

Abstract
Objective
To investigate the susceptibilities to and consequences of HIV-1 dual infection (DI).
Design
We compared clinical, virologic, and immunologic factors between participants who were dually infected with HIV-1 subtype B, and monoinfected (MI) controls who were matched by ongoing HIV risk factor.
Methods
The viral load and CD4 progressions of dually and singly infected participant groups were compared with linear mixed-effects models, and individual dynamics before and after superinfection were assessed with a structural change test (Chow test). Recombination breakpoint analysis (GARD), HLA frequency analysis, and cytotoxic T-lymphocyte (CTL) epitope mapping were also performed (HIV LANL Database).
Results
The viral loads of DI participants increased more over 3 years of follow-up than the viral loads of MI controls, while CD4 progressions of the two groups did not differ. Viral escape from CTL responses following superinfection was observed in two participants whose superinfecting strain completely replaced the initial strain. This pattern was not seen among participants whose superinfecting virus persisted in a recombinant form with the initial virus or was only detected transiently. Several HLA types were overrepresented in DI participants as compared to MI controls.
Conclusions
These results identify potential factors for DI susceptibility and further define its clinical consequences.
Keywords: HIV-1 dual infection, viral load, CD4 count, HLA, CTL
Introduction
Given the multitude of circulating and unique recombinant forms of HIV-1 [1], dual infection (DI) with two or more distinct viral strains has been relatively frequent since the beginning of the epidemic. Dual infection can be categorized either as interclade, when the two infecting strains originate from different clades, or intraclade otherwise. DI can be further classified as either: (i) coinfection (CI), when infection with the second strain occurs before an immune response to the first strain has been mounted, or (ii) superinfection (SI), when infection with the second strain occurs after an immune response to the first strain has been mounted [2].
Although 16 years have passed since the first cases of HIV-1 DI were documented [3–9], many questions remain about its clinical, virologic, and immunologic correlates. Clinically, DI has been associated with accelerated disease progression with a more rapid decline in CD4+ T-lymphocytes, and a shorter time to AIDS diagnosis [10]. Also, SI has been associated with an increase in viral load after SI [11–13]. Most previous reports have been anecdotal with SI identified using population-based sequencing of one HIV coding region within longitudinally collected samples. However, the detection of DI may be obscured by recombination when examining only one HIV-1 coding region [14], or if one of the strains is present at only low or transient levels [15, 16].
To further investigate DI, we performed a case-control study among well-characterized HIV-infected men who have sex with men (MSM) followed since primary infection. We screened longitudinal samples collected from cohort participants for DI with ultra-deep sequencing (UDS) of circulating viral populations in three HIV-1 coding regions. Within this case-control study, we investigated the clinical, virologic, and immunologic correlates associated with the presence of DI, including CI and SI. We report that intraclade subtype B DI is often associated with changes in viral load, and that certain HLA haplotypes are likely associated with SI. This report also characterizes a variety of virologic outcomes of intraclade DI, including complete replacement of one viral strain by the other, transient presence of a second strain, low-level persistence of one strain, and the production of recombinant viral populations. Taken together, these data may be of interest to patients and their health care providers concerning the susceptibilities and risks of DI.
Methods
This study was approved by UCSD Human Research Protection Program. Written, informed consent was obtained from all participants and guidelines from the U.S. Department of Health and Human Services were followed.
Study Participants, Demographics and HLA
All participants of the San Diego Primary HIV Infection Program between January 1998 and January 2007 who deferred antiretroviral therapy (ART) for at least the first 6 months, and had at least two blood samples available, were included. All received baseline drug resistance testing (Geneseq, Monogram Biosciences, South San Francisco, CA, USA), and estimated duration of infection (EDI) was calculated, per established protocols [16]. HLA data were collected for the HLA A, HLA B, HLA C and HLA DRB1 loci [17]. For participants with HLA B35, four-digit allele resolution was subsequently obtained.
For the case-control design, we matched mono-infected (MI) controls to participants who were classified as CI or SI based on: 1) follow-up >6 months, 2) ART naïve, 3) men who reported sex with men as their HIV risk factor, 4) ultra-deep sequencing (UDS) or single genome sequencing (SGS) at a time point <12 months from last date of follow-up with sequencing in two or more coding regions (including env), and 4) absence of phylogenetic evidence of DI, see below. Participants who were not classified as CI, SI, or MI were assigned to undetermined infection (UI) class. Demographic and clinical data available from these UI participants were then compared to the cases (CI and SI) and the controls (MI) to offer some assessment of potential biases.
Sequencing Methods
Population-based HIV-1 pol sequences (HXB2 coordinates 2253–3554), SGS of env C2-V3 and pol RT, and UDS of gag p24 (HXB2 coordinates 1366–1619), pol RT (HXB2 coordinates 2708–3242), and env C2-V3 (HXB2 coordinates 6928–7344) were generated [18] from cryostored blood plasma. UDS runs were done on batches of 16 samples physically separated with rubber gaskets on a 454 GS FLX Titanium picoliter plate (454 Life Sciences, Roche, Branford, CT). Read alignment and filtering were performed using a bioinformatics pipeline for the HyPhy software package [19] that selected high-quality UDS reads, generated sample-specific consensus sequences, aligned reads to the consensus, and performed phylogenetic analysis of specific coding regions, as previously described [20]. All HIV-1 infections were subtyped using pol sequences and the SCUEAL tool [21].
