

SUMMARY
The adenylation (A) domains of nonribosomal peptide synthetases (NRPSs) activate aryl acids or amino acids to launch their transfer through the NRPS assembly line for the biosynthesis of many medicinally important natural products. In order to expand the substrate pool of NRPSs, we developed a method based on yeast cell surface display to engineer the substrate specificities of the A-domains. We acquired A-domain mutants of DhbE that have 11- and 6-fold increases in kcat/Km with nonnative substrates 3-hydroxybenzoic acid and 2-aminobenzoic acid, respectively and corresponding 3- and 33-fold decreases in kcat/Km values with the native substrate 2,3-dihydroxybenzoic acid, resulting in a dramatic switch in substrate specificity of up to 200-fold. Our study demonstrates that yeast display can be used as a high throughput selection platform to reprogram the “nonribosomal code” of A-domains.
INTRODUCTION
Nonribosomal peptide synthetases (NRPSs) are large multifunctional enzymes that synthesize peptide natural products known as nonribosomal peptides (NRPs), which are structurally diverse and possess many important medicinal activities (Cane et al., 1998; Clardy and Walsh, 2004). The anticancer agent bleomycin; the immunosuppresant cyclosporin; and the antibiotics vancomycin, daptomycin, and capreomycin are all examples of nonribosomal peptide drugs approved by the Food and Drug Administration. Since NRPSs do not use the mRNA-templated ribosomal machinery, they are not restricted to the 20 proteinogenic amino acids and often contain D-amino acids as well as unnatural α-amino acids. Other conspicuous features of NRPSs include N-methylation and cyclization of the peptide backbone, both of which serve to enhance proteolytic stability. NRPSs utilize a modular architecture where each module is responsible for the incorporation of one amino acid substrate into the final molecule (Fischbach and Walsh, 2006; Sieber and Marahiel, 2005). An NRPS extension module is composed of three core domains including a condensation (C) domain, an adenylation (A) domain and a peptidyl carrier protein (PCP) domain, while the initial loading module requires only the A and PCP domains. The A-domain is responsible for the selection, activation, and loading of its cognate amino acid substrate onto the downstream PCP-domain where it is covalently attached via a thioester linkage (Figure 1A). The thiol moiety from the PCP-domain is not from cysteine but rather from the terminal thiol moiety of an approximately 20 Å long phosphopantetheine (Ppant) cofactor moiety that modifies a conserved serine residue of the PCP-domain. Movements of the PCP-domain and the Ppant arm deliver the amino acid substrates to the C-domain that catalyzes peptide bond coupling between substrate molecules loaded on neighboring modules. This leads to the elongation of the peptide chain in the N → C direction. NRPSs thus work in an assembly-line fashion with the growing peptide chain being passed downstream from one module to the next until it reaches the last module. The full-length peptide chain is released by a thioesterase domain, usually via macrocyclization, to afford the final product.
Figure 1. DhbE Catalyzed Aryl Acid Activation and the Yeast Selection Scheme to Change the Substrate Specificity of DhbE.
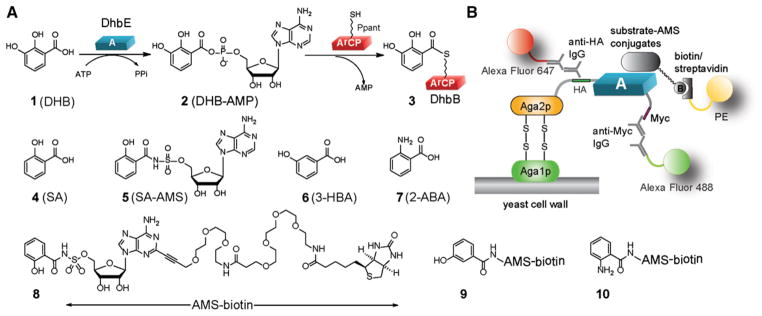
(A) DhbE catalyzes the condensation of DHB 1 with ATP to form DHB-AMP 2. The activated DHB is then transferred to the ArCP domain of DhbB to form a thioester conjugate 3 with the Ppant group of ArCP. DhbE can also activate SA 4. SA-AMS conjugate 5 is a bisubstrate inhibitor of DhbE. Structures of 3-HBA 6 and 2-ABA 7 as examples of nonnative substrates of DhbE are also shown.
(B) Selection of the A-domain library displayed on the surface of yeast cells. AMS-biotin conjugated SA (8) is used in the model selection to test the binding of wtDhbE on yeast cell surface with substrate-AMS conjugate. Compounds 9 and 10 have nonnative substrates 3-HBA 6 and 2-ABA 7 conjugated to AMS-biotin. They were used in the selection of DhbE mutants by yeast cell surface display.
See also Figures S1–S14 and Tables S1 and S3.
A-domains are the gatekeepers of NRPSs since they are responsible for selection of the appropriate carboxylic acid substrate, which is usually an amino acid but can also be an aryl acid as found in siderophore NRPs, a fatty acid for lipopeptide NRPs, or a hydroxy acid for peptide ester containing NRPs (Gulick, 2009; Sieber and Marahiel, 2005). A-domains are 55–60 kDa and contain a large N-terminal subdomain and a small C-terminal subdomain with the active site located at the domain interface. These proteins are conformationally dynamic and exist in an open unliganded conformation as well as two different closed ligand-bound conformations. The carboxylic acid substrate binding pocket is lined by 10 residues that comprise the first-shell interactions with the substrate and define the “nonribosomal codes” that enable the in silico prediction of A-domain substrate specificity (Challis et al., 2000; Stachelhaus et al., 1999; von Döhren et al., 1999). A-domains catalyze a two-step adenylation-thioesterification reaction; in the first step, they bind the substrate acid and ATP to afford a ternary complex and then catalyze their condensation to afford an intermediate acyl-adenylate (Sikora et al., 2010) (Figure 1A). Following release of pyrophosphate, the C terminus of the A-domain undergoes an approximately 140° rigid body rotation, which enables binding of the downstream PCP-domain and insertion of the Ppant cofactor arm into the A-domain active site (Gulick, 2009). Acylation of the Ppant moiety followed by release of the thioacylated PCP and AMP completes the catalytic cycle.
Several strategies have been explored to modify the substrate specificity of A-domains in NRPS assembly lines to incorporate nonnative building blocks to synthesize analogs with improved or alternate biological activities (Cane et al., 1998; Nguyen et al., 2006). Domain swapping to produce chimeric proteins was first used to alter A-domain specificity; however, this approach suffers from nonoptimal interactions between noncognate protein domains that often result in drastically reduced catalytic turnover, as well as premature truncation of the nascent peptide (Baltz, 2008; Fischbach et al., 2007; Robbel and Marahiel, 2010; Stachelhaus et al., 1995). In an effort to alleviate these problems, Walsh and coworkers used directed evolution to improve the activity of chimeric assembly lines, which led to nearly 10-fold improvements in catalytic activity (Fischbach et al., 2007). A complimentary approach to reengineer A-domain specificity preserving the native protein-protein interactions within an NRPS module has been to use the nonribosomal peptide code and, more recently, computational structure-based redesign to rationally mutate residues important for substrate recognition (Chen et al., 2009; Eppelmann et al., 2002; Thirlway et al., 2012). In order to screen potential mutants more comprehensively, Kelleher and co-workers developed a novel workflow involving directed evolution to create a library of ~1,000 mutants based on the “nonribosomal peptide code” and mass spectrometry to measure the ability of the resulting mutants to support production of novel NRPs (Evans et al., 2011). In this study, we report a method to engineer the substrate specificity of A-domains by yeast cell surface display that takes advantage of high throughput fluorescence-activated cell sorting (FACS) for iterative rounds of selection of millions of A-domain mutants.
