythmogenic right ventricular cardiomyopathy: an electron microscopy investigation on endomyocardial biopsies. Eur Heart J. 2006;27:1847–1854. doi: 10.1093/eurheartj/ehl095. [DOI] [PubMed] [Google Scholar]
- 52.Bhuiyan Z.A., Jongbloed J.D., van der Smagt J., Lombardi P.M., Wiesfeld A.C., Nelen M. Desmoglein-2 and desmocollin-2 mutations in dutch arrhythmogenic right ventricular dysplasia/cardiomypathy patients: results from a multicenter study. Circ Cardiovasc Genet. 2009;2:418–427. doi: 10.1161/CIRCGENETICS.108.839829. [DOI] [PubMed] [Google Scholar]
- 53.Pilichou K., Nava A., Basso C., Beffagna G., Bauce B., Lorenzon A. Mutations in desmoglein-2 gene are associated with arrhythmogenic right ventricular cardiomyopathy. Circulation. 2006;113:1171–1179. doi: 10.1161/CIRCULATIONAHA.105.583674. [DOI] [PubMed] [Google Scholar]
- 54.Xu T., Yang Z., Vatta M., Rampazzo A., Beffagna G., Pillichou K. Compound and digenic heterozygosity contributes to arrhythmogenic right ventricular cardiomyopathy. J Am Coll Cardiol. 2010;55:587–597. doi: 10.1016/j.jacc.2009.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bauce B., Rampazzo A., Basso C., Mazzotti E., Rigato I., Steriotis A. Clinical phenotype and diagnosis of arrhythmogenic right ventricular cardiomyopathy in pediatric patients carrying desmosomal gene mutations. Heart Rhythm. 2011;8:1686–1695. doi: 10.1016/j.hrthm.2011.06.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pilichou K., Remme C.A., Basso C., Campian M.E., Rizzo S., Barnett P. Myocyte necrosis underlies progressive myocardial dystrophy in mouse dsg2-related arrhythmogenic right ventricular cardiomyopathy. J Exp Med. 2009;206:1787–1802. doi: 10.1084/jem.20090641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rizzo S., Lodder E.M., Verkerk A.O., Wolswinkel R., Beekman L., Pilichou K. Intercalated disc abnormalities, reduced Na(+) current density, and conduction slowing in desmoglein-2 mutant mice prior to cardiomyopathic changes. Cardiovasc Res. 2012;95:409–418. doi: 10.1093/cvr/cvs219. [DOI] [PubMed] [Google Scholar]
- 58.Kant S., Krull P., Eisner S., Leube R.E., Krusche C.A. Histological and ultrastructural abnormalities in murine desmoglein 2-mutant hearts. Cell Tissue Res. 2012;348:249–259. doi: 10.1007/s00441-011-1322-3. [DOI] [PubMed] [Google Scholar]
- 59.den Haan A.D., Tan B.Y., Zikusoka M.N., Llado L.I., Jain R., Daly A. Comprehensive desmosome mutation analysis in North Americans with arrhythmogenic right ventricular dysplasia/cardiomyopathy. Circ Cardiovasc Genet. 2009;2:428–435. doi: 10.1161/CIRCGENETICS.109.858217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yu C.C., Yu C.H., Hsueh C.H., Yang C.T., Juang J.M., Hwang J.J. Arrhythmogenic right ventricular dysplasia: clinical characteristics and identification of novel desmosome gene mutations. J Formos Med Assoc. 2008;107:548–558. doi: 10.1016/S0929-6646(08)60168-0. [DOI] [PubMed] [Google Scholar]
- 61.De Bortoli M., Beffagna G., Bauce B., Lorenzon A., Smaniotto G., Rigato I. The p.A897KfsX4 frameshift variation in desmocollin-2 is not a causative mutation in arrhythmogenic right ventricular cardiomyopathy. Eur J Hum Genet. 2010;18:776–782. doi: 10.1038/ejhg.2010.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Chitaev N., Averbakh A., Troyanovsky R., Troyanovsky S. Molecular organization of the desmoglein–plakoglobin complex. J Cell Sci. 1998;111:1941–1949. doi: 10.1242/jcs.111.14.1941. [DOI] [PubMed] [Google Scholar]