Dual Infection Screening and Confirmation
HIV-1 DI was suspected in a sample when the estimated lower 2.5% confidence bound of nucleotide divergence of at least one coding region’s SGS and/or UDS reads exceeded 5% for env and 2.5% for pol and gag, and when the phylogenetic structure of that region included two branches separated with bootstrap support >95% [18]. Based on timing of DI, participants with DI were then classified as CI (i.e. two distinct viral populations at baseline) or SI (i.e. one distinct viral population at baseline but two distinct populations in a subsequent sample at least three months beyond EDI). The algorithm for MI and DI determinations is diagrammed in Fig. 1. Participants who were classified as ambiguous or did not complete full sequencing evaluations were classified as UI and excluded from the case-control analysis.
Figure 1. Algorithm for characterizing DI status.
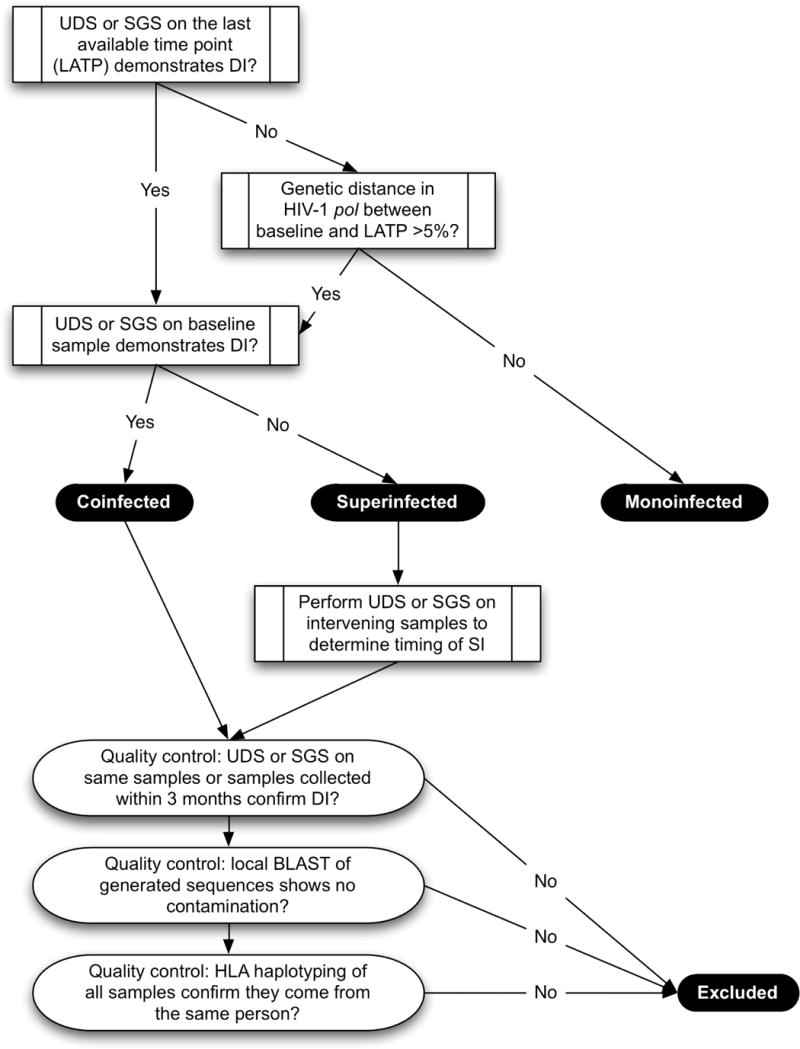
Legend: The study flowchart for the algorithm to determine DI status.
Clinical Correlates Analysis
Blood plasma was collected from participants at a median of 1.1 month intervals (IQR 0.7–2.1 months), and viral loads were measured at each time point (Amplicor v. 1.5, Roche, Indianapolis, IN) with a lower level of detection at 50 HIV-1 RNA copies/ml and CD4 counts measured by flow cytometry. To analyze the viral load and CD4 count dynamics with DI, we transformed viral load (log10) and CD4 counts (square root) and plotted these values versus time since EDI for each participant (Supp. Figs. 1–4). Linear mixed-effects models assessed transformed viral load and CD4 dynamics of each group (MI, CI, and SI) (nlme package in R). Each group was included as an indicator variable, and both intercepts and slopes were estimated. Each participant with SI was examined for structural change in viral load or CD4 count dynamics following SI using the Chow test [22]. A Kaplan-Meier analysis was performed for CD4 count persistence >400 cells/ul within MI, CI, and SI groups, and significance determined by Cox proportional hazards model.
Virologic and Immunologic Correlates Analysis
Recombination and Selection Analysis
Recombination breakpoint analysis of complete RT and C2V3 sequence alignments for each sample was performed using GARD [23]. Since the power of GARD to detect recombination is reduced if many highly similar sequences are present in an alignment, we represented each group of sequences that were all ≤ 1.0% distant in pol and ≤ 2.0 % distant in env from each other by a single randomly chosen clone. Pairwise genetic distances were obtained by maximum likelihood under the GTR model of nucleotide substitution [24]. We screened all sequences for each participant/coding region combination for evidence of positive selection using PARRIS [25], which corrects for possible confounding effects of recombination, using partitions produced by GARD as input.
Epitope mapping
To study the CTL epitopes inside pol and env sequences, we used available clade B HIV-1 CTL epitope maps for pol, RT, and env[ 26]. For participants with SI, we evaluated SGS generated from baseline samples and samples with detectable SI. For participants with CI, we evaluated SGS generated from baseline samples. We compared available sequences to CTL maps and predicted all epitopes targeted by CTL based on the two-digit HLA haplotype of each participant. To assess the HLA allele frequencies in the MI, CI, and SI groups, we used the online Los Alamos HLA Frequency Analysis tool, which reports both p-values and q-values (the false discovery rate) [26].