As a first step to test our method, we selected the A-domain of DhbE as a model system due to the availability of its 2.15 Å X-ray cocrystal structure with bound acyl-adenylate (May et al., 2002), availability of active-site directed inhibitors (Miethke et al., 2006), and detailed knowledge of the kinetic mechanism (Sikora et al., 2010). DhbE is part of a three-module NRPS assembly line comprising DhbE, DhbB, and DhbF responsible for the synthesis of the siderophore bacillibactin in Bacillus subtilis (May et al., 2001). DhbE activates 2,3-dihydroxybenzoic acid (DHB, 1) to form the DHB-AMP acyl-adenylate 2 and transfers it to the aryl carrier protein (ArCP) domain of DhbB (Figure 1A). Another distinct advantage of DhbE is that it is not embedded in a multi-domain NRPS but is a stand-alone protein, which loads the carrier protein domain in DhbB in an intermolecular fashion. Consequently, DhbE is amenable to kinetic characterization of the entire adenylation-thioesterification reaction as one can use stoichiometric amounts of its cognate ArCP domain of DhbB to obtain catalytic turnover. By contrast, most previous reports for A-domain engineering where kinetic data are given have only measured the adenylation partial reaction, as catalytic turnover was not possible with the given multidomain NRPS protein, and thus do not report on the kinetically and functionally relevant overall reaction (Chen et al., 2009; Eppelmann et al., 2002).
RESULTS
Engineer A-Domain Specificity by Yeast Cell Surface Display
Yeast cell surface display has been extensively used to engineer the binding specificity of antibodies (Chao et al., 2006; Miller et al., 2008). The yeast vector pCTCON2 expresses the antibody library as a fusion to the yeast agglutinin protein Aga2p that is attached through disulfide bonds to Aga1p protein as part of the yeast cell wall (Figure 1B). The yeast cell library is then incubated with a fluorescently labeled antigen to allow the binding of antigen molecules to the antibody displayed on the yeast surface. FACS is then used to isolate yeast cells displaying antibody mutants with high affinities with the antigen. To test if yeast selection can be used to engineer the substrate specificity of the A-domains, we cloned DhbE into the pCTCON2 vector to display the DhbE enzyme on the yeast cell surface. The Aga2p-DhbE domain fusion also has a hemagglutinin (HA) tag and a Myc tag at the N and C termini, respectively, of the A-domain to enable the detection of A-domain displayed on the cell surface (Figure 1B). After inducing the yeast cell to express the Aga2p-DhbE fusion, we incubated the cells with a mouse anti-HA antibody and a chicken anti-Myc antibody so that the antibodies would bind to the peptide tags flanking DhbE on the cell surface. Cells were then washed and incubated with a mixture of goat antimouse antibody conjugated with Alexa Fluor 647 and goat antichicken antibody conjugated with Alexa Fluor 488 to label DhbE displayed on the cell surface with fluorophores. Flow cytometry analysis of the yeast cells showed that more than 30% of the cells were doubly labeled with Alexa Fluor 647 and 488 fluorophores, indicating efficient display of DhbE on the yeast cell surface (Figure S1A available online).
Next, we needed to develop a method to fluorescently label the yeast cells displaying A-domain mutants with desired substrate specificity in order to select these cells from the A-domain library by FACS. Given the relatively low affinity of A-domains for their substrate acids (1 μM–1 mM) and inability to attach a biotin moiety conveniently without drastically affecting substrate binding affinity, we elected to design a chemical probe to report substrate recognition of the A-domains on the yeast cell surface that exploits the following: (1) the high affinity of A-domains for their intermediate acyl-adenylate (acyl-AMP) as a result of the bisubstrate nature of this intermediate that interacts with both the acid and ATP substrate binding pockets, (2) the ability to mimic the acyl-AMP and therefore generate a chemically stable probe by isosteric replacement of the phosphate moiety for a sulfamate (acyl-AMS probe, wherein AMS denotes adenosine monosulfamate, an isostere of AMP) (Ferreras et al., 2005; Finking et al., 2003; Miethke et al., 2006; Somu et al., 2006), (3) the potential to modify acyl-AMS probes at their C-2 position for incorporation of a biotin moiety without compromising binding affinity (Neres et al., 2008), and (4) the high discrimination of acyl-AMS probes for their cognate A-domain (Qiao et al., 2007). Based on these design principles we prepared salicyl-AMS probe 8 with biotin attached to the C-2 atom of the purine base through a long and flexible linker (Figure 1A; for synthesis, see the Supplemental Information, Figures S5–S14, and Table S3) to report the binding to DhbE on the surface of yeast cells. Probe 8 contains salicylic acid (SA) rather than DHB due to the oxidative instability of the catechol in DHB. We incubated 8 and chicken anti-Myc antibody with yeast cells to allow their binding with wtDhbE on the cell surface. After washing the cell, a mixture of streptavidin-conjugated phycoerythrin (PE) and antichicken antibody conjugated with Alexa Fluor 488 were added to the cells to detect the binding of 8 and the anti-Myc antibody to wtDhbE. Flow cytometry showed that more than 17% of the cell population was double positive with labeling of both fluorophores, indicating that wtDhbE expressed on the cell surface was able to bind 8 (Figure S1B). Thus yeast cell surface display can be used to select A-domains based on their affinity with acyl-AMS probes.
Construct an A-Domain Library of DhbE for Yeast Selection
To assess the substrate specificity of DhbE, we measured the steady-state kinetics of the complete adenylation-thioesterfication reaction catalyzed by DhbE using saturating concentrations of the ArCP domain of DhbB in a coupled assay that measures release of pyrophosphate (PPi). During this assay, PPi generated in the DhbE-catalyzed condensation reaction between the aryl substrate and ATP is converted to Pi by inorganic pyrophosphatase (Ehmann et al., 2000; Webb, 1992). Subsequently, purine nucleoside phosphorylase catalyzes the phosphorolysis of the chromogenic substrate 7-methylthioguanosine that can be monitored at 360 nm. After catalyzing the first round of aryl-AMP formation, DhbE remains inactive until the aryl-AMP intermediate dissociates from the enzyme or is broken down by the transfer to DhbB. Consequently, the rate of PPi release catalyzed by DhbE is a measure of the combined rate of aryl-AMP dissociation from the enzyme and aryl transfer to ArCP. A previous study on EntE, a DhbE homolog in Escherichia coli, which also activates DHB and loads it onto an ArCP, showed that the rate of DHB transfer to ArCP is more than 100-fold faster than the dissociation of DHB-AMP from the enzyme, due to the tight-binding nature of the intermediate acyl-adenylate (Ehmann et al., 2000). We can therefore approximate the rate of PPi release in the assay to that of substrate transfer to the ArCP domain catalyzed by DhbE. Using the PPi release assay, we determined the specificity constants (kcat/Km) for 2,3-dihydroxybenzoic acid (DHB, 1), 2-hydroxybenzoic (SA, 4), 3-hydroxybenzoic acid (3-HBA, 6), and 2-aminobenzoic acid (2-ABA, 7) as 1,100, 140, 22, and 5.5 min−1mM−1, respectively (Table 1). 3-HBA and 2-ABA exhibit 2% and 0.5% activity, respectively, relative to the native substrate DHB. Based on these results, we decided to use yeast cell surface display to engineer DhbE so that it can preferentially activate 3-HBA and 2-ABA over DHB and accordingly synthesized the corresponding acyl-AMS probes 9 and 10 (Figure 1A; see Supplemental Information for synthesis). For comparative purposes, we also measured the steady-state kinetic parameters of DhbE using the conventional ATP-PPi exchange assay that only measures the adenylation partial reaction. We demonstrated DhbE adenylates 3-HBA and 2-ABA, but at much lower rates than its native substrate DHB, and the relative trend in activity was identical to that observed with the PPi release assay (Figure S2A).
Table 1.
