

Summary
RNase P is the endonuclease that removes 5′ extensions from tRNA precursors. In its best-known form, the enzyme is composed of a catalytic RNA and a protein moiety variable in number and mass. This ribonucleoprotein enzyme is widely considered ubiquitous and apparently reached its highest complexity in the eukaryal nucleus, where it is typically composed of at least ten subunits. Here, we show that in the protist Trypanosoma brucei, two proteins are the sole forms of RNase P. They localize to the nucleus and the mitochondrion, respectively, and have RNase P activity each on their own. The protein-RNase P is, moreover, capable of replacing nuclear RNase P in yeast cells. This shows that complex ribonucleoprotein structures and RNA catalysis are not necessarily required to support tRNA 5′ end formation in eukaryal cells.
Graphical Abstract

Highlights
► Nuclear and mitochondrial RNase P in trypanosomatids are each built of a single protein ► T. brucei protein can replace yeast nuclear RNase P, a multicomponent RNA-protein complex ► Complex ribonucleoprotein structures and RNA catalysis are not required to build a nuclear RNase P
RNase P is the endonuclease that removes 5′ extensions from tRNA precursors. In the nucleus of man and yeast, RNase P is composed of RNA and protein components. Rossmanith and colleagues now show that in Trypanosoma brucei, nuclear and mitochondrial RNase P are each built of a single protein only. Remarkably, this protein-RNase P was capable of replacing yeast nuclear RNase P. Thus, complex ribonucleoprotein structures and RNA catalysis are not required to build a nuclear RNase P.
Introduction
RNase P is the endonuclease responsible for the removal of extraneous nucleotides from the 5′ end of tRNA precursors, an early and essential step in tRNA biogenesis (Phizicky and Hopper, 2010; Liu and Altman, 2010). Two basically different forms of RNase P have been identified. One, apparently more ancient and widespread, is based on a structurally conserved RNA molecule forming the catalytic core of the enzyme (reviewed by Ellis and Brown, 2009; Hartmann et al., 2009; Lai et al., 2010b; or Liu and Altman, 2010). This RNA is catalytically active on its own in vitro and hypothesized to be the relic of a prebiotic RNA world. In the evolved, modern RNase P enzymes, the RNA nevertheless depends on protein to fulfill its cellular function. This RNA-based form of RNase P is found in all domains of life, but there is an apparent trend from RNA to protein predominance in the overall composition and functioning of these ribonucleoproteins (RNPs) from Bacteria to Eukarya. RNase P of the former is built from a catalytically proficient RNA and a single small protein only. RNase P RNA of Archaea is a less-efficient catalyst in vitro and associates with five proteins, none of which is related to the bacterial protein. Yet, they are homologous to five of the ten proteins found in human nuclear RNase P, the RNA component of which is barely active on its own. By the mere number of components, the nuclear enzyme also appears to be the most complex form of RNase P, a fact that has been attributed to the need for increased flexibility in substrate recognition due to a presumably greater variety of additional, non-tRNA substrates, the possible need for coordination with other molecular machines, and/or the acquisition of further, unrelated functions (Marvin and Engelke, 2009; Jarrous and Gopalan, 2010; Walker et al., 2010; Chen et al., 2012). Unfortunately, this view of eukaryal nuclear RNase P is almost exclusively built on studies of yeast and human cells, and thus, little is known about nuclear RNase P in eukaryal branches other than opisthokonts/unikonts. In fact homologs of (some) yeast/human nuclear RNase P proteins seem to be absent from many eukaryal genomes, and an RNase P RNA has not been identified in the genome of plants, algae, and some protist groups (Hartmann and Hartmann, 2003; Piccinelli et al., 2005; Rosenblad et al., 2006).
Another entirely different form of RNase P, apparently not containing RNA, was initially observed in the organelles of different Eukarya (Wang et al., 1988; Thomas et al., 1995; Rossmanith and Karwan, 1998; Salavati et al., 2001). The identification of its components made clear that this form of RNase P does indeed not contain any RNA but is composed of protein only (Holzmann et al., 2008). Its key component is an ∼60 kDa protein characterized by an NYN metallonuclease domain, a CXXC zinc finger-like motif, and two in-tandem pentatricopeptide repeats (Rossmanith, 2012). The first-identified, human protein was originally termed “mitochondrial RNase P protein 3” (MRPP3), but meanwhile, we have coined the more generally applicable name “proteinaceous RNase P” (PRORP). Although human PRORP makes use of two further proteins for mitochondrial RNase P function, its plant mitochondrial/plastidial homolog does not require a partner for RNase P activity (Gobert et al., 2010). PRORP homologs are not found in Archaea and Bacteria, but in a wide range of eukaryal branches, suggesting an origin at the root of the eukaryal tree.
The constraints that preserved a catalytic RNA for RNase P function throughout evolution in a cellular biochemistry so much dominated by protein catalysts are not clear, and its presence in modern life is all the more surprising now that it is known that a single protein is able to catalyze the same, specific hydrolytic reaction. Given that PRORP enzymes have so far only been found responsible for organellar tRNA processing, it was suggested that the RNA-based enzymes, for reasons yet unclear, would allow more flexibility with respect to, e.g., substrate recognition, and that PRORPs therefore remained restricted to the simpler organellar systems (Esakova and Krasilnikov, 2010; Marvin and Engelke, 2009; Walker and Engelke, 2008). Bioinformatic evidence for PRORP homologs in organisms lacking RNase P RNA has, however, challenged this view. Here, we report a study of RNase P in the model organism Trypanosoma brucei. Notably, the genomes of trypanosomatids lack evidence for genes related to RNA-based RNase P (Piccinelli et al., 2005; Rosenblad et al., 2006), but they encode two homologs of human and plant PRORP genes (Holzmann et al., 2008; Gobert et al., 2010). We were most intrigued whether a “simple” PRORP could accomplish the role of nuclear RNase P, traditionally ascribed to highly complex, multicomponent RNP enzymes.
Results
Two presumptive PRORP genes (PRORP1, PRORP2) were identified in all available trypanosomatid genomes. Although the overall similarity between the two (∼30% amino acid identity) is no more than their similarity to plant PRORPs, they are nevertheless distinguished from plant and animal sequences by an insertion of 35–41 amino acids in the NYN metallonuclease domain (Figure S1). A second, distinctive insertion of 13–18 amino acids is found in the C-terminal part of the NYN domain of trypanosomatid PRORP2 only.
Figure S1.

Multiple Sequence Alignment of Trypanosomatid PRORP Sequences to Human PRORP and A. thaliana PRORP1, Related to Figure 1
ClustalW multiple sequence alignment of PRORP sequences retrieved from T. brucei, T. cruzi, and L. major genomes to human PRORP (MRPP3) and A. thaliana PRORP1; official/alternative gene names and RefSeq accession number listed in the first line in parentheses. Only the more conserved C-terminal part encompassing the conserved zinc-finger-like CXXC motif (green bar) and the NYN metallonuclease domain (red bar) is shown; two insertions within the NYN domain are indicated (blue bars); amino acid positions are indicated at the beginning and end of each line. Dark and light gray shading indicate identities and similarities in at least 6 of the 8 aligned sequences, respectively.