Results
Study Participants
After screening 110 male, ART-naïve participants, 18 were classified as MI (16.4%), 7 as SI (0.06%), 4 as CI (0.04%) and 81 as undetermined infection (UI) (73.6%). The UI participants were used as a comparison population. To lessen confounding by risk factor, all selected participants reported being MSM as their initial and on-going HIV risk factor. The median age at enrollment was 31 years (range: 19–58 years), and most participants were white (83%). There was no difference in baseline CD4 count between the CI, SI, MI, and UI groups (Kruskal-Wallis test p=0.68), but there was a trend for the SI group to have a lower baseline viral load (Kruskal-Wallis test p=0.08) (Table 1).
Table 1.
Study cohort.
| Groups | |||||
|---|---|---|---|---|---|
| Factor | Monoinfection (N=18) | Coinfection (N=4) | Superinfection (N=7) | Unknown infections (N=81) | |
| Age at enrollment: median years (range) | 28 (19–57) | 30 (20–41) | 30 (21–35) | 32 (21–58) | |
| EDI at enrollment: median days (range) | 85 (21–133) | 65 (45–85) | 85 (45–85) | 85 (21–365) | |
| Sex: N (%) | Male | 18 (100%) | 4 (100%) | 7 (100%) | 81 (100%) |
| Race: N (%) | White | 14 (78%) | 4 (100%) | 6 (86%) | 67 (83%) |
| Black or African American | 2 (11%) | 0 (0%) | 1 (14%) | 4 (5%) | |
| American Indian | 2 (11%) | 0 (0%) | 0 (0%) | 7 (9%) | |
| Ethnicity: N (%) | Hispanic or Latino | 3 (17%) | 1 (25%) | 1 (14%) | 15 (19%) |
| Not Hispanic or Latino | 7 (39%) | 2 (50%) | 3 (43%) | 32 (39%) | |
| Unknown | 8 (44%) | 1 (25%) | 3 (43%) | 34 (42%) | |
| HIV risk factors: N (%) | MSM | 18 (100%) | 4 (100%) | 7 (100%) | 81 (100%) |
| HIV viral load at enrollment: median log10 copies/mL (range) | 4.48 (1.70–7.14) | 3.85 (2.89–5.42) | 3.38 (2.25–6.36) | 4.81 (1.74–7.65) | |
| CD4 at enrollment: median cells/uL (range) | 551 (210–1119) | 562 (382–744) | 605 (321–866) | 526 (162–1193) | |
| Presence of HLA B35: N (%) | 5 (28%) | 0 (0%) | 5 (71%) | 17 (22%) * | |
| Presence of HLA A29+C16: N (%) | 0 (0%) | 2 (50%) | 2 (28%) | 6 (8%) * | |
Legend
3 out of 81 UI participants were not HLA haplotyped, so the HLA percentages are out of 77 rather than 81.
Clinical Correlates
Longitudinal viral loads of participants were plotted over three years of follow-up for the MI, CI, and SI groups (Fig. 2A). The SI group had a lower median viral load at baseline (3.38 vs. 3.85 and 4.48 log10HIV RNA copies/ml), and at one year of follow-up the viral load of the SI group (median 4.26, IQR 3.78–4.75 log10HIV RNA copies/ml) had overtaken that of the MI group (median 3.20, IQR 2.07–4.46 log10HIV RNA copies/ml). Compared to the MI group, the SI group had a significantly faster viral load increase over time according to the mixed effects model (p<0.001). The CI group also had a trend for a faster viral load increase over time than the MI group (p=0.09). These observations were consistent with structural changes in viral load after SI using the Chow test (Fig. 3A).
Figure 2. (A) Viral load and (B) CD4 progressions of the 3 infection groups.
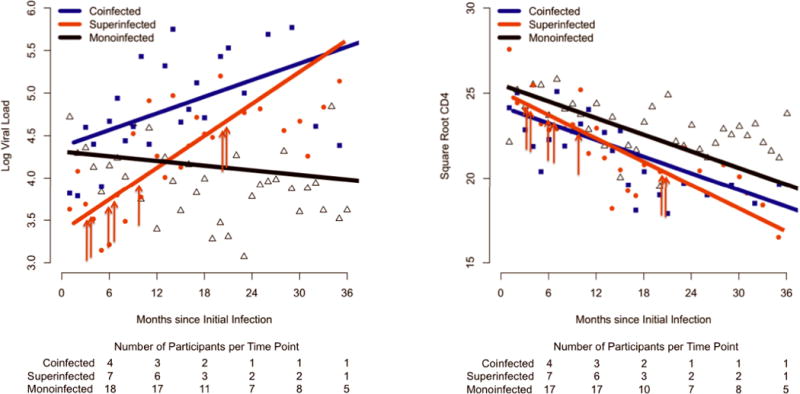
Legend: Mean VL and mean square root CD4 per month of infection for each group are plotted, and the linear mixed model fit is shown for each group. Red arrows indicate the timing of SI for each of the 7 SI participants.
Figure 3. Temporal dynamics of (A) viral loads and (B) CD4 counts in the seven superinfected (SI) patients.