PPi Release Rate of the Aryl Acid Adenylation Reaction Catalyzed by wtDhbE and Mutants
| Enzyme and Substrate | Km (μM) | kcat (min−1) | kcat/Km (min−1mM−1) | Ratio of kcat/Km with the Same Substrate (Mutant/wtDhbE) |
|---|---|---|---|---|
| wtDhbE | ||||
| DHB | 4.3 ± 0.4 | 4.61 ± 0.09 | 1,100 | |
| SA | 14 ± 2.2 | 1.90 ± 0.07 | 140 | |
| 3-HBA | 25 ± 7.7 | 0.56 ± 0.02 | 22 | |
| 2-ABA | 62 ± 17 | 0.34 ± 0.03 | 5.5 | |
| KZ4(Trp234His) | ||||
| DHB | 3.5 ± 0.8 | 1.45 ± 0.08 | 410 | 0.37 |
| SA | 46 ± 4.8 | 0.84 ± 0.01 | 18 | 0.13 |
| 3-HBA | 4.8 ± 0.7 | 1.14 ± 0.02 | 240 | 11 |
| KZ12(Trp234His) | ||||
| DHB | 38 ± 12 | 1.25 ± 0.14 | 33 | 0.03 |
| SA | 56 ± 14 | 0.37 ± 0.03 | 6.6 | 0.047 |
| 2-ABA | 3.5 ± 0.3 | 0.12 ± 0.01 | 34 | 6.2 |
See also Table S1.
Figure 2. Substrate Binding Site of DhbE.
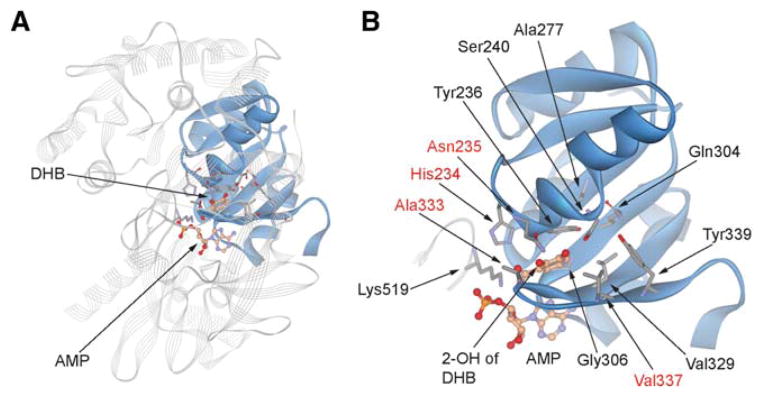
(A) Crystal structure of DhbE with the binding pocket of DHB shown in solid ribbons and the rest of the protein in line ribbons (Protein Data Bank ID 1MD8) (May et al., 2002).
(B) Detailed structure of DHB binding site showing key active-site residues as the nonribosomal code for DHB recognition. Names of the residues randomized in the DhbE library are in red.
See also Figure S4.
The crystal structure of the DhbE A-domain in complex with DHB has been solved (Figure 2A), and it reveals a set of 10 active-site residues of DhbE that serve as the specificity conferring nonribosomal code for DHB recognition: Asn235, Tyr236, Ser240, Ala277, Gln304, Gly306, Val329, Val337, Tyr 339, and Lys519 (Figure 2B) (May et al., 2002). Among them, the amide moiety of the Asn235 side chain is engaged in hydrogen-bonding interactions with the 2-OH and 3-OH groups of DHB. The side chain of Val337 is also in close distance (5.1 Å) with the 2-OH group of DHB. Outside the residues involved in the nonribosomal code, the imidazole nitrogen of His234 is within hydrogen-bonding distance with both the carboxylate and 2-OH groups of DHB, and the methyl side chain of Ala333 is in close proximity (3.5 Å) to the 2-OH of DHB (Figure 2B). Since we intended to engineer DhbE to be specific for 3-HBA and 2-ABA, in which the 2-OH substitution of DHB is replaced with a hydrogen atom or an amino group, we focused on improving the interactions between DhbE and the C-2 functional groups in the aryl acid substrates. We thus randomized His234, Asn235, Ala333, and Val337 in DhbE to construct an A-domain library with a size of 5 × 106, large enough to cover all the mutants in a library with four randomized residues (1.6 × 105) (Table S2).
Yeast Cell Surface Display to Identify DhbE Mutants that Are Specific for 3-HBA Activation
We displayed the library on the surface of yeast cells and carried out iterative rounds of selection with acyl-AMS probe 9 containing 3-HBA as the acyl moiety (Figure 1B). The yeast selection followed the protocol developed by Wittrup and Miller with some modifications (Chao et al., 2006; Miller et al., 2008). In the first round of selection we incubated the yeast cell library with 3 μM 9. After washing the cells to remove unbound probes, yeast cells labeled with biotin were collected by magnetic beads coated with streptavidin. The use of magnetic-activated cell sorting (MACS) allowed selection of a large quantity of yeast cells (~1 × 1010) by binding to the biotinylated probe. The first round of selection was to identify yeast cells based on the affinity of probe 9 with the DhbE mutants displayed on the cell surface. In the second round of selection, we bound the cells to anti-HA and anti-Myc antibodies to enrich cells displaying full-length DhbE in the library. Our DhbE was flanked by an N-terminal HA tag and a C-terminal Myc tag when it was displayed as a fusion with the Aga2 protein on the surface of yeast cells. Full-length DhbE should have both tags intact and be bound to both anti-HA and anti-Myc antibodies (Figure 1B). The two types of antibodies on yeast cell surface were detected by an antichicken antibody conjugated to Alexa Fluor 488 and an antimouse antibody conjugated to Alexa Fluor 647. We used FACS to collect the top 20% of cells that were doubly labeled by the two fluorophores to eliminate cells displaying DhbE with N- or C-terminal truncations in the library (Figure 3B). In subsequent rounds of selection and FACS sorting, library cells were bound to probe 9 and chicken anti-Myc antibody as the primary reagents, and streptavidin-PE and antichicken antibody Alexa Fluor 488 as secondary reagents. Sort gate covered the range of cells that were the top 0.1%–1.0% in brightness for binding to 9 and the anti-Myc antibody (Figures 3C–3E). We also increased the selection stringency by decreasing the concentration of 9 from 1 μM in round 3 to 0.1 μM in round 5. We observed a steady increase in the yeast cell population appearing at the diagonal of the flow cytometry plot corresponding to the enrichment of doubly labeled cells that displayed DhbE mutants with high affinity to probe 9 (Figure 3).
Figure 3. Sorting of the DhbE Library Displayed on the Yeast Cell Surface by Binding to Biotin-Linked 3-HBA-AMS 9.
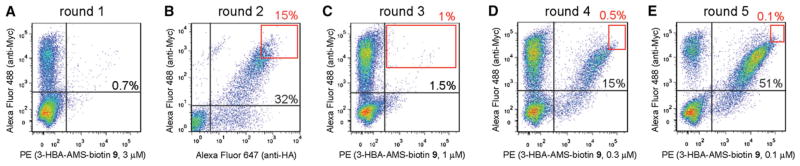
(A) Sorting with magnetic beads coated with streptavidin. Binding of cells to 9 and an anti-Myc antibody was analyzed by flow cytometry.
(B) FACS sorting by antibody binding to the HA and Myc tags flanking DhbE on the cell surface.
(C–E), FACS by binding to 9 and an anti-Myc antibody. Percentages of doubly labeled cells were shown in the flow cytometry plots. Red frames in the plots represent the sorting gates used for the collection of yeast cells.