In T. brucei the transcript copy number of PRORP1 (locus tag Tb09.211.0870) and PRORP2 (Tb11.02.0510) was estimated to be ∼2 mRNA molecules per cell (insect form) each in a recent transcriptome study by Kolev et al. (2010). Similar mRNA levels were reported for several genes encoding putative homologs of other tRNA-processing and -modification enzymes in the same study, though finally 75% of all T. brucei mRNAs were estimated in the range of one to ten (median three) molecules per cell. None of the two genes appears to be regulated at the RNA level in life-cycle development or during cell cycle (Archer et al., 2011; Jensen et al., 2009; Nilsson et al., 2010; Siegel et al., 2010; Veitch et al., 2010).
T. brucei PRORP1 and PRORP2 Have RNase P Activity
Recombinant, tagged proteins were purified to near homogeneity by one- or two-step affinity chromatography and subsequent size exclusion chromatography (Figure 1A). Compared to standards, PRORP1 and PRORP2 eluted close to their calculated molecular weight at ∼60–65 kDa, indicating that they do not form dimers or higher-order oligomers (Figure 1B). Both recombinant proteins cleaved different T. brucei tRNA precursors as well as two commonly used bacterial model substrates at the same site as E. coli RNase P (Figures 1C–1G; canonical cleavage, immediately upstream of the first nucleotide of the tRNA structure, was also confirmed by sizing along nuclease ladders in high-resolution gel electrophoresis [data not shown]). Thus, similar to Arabidopsis PRORP1 (Gobert et al., 2010) and despite their divergence in nuclease domain primary structure, both T. brucei PRORPs have RNase P activity on their own and, unlike human PRORP, do not require additional proteins (Holzmann et al., 2008).
Figure 1.

Purification and tRNA-Processing Activity of Recombinant T. brucei PRORP1 and PRORP2
(A) SDS-PAGE of recombinant PRORP1 and PRORP2 purified by affinity chromatography (AC), or affinity chromatography with subsequent size exclusion chromatography (SEC) is shown; molecular weight of marker proteins indicated in kDa.
(B) Size exclusion chromatography profile of purified recombinant PRORP1 (blue) and PRORP2 (red) is illustrated. The peak at about 8 ml represents aggregated protein eluting in the void volume. The peak positions of molecular weight marker proteins resolved under identical conditions are indicated.
(C) RNase P activity of recombinant PRORP1 and PRORP2 is presented. A T. brucei tRNAiMet precursor was incubated with PRORP1, PRORP2, or E. coli RNase P. 5′ end-labeled substrate RNA and cleavage product (indicated by icons to the right) were resolved by denaturing PAGE.
(D) Same as (C), but T. brucei tRNAPhe precursor used as substrate is shown.
(E) Same as (C), but T. brucei tRNAHis precursor used as substrate is presented.
(F) Same as (C), but E. coli tRNATyrsu3+ precursor used as substrate is illustrated.
(G) Same as (C), but tRNAGly precursor from Thermus thermophilus used as substrate is demonstrated.
See Figure S1 for alignment of trypanosomatid PRORP sequences to human and plant PRORP.
PRORP1 Localizes to the Nucleus/Nucleolus and PRORP2 to the Mitochondrion
Eukaryal cells require RNase P in any tRNA-synthesizing compartment, i.e., nucleus, mitochondria, and chloroplasts if applicable. An RNase P activity was previously purified from T. brucei mitochondria by Salavati et al. (2001). Although the responsible enzyme was not identified, its apparent lack of an RNA component and a molecular weight of ∼70 kDa would both be consistent with this activity being derived from either PRORP1 or PRORP2. In fact PRORP2 is predicted to be mitochondrial and to have a cleavable N-terminal targeting sequence. PRORP1, conversely, seems to harbor a nuclear localization signal.
To directly determine the localization of PRORP1 and PRORP2, we expressed both proteins with a C-terminal YFP tag in T. brucei cells (insect form) and alternatively also localized the endogenous proteins by immunofluorescence using antibodies raised against the recombinant proteins. PRORP1-YFP overexpression gave rise to nuclear fluorescence only (Figure 2A). In contrast to the apparently homogeneous nuclear distribution of PRORP1-YFP, immunostaining of endogenous PRORP1 was largely confined to the single central nucleolus of T. brucei nuclei and only weak in the nucleoplasm (Figure 2B), a discrepancy possibly due to the overexpression of PRORP1-YFP from the strong PARP promoter. PRORP2 was localized to the branched trypanosomatid mitochondrion by YFP tag or immunofluorescence, coinciding with MitoTracker staining (Figures 2C and 2D). Hence, like other eukaryal cells T. brucei has two RNase P activities: one present in the nucleus, and a different one in its mitochondrion.
Figure 2.

Subcellular Localization of PRORP1 and PRORP2
Analysis of the subcellular localization in procyclic T. brucei Lister 427 cells by expression of C-terminally YFP-tagged proteins (YFP) or by immunofluorescence (IF) of endogenous proteins is presented. Mitochondria were stained by MitoTracker red (MT), nuclei and kinetoplasts by Hoechst (H); cells are also shown in bright-field view (BF). Pictures were taken by epifluorescence microscopy. Scale bars, to 2 μm.
(A) Expression of YFP-tagged PRORP1 is shown.
(B) Immunofluorescence of endogenous PRORP1 is demonstrated; overlay of immunofluorescence and Hoechst staining (IF/H) is illustrated.
(C) Expression of YFP-tagged PRORP2 is shown.
(D) Immunofluorescence of endogenous PRORP2 is demonstrated.