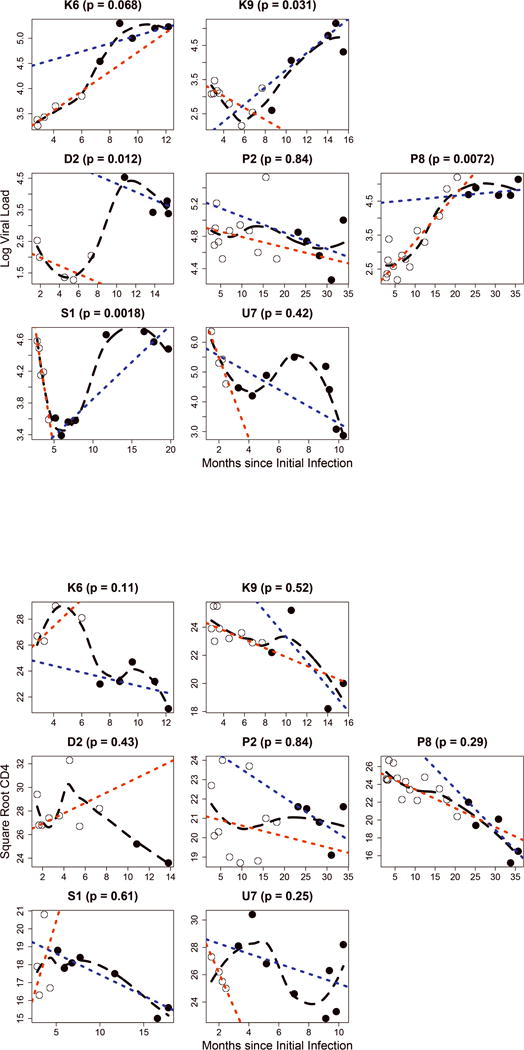
Legend: p – significance level for a structural shift in dynamics following SI (Chow’s test); open circles – samples preceding SI; filled circles – samples following SI; black dashed line – loess fit; red dotted line – linear fit to pre-SI points; blue dotted line – linear fit to post-SI points. EDI: estimated date of infection.
To assess the dynamics of CD4 cell counts, longitudinal counts were plotted over three years of follow-up (Fig. 2B). Compared to the MI group, neither the CI nor the SI groups had a significantly faster CD4 count decline (p>0.05), and Kaplan-Meier analysis showed no significant differences in CD4 count persistence above 400 cells/ul among the groups (data not shown). Unlike the changes with viral loads, none of the SI cases had a significant structural change in CD4 count dynamics (p > 0.05, Chow’s test) (Fig. 3B). We also investigated time to the start of ART, and although a higher proportion of SI compared to MI participants started ART within the 3-year period (7/7 SI vs. 11/18 MI), the difference was not statistically significant (p = 0.13, Fisher’s Exact Test). Overall, there were no significant differences between MI, UI, CI, SI, and DI groups for the initiation of ART during the 3 years (all p > 0.05, Fisher’s Exact Test).
Virologic Correlates during DI
Similar to previous reports, the ability to detect DI was increased by sequencing more than one coding region [14, 27], and together, UDS and single genome sequencing (SGS) identified CI in 4 and SI in 7 participants; UDS and SGS agreed on all samples sequenced with both methods. DI was detected in the env C2V3 coding region at ≥1 time point for all 11 DI participants, in 8/11 DI participants in RT, and in 2/8 (8 of the 11 were sequenced) in gag p24. Of the seven participants with SI, five acquired the second strain during their first year of infection and two during their second year. Two individuals (K6 and K9), previously characterized with DI [13], demonstrated complete replacement of the original strain in RT and C2V3, while in the remaining 5 cases, superinfecting viruses did not completely replace the original population in one or more HIV-1 coding regions (Supp. Table 1).
Since HIV-1 can recombine during DI [28], we screened C2V3 and RT sequences generated before and after DI for recombination [23]. Significant evidence for recombination was detected in C2V3 for participant P2 (Supp. Table 1) and in RT for two participants (D2 and S1) (p ≤ 0.05 Kishino-Hasegawa test [29]). Representative samples from initial, superinfecting, and recombinant populations of SI participant D2 are shown in Supp. Fig. 5.
Immunologic Correlates
If CTL responses to the initially infecting strain influence resistance to SI, then superinfecting strains may display signatures of escape in putative CTL epitopes. For the five SI cases that demonstrated incomplete or transient changes in their viral population after SI, amino acid differences between the initial and subsequent populations were no more likely to be within putative CTL epitopes than outside CTL epitopes (Table 2, permutation test p > 0.05). However, two SI participants (K6 and K9) demonstrated complete replacement of the pol and C2V3 coding regions following SI. For both of these cases, amino acid residues conferring putative CTL escape (Table 2) were observed in the later population of SI virus.
Table 2.
CTL escape by superinfecting viruses.
| Subject | HLA-A | HLA-B | C2V3 | RT | pol |
|---|---|---|---|---|---|
| K6 | 23, 29 | 44, 44 | In=Out | In>Out | In>Out |
| K9 | 03, 29 | 44, 57 | Out>In | In=Out | In>Out |
| D2 | 03, 32 | 35, 47 | Out>In | Out>In | NA |
| P2 | 01, 68 | 35, 57 | In=Out | NA | NA |
| P8 | 24, 31 | 35, 41 | In=Out | In=Out | NA |
| S1 | 24, 66 | 35, 41 | In=Out | In=Out | NA |
| U7 | 01, 03 | 08, 35 | In=Out | In=Out | NA |
Legend: For the coding regions C2V3, RT, and pol, the likelihood comparisons of amino acid differences between initial and subsequent populations to be inside vs. outside CTL epitopes are shown.