After five rounds of selection with 9, 30 clones were sequenced. Alignment with wtDhbE showed that clone KZ2 is the dominant clone after yeast selection with 14 appearances among the 30 sequenced clones (Table 2). All selected DhbE clones have His234 mutated to a Trp residue, while Val337 is invariable and unchanged from the wild-type sequence. There is also a tendency for Ala333 to be unchanged, while it is replaced by Ser and Thr residues in some of the selected clones. Asn235 is more flexible, with Ala in KZ2 as the most preferred mutation. Other residues such as Ser, Thr, Cys, Val and Gln were also selected at position 235 to bind to 9. We expressed the DhbE mutants (KZ1–4) that appeared multiple times among the sequenced clones and assayed their catalytic activities. The ATP-PPi exchange assay showed that DhbE mutants KZ1–3 have similar activities as wtDhbE for activation of 3-HBA to form 3-HBA-AMP while KZ4 catalyzes 3-HBA activation at a rate about 10-fold higher than wtDhbE and mutants KZ1–3 (Figure S2B). Also, KZ4 has a Km of 4 μM with 3-HBA, only slightly larger than the Km of wtDhbE with the native substrate DHB (2.5 μM) (Table S1). We then tested the transfer of 3-HBA to the ArCP domain of DhbB catalyzed by the DhbE mutants. To our surprise, while wtDhbE gave about 10% loading of the ArCP domain with 3-HBA after 30 min (Figure 4A), none of the mutants could catalyze the loading of ArCP with 3-HBA (Figure 4B). These results confirm that the DhbE mutants from yeast selection can recognize 3-HBA for the adenylation reaction but that the mutants are unable to transfer the activated aryl substrate to the ArCP.
Table 2.
Alignment of DhbE Mutants Selected by Yeast Cell Surface Display with AMS Conjugated Probes 9 and 10
| wtDhbE | Number of Times Selected | Residue Number
|
|||
|---|---|---|---|---|---|
| 234
|
235
|
333
|
337
|
||
| H | N | A | V | ||
| Clones selected by 9 | |||||
|
| |||||
| KZ1 | 6 | W | S | A | V |
|
| |||||
| KZ2 | 14 | W | A | A | V |
|
| |||||
| KZ3 | 4 | W | N | S | V |
|
| |||||
| KZ4 | 2 | W | Q | T | V |
|
| |||||
| KZ5 | 2 | W | Q | S | V |
|
| |||||
| KZ6 | 1 | W | T | A | V |
|
| |||||
| KZ7 | 1 | W | C | A | V |
|
| |||||
| KZ8 | 1 | W | V | A | V |
|
| |||||
| Clones selected by 10 | |||||
|
| |||||
| KZ11 | 10 | W | D | T | R |
|
| |||||
| KZ12 | 9 | W | D | T | K |
See also Table S2.
Figure 4. MALDI Analysis of Transfer of 3-HBA Substrate to the ArCP Domain Catalyzed by wtDhbE and the KZ4 Mutants.
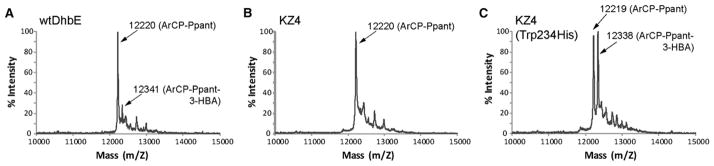
(A–C) Reactions were allowed to proceed for 30 min at room temperature. (A) wtDhbE has some activity of loading 3-HBA to the ArCP domain of DhbB. (B) KZ4 mutant is defective in 3-HBA loading to ArCP. (C) The KZ4 (Trp234His) mutant can efficiently load 3-HBA to ArCP.
We speculated that the His234Trp mutation, persistent in mutants KZ1–4, might have a detrimental effect on substrate loading to the ArCP. The crystal structure of DhbE in complex with DHB and AMP shows the imidazole side chain of His234 directly interacting with the DHB carboxylate and the 2-OH group (Figure 2B) (May et al., 2002). Furthermore, the His234 side chain shields the DHB substrate from a tunnel through which the Ppant group of PCP may approach the DHB-AMP intermediate for thioester formation. While the His234Trp mutation was selected because it may improve the binding between the DhbE mutants and the aryl acid substrate, the Trp side chain may block access of the Ppant thiol to the DHB adenylate due to its considerably larger size relative to His. To recover the ArCP loading activity of the DhbE mutants, we mutated Trp234 in KZ1–4 back to His and generated mutants KZ1–4(Trp234His). The ATP-PPi exchange assay showed that all four mutants catalyze 3-HBA adenylate formation, with KZ4(Trp234His) still possessing the strongest activity among the mutant clones (Figure S2C). A MALDI assay showed that within 30 min, the KZ4(Trp234His) mutant can load 60% of the ArCP with 3-HBA, while other mutants and wtDhbE can load less than 10% of the ArCP (Figure 4C). These results show that the Trp234His reverse mutation enabled substrate transfer from mutant DhbE to the ArCP domain of DhbB, and the KZ4 mutant with Trp234His mutation is significantly more active in loading 3-HBA onto ArCP than wtDhbE.
We then used the PPi release assay to characterize the kinetics of KZ4 (Trp234His) for catalyzing 3-HBA activation and transfer to the ArCP (Figure 5; Table 1). We found that KZ4 (Trp234His) has a Km of 4.8 μM and a kcat of 1.14 min−1 with 3-HBA. In comparison, wtDhbE has a Km of 25 μM with 3-HBA, 5.2-fold higher than KZ4 (Trp234His); and a kcat of 0.56 min−1, 2-fold less than KZ4 (Trp234His) with 3-HBA. This suggests that DhbE mutants enriched by yeast selection with the 3-HBA-AMS probe 9 acquired higher binding affinity with the nonnative substrate 3-HBA. As a result, the specificity constant (kcat/Km) of KZ4 (Trp234His) with 3-HBA is 240 min−1mM−1, which is 11-fold higher than wtDhbE with 3-HBA. The kcat/Km of KZ4 (Trp234His) with the native substrate DHB is 410 min−1mM−1, which is nearly 3-fold lower than wtDhbE with DHB (Table 1). Thus, KZ4 (Trp234His) makes a nearly 30-fold switch in substrate specificity.
Figure 5. Kinetic Analysis of DHB and 3-HBA Transfer to ArCP by wtDhbE and KZ4 (Trp234His) Based on the PPi Release Assay.
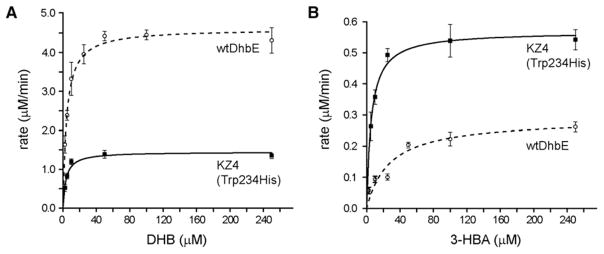
Michaelis-Menten plot to compare the initial rates of PPi release catalyzed by wtDhbE and KZ4(Trp234His) at varying concentrations of native substrate DHB (A) and nonnative substrate 3-HBA (B). Data are means ± ranges for at least three determinations.
Yeast Cell Surface Display to Identify DhbE Mutants Specific for 2-ABA Activation
We also carried out selection with the same DhbE library displayed on yeast cell surface and the acyl-AMS probe 10 containing 2-ABA as the acyl moiety (Figure 1). After five rounds of selection following the same protocol as employed with probe 9, we found that the yeast library converged to two very similar DhbE mutants: KZ11 and KZ12 (Figure S3; Table 2). Both mutants have His234 replaced by Trp, the same as the mutants from the selection with probe 9. The two mutants also have identical Asn235Asp and Ala333Thr mutations. The only difference between these two mutants is that Val337 is mutated to Arg in KZ11 and to Lys in KZ12. Initially selected KZ11 and KZ12 are not active in transferring 2-ABA substrate to the ArCP domain of DhbB, similar to mutants KZ1–4. However, when we mutated Trp234 back to His, KZ11(Trp234His) and KZ12(Trp234His) became catalytically active for loading 2-ABA to the ArCP.
