

Abstract
The Suzuki-Miyaura cross-coupling of unprotected, nitrogen-rich heterocycles using precatalysts P1 or P2 is reported. The procedure allows for the reaction of variously substituted indazole, benzimidazole, pyrazole, indole, oxindole and azaindole halides under mild conditions in good to excellent yields. Additionally, the mechanism behind the inhibitory effect of unprotected azoles on Pd-catalyzed cross-coupling reactions is described based on evidence gained through experimental, crystallographic, and theoretical investigations.
1. Introduction
The ability to combine aromatic heterocyclic fragments is an important and challenging subset of the field of C-C cross-coupling reactions with implications both in academic and industrial settings.1 Heterocyclic structures are often key fragments of biologically active molecules and many drugs contain nitrogen-rich heterocyclic moieties such as pyrimidines, piperazines or benzimidazoles, as well as indoles and indazoles.2 Of these, annulated azoles bearing relatively acidic NH groups are key components in several important compounds (see Figure 1).
Figure 1.
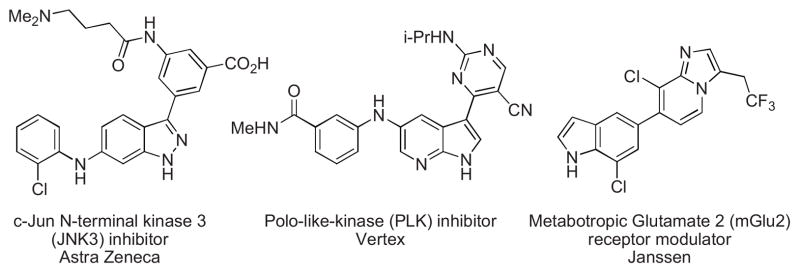
Bioactive molecules containing free N-H azole moieties.3
Unfortunately, many of the standard protocols for palladium-catalyzed C-C bond-forming cross-coupling reactions fail in the presence of substrates bearing free N-H groups.4 Instead, such molecules are obtained either via construction of the heterocyclic rings with the desired residues already attached,5 or via functionalization using the corresponding N-protected analogues.6 While these routes have merit, they are often limited in substrate scope and/or necessarily require extra protection/deprotection steps. Thus, access to facile methods for the direct use of these acidic, nitrogen-rich heterocycles in palladium-catalyzed C-C cross-coupling processes is highly desirable.7
The Suzuki-Miyaura coupling is arguably the most important C-C-coupling method practiced today due to its broad substrate scope, high level of functional group tolerance, and, with modern catalysts, high turnover rates.8 However, to date there are only a few examples of Suzuki-Miyaura reactions of substrates containing an unprotected azole,9,10 and these typically employ costly starting materials and require strong bases and/or high catalyst loadings. Though these heterocycles are common amongst biologically-active compounds, the reasons for the generally observed low yields of these substrates have so far not been examined in detail. Either the low reactivity of the corresponding oxidative addition complex or an inhibition of Pd(II) by starting material or product was proposed, but not further investigated.11
Herein, we report a method for the Suzuki-Miyaura coupling of a wide range of unprotected azoles, such as indazoles, benzimidazoles, pyrazoles, indoles, oxindoles, and azaindoles, under relatively mild conditions. Additionally, we provide insight into the mechanism behind the inhibitory effects of acidic NH groups on Pd-catalyzed cross-coupling reactions.
2. Results and Discussion
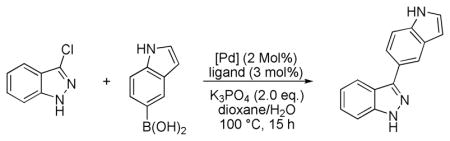
We began our study by evaluating the coupling of 3-chloroindazole both unprotected (1a) and protected with an N-benzyl group (1b) using conditions that we have previously reported (Scheme 1).12
Scheme 1.

Suzuki-Miyaura cross-coupling of unprotected and Bn-protected 3-chloroindazole (1a,b).
For the protected chloroindazole 1b the desired product was obtained in high yield. In contrast, for unprotected 1a no product was observed. We found that by increasing the reaction temperature to 100 °C and by employing 2.0 equivalents of boronic acid, a modest yield of coupled product could be obtained for 1a. An examination of different palladium sources and ligands was carried out (Table 1) and we found that protocols that employed SPhos and XPhos provided the coupled product in the highest yields.13 While the use of Pd2dba3 as the palladium source provided the product in up to 56% yield, the use of our second generation precatalysts P1 and P2 provided the best results; with P2 the product was formed in 80% yield. Additionally, a higher turnover rate and thus less protodeboronation was observed in the presence of SPhos precatalyst P2 as opposed to when Pd(OAc)2 or Pd2dba3 were employed. In the absence of ligand, no reaction occurred.14
Table 1.
Influence of ligands and Pd sources on the Suzuki-Miyaura cross-coupling of 3-chloroindazole.[a]
| ||||
|---|---|---|---|---|
| [Pd] | Ligand | Conversion [%][b] | Yield [%][c] | |
| 1 | Pd2dba3 | XPhos | 75 | 56 |
| 2 | Pd2dba3 | SPhos | 71 | 52 |
| 3 | Pd2dba3 | RuPhos | 59 | 40 |
| 4 | Pd2dba3 | Pt-Bu3 | 23 | 10 |
| 5 | Pd2dba3 | none | <5 | 0 |
| 6 | Pd(OAc)2 | XPhos | 68 | 49 |
| 7 | Pd(OAc)2 | SPhos | 65 | 47 |
| 8 | P1 | XPhos | 87 | 69 |
| 9 | P2 | SPhos | 97 | 80 |
| 10 | P2 | SPhos | 100 | 90[d] |
Reaction conditions: 3-chloroindazole (0.25 mmol), 5-indole boronic acid (0.50 mmol), Pd source (2 mol%), ligand (3 mol%), K3PO4 (0.50 mmol), dioxane (1 mL), H2O (0.2 mL), 100 °C, 15 h.
Determined by GC.
Determined by HPLC.
2.5 mol% of P2 was used.