- 63.Troyanovsky S.M., Troyanovsky R.B., Eshkind L.G., Krutovskikh V.A., Leube R.E., Franke W.W. Identification of the plakoglobin-binding domain in desmoglein and its role in plaque assembly and intermediate filament anchorage. J Cell Biol. 1994;127:151–160. doi: 10.1083/jcb.127.1.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Gehmlich K., Asimaki A., Cahill T.J., Ehler E., Syrris P., Zachara E. Novel missense mutations in exon 15 of desmoglein-2: role of the intracellular cadherin segment in arrhythmogenic right ventricular cardiomyopathy? Heart Rhythm. 2010;7:1446–1453. doi: 10.1016/j.hrthm.2010.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kami K., Chidgey M., Dafforn T., Overduin M. The desmoglein-specific cytoplasmic region is intrinsically disordered in solution and interacts with multiple desmosomal protein partners. J Mol Biol. 2009;386:531–543. doi: 10.1016/j.jmb.2008.12.054. [DOI] [PubMed] [Google Scholar]
- 66.Rutman A.J., Buxton R.S., Burdett I.D. Visualisation by electron microscopy of the unique part of the cytoplasmic domain of a desmoglein, a cadherin-like protein of the desmosome type of cell junction. FEBS Lett. 1994;353:194–196. doi: 10.1016/0014-5793(94)01049-8. [DOI] [PubMed] [Google Scholar]
- 67.Chen J., Nekrasova O.E., Patel D.M., Klessner J.L., Godsel L.M., Koetsier J.L. The C-terminal unique region of desmoglein 2 inhibits its internalization via tail–tail interactions. J Cell Biol. 2012;199:699–711. doi: 10.1083/jcb.201202105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Tan B.Y., Jain R., den Haan A.D., Chen Y., Dalal D., Tandri H. Shared desmosome gene findings in early and late onset arrhythmogenic right ventricular dysplasia/cardiomyopathy. J Cardiovasc Transl Res. 2010;3:663–673. doi: 10.1007/s12265-010-9224-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Christensen A.H., Benn M., Bundgaard H., Tybjaerg-Hansen A., Haunso S., Svendsen J.H. Wide spectrum of desmosomal mutations in Danish patients with arrhythmogenic right ventricular cardiomyopathy. J Med Genet. 2010;47:736–744. doi: 10.1136/jmg.2010.077891. [DOI] [PubMed] [Google Scholar]
- 70.Cirillo N., Lanza A., Prime S.S. Induction of hyper-adhesion attenuates autoimmune-induced keratinocyte cell–cell detachment and processing of adhesion molecules via mechanisms that involve PKC. Exp Cell Res. 2010;316:580–592. doi: 10.1016/j.yexcr.2009.10.005. [DOI] [PubMed] [Google Scholar]
- 71.Kimura T.E., Merritt A.J., Garrod D.R. Calcium-independent desmosomes of keratinocytes are hyper-adhesive. J Invest Dermatol. 2007;127:775–781. doi: 10.1038/sj.jid.5700643. [DOI] [PubMed] [Google Scholar]
- 72.Sugiyama N., Masuda T., Shinoda K., Nakamura A., Tomita M., Ishihama Y. Phosphopeptide enrichment by aliphatic hydroxy acid-modified metal oxide chromatography for nano-LC-MS/MS in proteomics applications. Mol Cell Proteomics. 2007;6:1103–1109. doi: 10.1074/mcp.T600060-MCP200. [DOI] [PubMed] [Google Scholar]
- 73.Amanchy R., Kalume D.E., Iwahori A., Zhong J., Pandey A. Phosphoproteome analysis of HeLa cells using stable isotope labeling with amino acids in cell culture (SILAC) J Proteome Res. 2005;4:1661–1671. doi: 10.1021/pr050134h. [DOI] [PubMed] [Google Scholar]
- 74.Olsen J.V., Blagoev B., Gnad F., Macek B., Kumar C., Mortensen P. Global, in vivo, and site-specific phosphorylation dynamics in signaling networks. Cell. 2006;127:635–648. doi: 10.1016/j.cell.2006.09.026. [DOI] [PubMed] [Google Scholar]
- 75.Wallis S., Lloyd S., Wise I., Ireland G., Fleming T.P., Garrod D. The alpha isoform of protein kinase C is involved in signaling the response of desmosomes to wounding in cultured epithelial cells. Mol Biol Cell. 2000;11:1077–1092. doi: 10.1091/mbc.11.3.1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Payne A.S., Hanakawa Y., Amagai M., Stanley J.R. Desmosomes and disease: pemphigus and bullous impetigo. Curr Opin Cell Biol. 2004;16:536–543. doi: 10.1016/j.ceb.2004.07.006. [DOI] [PubMed] [Google Scholar]