The PPi release assay showed that KZ12 (Trp234His) has a Km of 3.5 μM with the 2-ABA substrate, 18-fold lower than that of wtDhbE with 2-ABA (62 μM) (Table 1). Due to the lower Km, the kcat/Km of KZ12 (Trp234His) with 2-ABA (34 min−1mM−1) is 6.2-fold higher than that of wtDhbE with 2-ABA. The kcat/Km of KZ12 (Trp234His) with the native substrate DHB is 33 min−1mM−1, which is nearly 33-fold lower than wtDhbE with DHB (Table 1). As a result, KZ12 (Trp234His) makes a 206-fold switch in substrate specificity. Kinetic analysis of KZ12 (Trp234His) showed that the mutant has a kcat of 0.12 min−1, similar to that of wtDhbE-catalyzed 2-ABA activation (0.34 min−1) (Table 1); however this kcat value is considerably lower than the kcat of wtDhbE with its native substrate, DHB (4.6 min−1). This suggests that, while yeast selection based on affinity binding with bisubstrate analog probes may improve binding interactions between the engineered enzyme and nonnative substrates to give a lower Km, such affinity-based selection may not be able to improve the kcat of the engineered enzyme with nonnative substrates.
DISCUSSION
In this study, we performed yeast selection on an A-domain library to change the substrate specificities of the A-domains. The yeast selection enriched A-domain mutants with high binding affinities with the acyl-AMS probes. In this way, the selected A-domain mutants may have better recognition of the nonnative substrates and enable their loading onto the ArCP domains in the NRPS assembly line. We demonstrated that yeast cell surface display is a very efficient platform to identify A-domain mutants with altered binding specificities using nonnative substrates conjugated to AMS. In just five rounds of selection of a yeast library of 5 × 106 clones, we were able to identify distinct sets of DhbE mutants that can bind to probes 9 and 10, containing 3-HBA and 2-ABA as the acyl moieties, respectively.
We also found that the DhbE mutants from yeast selection have Km values 5- to 18-fold lower than wtDhbE for binding to the nonnative substrates 3-HBA and 2-ABA (Table 1). By contrast, the kcat values of the DhbE mutants change very little with KZ4 catalyzing 3-HBA transfer with a kcat 2-fold higher than wtDhbE, and KZ12 catalyzing 2-ABA transfer with a kcat 2-fold lower than wtDhbE. These results suggest that the improvement in catalytic efficiency (kcat/Km) of the A-domain mutants with the nonnative substrates is mainly due to the decrease in Km, an indication of better recognition of the nonnative substrates by the enzyme active site. These results agree with previous work on engineering catalytic antibodies that showed tighter binding with the transition state analog by the antibody does not lead to a higher turnover rate (kcat) of the catalytic reactions (Baca et al., 1997).
Our work on DhbE engineering also shed light on the mechanism of substrate transfer catalyzed by the A-domain. Yeast selection of the A-domain library identified a His234Trp mutation present in all the mutants (Table 2). The crystal structure of DhbE in complex with DHB and AMP suggested that the Trp residue at this position may provide better stacking interactions between the indole ring of the Trp side chain and the carboxylate and sulfonamide groups of the aryl-AMS conjugate (Figure 2B). However, the His234Trp mutation may also block the access of the Ppant group from the ArCP to the aryl-AMP formed at the enzyme active site. This notion is supported by the recently reported crystal structure of EntE, a homologous enzyme of DhbE from E. coli., covalently tethered to the ArCP domain of EntB, equivalent to the ArCP domain of DhbB, through a mechanism-based inhibitor (Mitchell et al., 2012; Sundlov et al., 2012). In the EntE-EntB structure, the Ppant arm of the EntB ArCP is inserted into the substrate binding site of the EntE A-domain in order to form a covalent bond with the inhibitor molecule bound to the A-domain (Figure S4) (Sundlov et al., 2012). The side chain of His234 from EntE has to adopt a different conformation from the corresponding His234 in the DhbE structure to allow the Ppant group to extend into the EntE active site. A His234Trp mutation places a large indole ring in the path of Ppant that may block the entry of the Ppant group. Indeed, none of the mutants we assayed with the His234Trp mutation could load the aryl acid substrate onto the ArCP. We were able to recover the transfer activity of the DhbE mutant by introducing a reverse mutation to put His234 residue back in the DhbE mutants (Figure 4C; Table 1). This suggests that yeast selection is best utilized to improve substrate recognition at the A-domain residues devoted to substrate binding, but is less useful in optimizing A-domain kinetics. More structural and mechanistic insights on A-domain catalysis would certainly guide the future library design and selection.
Yeast selection of an A-domain library also provided new insights on DhbE domain recognition of aryl acid substrates (Ames and Walsh, 2010; Stachelhaus et al., 1999). Asn235 and Val337 are the nonribosomal code residues that were randomized in the DhbE library. When the yeast library was screened against probe 9 for 3-HBA activation, KZ4 with Asn235 replaced by a Gln residue showed higher efficiency in adenylate formation with 3-HBA (Table 2; Table S1). The Gln side chain is one CH2 group longer than the Asn side chain in wtDhbE. It may extend further into the substrate binding pocket to fill the space that used to be occupied by the 2-OH substitute of DHB. The amide NH2 of the Gln residue may also form hydrogen bonds with the 3-OH group in 3-HBA (Figure 2B). Yeast selection with probe 9 also showed that Val337 is indispensable for binding to 3-HBA. When the yeast library was selected for binding to probe 10, Val337 is replaced by Lys or Arg with longer and positively charged side chains (Table 2). The Lys and Arg residues at this position may interact with the 2-NH2 group of 2-ABA. Ala333 was not assigned as a nonribosomal code residue for A-domain binding with the aryl acid substrates (Ames and Walsh, 2010). After yeast selection with AMS conjugates to 3-HBA or 2-ABA, Ala333 was replaced with Ser or Thr residues with hydroxyl side chains that may form hydrogen bonds with the 3-HBA and 2-ABA substrates. Overall, our results demonstrate that A-domains can be reprogrammed by protein engineering to recognize nonnative substrates. It would be interesting to see if the A-domain mutants we acquired in this study incorporate 3-HBA or 2-ABA substrates into the bacillibactin scaffold.
Once the A-domain is engineered to incorporate nonnative aryl or amino acid substrates into the NRPS assembly line, nonnative substrates could be used for the synthesis of “unnatural natural products.” A correlation could be drawn between engineering NRPS A-domains and the engineering of tRNA synthetases, which are also adenylation domains for amino acid activation (Wang et al., 2006). When tRNA synthetases are engineered to load unnatural amino acids on tRNA, they open the gate for the incorporation of unnatural building blocks into proteins. NRPS enzymes seem to be more discerning of nonnative substrates than the ribosome for protein biosynthesis. While early work showed that downstream modules may block the elongation of peptide intermediates with the incorporation of nonnative substrates (Pavela-Vrancic et al., 1999; Uguru et al., 2004), more recent studies have successfully demonstrated the ability of reprogrammed A-domains to incorporate nonnative substrates into the final NRPs (Evans et al., 2011; Fischbach et al., 2007; Thirlway et al., 2012).
The structural complexity of NRP natural products in many cases is restrictive to the production of analogs with improved medicinal activities by traditional synthetic chemistry. Historically, natural product analogs were generated by derivatization of the parent natural product. Combinatorial biosynthesis offers a complimentary method to generate analogs through genetic engineering of the biosynthetic genes. Addition, deletion, or replacement of genes in NRPS gene clusters has been used to reprogram NRPS enzymatic assembly lines to produce new analogs of the antibiotics surfactin, daptomycin, and echinomycin (Nguyen et al., 2006; Stachelhaus et al., 1995; Watanabe et al., 2009). Similar approaches have also been used to reconfigure closely related gene clusters of polyketide synthases to generate analogs of the antibiotic erythromycin (McDaniel et al., 1999; Pfeifer et al., 2001). We expect our method for engineering NRPS A-domains should greatly expand the scope of the chemical building blocks for the combinatorial biosynthesis of NRPs. Engineered nonribosomal peptide synthesis can be used in parallel with engineered ribosomal peptide synthesis (Yamagishi et al., 2011) to generate peptide libraries of diverse structures.
