

Abstract
Eosinophils are major effector cells in type 2 inflammatory responses and become activated in response to IL-4 and IL-33, yet the molecular mechanisms and cooperative interaction between these cytokines remain unclear. Our objective was to investigate the molecular mechanism and cooperation of IL-4 and IL-33 in eosinophil activation. Eosinophils derived from bone marrow or isolated from Il5-transgenic mice were activated in the presence of IL-4 or IL-33 for 1 or 4h and the transcriptome was analysed by RNA-sequencing. The candidate genes were validated by qPCR and ELISA. We first demonstrated that murine cultured eosinophils respond to IL-4 and IL-33 by phosphorylation of STAT-6 and NFκB, respectively. RNA sequence analysis of murine cultured eosinophils indicated that IL-33 induced 519 genes; whereas, IL-4 induced only 28 genes, including 19 IL-33-regulated genes. Interestingly, IL-33 induced eosinophil activation via two distinct mechanisms, IL-4 independent and IL-4 secretion/auto-stimulation dependent. Anti-IL-4 or anti-IL-4Rα antibody-treated cultured and mature eosinophils, as well as Il4- or Stat6-deficient cultured eosinophils, had attenuated protein secretion of a subset of IL-33-induced genes, including Retnla and Ccl17. Additionally, IL-33 induced the rapid release of pre-formed IL-4 protein from eosinophils by an NFκB-dependent mechanism. However, the induction of most IL-33-regulated transcripts (e.g. Il6 and Il13) was IL-4 independent and blocked by NFκB inhibition. In conclusion, we have identified a novel activation pathway in murine eosinophils that is induced by IL-33 and differentially dependent upon an IL-4 auto-amplification loop.
Introduction
Interleukin (IL)-33 is a member of the IL-1 family, which includes IL-1 and IL-18, and binds to its heterodimeric receptor, IL-33R, consisting of the ST2 molecule and IL-1 receptor accessory protein (IL-1RAcP). Binding of IL-33 to its receptor typically triggers NFκB phosphorylation and translocation to the nucleus to activate transcription of target genes. However, a recent study in fibroblasts overexpressing ST2 has demonstrated that IL-33 can signal through phospho-ERK, independently of NFκB, indicating two distinct pathways of activation (1). Whether this pathway is operational in other cells remains unclear. IL-33 potently polarizes T helper lymphocytes type 2 (Th2) (2) and activates mast cells (3), basophils (4), and alternatively activated M2 macrophages (5). Recently, a pathogenic role of IL-33 in inflammatory airway disease has been reported [for review (6)]. Notably, asthmatic patients exhibit high levels of IL-33 in bronchoalveolar lavage fluid and in bronchial epithelial and airway smooth muscle cells (7, 8). Interestingly, intranasal administration of IL-33 in naïve mice induces inflammatory airway responses (5) and exacerbates eosinophil-mediated airway inflammation (9), suggesting a key role of IL-33 in the development of allergic disease pathogenesis. Although eosinophils have been shown to respond to IL-33 (9), little is known about the molecular mechanism involved.
During immune responses, eosinophils are recruited from the circulation to inflammatory sites, rich in cytokines (e.g. IL-4, IL-13, and IL-33) and chemokines (e.g. CCL11), become activated and modulate the response through diverse mechanisms. Upon activation, eosinophils undergo degranulation by releasing cationic proteins, which consequently induce cell damage and dysfunction (10). In vitro studies have shown that eosinophil granule constituents are toxic to a variety of tissues such as the intestine, skin,and trachea (10). In addition, activated eosinophils secrete an array of cytokines (e.g. IL-6, IL-10, and IL-13) and chemokines (e.g. CCL3, CCL5, and CCL17) capable of activating T cells (9, 10). Moreover, eosinophils have a profibrogenic role by producing transforming growth factor (TGF)-α/β, resistin-like molecule-α (RELM-α), metalloproteinase (MMP)-9, and fibroblast growth factor (FGF)-2 (11–13). Notably, eosinophils have the capacity to auto-activate themselves as they express and respond to granulocyte-macrophage colony stimulating factor (GM-CSF), which promotes their survival (14, 15). It has not yet been determined whether similar auto-activation loops may also apply to other eosinophil-derived cytokines, especially IL-4, as eosinophils are often a chief source of IL-4 (16–18).
In allergic airway inflammation, the overexpression of IL-4 and IL-13 has been demonstrated to have a role in the development of pulmonary eosinophilia (19, 20). IL-4 mediates its effects through either the type I IL-4 receptor (IL-4R) (composed of IL-4Rα and the common γ-chain) or the type II IL-4R (composed of IL-4Rα and IL-13Rα1), which can also mediate IL-13 signalling (21–23). Engagement of both receptors induces the phosphorylation of signal transducer and activator of transcription (STAT)-6, which subsequently dimerizes and translocates to the nucleus to induce transcription of specific genes (22, 23). Despite IL-4 and IL-13′s similar intracellular cascades, IL-4 can mediate specific signals independently of IL-13 (24). In asthma, IL-13Rα1 has been highlighted as having a critical role in the pathogenesis of allergic lung responses, regulating different subsets of genes according to the stimulus (IL-4, IL-13, or allergen) (25).
In this study, we demonstrated that murine eosinophils directly respond to IL-4and IL-33, but not IL-13, by the phosphorylation of STAT-6 and NFκB, respectively. Using transcriptome analysis, we identified the genes associated with the IL-33 and IL-4 pathways. We found that IL-33 upregulated 519 genes; whereas, IL-4 only induced 28 genes, including 19 genes that were also IL-33-induced. Interestingly, IL-33 induced eosinophil activation via two distinct mechanisms depending upon IL-4 secretion and auto-stimulation. Anti-IL-4 or anti-IL-4Rαantibody-treated cultured and mature eosinophils, as well as Il4- or Stat6-deficient cultured eosinophils, had attenuated protein secretion of a subset of IL-33-induced genes, including Retnla and Ccl17. However, the induction of most IL-33-regulated transcripts (e.g. Il6 and Il13) was independent of IL-4 and blocked by an inhibitor of NFκB. Our study provides evidence for a novel eosinophil activation pathway that is triggered by IL-33 involving an IL-4/STAT-6 auto-amplification loop or an IL-4-independent/NFκB-dependent pathway.