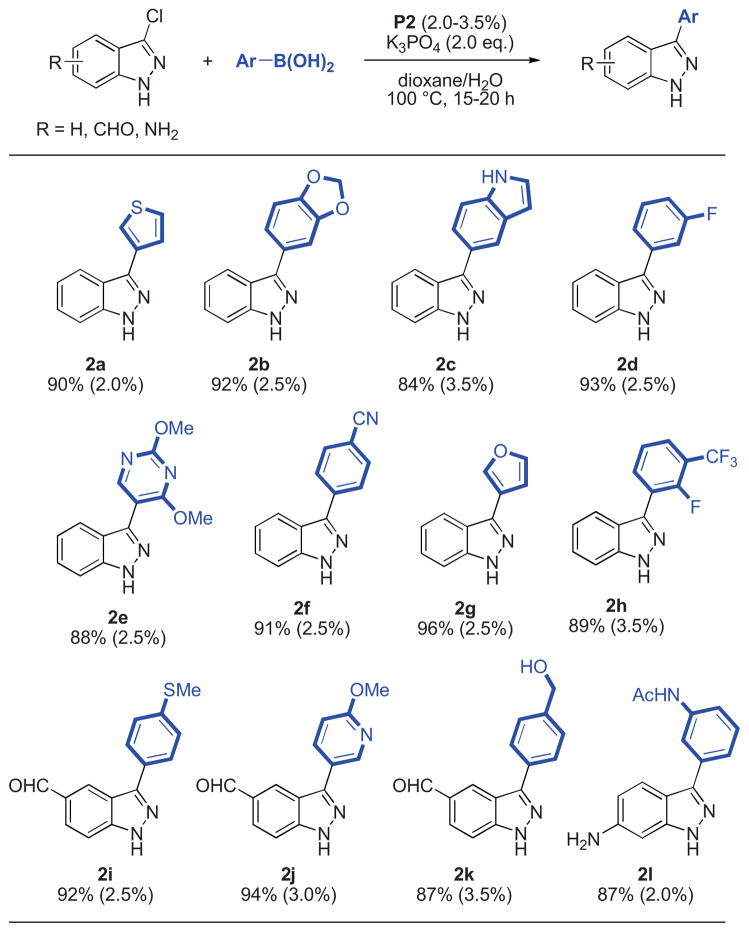
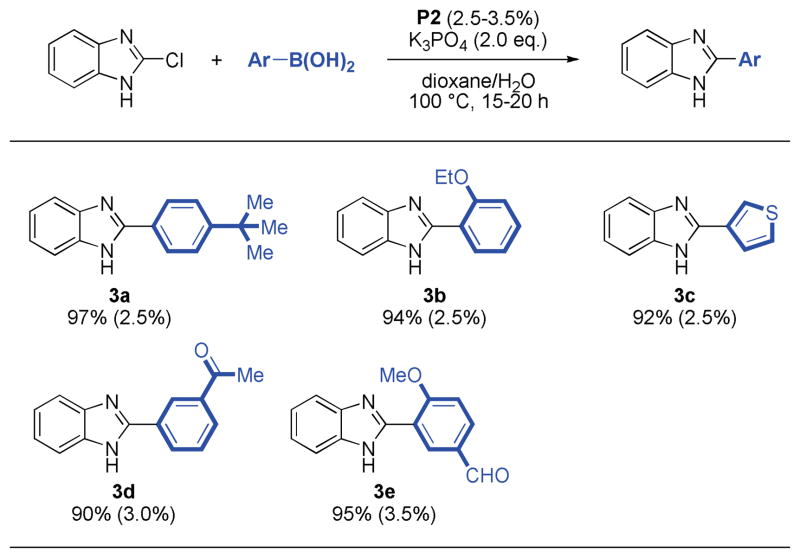
Having identified a successful method,15 a variety of aryl and heteroaryl boronic acids were successfully coupled with substituted indazole and benzimidazole chlorides (Table 2, 3). All reactions reached full conversion within 15–20 hours and gave the corresponding products in good to excellent yields. A diverse set of heterocyclic structures and sensitive functional groups were tolerated. In general, moderate catalyst loadings were sufficient for reactions employing both electron-rich aryl halides or electron-rich boronic acids.
Table 2.
|
Reaction conditions: aryl halide (1.00 mmol), boronic acid (2.00 mmol), P2 (2.0–3.5 mol%), K3PO4 (2.00 mmol), dioxane (4 mL), H2O (1 mL), 100 °C, 15–20 h.
Isolated yields represent the average of two runs.
Table 3.
|
Reaction conditions: aryl halide (1.00 mmol), boronic acid (2.00 mmol), P2 (2.5–3.5 mol%), K3PO4 (2.00 mmol), dioxane (4 mL), H2O (1 mL), 100 °C, 15–20 h.
Isolated yields represent the average of two runs.
It was also possible to couple 3- and 4-bromopyrazole,16 although the reactions required a slightly increased quantity of catalyst and the addition of another equivalent of ligand relative to palladium. In these cases the use of XPhos-derived precatalyst P1 provided higher yields of products. Thus, 4-aryl and 3-aryl pyrazoles were isolated in good to very good yields (61–86%, Table 4). Generally, 3-bromopyrazoles tend to react faster than 4-bromopyrazoles. The reaction of 4-bromoimidazole and phenylboronic acid was also attempted under these reaction conditions but provided the product only in low yield (31%) due to incomplete conversion of the heteroaryl bromide.
Table 4.
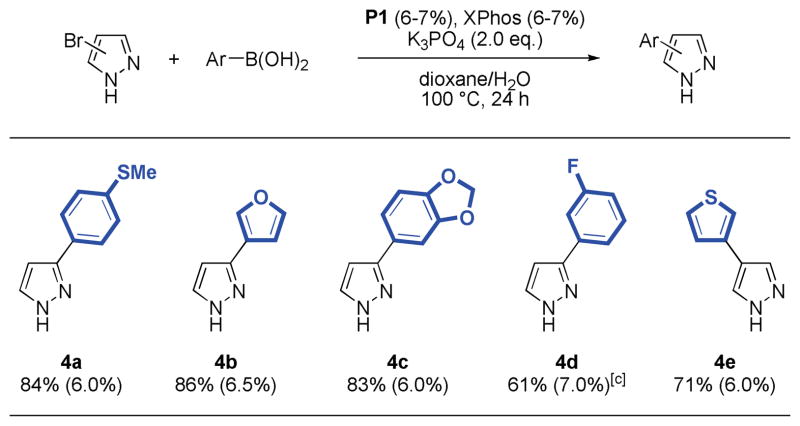
|
Reaction conditions: aryl halide (1.00 mmol), boronic acid (2.00 mmol), P1 (6–7 mol%), K3PO4 (2.00 mmol), dioxane (4 mL), H2O (1 mL), 100 °C, 24 h.
Isolated yields represent the average of two runs.