PRORP1 and PRORP2 Are the Sole Forms of RNase P in Trypanosomatids
With the exception of a parasitic archaeon that dispensed with RNase P by making leaderless tRNAs (Randau et al., 2008), the RNA-based form of RNase P is widely considered a ubiquitous RNP machine, indispensable for life, reminiscent of the ribosome (Altman, 2010). Previous bioinformatic studies did not find any possible RNase P RNA candidate sequences in trypanosomatid genomes (Piccinelli et al., 2005), nor the commonly associated protein subunits (with the exception of a possible RPP25/POP6 homolog; Rosenblad et al., 2006). However, RNase P RNAs are notoriously difficult to identify, and structurally more divergent variants might have escaped bioinformatic analyses so far. The recent case of Pyrobaculum demonstrated that RNase P RNAs (and associated proteins) can diverge considerably from the structural consensus (Lai et al., 2010a). Likewise, the structurally degenerate fungal mitochondrial RNase P RNAs seem to have recruited an entirely new protein moiety even more than once during evolution (Rossmanith, 2012). Because life solely built on the proteinaceous form of RNase P is in fact unprecedented, we investigated the possibility of the simultaneous presence of a further, possibly RNA-based form of RNase P in T. brucei. We prepared whole-cell extracts by a combination of hypotonic swelling/mechanical disruption and lysis with a nonionic detergent. Similar extraction procedures have been used previously to isolate RNase P enzymes from various sources, including the fragile human nuclear and mitochondrial enzymes (Rossmanith et al., 1995). The T. brucei-cell extraction procedure was optimized to yield a maximal amount of RNase P activity in the crude whole-cell extract. We then subjected the extract to immunodepletion using PRORP1 and PRORP2 antisera. Depletion of either PRORP1 or PRORP2 led to a reduction of RNase P activity, but the depletion of both proteins eliminated all activity (Figures 3 and S2). Thus, apart from PRORP1 and PRORP2, T. brucei apparently contains no other kind of RNase P.
Figure 3.
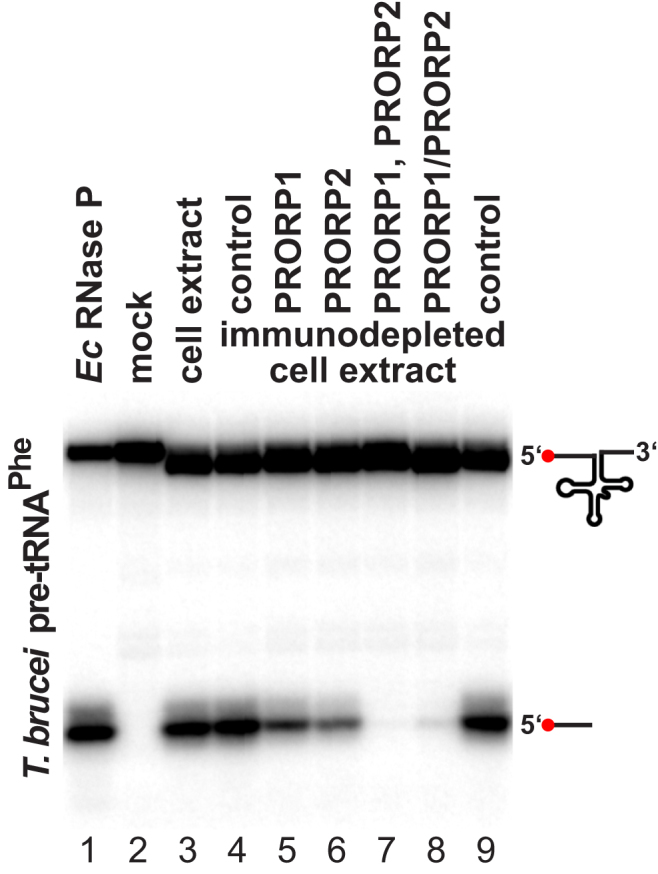
Immunodepletion of RNase P Activity from T. brucei Whole-Cell Extract by PRORP1 and PRORP2 Antibodies
RNase P activity in procyclic T. brucei Lister 427 whole-cell extract (lane 3) and extract depleted of PRORP1 or/and PRORP2 by immunoprecipitation (lanes 5–8) is presented. Control immunodepletions were carried out with mixed PRORP1 and PRORP2 preimmune sera (lanes 4 and 9). Double immunodepletion was carried out either in sequence, i.e., PRORP2 depletion of a PRORP1-depleted extract (lane 7), or simultaneously by mixing the two antisera (lane 8). RNase P activity was assayed using the T. brucei tRNAPhe precursor as substrate. E. coli RNase P was used to verify the cleavage site (lane 1). 5′ end-labeled substrate RNA and cleavage product (indicated by icons to the right) were resolved by denaturing PAGE. Note that a few nucleotides were removed from the 3′ trailer of the tRNA precursor by incubation in the whole-cell extract.
See also Figure S2.
Figure S2.
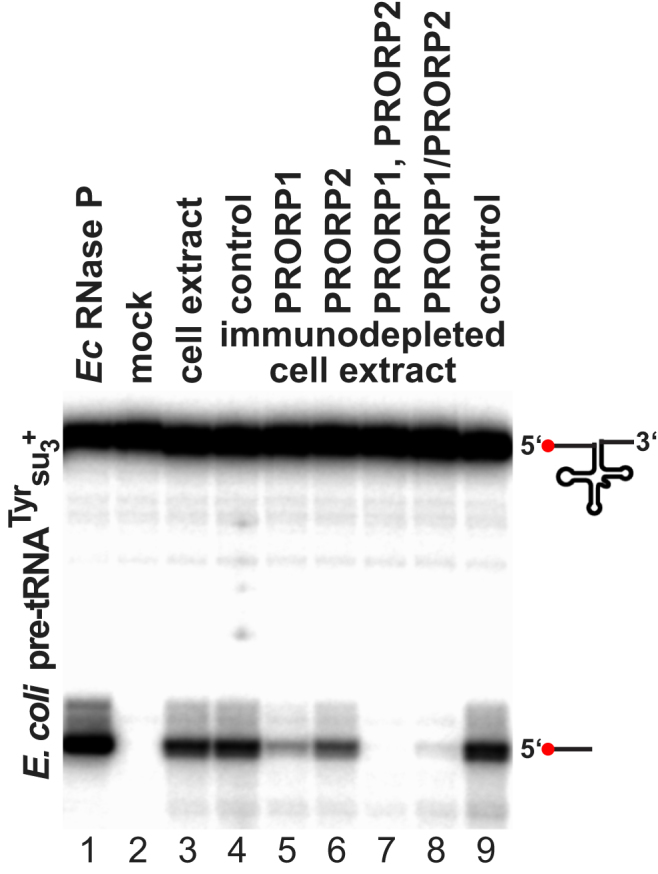
RNase P Activity of PRORP1- and PRORP2-Depleted T. brucei Whole-Cell Extract Using a Bacterial tRNA Precursor Model Substrate, Related to Figure 3
RNase P activity in procyclic T. brucei Lister 427 whole-cell extract (lane 3) and extract depleted of PRORP1 or/and PRORP2 by immunoprecipitation (lanes 5–8). Control immunodepletions were carried out with mixed PRORP1 and PRORP2 pre-immune sera (lanes 4 and 9). Double immunodepletion was carried out either in sequence, i.e., PRORP2 depletion of a PRORP1 depleted extract (lane 7), or simultaneously by mixing the two antisera (lane 8). RNase P activity was assayed using the E. coli tRNATyrsu3+ precursor, a substrate that has been employed in studies of RNase P from a wide range of organisms. E. coli RNase P was used to verify the cleavage site (lane 1). 5′ end-labeled substrate RNA and cleavage product (indicated by icons to the right) were resolved by denaturing PAGE.