Materials and Methods
Mice
BALB/c wild-type, Il13ra1−/− (IL-13Rα1), Il4−/− (IL-4), Il5 (IL-5)-CD2-transgenic or Stat6−/− (STAT-6) mice were analyzed at 4 to 8 weeks of age. All mice were housed under specific pathogen-free conditions and treated according to institutional guidelines.
Bone marrow-derived eosinophil culture
Bone marrow-derived eosinophil culture was modified from Dyer et al. (26). Briefly, total bone marrow cells were isolated, and erythrocytes were lysed by red blood cell lysis buffer (Sigma-Aldrich®). After a density gradient of Histopaque 1083 (Sigma-Aldrich®), the low-density bone marrow (LDBM) cells were collected and plated at 1×106 cells/ml in IMDM (Gibco®) supplemented with 10% FBS (Hyclone), penicillin-streptomycin (Gibco®), L-glutamine 200 mM (Gibco®), and β-mercaptoethanol 55 μM (Sigma-Aldrich®). During the first 4 days, the medium also contained stem cell factor (SCF) (PeproTech®) and Fms-like tyrosine kinase 3 (FLT3)-ligand (PeproTech®) at 100 ng/ml each. From day 4 to day 14, the cells were cultured in medium containing 10 ng/ml IL-5 (PeproTech®). The medium was changed every 2 days until day 14.
For eosinophil activation, cells were collected, pooled and plated for at least 1 hour in a tissue culture dish, to remove any contaminating cells, such as stromal cells or macrophages. Then, the non-adherent cells were collected, washed, counted and incubated with different treatment, according to the experiments. Murine recombinant IL-4 and IL-13 were purchased from PeproTech®, and IL-33 was purchased from R&D Systems®. The NFκB inhibitor BAY 11-7082 was purchased from Santa Cruz Biotechnology® and was administered at 5 μM for all of the experiments in which it was used.
Eosinophils purification from Il5-transgenic mice
Briefly, spleen from Il5–transgenic mice were collected and crushed onto a cell strainer 40 μm (BD Falcon). After red blood cell lysis, the T and B cells were depleted by using microbeads anti-CD90.2 and anti-CD19 (MACS, Miltenyi Biotec). The cell suspension was incubated for 1 hour in a tissue culture dish to deplete the adherent cells (macrophages). Then, the cells were collected, plated at a density of 2.5×106 cells/ml and activated for 24 hours, in complete IMDM (see details in bone marrow-derived eosinophil culture section) supplemented with 10ng/ml of IL-5.
Extraction of mRNA and quantitative real-time (qRT)-PCR analysis
Total RNA was isolated with the RNeasy mini kit (QIAGEN®) according to the manufacturer’s protocol. cDNA was synthetized from 1 or 0.5 μg of RNA using the iScript™ synthesis kit (Bio-Rad®). qRT-PCR was performed using a 7900HT Fast Real-Time PCR system (Applied Biosystems™) with FastStart Universal SYBR Green Master mix (Roche). Primers will be provided upon request.
RNA sequencing analysis
FASTQ files from Illumina Pipeline were aligned by TopHat (version 1.4.1) (27), with -T and -G parameters and iGenome annotation from Illumina (08/30/11) for the mm9 genome. The -T parameter is used to align reads to the mouse transcriptome, and the -G parameter is used to provide transcriptome annotation. Produced .bam files were fed to cuffdiff (version 1.3.0) (27) annotated with the same GTF file. Only genes for which at least one isoform was significantly changed and had fragment per kilobase per million mapped reads (FPKM) greater than two in at least one condition were analyzed. Experimental conditions were compared to appropriate controls, and a total set of 1593 genes was generated. To generate heat maps, genes were clustered using Gene Cluster 3.0 (28) with the following parameters: clustering average linkage and correlation (centered) similarity. For visualizing of clustered data, Java Tree View(29) software was applied: http://jtreeview.sourceforge.net/. The database is available following this link: http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?token=nfelrwciwwesida&acc=GSE43660
Flow cytometry
Eosinophil markers were assessed by fluorescence-activated cell sorting (FACS) staining for CCR3 and Siglec-F (BD Biosciences) at day 14 of culture. For the detection of phospho-p38 or phospho-STAT-6, the cells were activated with IL-33 (100ng/ml) for 5 min, IL-4 or IL-13 (100ng/ml) for 30 min, washed with PBS, and then processed for fixation (1% formaldehyde) and permeabilization (90% methanol) and stained with 1 μg/ml of anti-mouse phospho-p38 (Cell Signaling Technology) or phospho-STAT-6 antibody (BD Biosciences) per 1×106 cells. For the intracellular staining, bone marrow derived cultured eosinophils, activated overnight by IL-4 or IL-33, were incubated for 4 hours with 5 μg/ml of brefeldin A (Sigma) to block the protein secretion. Cells were washed with PBS and then processed for the fixation, permeabilization and staining with anti-mouse Siglec-F PE (BD Biosciences), anti-mouse RELM-α (Peprotech®), anti-ST2 (R&D Systems), anti-IL-4Rα (BD Biosciences) antibodies and secondary antibody: goat anti-rabbit Alexa Fluor® 488 (Life Technologies, Invitrogen). All antibodies were used at 1 μg/ml per 106 cells. Flow cytometry was performed on a fluorescence activated cell sorting (FACSCalibur) and data were analyzed with the FlowJo software (Tree Star, Inc.).