P1:XPhos ratio is 1:1.5.
We also found that chloroindoles, oxindoles and azaindoles were excellent substrates under these conditions. In fact, these coupling reactions proceeded with lower quantities of catalyst (1.0–1.5 %), fewer equivalents of boronic acid (1.5 eq.), in shorter reaction times, and at lower temperatures (60 °C), while still providing the desired products in excellent yields (91–99%). These milder conditions even allowed for the use of less stable boronic acids such as 6-fluoro-3-pyridyl and 2-benzofuranyl boronic acid (5a, 5d), which are known to readily undergo protodeboronation.12a Precatalyst P1 gave superior results, while the use of P2 did not lead to significant formation of product (Table 5).
Table 5.
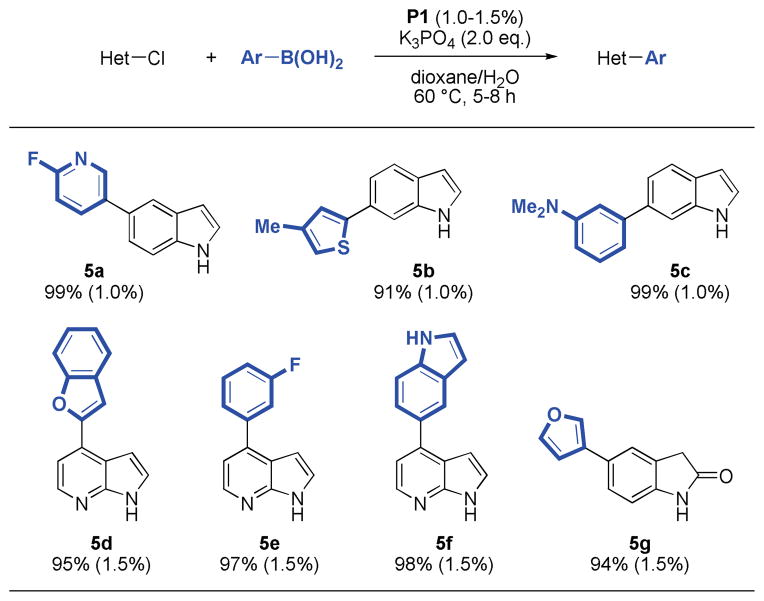
|
Reaction conditions: aryl halide (1.00 mmol), boronic acid (1.50 mmol), P1 (1.0–1.5 mol%), K3PO4 (2.00 mmol), dioxane (4 mL), H2O (1 mL), 60 °C, 5–8 h.
Isolated yields represent the average of two runs.
To illustrate the utility of our method we prepared the c-Jun N-terminal kinase 3 inhibitor 8 in two steps from commercially available indazole 6 (Scheme 2).17 Suzuki-Miyaura cross-coupling provided the 3-arylindazole 7 in excellent yield (95%). Intermediate 7 was then subjected to a Pd-catalyzed C-N bond-forming reaction to provide N-arylamine 8 in 82% yield over the two steps.18
Scheme 2.

Synthesis of JNK3-inhibitor 8.
While this new methodology allows for the direct formation of a wide range of biaryl-containing unprotected azoles, some limitations still remain with respect to substrate scope. For example, the presence of acidic functional groups on the boronic acid that can potentially bind to the palladium center (carboxylic acids, phenols, pyrazole) appears to inhibit the reaction. Additionally, the use of very unstable boronic acids (4-pyridyl, 2-benzothienyl) in conjunction with halo-pyrazole, indazole and benzimidazole coupling partners primarily resulted in protodeboronation and, subsequently, low product yields (<5%) due to the use of higher temperature (>60 °C) reaction conditions.
3. Mechanistic investigation
In order to understand the low reactivity of these substrates under classical cross-coupling conditions we chose to investigate their effect on the reaction mechanism. We reasoned that the most probable mechanisms for inhibition were: 1. complexation of the metal center and thus deactivation of the catalyst, 2. a high energy barrier for oxidative addition of ArX or 3. a high energy barrier for reductive elimination to form product. While the first possibility could be influenced by several factors, oxidative addition and reductive elimination are primarily dependent upon the structures and properties of the substrates and catalysts.
3.1 Experimental studies: metal complexation
Under standard conditions (as in Table 5) 6-chloroindole readily reacts with phenyl boronic acid to give the coupling product in 97% yield. However, upon addition of indazole (10a) or benzimidazole (11a), product formation is inhibited. Not surprisingly, the addition of indole (9a) itself has no such effect (Scheme 3).
Scheme 3.

Influence of additives on the Suzuki-Miyaura reaction of 6-chloroindole.
Comparing the reactivity with the acidity of the substrates is informative. Under typical Suzuki-Miyaura conditions, more acidic substrates often exist mainly in their deprotonated form. In particular, there is a significant decrease in the rates of reaction for indazole substrates as compared to indole ones, which correlates with a difference in the ratio of azole-H to azole-anion (based on their respective pKa values). While the less acidic indoles, oxindoles and azaindoles should be found mainly in their neutral form under the reaction conditions, the predominant state of indazoles, benzimidazoles, pyrazoles and imidazoles would likely be as the corresponding anions as their pKa values are less than that of H2O (Figure 2).19,20
Figure 2.
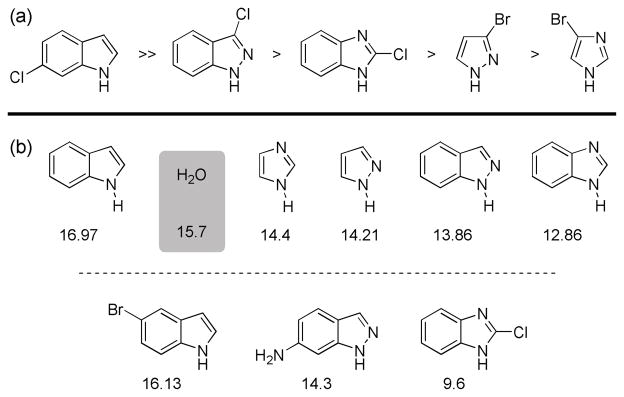
(a) Relative reactivity of heteroaryl halides in the Suzuki-Miyaura cross coupling and (b) pKa values (in H2O) of the parent compounds and relevant derivatives.19
Given this, we hypothesized that the formation of N-azolyl palladium complexes might account for the decreased reactivity of more acidic substrates.21 The presence of electron-donating groups increases the pKa along with the reactivity, electron-withdrawing groups display the opposite trend.20 The effect of the substituents on the pKa values of the parent heterocycles is likely due to an increased stabilization of the deprotonated form. While this similarity in trend between acidity and a decrease in reactivity holds true for annulated azoles, it does not account for the lower reactivity of pyrazole and imidazole halides when compared with indazole and benzimidazole derived substrates (see below for a discussion of other effects than acidity on the overall reactivity). Moreover, we could not completely rule out complexation of the basic imine-type nitrogen atoms as being the cause of reduced reaction rates. To this end, we examined the influence of the addition of several heterocycles as well as their N-methyl analogues on the yield of the cross-coupling between 6-chloroindole and phenylboronic acid. As depicted in Figure 2, in all cases the addition of the heterocycle led to a decreased yield (compared to the reaction with no additional heterocycle added) after a reaction time of 2 h. However, the influence of the N-methyl analogs is significantly smaller than that of the unprotected NH-heterocycles. The biggest difference was observed in the comparison of 1H- vs 1-Me-benzimidazole, showing a difference in inhibition of about two orders of magnitude (11a vs. 11b). Thus, while the imine-type basic nitrogen does seem to have an inhibitory effect, as especially evident in the case of 12b, the observed suppression of product formation is more strongly correlated with the presence of the acidic NH groups.