PRORP1 Is Able to Substitute for Yeast Nuclear RNase P In Vivo
In terms of number of components, the complexity of the trypanosomatid enzyme is reduced by one order of magnitude relative to yeast or human nuclear RNase P. In the tree of Eukarya, trypanosomatids (a kinetoplastid clade of euglenozoans) are placed in the supergroup of excavates and are thus only distantly related to animals and fungi (opisthokont/unikont supergroup). Notably, trypanosomatids differ from opisthokonts and most other Eukarya in several fundamental aspects of gene expression (Martínez-Calvillo et al., 2010). The seemingly simplified form of RNase P could be related to the peculiar genetic system of these protists, possibly being the result of reductive evolution, and accompanied by a reduced functionality of the enzyme. It seemed thus worthwhile to compare PRORP1, the nuclear RNase P of T. brucei, to the rather complex RNP form of RNase P found in the nucleus of animals or fungi.
Yeast nuclear RNase P is composed of nine proteins and an RNA considered to be the catalytic core (Walker et al., 2010). All subunits are essential for enzyme function in vivo, i.e., deletion of any of them is lethal. We tested whether T. brucei PRORP1 is able to rescue the deletion of RPR1, the gene encoding yeast nuclear RNase P RNA. First, RPR1 was confirmed to be essential using a standard gene disruption/tetrad analysis approach (Figure 4A). When we transformed the diploid RPR1/rpr1Δ::kanMX strain (one allele of RPR1 replaced by the selectable marker kanMX) with a plasmid for PRORP1 expression, meiotic tetrads could be dissected into three or four viable spores frequently (Figure 4B). Replica plating confirmed haploidy and the expected kanMX segregation among the spores. Analysis of plasmid-encoded leucine prototrophy revealed that the plasmid was frequently lost from RPR1 wild-type spores, but never from the rpr1Δ::kanMX mutant spores. Even after long-term culture in leucine-containing medium, rpr1Δ::kanMX cells invariably remained leucine prototroph, i.e., had kept the plasmid with the actual dispensable LEU2 marker; by providing vital RNase P function to the cells, plasmid-encoded T. brucei PRORP1 obviously became an essential gene in rpr1Δ::kanMX yeasts. The genotype of rpr1Δ::kanMX [PRORP1] strains was also verified at the molecular level, and multiple PCR analyses confirmed the loss of RPR1 from their genome (Figure 4C). Compared to the RPR1 wild-type, PRORP1 strains gave rise to smaller colonies indicating a slower growth rate (Figure 4D). Nevertheless, we have been able to perpetuate these RNase P-engineered yeast strains for an as yet unlimited number of generations, demonstrating that even in an entirely different cellular environment, T. brucei PRORP1 sufficiently supports all RNase P functions required for vitality.
Figure 4.

Rescuing the Deletion of Yeast Nuclear RNase P RNA by Expression of T. brucei PRORP1
(A) Cartoon of RPR1 gene disruption and tetrad analysis in the budding yeast S. cerevisiae is depicted. One copy of RPR1 was replaced by the selectable marker kanMX by homologous recombination (rpr1Δ::kanMX). Because RPR1 is an essential gene, dissection of meiotic tetrads yields only two viable spores.
(B) Rescue of RPR1 gene disruption by a plasmid expressing T. brucei PRORP1 is illustrated. Without selection for LEU2 the plasmid is frequently lost from wild-type RPR1 spores, but never from rpr1Δ::kanMX [PRORP1] complementation spores.
(C) Genotyping of haploid strains derived from tetrad dissections of BY4743 RPR1/rpr1Δ::kanMX [PRORP1] is detailed. The analysis of a wild-type (RPR1) and two complementation (PRORP1; genotype rpr1Δ::kanMX [PRORP1]) strains is shown. PCR screening for RPR1 by primer pairs spanning different parts of the gene, for correct integration of the kanMX disruption cassette into RPR1, for T. brucei PRORP1, and for NME1 (the gene encoding the RNA component of RNase MRP, unaffected by the genetic experiments, used as a control for DNA quality) is shown. The part of the gene interrogated by the genotyping PCR is indicated by a black bar in the gene cartoon to the right of each agarose gel panel. PCR primers are listed in Table S1.
(D) Growth of haploid strains derived from tetrad dissections of BY4743 RPR1/rpr1Δ::kanMX [PRORP1] as analyzed in (C) is presented. Log10 dilutions of a wild-type (RPR1) and two complementation (PRORP1; genotype rpr1Δ::kanMX [PRORP1]) strains were spotted on a YPD plate and incubated for 2 days at 30°C. Note the smaller colony size of PRORP1 strains.
Discussion
In contrast to the peculiarities found in the expression of trypanosomatid protein-coding genes, e.g., generalized polycistronic transcription and trans-splicing (Martínez-Calvillo et al., 2010), trypanosomatid tRNA biology appears generally orthodox. Gene organization, transcription, processing, and modification, and tRNA structure and function, appear all similar as in other eukaryal systems. Their genome encodes a homolog of RNase Z and a CCA-adding enzyme, and only their tRNA 5′ end-processing machinery seems to be unusual. So far, trypanosomatids are the sole eukaryal group identified that has lost all genes associated with or related to the RNA-based form of RNase P and, instead, uses the protein-only form of RNase P (PRORP) to process its tRNAs.
Trypanosomatid PRORP1 differs from any previously characterized nuclear RNase P. Still, the basic pathway of tRNA processing seems preserved in T. brucei, and like in yeast (Walker et al., 2010), 5′ end maturation primarily occurs in the nucleolus. Moreover, PRORP1's ability to replace yeast nuclear RNase P suggests that the inherently different physical qualities of the two enzyme forms are not reflected in a basically different functionality. The “protein-only” pathway of nuclear tRNA processing actually seems to be more widespread than hitherto anticipated. RNase P RNA appears to be absent from the entire plant supergroup (land plants, green and red algae) and from stramenopiles (Piccinelli et al., 2005), all of which have one or more PRORP homologs that could serve as nuclear RNase P (Holzmann et al., 2008; Gobert et al., 2010). Indeed, Gutmann et al. (2012) just recently reported that Arabidopsis PRORP2 and PRORP3 function as nuclear RNase P. Thus, also in plants, all cellular tRNA 5′ end maturation appears to be exclusively protein dependent (Gobert et al., 2010; Gutmann et al., 2012). Still, another recent report claimed the purification of a plant nuclear RNP complex with RNase P activity, resembling the unikont nuclear enzyme (Chen et al., 2012). However, none of the components of this presumptive RNP enzyme was identified, and it remains to be clarified if plant cells contain an as yet unrecognized RNase P-RNP in addition to PRORPs.