Previous studies have analyzed the association of HLA haplotypes with rates of HIV-1 disease progression. Although such analyses are influenced by many factors, including the ethnic makeup of study populations, HLA haplotypes B27, B57, and B58 have been associated with slower disease progression [30–32], and B7 and B35 with faster progression [30, 32, 33]. The timing of CTL epitope recognition during primary HIV-1 infection is also different among the HLA haplotypes [34]. A comparison of HLA frequencies in our study cohort revealed that the SI group had trends (Fisher’s Exact Test p-value range 0.07–0.11) for higher frequencies of A29, C16, and B35 and lower frequencies of DRB1–11 than the MI group (Supp. Table 2). In comparison, the CI group had higher frequencies of A29, C02, C16, B4, B39 and DRB1–08 and lower frequencies of C04 (p-value range 0.02–0.18) than the MI group (Supp. Table 2). Additionally, the CI group was enriched for the presence of the linked A29 and C16 alleles, as compared to the MI group (50% versus 0%), but this was not statistically significant (Table 1). The SI group had a trend for a higher frequency of HLA B35. This haplotype is associated with faster HIV disease progression [35] and targets epitopes less frequently during primary infection [34]. When evaluating for the frequency of having at least one copy of HLA B35 instead of its overall frequency, the prevalence of HLA B35 in the SI group was greater than in the MI group (71% versus 22%, p = 0.058 two-tailed Fisher’s Exact Test, Table 1). Similarly, the prevalence of HLA B35 allele was greater than would be expected for a similar population in the United States, independent of racial or ethnic group [36, 37]. Interestingly, the two cases of SI with complete replacement of the viral populations (K6 and K9) were the only two SI participants who lacked HLA B35. As the type (Px or Py) of HLA B35 has been associated with peptide binding specificity and HIV disease progression [35], we compared those in the SI and MI groups for presence of HLA B35 Px or Py using four-digit HLA haplotyping. Although these numbers are very small (3 out of 5 of MI and 2 out of 5 of the SI subjects had Px B35), we found no difference in the frequency of these types of HLA B35 associated with SI.
Discussion
The clinical, virologic, and immunologic correlates of DI are poorly understood, largely due to insufficient numbers of subjects screened and characterized for DI. Understanding these correlates of intraclade DI is important because it is likely more common than interclade DI, given the propensity of HIV-1 clades to be distributed unevenly throughout the world. For example, over 90% of HIV-infections in the US are with subtype B virus [38], so if an HIV-infected person is exposed to SI in the United States and SI occurs, then it will most likely be an acquisition of a second HIV-1 clade B virus.
The clinical consequences of DI are likely influenced by the immune capability and reactivity of the individual, and this study found that DI (both SI and CI) was associated with faster viral load increases than the viral load changes observed in MI controls. Unlike viral load dynamics, CD4 dynamics of the DI groups did not differ significantly from that of the MI group. In four out of seven individuals with SI, we could detect a significant structural shift in viral load dynamics, indicating that viral load progression frequently but not always changes following SI, and that other factors must impact viral load dynamics following SI. Interestingly, this SI group had lower baseline viral loads than both CI and MI groups, but the significance of this finding remains unclear. One possibility is that lower viral loads during initial infection may not be able to stimulate sufficient CTL or neutralizing antibody responses to protect against SI. Another is that the individuals with SI may have been initially infected with a less fit strain and became superinfected after exposure to a more fit strain.
As virus-specific CTL immune responses that develop during primary HIV infection are responsible for the earliest control of viral replication [34, 39–43] and viral set point [44], we investigated if there was evidence of CTL escape in new variants observed after DI. There was no detectable evidence for CTL pressure and viral escape in any of the participants demonstrating transient DI. However, potential CTL escape was identified in the two participants who had replaced the two evaluated coding regions (RT and C2V3) completely. Interestingly, the two participants with complete viral replacement were the only ones with SI who did not have HLA B35. Overall, this evidence suggests that CTL responses that develop during primary HIV infection may confer some degree of resistance to SI unless the host is exposed to a strain containing residues permitting CTL escape. Therefore, a superinfecting strain with CTL escape mutations possesses an immediate selective advantage over the incumbent strain and can quickly replace it. Individuals with HLA haplotypes that mount late or weak responses (e.g., HLA-B35) also seem to be more susceptible to SI; however, these early observations require evaluation in larger cohort studies.
There are several limitations to the current study. Since this study used an observational cohort to select cases and controls, the decisions to initiate ART by participants and their health care providers varied, which likely limited our ability to evaluate all clinical consequences of DI, as well as potentially confounding some results. Although unavoidable in a case-control study design, there could be a bias in selecting the MI controls, as it is possible that these controls do not adequately represent the natural history of HIV-1 MI. The inclusion criteria associated with the “confirmation” of MI using sequencing methods could cause a systematic selection bias in the selection of the controls, especially since it is impossible to rule out that DI never took place among the MI group. The confirmation of DI may also be biased towards the detection of those DI individuals who have distinct viral populations that comprise a certain level of co-circulation or where the viral population has been completely replaced over time. Additionally, in the absence of a gold standard method for determination of DI, our methods for confirmation of DI were used to limit the number of false positives for DI due to a laboratory mistake, i.e. sample mix-up or contamination. The resulting conservative bias may have limited our number of observed cases and subsequently our power to see an effect. A more liberal set of criteria for confirmation of DI would likely impact the results of the clinical progressions of different groups, as well as the HLA analysis. However, our very conservative criteria increase our confidence in the results that were derived.
Despite these limitations, this study represents a rigorous evaluation of HIV-1 intraclade DI and identifies many instances of transient DI during the first two years of initial infection. Transient DI seems to occur more often among individuals who develop mature CTL responses later in infection, like those with HLA-B35 haplotypes. However, when B35 is not present and there is complete replacement of the viral population during DI, DI is most often associated with viral genetic differences consistent with CTL escape. This study also identified that both CI and SI are associated with higher blood plasma viral loads, but faster loss of CD4 T-cell counts was not observed. Although it will need to be confirmed in larger studies, these results suggest that individuals with lower baseline viral loads may be more susceptible to SI. Taken together, these data may provide information for patients and their health care providers on potential SI susceptibilities (e.g. HLA B35 haplotype and low baseline viral load) and clinical consequences of SI (e.g. increased viral load) that can be important factors to consider in discussing the risks of repeat exposure to HIV-1 after initial infection.