The addition of more boronic acid can partially counteract this inhibitory effect, as can raising the reaction temperature (see Figures S1, S2 in the Supporting Information). The former likely rules out a direct complexation of the Pd(0) species by an azolyl anion as the reverse process should be insensitive to added boronic acid.
Efforts were undertaken to characterize potential intermediates in the catalytic cycle. To this end, we prepared and isolated [SPhos-Pd(Ph)Cl] and [SPhos-Pd(Ph)OH],24,25 as well as dimeric Pd(II) azolyl complex 14 with indazole as the bridging ligand. Whether the dimeric species 14 or the corresponding monomer would be favored in solution is not clear but likely depends on the solvent employed.26
To assess whether 14 could be formed from [SPhos-Pd(Ph)Cl] (15) or [SPhos-Pd(Ph)OH] (16), both complexes were allowed to react with potassium indazolide. After only 20 minutes at room temperature, 14 was formed in 63% and 82% yield, respectively, as evidenced by the characteristic chemical shift of the ligated phosphine (31P-NMR).27 The ease of formation of 14 from 15 and 16 is consistent with 14 being an off-cycle species, in which the catalyst mainly resides (Scheme 4).28
Scheme 4.
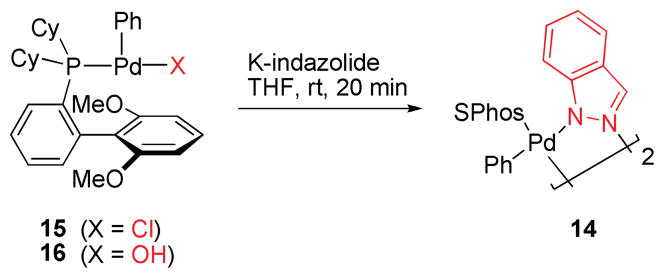
Formation of dimer 14 from 15 or 16.
Additionally, we tested how 14 would compare as catalyst in the Suzuki-Miyaura cross-coupling under our new conditions for indazole halides (Table 2) and under standard cross-coupling conditions.12a Interestingly, no difference in kinetic behavior was observed in the cross-coupling of 3-chloroindazole with 3-fluorophenylboronic acid, under our conditions for indazole halides (2.0 mol% catalyst, 2.00 eq. K3PO4, dioxane/H2O 4:1, 100 °C, 15 h), when either 14 or P2 was used as the precatalyst. In contrast, the reaction of 3-chloroanisole and 2,6-dimethylphenylboronic acid gave no product with 14 as catalyst under standard Suzuki-Miyaura conditions (2.0 mol% catalyst, 2.00 eq. K3PO4, dioxane/H2O 4:1, 25 °C, 18 h) but good conversion was achieved with 15 (see Figures S3, S4 in the Supporting Information).29,30 Even after three days at room temperature, complex 14 was the only species that could be observed by 31P-NMR spectroscopy in the reaction of 14 under the latter conditions.
We also briefly investigated the effect of the halide position (from ArX) on the overall reaction progress. As above, 3- and 4-bromoindazole were allowed to react with 3-fluorobenzeneboronic acid (Scheme 5). Unexpectedly, we found that the reaction employing 4-bromoindazole resulted in less product formation than the one using 3-bromoindazole. We thus hypothesized that the observed reactivity difference between the aryl halide regioisomers could be due to product inhibition. While the aryl halide substrates should display only minor differences in their steric properties, the products’ steric demand should be markedly different. We therefore examined the influence of the cross-coupling products and the parent heterocycles on the yield of the cross-coupling between 6-chloroindole and phenylboronic acid. As can be seen (Figure 5), while the effect of added 4-phenylindazole (10d) and indazole (10a) are comparable, 3-substituted azole 10c inhibits the reaction to a much lesser extent. The same decrease in inhibition can be observed when comparing benzimidazoles 11a and 11c. Again there is only small difference between the inhibitory effect of 4-phenylimidazole (12c) and imidazole (12a) itself, showing the difficulty of these substrates in cross-coupling reactions.31 We postulate that these trends can be attributed to the ability of the parent heterocycles, as well as some select coupling products, to form azole-bridged dimers or trimers, which most products cannot due to steric reasons (see Figure 6). Products that cannot form dimeric structures for steric reasons should inhibit the cross-coupling to a lesser degree, as has been observed, since dimer formation is potentially the major reason for the observed catalyst deactivation. Thus, it appears likely that the catalyst can mainly be found as dimeric complex 14 in the case of 1,2-azoles. It is not known whether a bridged 1,3-azolyl Pd(II) complex could also occur in reactions involving (benz)imidazole halides. However, such complexes are known.32 While for indole, indazole and benzimidazole the increased inhibition of the cross-coupling reaction can be correlated with the increased acidity of the NH functional group, pyrazole and imidazole inhibit reactions more strongly but, conversely, are less acidic than their annulated congeners. This is probably due to the increased basicity of the imine-type nitrogen in starting materials as well as products (cf. Figure 3) in conjunction with smaller steric demand.33 In addition, we cannot rule out an influence of the different electronic properties of the substrates or products, which could additionally explanation minor reactivity differences, e.g. between regioisomers.
Scheme 5.