For cell culture, samples were incubated with 10 μg/ml of anti-mouse IL-4 (eBioscience, Inc.) or anti-mouse IL-4Rα (BD Biosciences) antibody for 4 hours or 24 hours, depending on the experiment. Control IgG1 and IgG2a antibodies were purchased from BD Biosciences.
Western blot
Proteins extracted with RIPA buffer were loaded in equal amounts on 4–12% polyacrylamide gradient gels (Invitrogen™) and subjected to Western blot with anti-phospho NFκB p65 and anti-NFκB p65 antibodies (Cell Signaling Technology®).
ELISA
Murine IL-4 and IL-6 levels were analyzed by ELISA using a kit from Biolegend®. Murine IL-13 and CCL17 were measured by ELISA using the DuoSet® ELISA Development Systems from R&D Systems®. For detection of murine RELM-α, purified anti-RELM-α and biotinylated anti-RELM-α (PeproTech®) antibodies were used according to a protocol provided by the manufacturer. The lower detection limits for IL-4 and IL-13 were 1.95 pg/ml and 62.5 pg/ml, respectively, and for IL-6, CCL17, and RELM-α were 15.6 pg/ml.
Statistical analysis
Statistics were done by using the Student t-test or one-way ANOVA with Bonferroni’s correction. P-values < 0.05 were considered as significant.
Results
Gene expression profiles in murine eosinophils activated by IL-4 or IL-33
We first generated murine bone marrow-derived eosinophils, primarily as per Dyer et al. (26). After 14 days of culture, we confirmed that the cells were mature eosinophils, characterized by multi-lobed nuclei (Fig. 1A) and the expression of Siglec-F and CCR3 (Fig. 1B) and the expression of eosinophil granule proteins by staining for eosinophil peroxidase activity (data not shown). To minimize the possibility that non-eosinophils could be contaminating cells and responsible for the signaling induction by IL-4 or IL-33, adherent cells were removed prior to cellular activation and eosinophils represented ≥99% of the cells. IL-33 signaling triggered the phosphorylation of p65, a subunit of NF-κB, within 5 minutes; total NFκB (p65) did not change over time, demonstrating that eosinophils respond to IL-33 (Fig. 1C). We also confirmed that IL-33 induced the phosphorylation of p38 within 5 minutes of stimulation (Fig. 1D). Similarly, IL-4 induced STAT-6 phosphorylation in eosinophils within 30 minutes; whereas, IL-13 had no effect on STAT-6 phosphorylation (Fig. 1E).
Figure 1. Bone marrow-derived eosinophils respond to IL-33 and IL-4 but not IL-13.

Hematoxylin/eosin staining of bone marrow-derived eosinophils at Day 14 of culture (magnification x400) (A). Characterization of eosinophils by the co-expression of Siglec-F and CCR3, using flow cytometry analysis (B). Western blot of p-NFκB and NFκB from eosinophils (4x106/ml) treated with 100 ng/ml of IL-33 for 5 or 30 minutes (C). Flow cytometry of intracellular staining of p-p38 or p-STAT-6 from eosinophils (4×106/ml) treated with 100 ng/ml of IL-33 for 5 minutes (D) or 100ng/ml of IL-4 or IL-13 for 30 minutes (E). The results shown are representative of 3 independent experiments.
We generated genome-wide transcriptome data based on RNA sequence analysis from eosinophils activated with IL-4 or IL-33. We analyzed two time points, 1 and 4 hours, to determine early and late gene induction. We found that multiple genes were differentially expressed after 1 and 4 hours of IL-4 or IL-33 exposure (Fig. 2A). Each treatment induced a specific pattern of gene expression, with certain genes being regulated (≥ 2 fold-change) by IL-33 but not by IL-4 and vice versa (Fig. 2B). IL-33 exhibited a more profound effect compared with IL-4, inducing 519 genes. IL-4 induced 28 genes, with only 9 being specifically induced by IL-4. Notably, 19 genes were induced by both IL-33 and IL-4. Functional analysis revealed that the IL-33-induced genes were involved in cytokine production, immune response, and NFκB signaling; whereas, IL-4-induced genes were involved in cellular homeostasis and Jak/STAT signaling (data not shown) (http://toppgene.cchmc.org). Validating genes by qRT-PCR, we confirmed that Cxcl2, Cxcl10, Clec4e, Adora2b, Il6, and Il13 were specifically upregulated by IL-33, but not IL-4, after 1 and 4 hours of activation (Fig. 2C). We also confirmed that eosinophils upregulated Retnla mRNA after 1 and 4 hours of IL-4 exposure, but only after 4 hours of IL-33 exposure, and upregulated Tfec after 1 and 4 hours of IL-4 or IL-33 exposure.
Figure 2. Transcriptome analysis in eosinophils activated by IL-33 or IL-4.

Eosinophils were treated for 1 or 4 hours with IL-4 or IL-33 at 10 ng/ml. RNA was sequenced by RNA-sequencing technology. Heatmap represents the genes differentially regulated by IL-4 and IL-33 after 1 and 4 hours of exposure (A). Venn diagram represents the number of genes (with a fold change ≥ 2 in either direction) differentially regulated by IL-4 (red), IL-33 (green), or both (grey) (B). Validation by qRT-PCR analysis of genes identified by RNA sequencing as upregulated in eosinophils activated by IL-4 or IL-33 at 1 or 4 hours (C). Bars represent the mean of 2 wells and the error bars represent the SEM values. Data are representative of 3 independent experiments.
IL-33 is a potent activator of eosinophils
Since IL-33 induced Il6 and Il13 expression, we evaluated whether these two cytokines were released in response to 24-hours of exposure to different concentrations of IL-33. Eosinophil secretion of IL-6 and IL-13 increased in response to IL-33 in a dose-dependent manner (Fig. 3A). This response was specific to IL-33, as IL-4 had no effect. Similarly, CCL17 release increased in a dose-dependent manner in response to different doses of IL-33 but not IL-4 (Fig. 3B).