In mitochondria (and plastids), PRORP seems to be present in a majority of Eukarya, more frequently than the ancient RNA enzyme (Rossmanith, 2012). Paradoxically, it is for mitochondria that trypanosomatid tRNA biology becomes perplexing again: trypanosomatid mitochondrial genomes do not encode any tRNA genes, but a complete set of tRNAs is imported from the cytosol (Hancock and Hajduk, 1990; Tan et al., 2002). Although early in vitro studies suggested that tRNAs are translocated as precursors (Hancock et al., 1992), later work showed that tRNA import in vivo is independent of flanking sequences (Hauser and Schneider, 1995; Tan et al., 2002), suggesting that mature tRNAs are imported into the organelle. This appears also plausible in light of the nuclear localization of T. brucei PRORP1 and an essentially identical set of mitochondrial and cytosolic tRNAs. However, it also implies that PRORP2 might be the unique case of an RNase P activity not employed for its original role in tRNA 5′ end maturation but, instead, possibly in the processing of other mitochondrial RNA species only. Future genetic studies should allow determining the mitochondrial substrates and could also clarify if any unprocessed tRNAs are imported into the mitochondrion of T. brucei.
The apparent simplicity of T. brucei nuclear RNase P (a monomeric 64 kDa protein) is in striking contrast to the complexity of the ten-component RNP found in yeast nuclei (∼400 kDa; Walker et al., 2010). Finding that PRORP1 is able to functionally replace yeast nuclear RNase P was indeed most surprising. Although the enzyme swap resulted in a somewhat reduced growth rate, it nevertheless demonstrated that at least under standard growth conditions, PRORP1 is able to fulfill all the vital functions of yeast nuclear RNase P, including any essential, non-tRNA-processing functions. Considering this apparent exchangeability and the possibly more widespread use of PRORPs as nuclear RNase P in plants and algae, it is surprising that the RNP complex has not generally been replaced with PRORP in Eukarya. The wide phylogenetic distribution of PRORP orthologs suggests its emergence before the last common ancestor of Eukarya, where it must have been present together with the RNase P-RNP and RNase MRP. Reducing the number of enzyme subunits from ten to one should not only save costs in itself but also costs related to the coordination of expression and complex assembly. A one-protein enzyme might be physically more robust than a noncovalent ten-subunit assemblage in conditions of cellular stress and might have fewer constraints on its evolutionary adaptability. On the other hand the greater number of components might expand enzyme flexibility, provide more options of regulation, and could permit a more intricate integration with other cellular processes. In the end the reasons that prompted the final loss of the RNA-world relic in several eukaryal branches, while preventing its loss in others, are currently unclear. Our yeast RNase P complementation model, however, should allow to compare RNA-based and PRORP function in vivo in more detail to possibly find out what limited the spread of PRORP as nuclear RNase P and thereby shed light on the exceptional evolution of this highly diverse enzyme family.
Experimental Procedures
Expression and Purification of Recombinant Proteins
T. brucei PRORP1 fused to a C-terminal 6×His tag was expressed in E. coli and purified by immobilized-metal affinity chromatography (IMAC). PRORP2 (putative mitochondrial form) was fused to an N-terminal GST tag via a protease cleavage site, and to a C-terminal 6×His tag. The fusion protein was expressed in E. coli and purified by IMAC. After protease cleavage, uncleaved fusion protein and GST were removed by chromatography on glutathione Sepharose. Affinity-purified proteins were separated on a Superdex 200 gel filtration column. See Extended Experimental Procedures for details of constructs, expression, and purification.
Extended Experimental Procedures.
Bioinformatics
Trypanosomatid PRORP sequences were retrieved by iterative BLAST searches starting with human PRORP (MRPP3) as a query sequence (Camacho et al., 2009). The sequence alignment was generated with MacVector 6.5 (Oxford Molecular) using CLUSTAL W (Thompson et al., 1994). For the prediction of subcellular localization we used MitoProt II (Claros and Vincens, 1996) and PredictNLS (Cokol et al., 2000).
Expression and Purification of Recombinant Proteins
The complete coding sequence of T. brucei PRORP1 (locus tag Tb09.211.0870) was PCR amplified from Lister 427 genomic DNA and cloned into pET-28b(+) (Novagen). The protein was expressed in E. coli C41(DE3) (Miroux and Walker, 1996). Recombinant PRORP1 has a C-terminal 6 × His tag attached via a spacer of 3 amino acids.
The coding sequence of T. brucei PRORP2 (locus tag Tb11.02.0510; without the 30 N-terminal amino acids of the mitochondrial targeting sequence predicted to be removed after mitochondrial import) was PCR amplified from Lister 427 genomic DNA and cloned into pGEX-6P-1 (GE Healthcare). The protein was expressed in E. coli BL21(DE3). Recombinant PRORP2 starts with the N-terminal GST, the PreScission protease cleavage site, a spacer of 3 amino acids, 541 amino acids of PRORP2 (starting from amino acid 31), and a C-terminal 6 × His tag attached via a spacer of 2 amino acids.
Bacteria were broken by sonication and recombinant proteins purified on HisTrap HP columns using an ÄKTApurifier chromatography system (GE Healthcare); buffer A (150 mM NaCl, 50 mM Tris·Cl pH 7.4, 10% glycerol, 1 mM DTT), buffer A' (buffer A, but 1 M NaCl), buffer B (500 mM imidazole in buffer A). PRORP1 was loaded and washed with 5% buffer B, washed with buffer A', washed with 10% buffer B, and eluted with 100% buffer B. PRORP2 was loaded and washed with 10% buffer B, washed with buffer A', washed with 15% buffer B, and eluted with 100% buffer B. The purified GST-fusion protein was cleaved with PreScission protease (GE Healthcare) and passed twice over a GSTrap FF column (GE Healthcare) equilibrated with buffer A. Size exclusion chromatography was carried out in buffer A on a Superdex 200 10/300 GL column (GE Healthcare). The purity of the recombinant proteins was assessed by SDS-PAGE and Coomassie brilliant blue staining.
tRNA Precursor Substrates and RNase P Activity Assay
T. brucei tRNA genes with flanking sequences (tRNAiMet, nucleotides −7 to +79; tRNAPhe, −6 to +83; tRNAHis, −33 to +81) were PCR amplified and cloned into the EcoRI/XbaI sites of pGEM-1 (Promega). Plasmids were linearized by XbaI and in vitro transcribed with T7 RNA polymerase. The E. coli tRNATyrsu3+ and Thermus thermophilus tRNAGly precursor were described previously (Kirsebom and Altman, 1989; Busch et al., 2000). In vitro transcription, 5′ end labeling, and gel purification were carried out as previously described (Rossmanith et al., 1995).
RNase P activity assays were carried out as previously described (Holzmann et al., 2008; Rossmanith et al., 1995). Substrates at 2 nM were cleaved with recombinant enzymes at 200 nM. E. coli RNase P was reconstituted from in vitro transcribed M1 RNA and recombinant C5 protein (Vioque et al., 1988; Rivera-León et al., 1995).