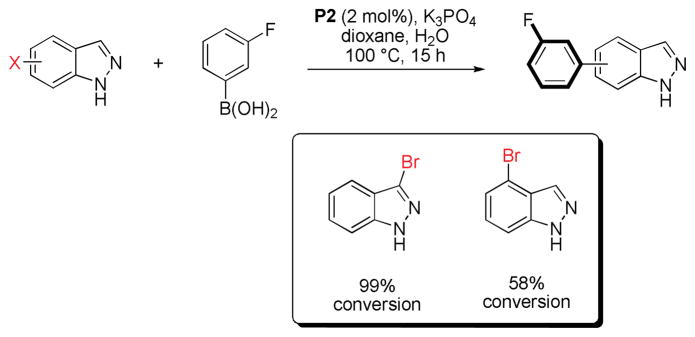
Influence of the halide substitution on the conversion of 3- and 4-haloindazoles.
Figure 5.

Effect of heterocyclic additives on the yield of the Suzuki-Miyaura coupling between 6-chloroindole and phenylboronic acid.22,23
Figure 6.
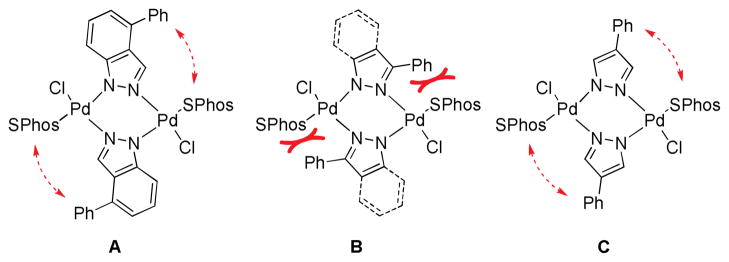
Potential steric interaction in the postulated dimers containing 10d (A), 10c or 13c (B) or 13d (C).
Figure 3.

Effect of 1H- and 1-methyl-heterocyclic additives on the yield of the Suzuki coupling between 6-chloroindole and phenylboronic acid.22,23
3.2 Computational studies: oxidative addition, reductive elimination and metal complexation
Besides complexation of the metal center, we additionally considered the oxidative addition and reductive elimination as being potentially rate-limiting. Thus, in addition to these experimental observations DFT calculations were undertaken to obtain the energy barriers for the oxidative addition as well as the reductive elimination of different aryl groups (Figure 7).24 Moreover, the relative energies of complexes 15 and 17 were investigated (see Supporting Information for details).
Figure 7.

Calculated energies in THF (B3LYP/6-31+G(d,p)) for the oxidative addition, reductive elimination and the equilibrium between 15 and 17. For oxidative addition, anionic aryl halides refer to the corresponding deprotonated azole chlorides.
The oxidative addition step was investigated for several aryl halides: phenyl, 5-indolyl, 3-indazolyl, 3-pyrazolyl and 4-imidazolyl chloride. The calculated activation energies for the neutral aryl halides range from 14.4 to 19.4 kcal/mol.34 For comparison, the energy barriers for some deprotonated aryl halides were also included,35 which show an increase in activation energy of about 4–6 kcal/mol. This effect mirrors generally observed reactivity trends as these substrates would be even more electron-rich. We also calculated the energy barrier in the reductive elimination step for these heteroaryl groups. The activation energy ranges from 5.3 to 6.9 kcal/mol, supporting the fact that biarylphosphine ligands allow for the ready reductive elimination in reactions of these substrate combinations.36 Overall, both in the case of the oxidative addition as well as the reductive elimination, these values are comparatively small and cannot explain the low reactivity of unprotected, acidic heterocycles observed in most cross-coupling reactions. For completeness, we also investigated the thermodynamic equilibrium between the chloro-Pd(II) complex 15 and the corresponding azolyl species 17. In all cases, the azolyl complex 17 is energetically favored but the energy difference is very small. From all of this data, we can conclude that primarily the innate inhibitory effect of the heterocycles, not their oxidative addition or reductive elimination energetics, determine the overall reactivity.
3.3 Proposed mechanism and nucleophile effect
Based on the experimental and theoretical data, we propose the following mechanism for the Suzuki-Miyaura cross-coupling in the presence of an inhibitory azole such as indazole (Scheme 6), which should also be applicable to other azoles as inhibitors. The presence of 1,2- or 1,3-azoles likely leads to the formation of monomeric or oligomeric Pd(II) intermediates (14, 17) that act as Pd reservoirs. For 3-chloroindazole and 2-chlorobenzimidazole as halides, the formation of dimers 20, 21 or the corresponding deprotonated azole-bridged complexes is also feasible.37 Depending on the reaction conditions, the transmetallation could either occur through [Pd-Cl] (15) or [Pd-OH] (16). However, a direct reaction of 17 with phenyl boronate cannot be ruled out.38 As was observed, the presence of additional boronic acid helps countermand the inhibitory effect by shifting the equilibrium back in favor of 15 or 16. Based on the calculated relative stabilities of 15 vs. 17, which only slightly favor the Pd-azolyl complexes, the rate determining step should therefore be the transmetallation.
Scheme 6.

Proposed catalytic cycle in the presence of indazole and potential intermediates 20, 21.
Indeed, when changing the nature of the nucleophile a drastic difference in reactivity can be observed, which can be roughly correlated with the relative nucleophilicity of the organometallic species.39 While boronic acids only react sluggishly, organozinc and especially organomagnesium nucleophiles are able to affect an efficient reaction turnover in the presence of indazole (Scheme 7).40
Scheme 7.
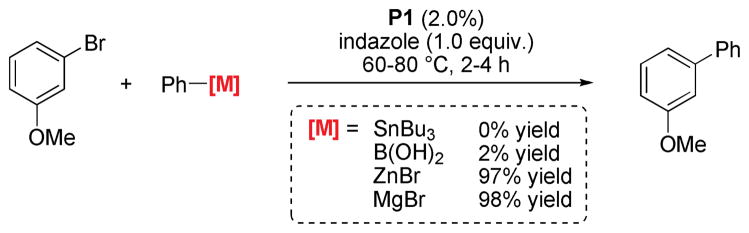
Comparison of different cross-coupling methods in the presence of unprotected indazole.