Figure 3. Effect of IL-33 and IL-4 on cytokine/chemokine expression by eosinophils.

Eosinophils (4×106/ml) were activated for 24 hours in the presence of the indicated concentrations of IL-33 or IL-4 (IL-4 at 100 ng/ml for A and B). Supernatants were collected and IL-6 and IL-13 (A), CCL17(B), IL-4 (C), and RELM-α (E) were measured by ELISA. qRT-PCR analysis represents the kinetics of Il6, Ccl17, and Il4 induction by IL-33 in eosinophils (D). Bars (A-C, E) or symbols (D) represent the mean of 2 wells, and error bars represent the SEM values. Flow cytometry on eosinophils after overnight incubation with the different cytokines at 100 ng/ml to evaluate SiglecF+/RELM-α+ cells (F). The percentage of cells in each quadrant is indicated (F). Data are representative of 3 independent experiments. ND, not detected. **p < 0.01, ***p < 0.001.
Eosinophils also secreted IL-4 in response to IL-33 in a dose-dependent manner (Fig. 3C). Notably, CCL17 and IL-4 release increased following IL-33 exposure, but IL-33 did not regulate Ccl17 or Il4 expression as determined by RNA sequencing. Indeed, Il4 mRNA was not induced by IL-33 at 2, 6, or 24 hours of exposure, suggesting that the protein is pre-formed in the cells and released when the eosinophils were activated (Fig. 3D). By ELISA, we detected IL-4 in a total protein lysate of unstimulated eosinophils compared to the negative control, Il4-deficient eosinophils (data not shown). In contrast, Ccl17 was increased after 6 hours of IL-33 treatment and decreased at 24 hours (Fig. 3D). For comparison, Il6 was induced at 2 and 6 hours and diminished at 24 hours. Finally, we observed that 24-hours of exposure to different concentrations of IL-4 or IL-33 induced significant, dose-dependent production of RELM-α by eosinophils (Fig. 3E). Additionally, we confirmed that IL-4 and IL-33-treated eosinophils did indeed express RELM-αby flow cytometry analysis (Fig. 3F).
IL-33 induces RELM-α and CCL17 expression through an IL-4 autocrine mechanism
Since eosinophils secreted RELM-αin response to IL-4 or IL-33 and released IL-4 after IL-33 exposure, we hypothesized that an IL-4 autocrine loop is involved in IL-33-induced RELM-α. We, first, generated Il4−/− and wild-type bone marrow derived-eosinophils. We monitored the cultures and did not observe any differences, in terms of proliferation, differentiation and morphology between the IL-4-deficient and wild-type eosinophils. In addition, IL-4-deficient eosinophils expressed CCR3, Siglec-F, IL-4Rα and ST2 at similar levels compared with wild-type eosinophils (data not shown). We, then, activated Il4−/− and wild-type bone marrow derived-eosinophils (obtained at day 14 of culture) for 24 hours with IL-4 or IL-33 and compared RELM-αas well as IL-6, IL-13, and CCL17 expression at the mRNA and protein levels. Notably, RELM-α was not induced in Il4−/− eosinophils stimulated with IL-33 compared to wild-type eosinophils;however, Il4−/− eosinophils responded to IL-4 by releasing RELM-α (Fig. 4A). Il4−/− eosinophils stimulated with IL-33 secreted IL-6 and IL-13 at similar levels to IL-33-stimulated wild-type eosinophils (Fig. 4B). Interestingly, the release of CCL17 was lower in IL-33-stimulated Il4−/− eosinophils in comparison to IL-33-stimulated wild-type eosinophils, suggesting a role for an IL-4 autocrine loop in CCL17 expression (Fig. 4C). However, in Figure 3B, the treatment by IL-4 did not induce CCL17 expression. Thus, we tested whether the addition of IL-4 with IL-33 could induce the expression of CCL17. Il4-deficient eosinophils treated with IL-33 and IL-4, produced CCL17 at similar levels compared with wild-type eosinophils (data not shown). However the co-treatment did not increase the expression of CCL17 in wild type eosinophils suggesting that the IL-4 has an additional but not synergistic effect. By qRT-PCR analysis, we confirmed that compared to IL-33-treated wild-type eosinophils, IL-33-treated Il4−/− eosinophils displayed decreased Retnla and a trend for decreased Ccl17 mRNA expression but unchanged Il6 and Il13 mRNA expression (Fig. 4D). Collectively, these data suggest the existence of an IL-4 autocrine loop that is responsible for IL-33-induced RELM-α, and to a lesser extent CCL17. In contrast, IL-6 and IL-13 are independent of the IL-4 autocrine loop and may be directly induced by IL-33.
Figure 4. IL-33 requires the IL-4 autocrine loop to induce RELM-αand CCL17.

Wild-type (WT) or Il4−/− eosinophils (4×106/ml) treated for 24 hours with IL-4 (10 ng/ml, A) or IL-33 (10 ng/ml, A–D). RELM-α, CCL17, IL-6, and IL-13 levels in the supernatants were determined by ELISA (A–C). qRT-PCR from wild-type (WT) or Il4−/− eosinophils (4×106/ml) incubated for 4 hours in the presence of IL-33 (10 ng/ml); graph represents the fold change of gene expression in IL-33-treated eosinophils compare to untreated (D). Eosinophils (4×106/ml) were activated for 4 hours (F) or 24 hours (E, G and H) by IL-33 (10 ng/ml) in the absence or presence of IgG1 (isotype control) or anti-IL-4 antibody (10 μg/ml). Gene expression of Cxcl2, Cxcl10, Clec4e, Il6, Il13, Adora2b, Ccl17, Tfec, and Retnla was analyzed by qRT-PCR (F). The levels of IL-4 (E), RELM-αand CCL17 (G), and IL-6 and IL-13 (H) in the supernatants were measured by ELISA. Bars represent the mean of 2 wells and the error bars represent the SEM values. Data are representative of 3 independent experiments. ND, not detected; ns, not significant. *p < 0.05, **p < 0.01, ***p < 0.001, § p=0.055 and # p=0.083.