Tissue Culture and Generation of PRORP-YFP Cell lines
Procyclic T. brucei Lister 427 were grown under standard conditions (SDM-79 supplemented with 20% FBS and 7 μg/ml hemin; 28°C). The complete coding sequence of PRORP1 or PRORP2 fused at their C terminus to eYFP was cloned into a derivative of pXS2 (Bangs et al., 1996; He et al., 2004). Cells were transfected with linearized constructs and stable lines selected as previously described (Klingbeil et al., 2002).
Antibodies
Custom rabbit polyclonal antibodies were raised against purified recombinant PRORP1 and PRORP2 (28-day protocol; Eurogentec). PRORP1 antibodies were affinity purified on immobilized recombinant PRORP1 using the Dynabeads co-immunoprecipitation kit (Invitrogen) according the manufacturers instructions. Crude PRORP2 antiserum was used. Antibody specificity was assessed by Western blotting. Pre-immune sera were used for controls.
Immunofluorescence and Staining
Where indicated, cells were incubated for 15 min with MitoTracker red 580 (Molecular Probes) at 1:2500 in PBS. Cells were spun onto coverslips and fixed with 4% paraformaldehyde. For immunofluorescence cells were permeabilized with 0.25% Triton X-100 (plus 0.002% SDS in the case of PRORP1). Coverslips were washed with PBS and blocked with FBS. Purified PRORP1 antibodies were used at ∼10 μg/ml and PRORP2 antiserum at 1:500, biotinylated rabbit IgG antibodies (GE Healthcare) at 1:100, and streptavidin Texas Red (GE Healthcare) or streptavidin Alexa Fluor 488 (Invitrogen) conjugates at 1:100 and 1:1000, respectively. PBS containing 0.3% Tween 20 was used for antibody dilutions and washing. Nuclei and kinetoplasts were stained with Hoechst 33342 and cells embedded in Kaiser's glycerol gelatine.
Preparation of Whole-Cell Extracts and Immunodepletion
PBS washed cells were swollen for 30 min in hypotonic buffer (1 mM Tris·Cl pH 7.4, 1 mM EDTA) on ice, and broken by passing them rapidly through a 27-gauge needle and extracted by addition of an equal volume of lysis buffer (300 mM NaCl, 50 mM Tris·Cl pH 7.4, 30% glycerol, 2% Triton X-100, 2 mM DTT, 1 mM EDTA, 0.4% proteinase inhibitor cocktail (Sigma P8340)). The lysate was cleared by centrifugation (20000 g) for 10 min. Immunodepletions were carried out essentially as previously described (Rossmanith and Karwan, 1993). Briefly, PRORP antisera or pre-immune serum were coupled to protein A sepharose (GE Healthcare), beads washed with IP buffer (150 mM NaCl, 50 mM Tris·Cl pH 7.4, 0.1% Triton X-100, 1 mM DTT, 0.2% proteinase inhibitor cocktail), and extract diluted in IP buffer incubated with the antibody coated beads. Extract and immunoprecipitation supernatants were analyzed for RNase P activity.
Yeast Genetics
S. cerevisiae strain BY4743 (Brachmann et al., 1998) and standard procedures of yeast genetics were used (Amberg et al., 2005). RPR1 was disrupted using a PCR cassette amplified from pFA6-kanMX4 (Wach et al., 1994) using primers ACAGTGGTAATTCCTACGATTAAGAAACCTGTTTACAGAAGGCGCGCCAGATCTGTT and CAGACCTTGACGCTCACGCCGTAGCGGGCGACAAGTCAAACTGGCGGCGTTAGTATCGAA. The complete coding sequence of PRORP1 was cloned into a derivative of the 2μ plasmid YEplac181 (Gietz and Sugino, 1988) containing the yeast ADH1 promoter (Huber et al., 2012). Transformation, selection, sporulation, tetrad dissection, replica plating to verify genetic markers and test mating type, were all carried out according to established standard procedures (Amberg et al., 2005).
Genomic DNA was directly extracted from yeast colonies (Lõoke et al., 2011) and genotypes determined by standard PCR using primers listed in Table S1.
tRNA Precursor Substrates and RNase P Activity Assay
Synthesis of labeled tRNA precursors and RNase P-processing reactions were carried out essentially as previously described by Holzmann et al. (2008) and Rossmanith et al. (1995). See Extended Experimental Procedures for details of constructs and processing reactions.
Subcellular Localization by YFP Tagging and Immunofluorescence
Stable cell lines overexpressing PRORP1 or PRORP2 fused to YFP at their C terminus were generated. Polyclonal antibodies were raised against purified recombinant PRORP1 and PRORP2. PRORP1 antibodies were affinity purified on immobilized recombinant PRORP1. Cells were stained with MitoTracker, fixed with paraformaldehyde (detergent permeabilized, subjected to standard immunofluorescent staining), and counterstained with Hoechst 33342. See Extended Experimental Procedures for details of tissue culture, YFP constructs, antibody production, immunofluorescence, and staining.
Preparation of Whole-Cell Extracts and Immunodepletion
Cells were swollen in a hypotonic buffer, mechanically broken by pushing through a needle, and extracted with 1% Triton X-100. The cleared lysate was subjected to immunodepletion essentially as previously described by Rossmanith and Karwan (1993). See Extended Experimental Procedures for details of antibody production and immunodepletion.
Yeast Genetics
S. cerevisiae strain BY4743 and standard methods of yeast genetics were used. RPR1 was disrupted using a kanMX PCR cassette. PRORP1 was expressed from a 2μ plasmid under the control of the yeast ADH1 promoter. See Extended Experimental Procedures for details of constructs and strain genotyping.
Acknowledgments
We would like to thank Christopher L. de Graffenried and Graham Warren for materials, advice, and support concerning work with T. brucei, and Elisa Vilardo for discussions and comments on the manuscript. This work was supported by the Austrian Science Fund (FWF), Grant I299 to W.R., and the German Research Foundation (DFG), Grant HA 1672/17-1 to R.K.H.
Published online: June 28, 2012
Footnotes
Supplemental Information includes Extended Experimental Procedures, two figures, and one table and can be found with this article online at http://dx.doi.org/10.1016/j.celrep.2012.05.021.
This is an open-access article distributed under the terms of the Creative Commons Attribution-Noncommercial-No Derivative Works 3.0 Unported License (CC-BY-NC-ND; http://creativecommons.org/licenses/by-nc-nd/3.0/legalcode).