Supplementary Material
Legend: Log viral load progressions are plotted over the length of follow-up. Filled circles indicate dual infection.
Legend: Log viral load progressions are plotted over the length of follow-up. Open circles indicate monoinfection.
Legend: Square root CD4 progressions are plotted over the length of follow-up. Filled circles indicate dual infection.
Legend: Square root CD4 progressions are plotted over the length of follow-up. Open circles indicate monoinfection.
Legend: clones within 1% of depicted sequences are not shown for clarity. The recombination breakpoint and corresponding trees are inferred by GARD. Interior branches with significant bootstrap support (Neighbor Joining, Tamura-Nei 93 distance, 1000 replicates) are accordingly annotated. Note that the viral population sampled in 01/1999 consists exclusively of RT recombinants before the original (open squares) and superinfecting (divergent triangle) sequences.
Supp. Table 1. Viral population dynamics of superinfected participants.
Supp. Table 2. Population HLA Frequency Comparisons.
Acknowledgments
This work was supported by National Institutes of Health grants MH083552, AI077304, AI69432, MH62512, AI27670, AI38858, AI43638, AI43752, AI047745, NS51132, GM093939, UCSD Centers for AIDS Research Translational Virology, Molecular Biology and Bioinformatics Evolutionary Analysis and Statistics Cores (AI36214), AI29164, AI47745, AI 064086, AI57167, GM093939, the International AIDS Vaccine Initiative, the Veterans Affairs Healthcare System, and the James B. Pendleton Charitable Trust, and the California HIV/AIDS Research Program D08-SD-316.
We would like to thank David Butler, Anya Umlauf, and Levon Budagyan for their insightful comments and Demetrius dela Cruz for his administrative assistance.
Footnotes
Conflicts of Interest and Source of Funding:
Sergei Kosakovsky Pond: has consulted for Monogram Biosciences and Gen-Probe.
Douglas D. Richman: has consulted for Biota, Chimerix, Gen-Probe, Merck, Bristol Myers Squibb, Gilead, Idenix, Monogram Biosciences, and Vertex.
Davey M. Smith: has received grant support from Pfizer and has consulted for Gen-Probe. For the remaining authors, none were declared.
Author contributions:
Mary Pacold: assisted in study design, assisted in the collection, analysis and interpretation of data, and in the writing of the report.
Sergei Kosakovsky Pond: assisted in the analysis and interpretation of data, and in the writing of the report.
Gabriel Wagner: assisted in the analysis and interpretation of data, and in the writing of the report.
Wayne Delport: assisted in study design, assisted in the analysis and interpretation of data, and in the writing of the report.
Susan J. Little: assisted in study design, assisted in the collection and interpretation of data, and in the writing of the report.
Douglas D. Richman: assisted in study design, assisted in the collection and interpretation of data, and in the writing of the report.
Davey M. Smith: conceived study design, assisted in the collection, analysis and interpretation of data, and in the writing of the report.
References
- 1.McCutchan FE. Global epidemiology of HIV. J Med Virol. 2006;78(Suppl 1):S7–S12. doi: 10.1002/jmv.20599. [DOI] [PubMed] [Google Scholar]
- 2.Smith D, Richman D, Little S. HIV superinfection. J Infect Dis. 2005;192:438–444. doi: 10.1086/431682. [DOI] [PubMed] [Google Scholar]
- 3.Artenstein A, VanCott T, Mascola J, Carr J, Hegerich P, Gaywee J, et al. Dual infection with human immunodeficiency virus type 1 of distinct envelope subtypes in humans. J Infect Dis. 1995;171:805–810. doi: 10.1093/infdis/171.4.805. [DOI] [PubMed] [Google Scholar]
- 4.Diaz R, Sabino E, Mayer A, Mosley J, Busch M. Dual human immunodeficiency virus type 1 infection and recombination in a dually exposed transfusion recipient. The Transfusion Safety Study Group. J Virol. 1995;69:3273–3281. doi: 10.1128/jvi.69.6.3273-3281.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pieniazek D, Janini LM, Ramos A, Tanuri A, Schechter M, Peralta JM, et al. HIV-1 patients may harbor viruses of different phylogenetic subtypes: implications for the evolution of the HIV/AIDS pandemic. Emerging Infect Dis. 1995;1:86–88. doi: 10.3201/eid0103.950303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sala M, Pelletier E, Wain-Hobson S. HIV-1 gp120 sequences from a doubly infected drug user. AIDS Res Hum Retroviruses. 1995;11:653–655. doi: 10.1089/aid.1995.11.653. [DOI] [PubMed] [Google Scholar]