- 77.Amagai M., Tsunoda K., Suzuki H., Nishifuji K., Koyasu S., Nishikawa T. Use of autoantigen-knockout mice in developing an active autoimmune disease model for pemphigus. J Clin Invest. 2000;105:625–631. doi: 10.1172/JCI8748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Tsunoda K., Ota T., Aoki M., Yamada T., Nagai T., Nakagawa T. Induction of pemphigus phenotype by a mouse monoclonal antibody against the amino-terminal adhesive interface of desmoglein 3. J Immunol. 2003;170:2170–2178. doi: 10.4049/jimmunol.170.4.2170. [DOI] [PubMed] [Google Scholar]
- 79.Payne A.S., Ishii K., Kacir S., Lin C., Li H., Hanakawa Y. Genetic and functional characterization of human pemphigus vulgaris monoclonal autoantibodies isolated by phage display. J Clin Invest. 2005;115:888–899. doi: 10.1172/JCI24185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Sharma P.M., Choi E.J., Kuroda K., Hachiya T., Ishii K., Payne A.S. Pathogenic anti-desmoglein MAbs show variable ELISA activity because of preferential binding of mature versus proprotein isoforms of desmoglein 3. J Invest Dermatol. 2009;129:2309–2312. doi: 10.1038/jid.2009.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Di Zenzo G., Di Lullo G., Corti D., Calabresi V., Sinistro A., Vanzetta F. Pemphigus autoantibodies generated through somatic mutations target the desmoglein-3 cis-interface. J Clin Invest. 2012;122:3781–3790. doi: 10.1172/JCI64413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Chan P.T., Ohyama B., Nishifuji K., Yoshida K., Ishii K., Hashimoto T. Immune response towards the amino-terminus of desmoglein 1 prevails across different activity stages in nonendemic pemphigus foliaceus. Br J Dermatol. 2010;162:1242–1250. doi: 10.1111/j.1365-2133.2010.09696.x. [DOI] [PubMed] [Google Scholar]
- 83.Ohyama B., Nishifuji K., Chan P.T., Kawaguchi A., Yamashita T., Ishii N. Epitope spreading is rarely found in pemphigus vulgaris by large-scale longitudinal study using desmoglein 2-based swapped molecules. J Invest Dermatol. 2012;132:1158–1168. doi: 10.1038/jid.2011.448. [DOI] [PubMed] [Google Scholar]
- 84.Li N., Aoki V., Hans-Filho G., Rivitti E.A., Diaz L.A. The role of intramolecular epitope spreading in the pathogenesis of endemic pemphigus foliaceus (fogo selvagem) J Exp Med. 2003;197:1501–1510. doi: 10.1084/jem.20022031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Calkins C.C., Setzer S.V., Jennings J.M., Summers S., Tsunoda K., Amagai M. Desmoglein endocytosis and desmosome disassembly are coordinated responses to pemphigus autoantibodies. J Biol Chem. 2006;281:7623–7634. doi: 10.1074/jbc.M512447200. [DOI] [PubMed] [Google Scholar]
- 86.Syed S.-e.-H., Trinnaman B., Martin S., Major S., Hutchinson J., Magee A.I. Molecular interactions between desmosomal cadherins. Biochem J. 2002;362:317–327. doi: 10.1042/0264-6021:3620317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Heupel W.M., Zillikens D., Drenckhahn D., Waschke J. Pemphigus vulgaris IgG directly inhibit desmoglein 3-mediated transinteraction. J Immunol. 2008;181:1825–1834. doi: 10.4049/jimmunol.181.3.1825. [DOI] [PubMed] [Google Scholar]
- 88.Waschke J., Bruggeman P., Baumgartner W., Zillikens D., Drenckhahn D. Pemphigus foliaceus IgG causes dissociation of desmoglein 1-containing junctions without blocking desmoglein 1 transinteraction. J Clin Invest. 2005;115:3157–3165. doi: 10.1172/JCI23475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Barahona-Dussault C., Benito B., Campuzano O., Iglesias A., Leung T.L., Robb L. Role of genetic testing in arrhythmogenic right ventricular cardiomyopathy/dysplasia. Clin Genet. 2010;77:37–48. doi: 10.1111/j.1399-0004.2009.01282.x. [DOI] [PubMed] [Google Scholar]