IL-33 induces eosinophil activation through IL-4-dependent and -independent pathways
To further investigate the role of IL-4 in mediating activation of eosinophils by IL-33, we examined the effect of IL-4-specific neutralizing antibody. Eosinophils were incubated for 24 hours with anti-IL-4 antibody in the presence of IL-33; IL-4 was not detected (by ELISA) following anti-IL-4 treatment (Fig. 4E). Anti-IL-4 antibody decreased the IL-33-induced secretion and transcription of RELM-α and CCL17 (Fig. 4F and G). Notably, cells incubated with IL-33 and IgG1 control antibody produced RELM-α at similar levels to cells incubated with IL-33 alone, indicating the specificity of the IL-4 neutralizing antibody in inhibiting IL-33-induced RELM-α expression. The expression and secretion of IL-6 and IL-13 were not affected by anti-IL-4 treatment (Fig. 4F and H). In addition, the expression of Cxcl2, Cxcl10, Clec4e, and Adora2b was not decreased with anti-IL-4 treatment, indicating that, similar to IL-6 and IL-13, these genes are not dependent on the IL-4 pathway (Fig. 4F). However, we observed a trend for Tfec mRNA to be decreased in the presence of anti-IL-4 antibody (Fig. 4F), suggesting that the IL-4 autocrine loop is involved in the regulation of Tfec expression. We validated our system with an ex vivo culture of mature eosinophils from Il5-transgenic mice. Figure 5 shows that mature eosinophils respond to IL-33 by releasing IL-4, RELM-α, CCL17, IL-6 and IL-13. However in the presence of the anti-IL-4 antibody, the RELM-αand CCL17 secretion (unlike IL-6 and IL-13) were significantly decreased.
Figure 5. Mature eosinophils respond to IL-33 and require IL-4 to induce RELM-αand CCL17.

Eosinophils from Il5-transgenic mice were activated 24 hours with IL-33 in the presence of anti-IL-4 antibody or isotype control (IgG1). IL-4, RELM-α, CCL17, IL-6 and IL-13 released in the supernatants were measured by ELISA. *p < 0.05, **p<0.001, ns, not significant.
IL-33 requires type I IL-4R to activate eosinophils
Next, we aimed to determine whether IL-4R type I and/or II are required for eosinophil activation. We first treated eosinophils with IL-33 for 24 hours in the presence of anti-IL-4Rαneutralizing antibody or IgG2a, an isotype control. Neutralizing IL-4Rαinhibited the IL-33-induced expression and production of RELM-αby eosinophils (Fig. 6A and 6B). Although the IL-33-induced expression of Ccl17 mRNA was not significantly decreased by anti-IL-4Rαtreatment (Fig. 6A), the production of CCL17 protein after 24 hours of IL-33 and anti-IL-4Rαtreatment was dramatically decreased in comparison to IL-33 and IgG2a treatment (Fig. 6B). Treatment with anti-IL-4Rαantibody did not change the production of IL-4 induced by IL-33 (Fig. 6B). In addition, IL-33-induced IL-6 and IL-13 expression and production were not affected by the anti-IL-4Rαor IgG2a antibody treatments (Fig. 6A and 6B). Similarly, the IL-33 induced expression of Cxcl2, Cxcl10, Clec4e, and Adora2b was not affected by the treatment with the anti-IL-4Rαantibody, indicating that these genes are directly regulated by IL-33 (Fig. 6A). IL-33-induced Tfec expression trended to be decreased in the presence of the IL-33 and anti-IL-4Rαantibody compared to IL-33 and IgG2a (Fig. 6A), indicating that this gene may be dependent on the IL-4 pathway. In summary, we found that IL-33 requires IL-4Rαto induce RELM-αand CCL17 secretion.
Figure 6. IL-4Rα, but not IL-13Rα1, is involved in IL-33-induced eosinophil activation.

Eosinophils (4×106/ml) activated for 4 hours (A) or 24 hours (B) by IL-33 (10 ng/ml) in the absence or presence of IgG2a (isotype control) or anti-IL-4Rαantibody, at 10 μg/ml. Gene expressions of Cxcl2, Cxcl10, Clec4e, Il6, Il13, Adora2b, Ccl17, Tfec, and Retnla were analyzed by qRT-PCR (A). The levels of RELM-α, CCL17, IL-4, IL-6, and IL-13 (B) in the supernatants were measured by ELISA. Wild-type (WT) or Il13ra1−/− eosinophils (4×106/ml) were treated for 24 hours in the presence of IL-33 (10ng/ml), and the levels of RELM-α, CCL17, IL-13, and IL-4 in the supernatants were measured by ELISA (C). Bars represent the mean of 2 wells and the error bars represent the SEM values. Data are representative of 3 independent experiments. ND, not detected; ns, not significant. *p < 0.05, **p < 0.01, ****p < 0.0001.
We then tested whether IL-4R type II was required for eosinophil activation. Since anti-mouse IL-13Rα1 neutralizing antibody was not commercially available, we generated eosinophils from Il13ra1-deficient mice. Wild-type and Il13ra1-deficient eosinophils were treated for 24 hours with IL-33. IL-33 induced RELM-α, CCL17, IL-4, and IL-13 secretion in Il13ra1−/− eosinophils at similar levels compared with wild-type eosinophils (Fig. 6C). Overall, these results demonstrated that IL-4Rα, but not IL-13Rα1, is required for the IL-33-mediated release of RELM-αand CCL17.