EXPERIMENTAL PROCEDURES
See the Supplemental Experimental Procedures for full synthetic procedures, cloning, and kinetic characterization of the A-domain mutants.
Yeast Display of the DhbE Library
DhbE library in pCTCON2 was chemically transformed into YB100 yeast cells following published protocols with some modifications (Gietz and Schiestl, 1991; Gietz and Woods, 2002). Briefly, yeast cells were first cultured in 200 ml YPD (20 g dextrose, 20 g peptone, and 10 g yeast extract in 1 l deionized H2O, sterilized by filtration) to an optical density 600 (OD600) around 0.5 at 30°C. The cells were then pelleted at 3,500 rpm for 5 min. Cells were subsequently washed by 20 ml TE (100 mM Tris base, 10 mM EDTA, pH 8.0) and 20 ml LiOAc-TE (100 mM lithium acetate in TE), before resuspension in approximately 800 μl LiOAc-TE. A typical transformation reaction contained a mixture of 1 μg pCTCON2 plasmid DNA, 2 μl denatured single-stranded carrier DNA from salmon testes (Sigma Aldrich), 25 μl resuspended yeast competent cells, and 300 μl polyethylene glycol (PEG) solution (40% [w/v] PEG 3350 in LiOAc-TE). In order to achieve a library size of 106, 30 transformations were set up in parallel. A control was also prepared in which the pCTCON2 plasmid was excluded. Both the transformation reactions and the control were incubated at 30°C for 1 hr and then at 42°C for 20 min. Cells in each transformation were pelleted by centrifuging at 13,000 rpm for 30 s and resuspended in 20 μl SDCAA medium (2% [w/v] dextrose, 6.7 g Difco yeast nitrogen base without amino acids, 5 g Bacto casamino acids, 50 mM sodium citrate, and 20 mM citric acid monohydrate in 1 l deionized H2O, sterilized by filtration). Yeast cells were resuspended, pooled together into 1 l SDCAA medium and allowed to grow at 30°C over a 2-day period to an OD600 above 5. For long-term storage of the yeast library, 20 ml of the yeast culture was aliquoted in 15% glycerol stock and stored at −80°C.
To titer transformation efficiency, 10 μl of the resuspended yeast transformants from either the library mix or control was serially diluted in SDCAA medium and plated on Trp- plates—20 g agar, 20 g dextrose, 5 g (NH4)2SO4, 1.7 g Difco yeast nitrogen base without amino acids, 1.3 g drop-out mix excluding Trp in 1 l deionized H2O, autoclaved. Yeast cells transformed with pCTCON2 plasmids were expected to appear within 2 days of incubation at 30°C.
Model Selection of Yeast Cells Displaying wtDhbE
Yeast cell EYB100 was transformed with wild-type DhbE (wtDhbE) in pCTCON2 and streaked on a Trp- plate. Yeast colonies grew up after two days of incubation at 30°C. Cells were scraped from the Trp- plate to inoculate a 5 ml SDCAA culture that was allowed to shake at 30°C to reach an initial OD600 of 0.5. Cells were centrifuged at 3,000 rpm for 5 min and induced for DhbE expression by resuspension in 5 ml SGCAA (2% [w/v] galactose, 6.7 g Difco yeast nitrogen base without amino acids, 5 g Bacto casamino acids, 38 mM Na2HPO4 and 62 mM NaH2PO4 · H2O in 1 l deionized H2O, sterilized by filtration). The yeast culture was shaken at 20°C for 16–24 hr.
For analysis of DhbE display on the surface of yeast cells, 106 cells were resuspended in 0.1 ml Tris-buffered saline (TBS) (25 mM Tris, pH 7.5, 150 mM NaCl) with 0.1% bovine serum albumin (BSA). Mouse anti-HA antibody (Santa Cruz Biotechnology, sc-7392) and chicken anti-c-myc antibody (Invitrogen, A-21281) were used as primary antibodies, and they were added to the cell suspension at a concentration of 10 μg/ml. The cells were incubated with antibodies for 45 min at 4°C. The cells were then washed twice with 0.1% BSA in TBS and stained with 5 μg/ml goat antimouse antibody conjugated with Alexa Fluor 647 (Invitrogen A-21235) and goat antichicken antibody conjugated with Alexa Fluor 488 (Invitrogen, A-11039) in 0.1 ml 0.1% BSA in TBS. The cell suspension was shielded from light and incubated at 4°C for 30 min. After washing twice with 0.1% BSA in TBS, cells were analyzed on a flow cytometer (LSRII, BD Biosciences) to count the number of cells that were labeled with both fluorophores. Cells were also analyzed from control labeling reactions in which primary antibodies were either excluded from the reaction or cells were only labeled with primary and secondary antibodies for just one of the affinity tags.
Cells displaying wtDhbE were also assayed by binding to acyl-AMS probe 8. During the labeling with primary reagents, 10 nM 8 and 10 μg/ml chicken antic-myc antibody (Invitrogen, A-21281) were used. In the following step, 5 μg/ml streptavidin conjugated with PE (Invitrogen, S-32350) and 5 μg/ml goat anti-chicken antibody conjugated with Alexa Fluor 488 (Invitrogen, A-11039) were used as secondary reagents for the detection of wtDhbE on the cell surface and the binding of wtDhbE to 8.
Selection of the DhbE Library Displayed on the Yeast Cell Surface
The first round of selection of the yeast library was carried out with MACS. For subsequent rounds of selection, FACS was used to identify yeast clones displaying DhbE mutants that were bound to acyl-AMS probes 9 or 10 with high affinity. For MACS selection, approximately 8 × 107 yeast cells displaying the DhbE library were incubated with 9 or 10 in a total volume of 600 μl 0.1% BSA in TBS. After 45 min at 4°C with intermittent inversion to ensure thorough mixing, cells were pelleted by centrifugation. Cells were then resuspended in fresh 0.1% BSA in TBS and pelleted again. This procedure was repeated twice to remove biotin-linked affinity probe that was not bound to the yeast cells. After washing, cells were mixed with 90 μl of streptavidin-coated microbeads provided by the μMACS Streptavidin Starting Kit (Miltenyi Biotec, 130-091-287) in a total volume of 800 μl of 0.1% BSA in TBS. Cell suspension was shielded from light and incubated at 4°C for 30 min. After the labeling reaction was finished, the cells and magnetic beads were added to 30 ml of 0.1% BSA in TBS. The cell suspension was pelleted by centrifugation at 500 g for 10 min. The supernatant was aspirated, and the cell pellet including the magnetic beads was resuspended in 500 μl 0.1% BSA-TBS. Yeast cells bound to l magnetic beads by biotin-streptavidin interaction were then captured by a magnet according to manufacturer’s instructions, and the beads were washed with 0.1% BSA in TBS. Cells bound to the magnetic beads were eluted into 5 ml SDCAA medium supplemented with 100 μg/ml ampicillin and 50 μg/ml kanamycin and were cultured at 30°C overnight. In parallel, library cells were bound to primary and secondary antibodies to evaluate the display of DhbE mutants on the yeast cell surface.
The second-round selection of the yeast library was to enrich cells displaying full-length DhbE mutants. The library cells amplified from the first round of selection were labeled with 10 μg/ml mouse anti-HA antibody (Santa Cruz Biotechnology, sc-7392) and chicken anti-c-myc antibody (Invitrogen, A-21281). After washing the cells, secondary antibodies of goat antimouse antibody conjugated with Alexa Fluor 647 (Invitrogen, A-21235) and goat anti-chicken antibody conjugated with Alexa 488 (Invitrogen, A-11039) were added to the cells in 0.1% BSA in TBS. After incubation, cells were washed twice with 0.1% BSA in TBS. Cells with the top 15%–20% of brightness labeled by both fluorophores were collected by FACS.