- 90.Beffagna G., De Bortoli M., Nava A., Salamon M., Lorenzon A., Zaccolo M. Missense mutations in desmocollin-2 N-terminus, associated with arrhythmogenic right ventricular cardiomyopathy, affect intracellular localization of desmocollin-2 in vitro. BMC Med Genet. 2007;8:65. doi: 10.1186/1471-2350-8-65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Cox M.G., van der Zwaag P.A., van der Werf C., van der Smagt J.J., Noorman M., Bhuiyan Z.A. Arrhythmogenic right ventricular dysplasia/cardiomyopathy: pathogenic desmosome mutations in index-patients predict outcome of family screening: Dutch arrhythmogenic right ventricular dysplasia/cardiomyopathy genotype-phenotype follow-up study. Circulation. 2011;123:2690–2700. doi: 10.1161/CIRCULATIONAHA.110.988287. [DOI] [PubMed] [Google Scholar]
- 92.Gehmlich K., Syrris P., Reimann M., Asimaki A., Ehler E., Evans A. Molecular changes in the heart of a severe case of arrhythmogenic right ventricular cardiomyopathy caused by a desmoglein-2 null allele. Cardiovasc Pathol. 2012;21:275–282. doi: 10.1016/j.carpath.2011.09.005. [DOI] [PubMed] [Google Scholar]
- 93.Elliott P., O'Mahony C., Syrris P., Evans A., Rivera Sorensen C., Sheppard M.N. Prevalence of desmosomal protein gene mutations in patients with dilated cardiomyopathy. Circ Cardiovasc Genet. 2010;3:314–322. doi: 10.1161/CIRCGENETICS.110.937805. [DOI] [PubMed] [Google Scholar]
- 94.Huber A.H., Weis W.I. The structure of the beta-catenin/E-cadherin complex and the molecular basis of diverse ligand recognition by beta-catenin. Cell. 2001;105:391–402. doi: 10.1016/s0092-8674(01)00330-0. [DOI] [PubMed] [Google Scholar]
- 95.Choi H.-J., Gross J.C., Pokutta S., Weis W.I. Interactions of plakoglobin and beta-catenin with desmosomal cadherins: basis of selective exclusion of alpha- and beta-catenin from desmosomes. J Biol Chem. 2009;284:31776–31788. doi: 10.1074/jbc.M109.047928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Witcher L.L., Collins R., Puttagunta S., Mechanic S.E., Munson M., Gumbiner B. Desmosomal cadherin binding domains of plakoglobin. J Biol Chem. 1996;271:10904–10909. doi: 10.1074/jbc.271.18.10904. [DOI] [PubMed] [Google Scholar]
- 97.Bonné S., Gilbert B., Hatzfeld M., Chen X., Green K.J., van Roy F. Defining desmosomal plakophilin-3 interactions. J Cell Biol. 2003;161:403–416. doi: 10.1083/jcb.200303036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Chen X., Bonne S., Hatzfeld M., van Roy F., Green K.J. Protein binding and functional characterization of plakophilin 2. Evidence for its diverse roles in desmosomes and beta -catenin signaling. J Biol Chem. 2002;277:10512–10522. doi: 10.1074/jbc.M108765200. [DOI] [PubMed] [Google Scholar]
- 99.Kowalczyk A.P., Bornslaeger E.A., Borgwardt J.E., Palka H.L., Dhaliwal A.S., Corcoran C.M. The amino-terminal domain of desmoplakin binds to plakoglobin and clusters desmosomal cadherin–plakoglobin complexes. J Cell Biol. 1997;139:773–784. doi: 10.1083/jcb.139.3.773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Gaudry C.A., Palka H.L., Dusek R.L., Huen A.C., Khandekar M.J., Hudson L.G. Tyrosine-phosphorylated plakoglobin is associated with desmogleins but not desmoplakin after epidermal growth factor receptor activation. J Biol Chem. 2001;276:24871–24880. doi: 10.1074/jbc.M102731200. [DOI] [PubMed] [Google Scholar]