Supplemental Information
References
- Altman S. History of RNase P and overview of its catalytic activity. In: Liu F., Altman S., editors. Ribonuclease P. Springer; New York: 2010. pp. 1–15. [Google Scholar]
- Archer S.K., Inchaustegui D., Queiroz R., Clayton C. The cell cycle regulated transcriptome of Trypanosoma brucei. PLoS One. 2011;6:e18425. doi: 10.1371/journal.pone.0018425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen W.Y., Singh D., Lai L.B., Stiffler M.A., Lai H.D., Foster M.P., Gopalan V. Fidelity of tRNA 5′-maturation: a possible basis for the functional dependence of archaeal and eukaryal RNase P on multiple protein cofactors. Nucleic Acids Res. 2012;40:4666–4680. doi: 10.1093/nar/gks013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellis J.C., Brown J.W. The RNase P family. RNA Biol. 2009;6:362–369. doi: 10.4161/rna.6.4.9241. [DOI] [PubMed] [Google Scholar]
- Esakova O., Krasilnikov A.S. Of proteins and RNA: the RNase P/MRP family. RNA. 2010;16:1725–1747. doi: 10.1261/rna.2214510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gobert A., Gutmann B., Taschner A., Gössringer M., Holzmann J., Hartmann R.K., Rossmanith W., Giegé P. A single Arabidopsis organellar protein has RNase P activity. Nat. Struct. Mol. Biol. 2010;17:740–744. doi: 10.1038/nsmb.1812. [DOI] [PubMed] [Google Scholar]
- Gutmann B., Gobert A., Giegé P. PRORP proteins support RNase P activity in both organelles and the nucleus in Arabidopsis. Genes Dev. 2012;26:1022–1027. doi: 10.1101/gad.189514.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hancock K., Hajduk S.L. The mitochondrial tRNAs of Trypanosoma brucei are nuclear encoded. J. Biol. Chem. 1990;265:19208–19215. [PubMed] [Google Scholar]
- Hancock K., LeBlanc A.J., Donze D., Hajduk S.L. Identification of nuclear encoded precursor tRNAs within the mitochondrion of Trypanosoma brucei. J. Biol. Chem. 1992;267:23963–23971. [PubMed] [Google Scholar]
- Hartmann E., Hartmann R.K. The enigma of ribonuclease P evolution. Trends Genet. 2003;19:561–569. doi: 10.1016/j.tig.2003.08.007. [DOI] [PubMed] [Google Scholar]
- Hartmann R.K., Gössringer M., Späth B., Fischer S., Marchfelder A. The making of tRNAs and more—RNase P and tRNase Z. Prog Mol Biol Transl Sci. 2009;85:319–368. doi: 10.1016/S0079-6603(08)00808-8. [DOI] [PubMed] [Google Scholar]
- Hauser R., Schneider A. tRNAs are imported into mitochondria of Trypanosoma brucei independently of their genomic context and genetic origin. EMBO J. 1995;14:4212–4220. doi: 10.1002/j.1460-2075.1995.tb00095.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holzmann J., Frank P., Löffler E., Bennett K.L., Gerner C., Rossmanith W. RNase P without RNA: identification and functional reconstitution of the human mitochondrial tRNA processing enzyme. Cell. 2008;135:462–474. doi: 10.1016/j.cell.2008.09.013. [DOI] [PubMed] [Google Scholar]
- Jarrous N., Gopalan V. Archaeal/eukaryal RNase P: subunits, functions and RNA diversification. Nucleic Acids Res. 2010;38:7885–7894. doi: 10.1093/nar/gkq701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen B.C., Sivam D., Kifer C.T., Myler P.J., Parsons M. Widespread variation in transcript abundance within and across developmental stages of Trypanosoma brucei. BMC Genomics. 2009;10:482. doi: 10.1186/1471-2164-10-482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolev N.G., Franklin J.B., Carmi S., Shi H., Michaeli S., Tschudi C. The transcriptome of the human pathogen Trypanosoma brucei at single-nucleotide resolution. PLoS Pathog. 2010;6:e1001090. doi: 10.1371/journal.ppat.1001090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai L.B., Chan P.P., Cozen A.E., Bernick D.L., Brown J.W., Gopalan V., Lowe T.M. Discovery of a minimal form of RNase P in Pyrobaculum. Proc. Natl. Acad. Sci. USA. 2010;107:22493–22498. doi: 10.1073/pnas.1013969107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai L.B., Vioque A., Kirsebom L.A., Gopalan V. Unexpected diversity of RNase P, an ancient tRNA processing enzyme: challenges and prospects. FEBS Lett. 2010;584:287–296. doi: 10.1016/j.febslet.2009.11.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu F., Altman S., editors. Ribonuclease P. Springer; New York: 2010. [Google Scholar]
- Martínez-Calvillo S., Vizuet-de-Rueda J.C., Florencio-Martínez L.E., Manning-Cela R.G., Figueroa-Angulo E.E. Gene expression in trypanosomatid parasites. J. Biomed. Biotechnol. 2010;2010:525241. doi: 10.1155/2010/525241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marvin M.C., Engelke D.R. RNase P: increased versatility through protein complexity? RNA Biol. 2009;6:40–42. doi: 10.4161/rna.6.1.7566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilsson D., Gunasekera K., Mani J., Osteras M., Farinelli L., Baerlocher L., Roditi I., Ochsenreiter T. Spliced leader trapping reveals widespread alternative splicing patterns in the highly dynamic transcriptome of Trypanosoma brucei. PLoS Pathog. 2010;6:e1001037. doi: 10.1371/journal.ppat.1001037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phizicky E.M., Hopper A.K. tRNA biology charges to the front. Genes Dev. 2010;24:1832–1860. doi: 10.1101/gad.1956510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piccinelli P., Rosenblad M.A., Samuelsson T. Identification and analysis of ribonuclease P and MRP RNA in a broad range of eukaryotes. Nucleic Acids Res. 2005;33:4485–4495. doi: 10.1093/nar/gki756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Randau L., Schröder I., Söll D. Life without RNase P. Nature. 2008;453:120–123. doi: 10.1038/nature06833. [DOI] [PubMed] [Google Scholar]
- Rosenblad M.A., López M.D., Piccinelli P., Samuelsson T. Inventory and analysis of the protein subunits of the ribonucleases P and MRP provides further evidence of homology between the yeast and human enzymes. Nucleic Acids Res. 2006;34:5145–5156. doi: 10.1093/nar/gkl626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossmanith W. Of P and Z: mitochondrial tRNA processing enzymes. Biochim. Biophys. Acta. 2012 doi: 10.1016/j.bbagrm.2011.11.003. in press. Published online November 23, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossmanith W., Karwan R. Definition of the Th/To ribonucleoprotein by RNase P and RNase MRP. Mol. Biol. Rep. 1993;18:29–35. doi: 10.1007/BF01006892. [DOI] [PubMed] [Google Scholar]