This phenomenon indicates that the energetically unfavored reverse reaction of 17/14 to 15/16 is not the cause for the low reactivity observed in the case of the Suzuki and Stille cross-coupling reaction, but rather points again to transmetallation as the rate determining step.41 However, the existence of an equilibrium between 17/14 and 15/16 favoring the N-azolyl species additionally necessitates elevated temperatures of up to 100 °C and extended reaction times. Thus, when using strongly nucleophilic organometallic coupling partners (organomagnesium and organozinc nucleophiles), the transmetallation should be fast enough to ensure efficient reaction turnover and help push the equilibrium back from the off-cycle species 17 or 14 into the active transmetallating species (15 or 16, depending on the nucleophile and reaction conditions). Thus, when faced with unreactive substrate combinations, the use of alternative aryl nucleophiles could promote the formation of the desired cross-coupling products. Although not always applicable due to issues of functional group compatibility, this principle can aid in the selection of appropriate cross-coupling conditions in the presence of nitrogen-rich, unprotected heterocycles.
4. Conclusion
In conclusion, we have developed an operatively simple and efficient general procedure for the Suzuki-Miyaura cross-coupling of several classes of unprotected azole halides. We have demonstrated the utility of this method in a straightforward synthesis of kinase inhibitor 8 in two steps from commercially available starting materials. With this methodology in hand, arylated azoles may be selectively accessed in a direct manner without the need of N-protection. A Pd-azolyl intermediate has been isolated that we propose is the catalytic resting state in the cross-coupling reaction of nitrogen-rich, acidic heterocycles. The presented experimental and computational mechanistic work is consistent with the transmetallation as the rate determining step in the cross-coupling reaction. This knowledge should prove useful in selecting optimal conditions and nucleophile/electrophile combinations while circumventing catalyst deactivation.
Supplementary Material
Figure 4.

Crystal structure of indazole-bridged Pd complex 14.
Acknowledgments
We thank the National Institutes of Health for financial support of this project (GM46059). M.A.D. is indebted to the DFG for a postdoctoral fellowship. We thank H. Harbrecht and T. Kinzel for initial studies in the synthesis of 16, M. Su for computational and experimental assistance, G. Teverovskiy, E. Vinogradova and M. McGowan for helpful discussions and P. Müller for X-Ray structural analysis of 14 and 16.
Footnotes
Notes
The authors declare the following competing financial interest(s): MIT has patents on the ligands used in this work for which SLB receives royalty payments.
Supporting Information. Experimental, spectroscopic and computational details as well as X-ray structural parameters for 14 and 16. Cartesian coordinates of all calculated intermediates. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.(a) Kotschy A, Timári T. Heterocycles from Transition Metal Catalysis: Formation And Functionalization. Springer; Dordrecht: 2005. [Google Scholar]; (b) de Meijere A, Diederich F, editors. Metal-catalyzed cross-coupling reactions. 2. Wiley-VCH; Weinheim: 2004. [Google Scholar]; (c) Ackermann L, editor. Modern Arylation Methods. Wiley-VCH; Weinheim: 2009. [Google Scholar]; (d) Schröter S, Stock C, Bach T. Tetrahedron. 2005;61:2245–2267. [Google Scholar]
- 2.(a) Roughley SD, Jordan AM. J Med Chem. 2011;54:3451–3479. doi: 10.1021/jm200187y. [DOI] [PubMed] [Google Scholar]; (b) Cerecetto H, Gerpe A, Gonzalez M, Aran VJ, Ocariz CO. Mini-Rev Med Chem. 2005;5:869–878. doi: 10.2174/138955705774329564. [DOI] [PubMed] [Google Scholar]; (c) Bansal Y, Silakari O. Bioorg Med Chem. 2012;20:6208–6236. doi: 10.1016/j.bmc.2012.09.013. [DOI] [PubMed] [Google Scholar]
- 3.(a) Swahn BM, Huerta F, Kallin E, Malmström J, Weigelt T, Viklund J, Womack P, Xue Y, Öhberg L. Bioorg Med Chem Lett. 2005;15:5095–5099. doi: 10.1016/j.bmcl.2005.06.083. [DOI] [PubMed] [Google Scholar]; (b) Mortimore M, Young SC, Everitt SRL, Knegtel R, Pinder JL, Rutherford AP, Durrant S, Brenchley G, Charrier JD, O’Donnell M. WO 2008079346. Vertex, USA PCT Int Appl. 2008:A1.; (c) Trabanco AA, Tresadern G, Macdonald GJ, Vega JA, de Lucas AI, Matesanz E, García A, Linares ML, de Diego SAA, Alonso JM, Oehlrich D, Ahnaou A, Drinkenburg W, Mackie C, Andrés JI, Lavreysen H, Cid JM. J Med Chem. 2012;55:2688–2701. doi: 10.1021/jm201561r. [DOI] [PubMed] [Google Scholar]
- 4.For a general exception, cf.: Manolikakes G, Hernandez CM, Schade MA, Metzger A, Knochel P. J Org Chem. 2008;73:8422–8436. doi: 10.1021/jo8015852.Manolikakes G, Schade MA, Hernandez CM, Mayr H, Knochel P. Org Lett. 2008;10:2765–2768. doi: 10.1021/ol8009013.Manolikakes G, Dong Z, Mayr H, Li J, Knochel P. Chem Eur J. 2009;15:1324–1328. doi: 10.1002/chem.200802349.Yang Y, Oldenhuis NJ, Buchwald SL. Angew Chem Int Ed. 2013;52:615–619. doi: 10.1002/anie.201207750.