- 101.Miravet S., Piedra J., Castano J., Raurell I., Franci C., Dunach M. Tyrosine phosphorylation of plakoglobin causes contrary effects on its association with desmosomes and adherens junction components and modulates beta-catenin-mediated transcription. Mol Cell Biol. 2003;23:7391–7402. doi: 10.1128/MCB.23.20.7391-7402.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.McKoy G., Protonotarios N., Crosby A., Tsatsopoulou A., Anastasakis A., Coonar A. Identification of a deletion in plakoglobin in arrhythmogenic right ventricular cardiomyopathy with palmoplantar keratoderma and woolly hair (Naxos disease) Lancet. 2000;355:2119–2124. doi: 10.1016/S0140-6736(00)02379-5. [DOI] [PubMed] [Google Scholar]
- 103.Erken H., Yariz K.O., Duman D., Kaya C.T., Sayin T., Heper A.O. Cardiomyopathy with alopecia and palmoplantar keratoderma (CAPK) is caused by a JUP mutation. Br J Dermatol. 2011;165:917–921. doi: 10.1111/j.1365-2133.2011.10455.x. [DOI] [PubMed] [Google Scholar]
- 104.Pigors M., Kiritsi D., Krumpelmann S., Wagner N., He Y., Podda M. Lack of plakoglobin leads to lethal congenital epidermolysis bullosa: a novel clinico-genetic entity. Hum Mol Genet. 2011;20:1811–1819. doi: 10.1093/hmg/ddr064. [DOI] [PubMed] [Google Scholar]
- 105.Asimaki A., Syrris P., Wichter T., Matthias P., Saffitz J.E., McKenna W.J. A novel dominant mutation in plakoglobin causes arrhythmogenic right ventricular cardiomyopathy. Am J Hum Genet. 2007;81:964–973. doi: 10.1086/521633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Ly S., Marcus F.I., Xu T., Towbin J.A. A woman with incidental findings of ventricular aneurysms and a desmosomal cardiomyopathy. Heart Rhythm. 2008;5:1455–1457. doi: 10.1016/j.hrthm.2008.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Schmidt A., Langbein L., Rode M., Pratzel S., Zimbelmann R., Franke W.W. Plakophilins 1a and 1b: widespread nuclear proteins recruited in specific epithelial cells as desmosomal plaque components. Cell Tissue Res. 1997;290:481–499. doi: 10.1007/s004410050956. [DOI] [PubMed] [Google Scholar]
- 108.Mertens C., Kuhn C., Franke W.W. Plakophilins 2a and 2b: constitutive proteins of dual location in the karyoplasm and the desmosomal plaque. J Cell Biol. 1996;135:1009–1025. doi: 10.1083/jcb.135.4.1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Choi H.-J., Weis W.I. Structure of the armadillo repeat domain of plakophilin 1. J Mol Biol. 2005;346:367–376. doi: 10.1016/j.jmb.2004.11.048. [DOI] [PubMed] [Google Scholar]
- 110.Ishiyama N., Lee S.H., Liu S., Li G.Y., Smith M.J., Reichardt L.F. Dynamic and static interactions between p120 catenin and E-cadherin regulate the stability of cell–cell adhesion. Cell. 2010;141:117–128. doi: 10.1016/j.cell.2010.01.017. [DOI] [PubMed] [Google Scholar]
- 111.Hatzfeld M., Haffner C., Schulze K., Vinzens U. The function of plakophilin 1 in desmosome assembly and actin filament organization. J Cell Biol. 2000;149:209–222. doi: 10.1083/jcb.149.1.209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.McGrath J.A., McMillan J.R., Shemanko C.S., Runswick S.K., Leigh I.M., Lane E.B. Mutations in the plakophilin 1 gene result in ectodermal dysplasia/skin fragility syndrome. Nat Genet. 1997;17:240–244. doi: 10.1038/ng1097-240. [DOI] [PubMed] [Google Scholar]
- 113.Hall C., Li S., Li H., Creason V., Wahl J.K., III Arrhythmogenic right ventricular cardiomyopathy plakophilin-2 mutations disrupt desmosome assembly and stability. Cell Commun Adhes. 2009;16:15–27. doi: 10.1080/15419060903009329. [DOI] [PubMed] [Google Scholar]