- Rossmanith W., Karwan R.M. Characterization of human mitochondrial RNase P: novel aspects in tRNA processing. Biochem. Biophys. Res. Commun. 1998;247:234–241. doi: 10.1006/bbrc.1998.8766. [DOI] [PubMed] [Google Scholar]
- Rossmanith W., Tullo A., Potuschak T., Karwan R., Sbisà E. Human mitochondrial tRNA processing. J. Biol. Chem. 1995;270:12885–12891. doi: 10.1074/jbc.270.21.12885. [DOI] [PubMed] [Google Scholar]
- Salavati R., Panigrahi A.K., Stuart K.D. Mitochondrial ribonuclease P activity of Trypanosoma brucei. Mol. Biochem. Parasitol. 2001;115:109–117. doi: 10.1016/s0166-6851(01)00273-0. [DOI] [PubMed] [Google Scholar]
- Siegel T.N., Hekstra D.R., Wang X., Dewell S., Cross G.A.M. Genome-wide analysis of mRNA abundance in two life-cycle stages of Trypanosoma brucei and identification of splicing and polyadenylation sites. Nucleic Acids Res. 2010;38:4946–4957. doi: 10.1093/nar/gkq237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan T.H.P., Pach R., Crausaz A., Ivens A., Schneider A. tRNAs in Trypanosoma brucei: genomic organization, expression, and mitochondrial import. Mol. Cell. Biol. 2002;22:3707–3717. doi: 10.1128/MCB.22.11.3707-3716.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas B.C., Gao L., Stomp D., Li X., Gegenheimer P.A. Spinach chloroplast RNase P: a putative protein enzyme. Nucleic Acids Symp. Ser. 1995;33:95–98. [PubMed] [Google Scholar]
- Veitch N.J., Johnson P.C.D., Trivedi U., Terry S., Wildridge D., MacLeod A. Digital gene expression analysis of two life cycle stages of the human-infective parasite, Trypanosoma brucei gambiense reveals differentially expressed clusters of co-regulated genes. BMC Genomics. 2010;11:124. doi: 10.1186/1471-2164-11-124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker S.C., Engelke D.R. A protein-only RNase P in human mitochondria. Cell. 2008;135:412–414. doi: 10.1016/j.cell.2008.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker S.C., Marvin M.C., Engelke D.R. Eukaryote RNase P and RNase MRP. In: Liu F., Altman S., editors. Ribonuclease P. Springer; New York: 2010. pp. 173–202. [Google Scholar]
- Wang M.J., Davis N.W., Gegenheimer P. Novel mechanisms for maturation of chloroplast transfer RNA precursors. EMBO J. 1988;7:1567–1574. doi: 10.1002/j.1460-2075.1988.tb02981.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Supplemental References
- Amberg, D.C., Burke, D.J., and Strathern, J.N. (2005). Methods in Yeast Genetics: a Cold Spring Harbor Laboratory course manual, 2005 edn (Cold Spring Harbor, N.Y.: Cold Spring Harbor Laboratory Press).
- Bangs, J.D., Brouch, E.M., Ransom, D.M., and Roggy, J.L. (1996). A soluble secretory reporter system in Trypanosoma brucei. Studies on endoplasmic reticulum targeting. J. Biol. Chem. 271, 18387–18393. [DOI] [PubMed]
- Brachmann, C.B., Davies, A., Cost, G.J., Caputo, E., Li, J., Hieter, P., and Boeke, J.D. (1998). Designer deletion strains derived from Saccharomyces cerevisiae S288C: a useful set of strains and plasmids for PCR-mediated gene disruption and other applications. Yeast 14, 115–132. [DOI] [PubMed]
- Busch, S., Kirsebom, L.A., Notbohm, H., and Hartmann, R.K. (2000). Differential role of the intermolecular base-pairs G292-C(75) and G293-C(74) in the reaction catalyzed by Escherichia coli RNase P RNA. J. Mol. Biol. 299, 941–951. [DOI] [PubMed]
- Camacho, C., Coulouris, G., Avagyan, V., Ma, N., Papadopoulos, J., Bealer, K., and Madden, T.L. (2009). BLAST+: architecture and applications. BMC Bioinformatics 10, 421. [DOI] [PMC free article] [PubMed]
- Claros, M.G., and Vincens, P. (1996). Computational method to predict mitochondrially imported proteins and their targeting sequences. Eur. J. Biochem. 241, 779–786. [DOI] [PubMed]
- Cokol, M., Nair, R., and Rost, B. (2000). Finding nuclear localization signals. EMBO Rep. 1, 411–415. [DOI] [PMC free article] [PubMed]
- Gietz, R.D., and Sugino, A. (1988). New yeast-Escherichia coli shuttle vectors constructed with in vitro mutagenized yeast genes lacking six-base pair restriction sites. Gene 74, 527–534. [DOI] [PubMed]
- He, C.Y., Ho, H.H., Malsam, J., Chalouni, C., West, C.M., Ullu, E., Toomre, D., and Warren, G. (2004). Golgi duplication in Trypanosoma brucei. J. Cell Biol. 165, 313–321. [DOI] [PMC free article] [PubMed]
- Huber, A., Koch, J., Kragler, F., Brocard, C., and Hartig, A. (2012). A subtle interplay between three Pex11 proteins shapes de novo formation and fission of peroxisomes. Traffic 13, 157–167. [DOI] [PMC free article] [PubMed]
- Kirsebom, L.A., and Altman, S. (1989). Reaction in vitro of some mutants of RNase P with wild-type and temperature-sensitive substrates. J. Mol. Biol. 207, 837–840. [DOI] [PubMed]
- Klingbeil, M.M., Motyka, S.A., and Englund, P.T. (2002). Multiple mitochondrial DNA polymerases in Trypanosoma brucei. Mol. Cell 10, 175–186. [DOI] [PubMed]
- Lõoke, M., Kristjuhan, K., and Kristjuhan, A. (2011). Extraction of genomic DNA from yeasts for PCR-based applications. Biotechniques 50, 325–328. [DOI] [PMC free article] [PubMed]
- Miroux, B., and Walker, J.E. (1996). Over-production of proteins in Escherichia coli: mutant hosts that allow synthesis of some membrane proteins and globular proteins at high levels. J. Mol. Biol. 260, 289–298. [DOI] [PubMed]
- Rivera-León, R., Green, C.J., and Vold, B.S. (1995). High-level expression of soluble recombinant RNase P protein from Escherichia coli. J. Bacteriol. 177, 2564–2566. [DOI] [PMC free article] [PubMed]
- Thompson, J.D., Higgins, D.G., and Gibson, T.J. (1994). CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 22, 4673–4680. [DOI] [PMC free article] [PubMed]
- Vioque, A., Arnez, J., and Altman, S. (1988). Protein-RNA interactions in the RNase P holoenzyme from Escherichia coli. J. Mol. Biol. 202, 835–848. [DOI] [PubMed]
- Wach, A., Brachat, A., Pöhlmann, R., and Philippsen, P. (1994). New heterologous modules for classical or PCR-based gene disruptions in Saccharomyces cerevisiae. Yeast 10, 1793–1808. [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.