Role of STAT-6 in eosinophil activation
To further elucidate whether IL-4 signaling is involved in the expression of genes induced by IL-33, we generated Stat6-deficient bone marrow-derived eosinophils and examined their response to IL-33 or IL-4. We found that RELM-α production was not induced by either IL-4 or IL-33 in Stat6-deficient eosinophils compared to wild-type eosinophils (Fig. 7A), which is consistent with our previous observations that RELM-αexpression is IL-4/IL-4Rαdependent. In addition, IL-33 did not induce CCL17 production in Stat6−/− eosinophils (Fig. 7B). IL-4, IL-6, and IL-13 were secreted by wild-type and Stat6-deficient eosinophils after IL-33 exposure (Fig. 7C). Although Stat6-deficient eosinophils responded to IL-33 and expressed ST2 (data not shown), they secreted significantly less IL-4, IL-6, and IL-13 compared to wild-type eosinophils, suggesting a dysfunction in these cells, probably due to the loss of STAT-6.
Figure 7. Role of STAT-6 in eosinophil activation.
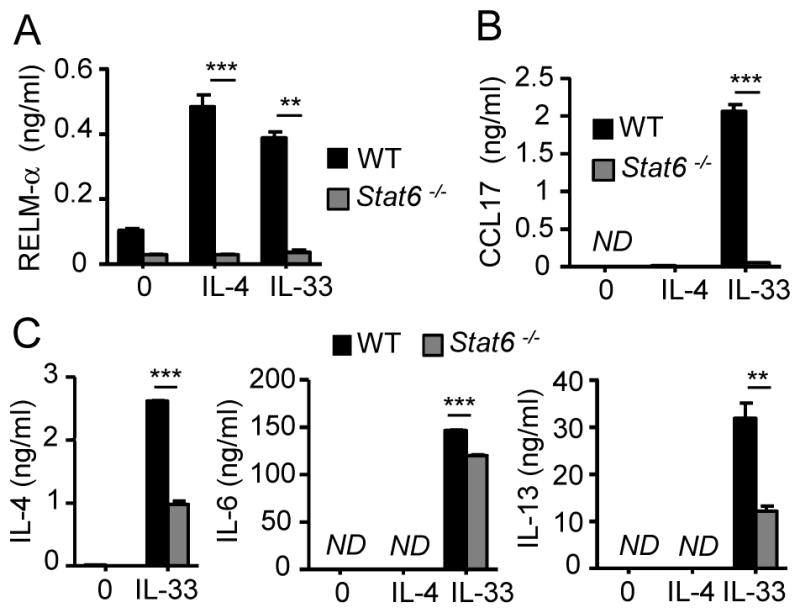
Wild-type (WT) and Stat6-deficient eosinophils were activated with IL-33 (100 ng/ml) or IL-4 (100 ng/ml). RELM-α (A), CCL17 (B), IL-4, IL-6, and IL-13 (C) released in the supernatants were measured by ELISA. Bars represent the mean of 2 wells and the error bars represent the SEM values. Data are representative of 3 independent experiments. ND, not detected. **p < 0.01, ***p < 0.001.
NFκB is involved in the expression of genes induced by IL-33
In order to determine whether the induction of genes by IL-33 occurs via NFκB signaling, eosinophils were pretreated for 1 hour with a NFκB inhibitor (BAY11-7082) and then activated for 4 hours with IL-33 or IL-4. Cxcl2, Cxcl10, Clec4e, Il6, Il13, Adora2b, Ccl17, Tfec, and Retnla displayed a decrease in their IL-33-induced expression when treated with the NFκB inhibitor, indicating that induction by IL-33 is regulated directly or indirectly by NFκB signaling (Fig. 8A). Notably, Cxcl2, Cxcl10, Clec4e, Il6, Il13, and Adora2b were not induced by IL-4, suggesting that these genes may be directly regulated by the binding of NFκB to the promoter region. This notion is supported by the presence of NFκB binding/consensus sites in the promoter regions of human IL6, IL13 and mouse Cxcl2, Cxcl10 (see link: http://bioinfo.lifl.fr/NF-KB/). We previously showed that Ccl17, Tfec, and Retnla were induced by IL-33 through an IL-4 autocrine loop. Consistent with this indirect mechanism, IL-4-induced Ccl17, Tfec, and Retlna expression were not affected by the NFκB inhibitor and the decrease in their IL-33-induced expression by NFκB inhibitor correlated to a decrease in IL-4 release (Fig. 8B). Indeed, our results indicate that the IL-4 autocrine loop requires IL-33-associated NFκB signaling.
Figure 8. Effect of NFκB inhibitor in the regulation in IL-33-induced gene expression.
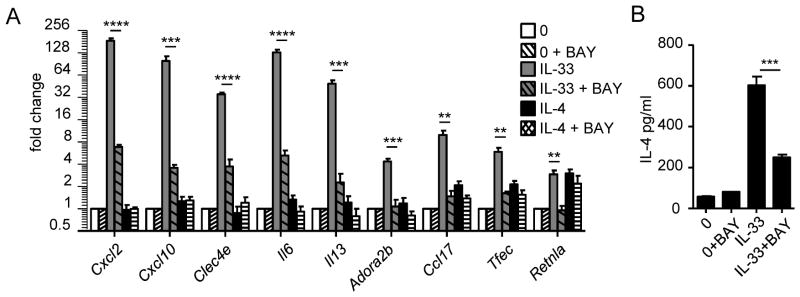
Eosinophils (4×106/ml) were pretreated for 1 hour with NFκB inhibitor (BAY11-7082 [BAY]) at 5 μM and then cultured for 4 hours in the presence of IL-33 (AB) or IL-4 (A) at 10 ng/ml. Gene expression of Cxcl2, Cxcl10, Clec4e, Il6, Il13, Adora2b, Ccl17, Tfec, and Retnla were analyzed by qRT-PCR (A), and IL-4 in the supernatant was measured by ELISA (B). Bars represent the mean of 2 wells and the error bars represent the SEM values. Data are representative of 3 independent experiments. **p < 0.01, ***p < 0.001, ****p < 0.0001 (A–B).