- 114.Kirchner F., Schuetz A., Boldt L.-H., Martens K., Dittmar G., Haverkamp W. Molecular insights into arrhythmogenic right ventricular cardiomyopathy caused by plakophilin-2 missense mutations. Circ Cardiovasc Genet. 2012;5:400–411. doi: 10.1161/CIRCGENETICS.111.961854. [DOI] [PubMed] [Google Scholar]
- 115.Kelley L.A., Sternberg M.J. Protein structure prediction on the Web: a case study using the Phyre server. Nat Protoc. 2009;4:363–371. doi: 10.1038/nprot.2009.2. [DOI] [PubMed] [Google Scholar]
- 116.Bass-Zubek A.E., Hobbs R.P., Amargo E.V., Garcia N.J., Hsieh S.N., Chen X. Plakophilin 2: a critical scaffold for PKC alpha that regulates intercellular junction assembly. J Cell Biol. 2008;181:605–613. doi: 10.1083/jcb.200712133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Mertens C., Hofmann I., Wang Z., Teichmann M., Chong S.S., Schnölzer M. Nuclear particles containing RNA polymerase III complexes associated with the junctional plaque protein plakophilin 2. Proc Natl Acad Sci U S A. 2001;98:7795–7800. doi: 10.1073/pnas.141219498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Stappenbeck T.S., Bornslaeger E.A., Corcoran C.M., Luu H.H., Virata M.L., Green K.J. Functional analysis of desmoplakin domains: specification of the interaction with keratin versus vimentin intermediate filament networks. J Cell Biol. 1993;123:691–705. doi: 10.1083/jcb.123.3.691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Stappenbeck T.S., Green K.J. The desmoplakin carboxyl terminus coaligns with and specifically disrupts intermediate filament networks when expressed in cultured cells. J Cell Biol. 1992;116:1197–1209. doi: 10.1083/jcb.116.5.1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Sonnenberg A., Rojas A.M., de Pereda J.M. The structure of a tandem pair of spectrin repeats of plectin reveals a modular organization of the plakin domain. J Mol Biol. 2007;368:1379–1391. doi: 10.1016/j.jmb.2007.02.090. [DOI] [PubMed] [Google Scholar]
- 121.Choi H.-J., Weis W.I. Crystal structure of a rigid four-spectrin-repeat fragment of the human desmoplakin plakin domain. J Mol Biol. 2011;409:800–812. doi: 10.1016/j.jmb.2011.04.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Al-Jassar C., Knowles T., Jeeves M., Kami K., Behr E., Bikker H. The nonlinear structure of the desmoplakin plakin domain and the effects of cardiomyopathy-linked mutations. J Mol Biol. 2011;411:1049–1061. doi: 10.1016/j.jmb.2011.06.047. [DOI] [PubMed] [Google Scholar]
- 123.Green K.J., Parry D.A., Steinert P.M., Virata M.L., Wagner R.M., Angst B.D. Structure of the human desmoplakins. Implications for function in the desmosomal plaque. J Biol Chem. 1990;265:11406–11407. [PubMed] [Google Scholar]
- 124.O'Keefe E.J., Erickson H.P., Bennett V. Desmoplakin I and desmoplakin II. Purification and characterization. J Biol Chem. 1989;264:8310–8318. [PubMed] [Google Scholar]
- 125.Choi H.J., Park-Snyder S., Pascoe L.T., Green K.J., Weis W.I. Structures of two intermediate filament-binding fragments of desmoplakin reveal a unique repeat motif structure. Nat Struct Mol Biol. 2002;9:612–620. doi: 10.1038/nsb818. [DOI] [PubMed] [Google Scholar]
- 126.Fontao L., Favre B., Riou S., Geerts D., Jaunin F., Saurat J.-H. Interaction of the bullous pemphigoid antigen 1 (BP230) and desmoplakin with intermediate filaments is mediated by distinct sequences within their COOH terminus. Mol Biol Cell. 2003;14:1978–1992. doi: 10.1091/mbc.E02-08-0548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Lapouge K., Fontao L., Champliaud M.-F., Jaunin F., Frias M.A., Favre B. New insights into the molecular basis of desmoplakinand desmin-related cardiomyopathies. J Cell Sci. 2006;119:4974–4985. doi: 10.1242/jcs.03255. [DOI] [PubMed] [Google Scholar]