In subsequent rounds of yeast selection, cells were labeled with affinity probes 9 or 10 and 4 μg/ml chicken anti-c-myc antibody (Invitrogen, A-21281) as the primary reagents, and 5 μg/ml streptavidin-PE (Invitrogen, S-32350) and 5 μg/ml goat antichicken antibody conjugated to Alexa Fluor 488 (Invitrogen, A-11039) as the secondary reagents. The labeling reaction was incubated at 4°C for 30 min; then cells were pelleted, washed twice, and resuspended in TBS with 0.1% BSA for sorting. The concentration of probe 9 or 10 was decreased from 1 μM in the third round of selection to 0.1 μM in the fifth round of selection. The sorting gate also became more stringent with the top 0.1% of doubly labeled cells collected in the fifth round of selection. After the fifth round of cell sorting, the collected cells were grown in an SDCCA medium to an OD600 around 0.5. Zymoprep II Yeast Plasmid Miniprep Kit (Zymo Research, D2004) was then used to extract pCTCON2 plasmid DNA from the yeast cells. The plasmids were then transformed into SS320 E. coli competent cells. Individual colonies were miniprepped, and the plasmid DNA was sequenced to reveal the selected mutations in the DhbE clones. The DNA sequences of the mutant DhbE clones were aligned by the ClustalW algorithm in the Lasergene MegAlign software (DNAStar).
Supplementary Material
SIGNIFICANCE.
We describe a powerful method based on yeast cell sorting to engineer A-domain substrate specificity employing acyl-AMS probes for affinity selection that enables one to screen millions of mutants, which is three orders of magnitude more than previously described. Using this method, we successfully switched the substrate specificity of DhbE for nonnative substrates. We envision that the yeast selection platform can also be used to engineer A-domain specificity with amino acids by selection with the corresponding aminoacyl-AMS probes. Our results also provided insight into the substrate transfer to the ArCP domain as the His234Trp mutation was catalytically incompetent in the thioesterification reaction, but fully active in the adenylation partial reaction. Activity could be fully recovered by introducing the reverse mutation, and these results are consistent with structural studies recently reported from Gulick and co-workers (Sundlov et al., 2012).
Acknowledgments
This work was supported by a lab startup grant from the University of Chicago (to J.Y.) and a grant from the National Institutes of Health (AI070219 to C.C.A.). We thank Professor K. Dane Wittrup of the Massachusetts Institute of Technology for providing the pCTCON2 plasmid and yeast strain EBY100. We also thank S. Annie Gai and Tiffany F. Chen of the Wittrup Group and Satoe Takahashi, Stephen Kron, Akiko Koide, and Shohei Koide of the University of Chicago for helpful discussions.
Footnotes
Supplemental Information includes fourteen figures, three tables, and Supplemental Experimental Procedures and can be found with this article online at http://dx.doi.org/10.1016/j.chembiol.2012.10.020.
References
- Ames BD, Walsh CT. Anthranilate-activating modules from fungal nonribosomal peptide assembly lines. Biochemistry. 2010;49:3351–3365. doi: 10.1021/bi100198y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baca M, Scanlan TS, Stephenson RC, Wells JA. Phage display of a catalytic antibody to optimize affinity for transition-state analog binding. Proc Natl Acad Sci USA. 1997;94:10063–10068. doi: 10.1073/pnas.94.19.10063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baltz RH. Biosynthesis and genetic engineering of lipopeptide antibiotics related to daptomycin. Curr Top Med Chem. 2008;8:618–638. doi: 10.2174/156802608784221497. [DOI] [PubMed] [Google Scholar]
- Cane DE, Walsh CT, Khosla C. Harnessing the biosynthetic code: combinations, permutations, and mutations. Science. 1998;282:63–68. doi: 10.1126/science.282.5386.63. [DOI] [PubMed] [Google Scholar]
- Challis GL, Ravel J, Townsend CA. Predictive, structure-based model of amino acid recognition by nonribosomal peptide synthetase adenylation domains. Chem Biol. 2000;7:211–224. doi: 10.1016/s1074-5521(00)00091-0. [DOI] [PubMed] [Google Scholar]
- Chao G, Lau WL, Hackel BJ, Sazinsky SL, Lippow SM, Wittrup KD. Isolating and engineering human antibodies using yeast surface display. Nat Protoc. 2006;1:755–768. doi: 10.1038/nprot.2006.94. [DOI] [PubMed] [Google Scholar]
- Chen CY, Georgiev I, Anderson AC, Donald BR. Computational structure-based redesign of enzyme activity. Proc Natl Acad Sci USA. 2009;106:3764–3769. doi: 10.1073/pnas.0900266106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clardy J, Walsh C. Lessons from natural molecules. Nature. 2004;432:829–837. doi: 10.1038/nature03194. [DOI] [PubMed] [Google Scholar]
- Ehmann DE, Shaw-Reid CA, Losey HC, Walsh CT. The EntF and EntE adenylation domains of Escherichia coli enterobactin synthetase: sequestration and selectivity in acyl-AMP transfers to thiolation domain co-substrates. Proc Natl Acad Sci USA. 2000;97:2509–2514. doi: 10.1073/pnas.040572897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eppelmann K, Stachelhaus T, Marahiel MA. Exploitation of the selectivity-conferring code of nonribosomal peptide synthetases for the rational design of novel peptide antibiotics. Biochemistry. 2002;41:9718–9726. doi: 10.1021/bi0259406. [DOI] [PubMed] [Google Scholar]
- Evans BS, Chen Y, Metcalf WW, Zhao H, Kelleher NL. Directed evolution of the nonribosomal peptide synthetase AdmK generates new andrimid derivatives in vivo. Chem Biol. 2011;18:601–607. doi: 10.1016/j.chembiol.2011.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreras JA, Ryu JS, Di Lello F, Tan DS, Quadri LE. Small-molecule inhibition of siderophore biosynthesis in Mycobacterium tuberculosis and Yersinia pestis. Nat Chem Biol. 2005;1:29–32. doi: 10.1038/nchembio706. [DOI] [PubMed] [Google Scholar]
- Finking R, Neumuller A, Solsbacher J, Konz D, Kretzschmar G, Schweitzer M, Krumm T, Marahiel MA. Aminoacyl adenylate substrate analogues for the inhibition of adenylation domains of nonribosomal peptide synthetases. Chembiochem: A European Journal of Chemical Biology. 2003;4:903–906. doi: 10.1002/cbic.200300666. [DOI] [PubMed] [Google Scholar]
- Fischbach MA, Walsh CT. Assembly-line enzymology for poly-ketide and nonribosomal Peptide antibiotics: logic, machinery, and mechanisms. Chem Rev. 2006;106:3468–3496. doi: 10.1021/cr0503097. [DOI] [PubMed] [Google Scholar]
- Fischbach MA, Lai JR, Roche ED, Walsh CT, Liu DR. Directed evolution can rapidly improve the activity of chimeric assembly-line enzymes. Proc Natl Acad Sci USA. 2007;104:11951–11956. doi: 10.1073/pnas.0705348104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gietz RD, Schiestl RH. Applications of high efficiency lithium acetate transformation of intact yeast cells using single-stranded nucleic acids as carrier. Yeast. 1991;7:253–263. doi: 10.1002/yea.320070307. [DOI] [PubMed] [Google Scholar]
- Gietz RD, Woods RA. Transformation of yeast by lithium acetate/single-stranded carrier DNA/polyethylene glycol method. Methods Enzymol. 2002;350:87–96. doi: 10.1016/s0076-6879(02)50957-5. [DOI] [PubMed] [Google Scholar]