Discussion
Although eosinophils have been shown to respond to IL-33 or IL-4 (9, 30–33), the regulation and role of both of these cytokines, as well as IL-13, in directly activating eosinophils has not been well characterized. Herein, we report RNA sequence analysis of murine eosinophils activated by IL-4 and IL-33. We demonstrate that IL-33 is a powerful activator of eosinophils; whereas, IL-4 and IL-13 induce modest and no activation of eosinophils, respectively. We demonstrate that IL-33 mediates its effects by two pathways, distinguished by their dependence on IL-4. IL-33 directly stimulates eosinophils to release pre-formed IL-4, which then auto-activates eosinophils via IL-4Rαand STAT-6; additionally, IL-33 directly induces transcription of a myriad of gene products via NFκB. Interestingly, functional analysis revealed that IL-33 mediates the expression of cytokine and chemokine gene products, such as IL-6, IL-13, CXCL2, and CXCL10; whereas, IL-4 induces genes enriched in the STAT-6 signaling pathway, such as RELM-αand TFEC, which are known to be STAT-6 dependent (34, 35). Figure 9 summarizes our findings, which support the ability of IL-33 to profoundly induce eosinophil activation, as measured by transcriptional induction and release of pleiotropic immunomodulatory mediators, and the induction of an IL-4-driven auto-inflammatory loop that is likely to contribute to a variety of Th2 and/or innate immune responses. While mature eosinophils are classically known as primarily granule protein secreting cells, there is emerging data that these cells remain transcriptionally active and secrete a number of gene products including IL-4 and RELM-αmRNA and protein (18, 36–38).
Figure 9. Proposed model summarizing our results.
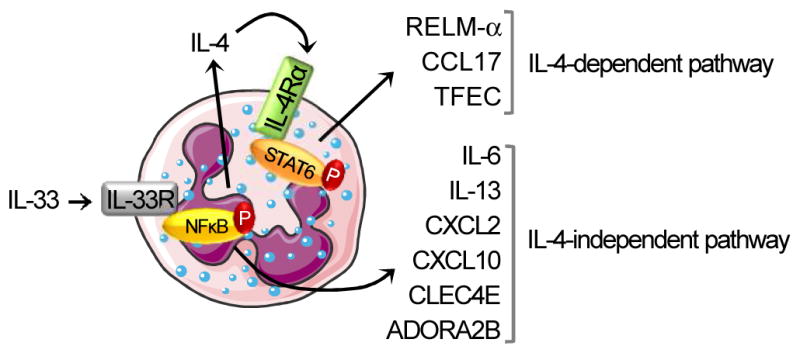
After binding to its receptor, IL-33 induces eosinophil activation, as measured by gene transcription and release of several mediators, through the phosphorylation of NFκB. Il6, Il13, Cxcl2, Cxcl10, and Clec4e are directly induced by IL-33 (IL-4-independent pathway). Although IL-4 transcription is not increased after IL-33 treatment, the protein release of preformed IL-4 is dependent on the IL-33/ST2 signaling pathway. Retnla, Ccl17 and Tfec induction by IL-33 is indirect and requires the activation of the IL-4/IL4Rα pathway (IL-4-dependent pathway) via the phosphorylation of STAT-6. This figure was produced using Servier Medical Art (www.servier.com).
We found that IL-33 induces IL-4 release by eosinophils. However, IL-4 mRNA did not increase after IL-33 treatment. Interestingly, we found that IL-4 was pre-stored in murine eosinophils under homeostatic conditions (at day 14 of culture) (data not shown). Similarly, human eosinophils have been demonstrated to contain pre-stored IL-4 in vesicles and to rapidly release IL-4 upon stimulation (39, 40). Previous studies have reported that IL-33 can induce IL-4 production by human basophils (4, 41) and murine splenic eosinophils when these cells are co-treated with IL-33 and IL-5 or GM-CSF (30). Moreover, a recent study showed that in DSS-induced colitis model, IL-33 exacerbates the disease via IL-4 (42). In addition, IL-33 has been demonstrated to enhance B-cell activation through IL-4 secreted by mast cells and eosinophils (43). Notably, IL-4-producing eosinophils have been demonstrated to have a role in the initiation of type 2 immune responses. In a helminth infection model, eosinophils were identified as one of the major innate IL-4-producing cells in the lung of infected mice (16, 17), whereas IL-33 is known to initiate the immune response (44). Interestingly, in adipose tissue, eosinophil-derived IL-4 has been demonstrated to have a role in glucose metabolism through the maintenance of adipose M2 macrophages, indicating an anti-inflammatory role of IL-4-secreting eosinophils (18). Moreover, two recent studies have demonstrated that after muscle or liver injury, eosinophil-derived IL-4 is required for tissue regeneration, through the regulation of the resident cells (45, 46). In addition, IL-33 protects mice from adipose tissue inflammation by increasing Th2 cells and macrophages (47). On the basis of this collective dataset as well as our data that IL-33 potently induces eosinophil production of a plethora of potent cytokines, we speculate that IL-33-activated eosinophils could be a key driving factor in the regulation of eosinophilic inflammatory responses. Our results also shed light on the potential function of eosinophil-derived IL-4; rather than strictly stimulating other cell types, eosinophil-derived IL-4 may have an essential role in auto-activating eosinophils, consistent with most studies that have shown that eosinophil-derived IL-4 is not essential for Th2 cell polarization (48–50). Recently, an in vitro study demonstrated that the activation of the IL-4/IL-4Rαaxis in murine eosinophils from IL-5 transgenic mice enhances eosinophil migration towards CCL11 (33), suggesting a role of IL-4 as a potent co-activator of eosinophils in allergic reactions.