- 128.Stappenbeck T.S., Lamb J.A., Corcoran C.M., Green K.J. Phosphorylation of the desmoplakin COOH terminus negatively regulates its interaction with keratin intermediate filament networks. J Biol Chem. 1994;269:29351–29354. [PubMed] [Google Scholar]
- 129.Armstrong D.K., McKenna K.E., Purkis P.E., Green K.J., Eady R.A., Leigh I.M. Haploinsufficiency of desmoplakin causes a striate subtype of palmoplantar keratoderma. Hum Mol Genet. 1999;8:143–148. doi: 10.1093/hmg/8.1.143. [DOI] [PubMed] [Google Scholar]
- 130.Whittock N.V., Ashton G.H., Dopping-Hepenstal P.J., Gratian M.J., Keane F.M., Eady R.A. Striate palmoplantar keratoderma resulting from desmoplakin haploinsufficiency. J Invest Dermatol. 1999;113:940–946. doi: 10.1046/j.1523-1747.1999.00783.x. [DOI] [PubMed] [Google Scholar]
- 131.Norgett E.E., Hatsell S.J., Carvajal-Huerta L., Cabezas J.C., Common J., Purkis P.E. Recessive mutation in desmoplakin disrupts desmoplakin-intermediate filament interactions and causes dilated cardiomyopathy, woolly hair and keratoderma. Hum Mol Genet. 2000;9:2761–2766. doi: 10.1093/hmg/9.18.2761. [DOI] [PubMed] [Google Scholar]
- 132.Whittock N.V., Wan H., Morley S.M., Garzon M.C., Kristal L., Hyde P. Compound heterozygosity for non-sense and mis-sense mutations in desmoplakin underlies skin fragility/woolly hair syndrome. J Invest Dermatol. 2002;118:232–238. doi: 10.1046/j.0022-202x.2001.01664.x. [DOI] [PubMed] [Google Scholar]
- 133.Jonkman M.F., Pasmooij A.M., Pasmans S.G., van den Berg M.P., Ter Horst H.J., Timmer A. Loss of desmoplakin tail causes lethal acantholytic epidermolysis bullosa. Am J Hum Genet. 2005;77:653–660. doi: 10.1086/496901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Uzumcu A., Norgett E.E., Dindar A., Uyguner O., Nisli K., Kayserili H. Loss of desmoplakin isoform I causes early onset cardiomyopathy and heart failure in a Naxos-like syndrome. J Med Genet. 2006;43:e5. doi: 10.1136/jmg.2005.032904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Yang Z., Bowles N.E., Scherer S.E., Taylor M.D., Kearney D.L., Ge S. Desmosomal dysfunction due to mutations in desmoplakin causes arrhythmogenic right ventricular dysplasia/cardiomyopathy. Circ Res. 2006;99:646–655. doi: 10.1161/01.RES.0000241482.19382.c6. [DOI] [PubMed] [Google Scholar]
- 136.Goldfarb L.G., Dalakas M.C. Tragedy in a heartbeat: malfunctioning desmin causes skeletal and cardiac muscle disease. J Clin Invest. 2009;119:1806–1813. doi: 10.1172/JCI38027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Bar H., Strelkov S.V., Sjoberg G., Aebi U., Herrmann H. The biology of desmin filaments: how do mutations affect their structure, assembly, and organisation? J Struct Biol. 2004;148:137–152. doi: 10.1016/j.jsb.2004.04.003. [DOI] [PubMed] [Google Scholar]
- 138.Lorenzon A., Beffagna G., Bauce B., De Bortoli M., Li Mura I.E., Calore M. Desmin mutations and arrhythmogenic right ventricular cardiomyopathy. Am J Cardiol. 2013;111:400–405. doi: 10.1016/j.amjcard.2012.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Klauke B., Kossmann S., Gaertner A., Brand K., Stork I., Brodehl A. De novo desmin-mutation N116S is associated with arrhythmogenic right ventricular cardiomyopathy. Hum Mol Genet. 2010;19:4595–4607. doi: 10.1093/hmg/ddq387. [DOI] [PubMed] [Google Scholar]
- 140.Clevers H., Nusse R. Wnt/beta-catenin signaling and disease. Cell. 2012;149:1192–1205. doi: 10.1016/j.cell.2012.05.012. [DOI] [PubMed] [Google Scholar]