- Gulick AM. Conformational dynamics in the Acyl-CoA synthetases, adenylation domains of non-ribosomal peptide synthetases, and firefly luciferase. ACS Chem Biol. 2009;4:811–827. doi: 10.1021/cb900156h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- May JJ, Wendrich TM, Marahiel MA. The dhb operon of Bacillus subtilis encodes the biosynthetic template for the catecholic siderophore 2,3-dihydroxybenzoate-glycine-threonine trimeric ester bacillibactin. J Biol Chem. 2001;276:7209–7217. doi: 10.1074/jbc.M009140200. [DOI] [PubMed] [Google Scholar]
- May JJ, Kessler N, Marahiel MA, Stubbs MT. Crystal structure of DhbE, an archetype for aryl acid activating domains of modular nonribosomal peptide synthetases. Proc Natl Acad Sci USA. 2002;99:12120–12125. doi: 10.1073/pnas.182156699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDaniel R, Thamchaipenet A, Gustafsson C, Fu H, Betlach M, Ashley G. Multiple genetic modifications of the erythromycin polyketide synthase to produce a library of novel “unnatural” natural products. Proc Natl Acad Sci USA. 1999;96:1846–1851. doi: 10.1073/pnas.96.5.1846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miethke M, Bisseret P, Beckering CL, Vignard D, Eustache J, Marahiel MA. Inhibition of aryl acid adenylation domains involved in bacterial siderophore synthesis. FEBS J. 2006;273:409–419. doi: 10.1111/j.1742-4658.2005.05077.x. [DOI] [PubMed] [Google Scholar]
- Miller KD, Pefaur NB, Baird CL. Construction and screening of antigen targeted immune yeast surface display antibody libraries. Curr Protoc Cytom. 2008;4(Unit 4.7) doi: 10.1002/0471142956.cy0407s45. [DOI] [PubMed] [Google Scholar]
- Mitchell CA, Shi C, Aldrich CC, Gulick AM. Structure of PA1221, a nonribosomal peptide synthetase containing adenylation and peptidyl carrier protein domains. Biochemistry. 2012;51:3252–3263. doi: 10.1021/bi300112e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neres J, Labello NP, Somu RV, Boshoff HI, Wilson DJ, Vannada J, Chen L, Barry CE, 3rd, Bennett EM, Aldrich CC. Inhibition of siderophore biosynthesis in Mycobacterium tuberculosis with nucleoside bisubstrate analogues: structure-activity relationships of the nucleobase domain of 5′-O-[N-(salicyl)sulfamoyl]adenosine. J Med Chem. 2008;51:5349–5370. doi: 10.1021/jm800567v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen KT, Ritz D, Gu JQ, Alexander D, Chu M, Miao V, Brian P, Baltz RH. Combinatorial biosynthesis of novel antibiotics related to daptomycin. Proc Natl Acad Sci USA. 2006;103:17462–17467. doi: 10.1073/pnas.0608589103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavela-Vrancic M, Dieckmann R, Döhren HV, Kleinkauf H. Editing of non-cognate aminoacyl adenylates by peptide synthetases. Biochem J. 1999;342:715–719. [PMC free article] [PubMed] [Google Scholar]
- Pfeifer BA, Admiraal SJ, Gramajo H, Cane DE, Khosla C. Biosynthesis of complex polyketides in a metabolically engineered strain of E. coli. Science. 2001;291:1790–1792. doi: 10.1126/science.1058092. [DOI] [PubMed] [Google Scholar]
- Qiao C, Gupte A, Boshoff HI, Wilson DJ, Bennett EM, Somu RV, Barry CE, 3rd, Aldrich CC. 5′-O-[(N-acyl)sulfamoyl]adenosines as antitubercular agents that inhibit MbtA: an adenylation enzyme required for siderophore biosynthesis of the mycobactins. J Med Chem. 2007;50:6080–6094. doi: 10.1021/jm070905o. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robbel L, Marahiel MA. Daptomycin, a bacterial lipopeptide synthesized by a nonribosomal machinery. J Biol Chem. 2010;285:27501–27508. doi: 10.1074/jbc.R110.128181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sieber SA, Marahiel MA. Molecular mechanisms underlying nonribosomal peptide synthesis: approaches to new antibiotics. Chem Rev. 2005;105:715–738. doi: 10.1021/cr0301191. [DOI] [PubMed] [Google Scholar]
- Sikora AL, Wilson DJ, Aldrich CC, Blanchard JS. Kinetic and inhibition studies of dihydroxybenzoate-AMP ligase from Escherichia coli. Biochemistry. 2010;49:3648–3657. doi: 10.1021/bi100350c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somu RV, Boshoff H, Qiao C, Bennett EM, Barry CE, 3rd, Aldrich CC. Rationally designed nucleoside antibiotics that inhibit siderophore biosynthesis of Mycobacterium tuberculosis. J Med Chem. 2006;49:31–34. doi: 10.1021/jm051060o. [DOI] [PubMed] [Google Scholar]
- Stachelhaus T, Schneider A, Marahiel MA. Rational design of peptide antibiotics by targeted replacement of bacterial and fungal domains. Science. 1995;269:69–72. doi: 10.1126/science.7604280. [DOI] [PubMed] [Google Scholar]
- Stachelhaus T, Mootz HD, Marahiel MA. The specificity-conferring code of adenylation domains in nonribosomal peptide synthetases. Chem Biol. 1999;6:493–505. doi: 10.1016/S1074-5521(99)80082-9. [DOI] [PubMed] [Google Scholar]
- Sundlov JA, Shi C, Wilson DJ, Aldrich CC, Gulick AM. Structural and functional investigation of the intermolecular interaction between NRPS adenylation and carrier protein domains. Chem Biol. 2012;19:188–198. doi: 10.1016/j.chembiol.2011.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thirlway J, Lewis R, Nunns L, Al Nakeeb M, Styles M, Struck AW, Smith CP, Micklefield J. Introduction of a non-natural amino acid into a nonribosomal peptide antibiotic by modification of adenylation domain specificity. Angew Chem Int Ed Engl. 2012;51:7181–7184. doi: 10.1002/anie.201202043. [DOI] [PubMed] [Google Scholar]
- Uguru GC, Milne C, Borg M, Flett F, Smith CP, Micklefield J. Active-site modifications of adenylation domains lead to hydrolysis of upstream nonribosomal peptidyl thioester intermediates. J Am Chem Soc. 2004;126:5032–5033. doi: 10.1021/ja048778y. [DOI] [PubMed] [Google Scholar]
- von Döhren H, Dieckmann R, Pavela-Vrancic M. The nonribosomal code. Chem Biol. 1999;6:R273–R279. doi: 10.1016/s1074-5521(00)80014-9. [DOI] [PubMed] [Google Scholar]
- Wang L, Xie J, Schultz PG. Expanding the genetic code. Annu Rev Biophys Biomol Struct. 2006;35:225–249. doi: 10.1146/annurev.biophys.35.101105.121507. [DOI] [PubMed] [Google Scholar]
- Watanabe K, Hotta K, Nakaya M, Praseuth AP, Wang CC, Inada D, Takahashi K, Fukushi E, Oguri H, Oikawa H. Escherichia coli allows efficient modular incorporation of newly isolated quinomycin biosynthetic enzyme into echinomycin biosynthetic pathway for rational design and synthesis of potent antibiotic unnatural natural product. J Am Chem Soc. 2009;131:9347–9353. doi: 10.1021/ja902261a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webb MR. A continuous spectrophotometric assay for inorganic phosphate and for measuring phosphate release kinetics in biological systems. Proc Natl Acad Sci USA. 1992;89:4884–4887. doi: 10.1073/pnas.89.11.4884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamagishi Y, Shoji I, Miyagawa S, Kawakami T, Katoh T, Goto Y, Suga H. Natural product-like macrocyclic N-methyl-peptide inhibitors against a ubiquitin ligase uncovered from a ribosome-expressed de novo library. Chem Biol. 2011;18:1562–1570. doi: 10.1016/j.chembiol.2011.09.013. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.