- 5.(a) Zeni G, Larock RC. Chem Rev. 2006;106:4644–4680. doi: 10.1021/cr0683966. [DOI] [PubMed] [Google Scholar]; (b) Müller TJJ. Synthesis. 2012;44:159–174. [Google Scholar]; (c) Platon M, Amardeil R, Djakovitch L, Hierso JC. Chem Soc Rev. 2012;41:3929–3968. doi: 10.1039/c2cs15350e. [DOI] [PubMed] [Google Scholar]; (d) Popowycz F, Mérour JY, Joseph B. Tetrahedron. 2007;63:8689–8707. [Google Scholar]; (e) Popowycz F, Routier S, Joseph B, Mérour JY. Tetrahedron. 2007;63:1031–1064. [Google Scholar]; (f) Song JJ, Reeves JT, Gallou F, Tan Z, Yee NK, Senanayake CH. Chem Soc Rev. 2007;36:1120–1132. doi: 10.1039/b607868k. [DOI] [PubMed] [Google Scholar]; (g) Schmidt A, Beutler A, Snovydovych B. Eur J Org Chem. 2008:4073–4095. [Google Scholar]
- 6.(a) Seregin IV, Gevorgyan V. Chem Soc Rev. 2007;36:1173–1193. doi: 10.1039/b606984n. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Knochel P, Schade MA, Bernhardt S, Manolikakes G, Metzger A, Piller FM, Rohbogner CJ, Mosrin M. Beilstein J Org Chem. 2011;7:1261–1277. doi: 10.3762/bjoc.7.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.(a) Baumann M, Baxendale IR, Ley SV, Nikbin N. Beilstein J Org Chem. 2011;7:442–495. doi: 10.3762/bjoc.7.57. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Meanwell NA. J Med Chem. 2011;54:2529–2591. doi: 10.1021/jm1013693. [DOI] [PubMed] [Google Scholar]; (c) Alvarez-Builla J, Vaquero JJ, Barluenga J. Modern Heterocyclic Chemistry. Wiley-VCH; Weinheim: 2011. [Google Scholar]
- 8.(a) Suzuki A. Angew Chem Int Ed. 2011;50:6722–6737. doi: 10.1002/anie.201101379. [DOI] [PubMed] [Google Scholar]; (b) Miyaura N, Suzuki A. Chem Rev. 1995;95:2457–2483. [Google Scholar]; (c) Martin R, Buchwald SL. Acc Chem Res. 2008;41:1461–1473. doi: 10.1021/ar800036s. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Fu GC. Acc Chem Res. 2008;41:1555–1564. doi: 10.1021/ar800148f. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Valente C, Çalimsiz S, Hoi KH, Mallik D, Sayah M, Organ MG. Angew Chem Int Ed. 2012;51:3314–3332. doi: 10.1002/anie.201106131. [DOI] [PubMed] [Google Scholar]
- 9.(a) Wu TYH, Schultz PG, Ding S. Org Lett. 2003;5:3587–3590. doi: 10.1021/ol035226w. [DOI] [PubMed] [Google Scholar]; (b) Kudo N, Perseghini M, Fu GC. Angew Chem Int Ed. 2006;45:1282–1284. doi: 10.1002/anie.200503479. [DOI] [PubMed] [Google Scholar]; (c) Billingsley K, Buchwald SL. J Am Chem Soc. 2007;129:3358–3366. doi: 10.1021/ja068577p. [DOI] [PubMed] [Google Scholar]; (d) Prieto M, Zurita E, Rosa E, Munoz L, Lloyd-Williams P, Giralt E. J Org Chem. 2004;69:6812–6820. doi: 10.1021/jo0491612. [DOI] [PubMed] [Google Scholar]; (e) Grob JE, Nunez J, Dechantsreiter MA, Hamann LG. J Org Chem. 2011;76:4930–4940. doi: 10.1021/jo2005928. [DOI] [PubMed] [Google Scholar]; (f) Lee DH, Choi M, Yu BW, Ryoo R, Taher A, Hossain S, Jin MJ. Adv Synth Catal. 2009;351:2912–2920. [Google Scholar]; (g) Capek P, Vrábel M, Hasník Z, Pohl R, Hocek M. Synthesis. 2006:3515–3526. [Google Scholar]; (h) Kwong FY, Lam WH, Yeung CH, Chan KS, Chan ASC. Chem Commun. 2004:1922–1923. doi: 10.1039/b407243j. [DOI] [PubMed] [Google Scholar]; (i) So CM, Chow WK, Choy PY, Lau CP, Kwong FY. Chem Eur J. 2010;16:7996–8001. doi: 10.1002/chem.201000723. [DOI] [PubMed] [Google Scholar]; (j) Kitamura Y, Sako S, Udzu T, Tsutsui A, Maegawa T, Monguchi Y, Sajiki H. Chem Commun. 2007:5069–5071. doi: 10.1039/b712207a. [DOI] [PubMed] [Google Scholar]; (k) Navarro O, Marion N, Mei J, Nolan SP. Chem Eur J. 2006;12:5142–5148. doi: 10.1002/chem.200600283. [DOI] [PubMed] [Google Scholar]; (l) Robbins DW, Hartwig JF. Org Lett. 2012;14:4266–4269. doi: 10.1021/ol301570t. [DOI] [PubMed] [Google Scholar]; (m) Bellina F, Cauteruccio S, Rossi R. J Org Chem. 2007;72:8543–8546. doi: 10.1021/jo701496p. [DOI] [PubMed] [Google Scholar]
- 10.Recently, the coupling of benzimidazole-derived potassium trifluoroarylborates was reported: Molander GA, Ajayi K. Org Lett. 2012;14:4242–4245. doi: 10.1021/ol301956p.
- 11.(a) Newman SG, Lautens M. J Am Chem Soc. 2010;132:11416–11417. doi: 10.1021/ja1052335. [DOI] [PubMed] [Google Scholar]; (b) Smith GB, Dezeny GC, Hughes DL, King AO, Verhoeven TR. J Org Chem. 1994;59:8151–8156. [Google Scholar]; (c) Burukin AS, Vasil’ev AA, Merkulova NL, Struchkova MI, Zlotin SG. Russ Chem Bull Int Ed. 2006;55:118–122. [Google Scholar]; (d) Slagt VF, de Vries AHM, de Vries JG, Kellogg RM. Org Process Res Dev. 2010;14:30–47. [Google Scholar]; (e) Beletskaya IP, Cheprakov AV. Organometallics. 2012;31:7753–7808. [Google Scholar]
- 12.(a) Kinzel T, Zhang Y, Buchwald SL. J Am Chem Soc. 2010;132:14073–14075. doi: 10.1021/ja1073799. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Bruno N, Tudge MT, Buchwald SL. Chem Sci. 2013;4:916–920. doi: 10.1039/C2SC20903A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.In the reaction using 5-chlorobenzotriazole as aryl halide which shows a comparable reactivity to pyrazole halides, we also tried several bidentate ligands, e.g. BINAP, XantPhos, DPEPhos and dppe. Only BINAP gave conversions nearly as good as SPhos. All other ligands fell short of SPhos. Also see Ref. (9m).
- 14.A possible catalysis by Pd nanoparticles thus seems unlikely, even if they are commonly found in substantial amount in Pd2dba3: Zalesskiy SS, Ananikov VP. Organometallics. 2012;31:2302–2309.
- 15.All chemicals can be weighed under air and stored on the benchtop.
- 16.Unsubstituted, unprotected 3-, 4- or 5-halopyrazoles have not been previously reported in literature to successfully undergo Suzuki-Miyaura, Negishi or Stille cross-coupling reactions. See ref. 7b.