- 141.Shimizu M., Fukunaga Y., Ikenouchi J., Nagafuchi A. Defining the roles of beta-catenin and plakoglobin in LEF/T-cell factor-dependent transcription using beta-catenin/plakoglobin-null F9 cells. Mol Cell Biol. 2008;28:825–835. doi: 10.1128/MCB.02375-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Li J., Swope D., Raess N., Cheng L., Muller E.J., Radice G.L. Cardiac tissue-restricted deletion of plakoglobin results in progressive cardiomyopathy and activation of {beta}-catenin signaling. Mol Cell Biol. 2011;31:1134–1144. doi: 10.1128/MCB.01025-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Asimaki A., Tandri H., Huang H., Halushka M.K., Gautam S., Basso C. A new diagnostic test for arrhythmogenic right ventricular cardiomyopathy. N Engl J Med. 2009;360:1075–1084. doi: 10.1056/NEJMoa0808138. [DOI] [PubMed] [Google Scholar]
- 144.Munkholm J., Christensen A.H., Svendsen J.H., Andersen C.B. Usefulness of immunostaining for plakoglobin as a diagnostic marker of arrhythmogenic right ventricular cardiomyopathy. Am J Cardiol. 2012;109:272–275. doi: 10.1016/j.amjcard.2011.08.044. [DOI] [PubMed] [Google Scholar]
- 145.Ross S.E., Hemati N., Longo K.A., Bennett C.N., Lucas P.C., Erickson R.L. Inhibition of adipogenesis by Wnt signaling. Science. 2000;289:950–953. doi: 10.1126/science.289.5481.950. [DOI] [PubMed] [Google Scholar]
- 146.Nakamura T., Sano M., Songyang Z., Schneider M.D. A Wnt- and beta-catenin-dependent pathway for mammalian cardiac myogenesis. Proc Natl Acad Sci U S A. 2003;100:5834–5839. doi: 10.1073/pnas.0935626100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Garcia-Gras E., Lombardi R., Giocondo M.J., Willerson J.T., Schneider M.D., Khoury D.S. Suppression of canonical Wnt/beta-catenin signaling by nuclear plakoglobin recapitulates phenotype of arrhythmogenic right ventricular cardiomyopathy. J Clin Invest. 2006;116:2012–2021. doi: 10.1172/JCI27751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Lombardi R., da Graca Cabreira-Hansen M., Bell A., Fromm R.R., Willerson J.T., Marian A.J. Nuclear plakoglobin is essential for differentiation of cardiac progenitor cells to adipocytes in arrhythmogenic right ventricular cardiomyopathy. Circ Res. 2011;109:1342–1353. doi: 10.1161/CIRCRESAHA.111.255075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Li D., Liu Y., Maruyama M., Zhu W., Chen H., Zhang W. Restrictive loss of plakoglobin in cardiomyocytes leads to arrhythmogenic cardiomyopathy. Hum Mol Genet. 2011;20:4582–4596. doi: 10.1093/hmg/ddr392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Kim C., Wong J., Wen J., Wang S., Wang C., Spiering S. Studying arrhythmogenic right ventricular dysplasia with patient-specific iPSCs. Nature. 2013;494:105–110. doi: 10.1038/nature11799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Fabritz L., Fortmuller L., Yu T.Y., Paul M., Kirchhof P. Can preload-reducing therapy prevent disease progression in arrhythmogenic right ventricular cardiomyopathy? Experimental evidence and concept for a clinical trial. Prog Biophys Mol Biol. 2012;110:340–346. doi: 10.1016/j.pbiomolbio.2012.08.010. [DOI] [PubMed] [Google Scholar]
- 152.Lorch J.H., Klessner J., Park J.K., Getsios S., Wu Y.L., Stack M.S. Epidermal growth factor receptor inhibition promotes desmosome assembly and strengthens intercellular adhesion in squamous cell carcinoma cells. J Biol Chem. 2004;279:37191–37200. doi: 10.1074/jbc.M405123200. [DOI] [PubMed] [Google Scholar]
- 153.Ferone G., Mollo M.R., Thomason H.A., Antonini D., Zhou H., Ambrosio R. p63 control of desmosome gene expression and adhesion is compromised in AEC syndrome. Hum Mol Genet. 2013;22:531–543. doi: 10.1093/hmg/dds464. [DOI] [PMC free article] [PubMed] [Google Scholar]