- 7.Sala M, Zambruno G, Vartanian J, Marconi A, Bertazzoni U, Wain-Hobson S. Spatial discontinuities in human immunodeficiency virus type 1 quasispecies derived from epidermal Langerhans cells of a patient with AIDS and evidence for double infection. J Virol. 1994;68:5280–5283. doi: 10.1128/jvi.68.8.5280-5283.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Xin KQ, Ma XH, Crandall KA, Bukawa H, Ishigatsubo Y, Kawamoto S, Okuda K. Dual infection with HIV-1 Thai subtype B and E. Lancet. 1995;346:1372–1373. doi: 10.1016/s0140-6736(95)92391-8. [DOI] [PubMed] [Google Scholar]
- 9.Zhu T, Wang N, Carr A, Wolinsky S, Ho D. Evidence for coinfection by multiple strains of human immunodeficiency virus type 1 subtype B in an acute seroconvertor. J Virol. 1995;69:1324–1327. doi: 10.1128/jvi.69.2.1324-1327.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gottlieb G, Nickle D, Jensen M, Wong K, Grobler J, Li F, et al. Dual HIV-1 infection associated with rapid disease progression. Lancet. 2004;363:619–622. doi: 10.1016/S0140-6736(04)15596-7. [DOI] [PubMed] [Google Scholar]
- 11.Altfeld M, Allen T, Yu X, Johnston M, Agrawal D, Korber B, et al. HIV-1 superinfection despite broad CD8+ T-cell responses containing replication of the primary virus. Nature. 2002;420:434–439. doi: 10.1038/nature01200. [DOI] [PubMed] [Google Scholar]
- 12.Jost S, Bernard M, Kaiser L, Yerly S, Hirschel B, Samri A, et al. A patient with HIV-1 superinfection. N Engl J Med. 2002;347:731–736. doi: 10.1056/NEJMoa020263. [DOI] [PubMed] [Google Scholar]
- 13.Smith D, Wong J, Hightower G, Ignacio C, Koelsch K, Daar E, et al. Incidence of HIV superinfection following primary infection. JAMA. 2004;292:1177–1178. doi: 10.1001/jama.292.10.1177. [DOI] [PubMed] [Google Scholar]
- 14.Piantadosi A, Ngayo M, Chohan B, Overbaugh J. Examination of a second region of the HIV type 1 genome reveals additional cases of superinfection. AIDS Res Hum Retroviruses. 2008;24:1221. doi: 10.1089/aid.2008.0100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yerly S, Jost S, Monnat M, Telenti A, Cavassini M, Chave J, et al. HIV-1 co/super-infection in intravenous drug users. AIDS. 2004;18:1413–1421. doi: 10.1097/01.aids.0000131330.28762.0c. [DOI] [PubMed] [Google Scholar]
- 16.Smith D, Strain M, Frost S, Pillai S, Wong J, Wrin T, et al. Lack of neutralizing antibody response to HIV-1 predisposes to superinfection. Virology. 2006;355:1–5. doi: 10.1016/j.virol.2006.08.009. [DOI] [PubMed] [Google Scholar]
- 17.Ahuja SK, Kulkarni H, Catano G, Agan BK, Camargo JF, He W, et al. CCL3L1-CCR5 genotype influences durability of immune recovery during antiretroviral therapy of HIV-1-infected individuals. Nat Med. 2008;14:413–420. doi: 10.1038/nm1741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pacold M, Smith D, Little S, Cheng PM, Jordan P, Ignacio C, et al. Comparison of Methods to Detect HIV Dual Infection. AIDS research and human retroviruses. 2010;26:1291–1298. doi: 10.1089/aid.2010.0042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pond S, Frost S, Muse S. HyPhy: hypothesis testing using phylogenies. Bioinformatics. 2005;21:676–679. doi: 10.1093/bioinformatics/bti079. [DOI] [PubMed] [Google Scholar]
- 20.Gianella S, Delport W, Pacold ME, Young JA, Choi JY, Little SJ, et al. Detection of Minority Resistance during Early HIV-1 Infection: Natural Variation and Spurious Detection Rather than Transmission and Evolution of Multiple Viral Variants. J Virol. doi: 10.1128/JVI.02582-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kosakovsky Pond SL, Posada D, Stawiski E, Chappey C, Poon AFY, Hughes G, et al. An evolutionary model-based algorithm for accurate phylogenetic breakpoint mapping and subtype prediction in HIV-1. PLoS Comput Biol. 2009;5:e1000581. doi: 10.1371/journal.pcbi.1000581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chow G. Tests of equality between sets of coefficients in two linear regressions. Econometrica: Journal of the Econometric Society. 1960 [Google Scholar]
- 23.Kosakovsky Pond S, Posada D, Gravenor M, Woelk C, Frost S. GARD: a genetic algorithm for recombination detection. Bioinformatics. 2006;22:3096–3098. doi: 10.1093/bioinformatics/btl474. [DOI] [PubMed] [Google Scholar]
- 24.Tavare S. Some Probabilistic and Statistical Problems in the Analysis of DNA Sequences. American Mathematical Society: Lectures on Mathematics in the Life Sciences: Amer Mathematical Society. 1986:57–86. [Google Scholar]
- 25.Scheffler K, Martin DP, Seoighe C. Robust inference of positive selection from recombining coding sequences. Bioinformatics (Oxford, England) 2006;22:2493–2499. doi: 10.1093/bioinformatics/btl427. [DOI] [PubMed] [Google Scholar]
- 26.http://www.hiv.lanl.gov/content/immunology/. In.