- 17.Siddiqui MA, Reddy PA. J Med Chem. 2010;53:3005–3012. doi: 10.1021/jm9003279. [DOI] [PubMed] [Google Scholar]
- 18.Henderson JL, Buchwald SL. Org Lett. 2010;12:4442–4445. doi: 10.1021/ol101929v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Catalan J, Abboud JLM, Elguero J. Adv Heterocycl Chem. 1987;41:187–274. [Google Scholar]
- 20.Only few pKa values for NH acidic heterocycles have been reported in H2O as opposed to DMSO as solvent. Please refer to reference 19 for an in-depth discussion of these compounds’ acidity.
- 21.The reaction of deprotonated starting material or product with the boronic acid could additionally lead to formation of a Lewis acid/base pair, potentially binding and deactivating the boronic acid:In the presence of potassium indazolide (2.0 eq.) in THF-d8 (BF3·OEt2 as standard), a distinct shift in the 11B-NMR spectrum of phenylboronic acid from 29.9 to 5.0 ppm was observed, which is indicative of a tetrahedral, anionic boronate species. The presence of indole led to no change, whereas indazole gave two signals at 31.9 (major) and 23.5 ppm (minor).
- 22.The overlaid curves in figure 3, 5 are only meant as visual guidance. They were not fitted but are simply smoothed using the B-Spline option, as implemented in Origin 7.0.
- 23.Some of the data points were taken twice by setting up the reaction a second time to insure reproducibility. In those cases, the measured yield was within ±1%.
- 24.Complex 15 has been previously isolated by our group. See: Barder TE, Biscoe MR, Buchwald SL. Organometallics. 2007;26:2183–2192.
- 25.(a) Matos K, Soderquist JA. J Org Chem. 1998;63:461–470. doi: 10.1021/jo971681s. [DOI] [PubMed] [Google Scholar]; (b) Amatore C, Jutand A, Le Duc G. Chem Eur J. 2011;17:2492–2503. doi: 10.1002/chem.201001911. [DOI] [PubMed] [Google Scholar]; (c) Amatore C, Jutand A, Le Duc G. Chem Eur J. 2012;18:6616–6625. doi: 10.1002/chem.201200516. [DOI] [PubMed] [Google Scholar]; (d) Carrow BP, Hartwig JF. J Am Chem Soc. 2011;133:2116–2119. doi: 10.1021/ja1108326. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Grushin VV, Alper H. Organometallics. 1996;15:5242–5245. [Google Scholar]; (f) Hayashi Y, Wada S, Yamashita M, Nozaki K. Organometallics. 2012;31:1073–1081. [Google Scholar]
- 26.One major signal and two minor ones could be observed by 31P-NMR spectroscopy in CD2Cl2. In THF-d8 as solvent, one major signal could be detected as well as 3 minor species.
- 27.The formation of Pd(II) pyrazolyl species has been previously reported starting both from [Pd-OH] and [Pd-I] complexes: Driver MS, Hartwig JF. Organometallics. 1997;16:5706–5715.
- 28.The reverse reaction starting from 14 to either 15 or 16 led to no product formation. Other examples of 1,2-azole bridged Pd(II) complexes have been previously reported: López G, Ruiz J, Garcia G, Vicente C, Casabó J, Molins E, Miravitlles C. Inorg Chem. 1991;30:2605–2610.Espinet P, Gallego AM, Martínez-Ilarduya JM, Pastor E. Inorg Chem. 2000;39:975–979. doi: 10.1021/ic9911062.de la Cruz R, Espinet P, Gallego AM, Martín-Alvarez JM, Martínez-Ilarduya JM. J Organomet Chem. 2002;663:108–117.
- 29.15 was chosen over P2 for comparing reactivity at room temperature to accommodate for a potential lag in activation of P2.
- 30.At 100 °C, no difference in reactivity was observed between 14 and 15 as catalyst in the reaction of 3-chloroanisole and 2,6-dimethylphenylboronic acid.
- 31.The observed product inhibition explains why 4-bromopyrazole generally reacts more slowly than its 3-bromo isomer, often resulting in lower yields.
- 32.Steffen A, Braun T, Neumann B, Stammler HG. Angew Chem Int Ed. 2007;46:8674–8678. doi: 10.1002/anie.200703393. [DOI] [PubMed] [Google Scholar]
- 33.The exact inhibition mechanism of compounds bearing basic imine-type nitrogen atoms was not further investigated but probably also involves complexation of Pd(II) intermediates.
- 34.Oxidative addition with SPhosPd(0) can occur even at −40 °C: Biscoe MR, Fors BP, Buchwald SL. J Am Chem Soc. 2008;130:6686–6687. doi: 10.1021/ja801137k.
- 35.The transition state of the oxidative addition for deprotonated 4-imidazole chloride was successfully computed in the gas phase but yielded two imaginary frequencies in THF.
- 36.Selected examples for challenging reductive eliminations using biarylphosphine ligands: Su M, Buchwald SL. Angew Chem Int Ed. 2012;51:4710–4713. doi: 10.1002/anie.201201244.Cho EJ, Senecal TD, Kinzel T, Zhang Y, Watson DA, Buchwald SL. Science. 2010;328:1679–1681. doi: 10.1126/science.1190524.Watson DA, Su M, Teverovskiy G, Zhang Y, Garcia-Fortanet J, Kinzel T, Buchwald SL. Science. 2009;325:1661–1664. doi: 10.1126/science.1178239.Salvi L, Davis NR, Ali SZ, Buchwald SL. Org Lett. 2012;14:170–173. doi: 10.1021/ol202955h. Also see ref 8c.
- 37.In the attempted formation of the oxidative addition complexes with 3-chloroindazole as electrophile, several phosphine species could be observed by 31P-NMR spectroscopy.
- 38.The formation of small quantitites of free ligand could be observed after several hours when monitoring 14 by 31P-NMR in the presence of PhMgBr at room temperature.
- 39.(a) Berionni G, Maji B, Knochel P, Mayr H. Chem Sci. 2012;3:878–882. [Google Scholar]; (b) Pratihar S, Roy S. Organometallics. 2011;30:3257–3269. [Google Scholar]
- 40.The chosen conditions were selected from published methods to minimize variation of the catalyst system (Pd/XPhos) and therefore allow a representative comparison. In the absence of indazole, all reactions gave the desired product in 91–99% yield. See the Supporting Information for details.
- 41.The proposed mechanism outlined in Scheme 7 should also be applicable to other cross-coupling reactions. Complex 16 should however only be relevant for Suzuki-Miyaura cross-coupling reactions.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.