- 27.Hoelscher M, Dowling W, Sanders-Buell E, Carr J, Harris M, Thomschke A, et al. Detection of HIV-1 subtypes, recombinants, and dual infections in east Africa by a multi-region hybridization assay. AIDS. 2002;16:2055–2064. doi: 10.1097/00002030-200210180-00011. [DOI] [PubMed] [Google Scholar]
- 28.Kijak G, McCutchan F. HIV diversity, molecular epidemiology, and the role of recombination. Curr Infect Dis Rep. 2005;7:480–488. doi: 10.1007/s11908-005-0051-8. [DOI] [PubMed] [Google Scholar]
- 29.Kishino H, Hasegawa M. Evaluation of the maximum likelihood estimate of the evolutionary tree topologies from DNA sequence data, and the branching order in hominoidea. J Mol Evol. 1989;29:170–179. doi: 10.1007/BF02100115. [DOI] [PubMed] [Google Scholar]
- 30.Lazaryan A, Song W, Lobashevsky E, Tang J, Shrestha S, Zhang K, et al. Human leukocyte antigen class I supertypes and HIV-1 control in African Americans. Journal of Virology. 2010;84:2610–2617. doi: 10.1128/JVI.01962-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tang J, Malhotra R, Song W, Brill I, Hu L, Farmer PK, et al. Human leukocyte antigens and HIV type 1 viral load in early and chronic infection: predominance of evolving relationships. PLoS ONE. 2010;5:e9629. doi: 10.1371/journal.pone.0009629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Trachtenberg E, Korber B, Sollars C, Kepler TB, Hraber PT, Hayes E, et al. Advantage of rare HLA supertype in HIV disease progression. Nature Medicine. 2003;9:928–935. doi: 10.1038/nm893. [DOI] [PubMed] [Google Scholar]
- 33.Carrington M, Nelson GW, Martin MP, Kissner T, Vlahov D, Goedert JJ, et al. HLA and HIV-1: heterozygote advantage and B*35-Cw*04 disadvantage. Science. 1999;283:1748–1752. doi: 10.1126/science.283.5408.1748. [DOI] [PubMed] [Google Scholar]
- 34.Streeck H, Jolin JS, Qi Y, Yassine-Diab B, Johnson RC, Kwon DS, et al. Human immunodeficiency virus type 1-specific CD8+ T-cell responses during primary infection are major determinants of the viral set point and loss of CD4+ T cells. Journal of Virology. 2009;83:7641–7648. doi: 10.1128/JVI.00182-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gao X, Nelson GW, Karacki P, Martin MP, Phair J, Kaslow R, et al. Effect of a single amino acid change in MHC class I molecules on the rate of progression to AIDS. N Engl J Med. 2001;344:1668–1675. doi: 10.1056/NEJM200105313442203. [DOI] [PubMed] [Google Scholar]
- 36.Maiers M, Gragert L, Klitz W. High-resolution HLA alleles and haplotypes in the United States population. Hum Immunol. 2007;68:779–788. doi: 10.1016/j.humimm.2007.04.005. [DOI] [PubMed] [Google Scholar]
- 37.Middleton D, Menchaca L, Rood H, Komerofsky R. New allele frequency database: http://www.allelefrequencies.net. Tissue Antigens. 2003;61:403–407. doi: 10.1034/j.1399-0039.2003.00062.x. [DOI] [PubMed] [Google Scholar]
- 38.Butler IF, Pandrea I, Marx PA, Apetrei C. HIV genetic diversity: biological and public health consequences. Curr HIV Res. 2007;5:23–45. doi: 10.2174/157016207779316297. [DOI] [PubMed] [Google Scholar]
- 39.Borrow P, Lewicki H, Hahn BH, Shaw GM, Oldstone MB. Virus-specific CD8+ cytotoxic T-lymphocyte activity associated with control of viremia in primary human immunodeficiency virus type 1 infection. J Virol. 1994;68:6103–6110. doi: 10.1128/jvi.68.9.6103-6110.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cao J, McNevin J, Malhotra U, McElrath MJ. Evolution of CD8+ T cell immunity and viral escape following acute HIV-1 infection. J Immunol. 2003;171:3837–3846. doi: 10.4049/jimmunol.171.7.3837. [DOI] [PubMed] [Google Scholar]
- 41.Cao J, McNevin J, Holte S, Fink L, Corey L, McElrath MJ. Comprehensive analysis of human immunodeficiency virus type 1 (HIV-1)-specific gamma interferon-secreting CD8+ T cells in primary HIV-1 infection. Journal of Virology. 2003;77:6867–6878. doi: 10.1128/JVI.77.12.6867-6878.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Koup RA, Safrit JT, Cao Y, Andrews CA, McLeod G, Borkowsky W, et al. Temporal association of cellular immune responses with the initial control of viremia in primary human immunodeficiency virus type 1 syndrome. J Virol. 1994;68:4650–4655. doi: 10.1128/jvi.68.7.4650-4655.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Oxenius A, Price DA, Trkola A, Edwards C, Gostick E, Zhang H-T, et al. Loss of viral control in early HIV-1 infection is temporally associated with sequential escape from CD8+ T cell responses and decrease in HIV-1-specific CD4+ and CD8+ T cell frequencies. J Infect Dis. 2004;190:713–721. doi: 10.1086/422760. [DOI] [PubMed] [Google Scholar]
- 44.Lichterfeld M, Yu XG, Mui SK, Williams KL, Trocha A, Brockman MA, et al. Selective depletion of high-avidity human immunodeficiency virus type 1 (HIV-1)-specific CD8+ T cells after early HIV-1 infection. Journal of Virology. 2007;81:4199–4214. doi: 10.1128/JVI.01388-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Legend: Log viral load progressions are plotted over the length of follow-up. Filled circles indicate dual infection.
Legend: Log viral load progressions are plotted over the length of follow-up. Open circles indicate monoinfection.
Legend: Square root CD4 progressions are plotted over the length of follow-up. Filled circles indicate dual infection.
Legend: Square root CD4 progressions are plotted over the length of follow-up. Open circles indicate monoinfection.
Legend: clones within 1% of depicted sequences are not shown for clarity. The recombination breakpoint and corresponding trees are inferred by GARD. Interior branches with significant bootstrap support (Neighbor Joining, Tamura-Nei 93 distance, 1000 replicates) are accordingly annotated. Note that the viral population sampled in 01/1999 consists exclusively of RT recombinants before the original (open squares) and superinfecting (divergent triangle) sequences.
Supp. Table 1. Viral population dynamics of superinfected participants.
Supp. Table 2. Population HLA Frequency Comparisons.