By dissecting the mechanism of IL-4 action on eosinophils, we discovered that IL-33 requires IL-4 signaling to induce RELM-αand CCL17. First, we found that IL-4 induces RELM-αproduction by eosinophils through the type I IL-4R. To further examine the specific role of IL-4 in IL-33-induced eosinophil responses, we generated eosinophils from Il4-deficient mice which demonstrated that eosinophils required IL-4 to induce RELM-αand CCL17 but not IL-6 and IL-13. In complementary experiments using anti-IL-4 or anti-IL-4Rαtreatment, we validated the IL-4/IL-4Rαdependency of IL-33-induced RELM-αand CCL17 expression. Interestingly, although single treatment by IL-4 did not induce the expression of CCL17 (Figure 3B), treatment with IL-4 and IL-33 increased CCL17 expression in Il-4 deficient eosinophils, suggesting that STAT-6 and NFκB signaling are both required for CCL17 production (data not shown). However in wild-type eosinophils, CCL17 expression was not increased by the co-treatment with IL-4 and IL-33 compared with IL-33 alone. These data indicate that CCL17 reached a maximal expression after IL-33 treatment or co-treatment with IL-4 and suggest that the signal cannot be amplified due to a saturation of the IL-4R by the IL-4. By using in vivo eosinophils from Il5-transgenic mice, we confirmed the in vitro findings showing that IL-33 elicits two activation pathways. Although, in vivo eosinophils respond to IL-33 and IL-4, we found that the levels of cytokines released are lower than bone marrow-derived eosinophils. In Il5-transgenic mice, the overexpression of IL-5 may pre-activate directly or indirectly the eosinophils. It is known that activated eosinophils are more sensitive to apoptosis. In addition, the eosinophil preparation process could also weaken the cells and alter their viability and so, their potential to respond to a stimulus.
Moreover, we showed that the IL-33-induced expression of Tfec was dependent on the IL-4 autocrine loop; whereas, the IL-33-mediated induction of Cxcl2, Cxcl10, Clec4e, and Adora2b transcripts was not affected by neutralizing the IL-4/IL-4Rαaxis. Although IL-4 displays a lower effect than IL-33 on eosinophil activation, these findings showed that IL-33 induces and amplifies eosinophil activation partially via IL-4. Overall, we suggest that IL-4 contributes to the IL-33-induced eosinophil activation by the release of CCL17, which enhances the recruitment of TCD4+ cells, and by producing RELM–α which promotes fibrogenic responses as well as eosinophil chemoattraction (51–53).
In agreement with a recent study (9), we showed that Il6 and Il13 were rapidly expressed in eosinophils and released within 24 hours of treatment with IL-33. Moreover, our study showed that IL-6 and IL-13 transcripts and proteins were not induced by IL-4 treatment alone. Using an NFκB inhibitor, we demonstrated that IL-33-induced Il6 and Il13 expression were dependent on NFκB signaling, suggesting that NFκB binds to Il6 and Il13 promoters and consequently induces their transcription. It is important to note that the IL-33-induced expression of some genes (e.g. Il6 or Il13) was not entirely abolished by NFκB inhibition, indicating that NFκB is not likely the only signaling molecule involved in IL-33′s activity; other mediators such as ERK, p38 or JNK could be involved as well.
In conclusion, we have demonstrated the ability of IL-33 to directly and profoundly induce eosinophil activation by an NFκB-dependent mechanism. Additionally, we have determined that IL-33 induces NFκB-dependent eosinophil release of IL-4. Further, we have identified an IL-33-induced IL-4-driven eosinophil autoinflammatory loop that likely contributes to a variety of Th2 and/or innate immune responses, involving both immature developing eosinophils as well as mature eosinophils. To date, few papers have demonstrated the effect of IL-33 on human eosinophils (4, 54, 55) and the transcriptome or gene expression profile has not been shown. Herein, we demonstrated that IL-6 is induced by IL-33 in mouse eosinophils similar to reported papers on human eosinophils (55). We also found that Ccl2 and Icam1 are expressed in mouse eosinophils after IL-33 treatment which is consistent with protein expression in IL-33 activated human eosinophils (55).
Acknowledgments
Grant support: This work was funded by the NIH/NIAID (R01AI083450, R37AI045898), CURED Foundation, FARE, and Buckeye Foundation.
We thank Dr. Yui-Hsi Wang (Cincinnati Children’s Hospital Medical Center [CCHMC]) for the kind gift of the Stat6−/− and Il4−/− mice, Melissa Mingler for animal colony maintenance and Shawna Hottinger (Medical Writer, CCHMC) for editorial assistance.
Abbreviations
- IL-33
interleukin 33
- IL-4R
interleukin 4 receptor
- NFκB
nuclear factor kappa B
- RELM-α
resistin-like molecule alpha
- STAT-6
signal transducer and activator of transcription 6
- Th2
T helper lymphocyte type 2
References
- 1.Funakoshi-Tago M, Tago K, Hayakawa M, Tominaga S, Ohshio T, Sonoda Y, Kasahara T. TRAF6 is a critical signal transducer in IL-33 signaling pathway. Cell Signal. 2008;20:1679–1686. doi: 10.1016/j.cellsig.2008.05.013. [DOI] [PubMed] [Google Scholar]
- 2.Schmitz J, Owyang A, Oldham E, Song Y, Murphy E, McClanahan TK, Zurawski G, Moshrefi M, Qin J, Li X, Gorman DM, Bazan JF, Kastelein RA. IL-33, an interleukin-1-like cytokine that signals via the IL-1 receptor-related protein ST2 and induces T helper type 2-associated cytokines. Immunity. 2005;23:479–490. doi: 10.1016/j.immuni.2005.09.015. [DOI] [PubMed] [Google Scholar]
- 3.Allakhverdi Z, Smith DE, Comeau MR, Delespesse G. Cutting edge: The ST2 ligand IL-33 potently activates and drives maturation of human mast cells. J Immunol. 2007;179:2051–2054. doi: 10.4049/jimmunol.179.4.2051. [DOI] [PubMed] [Google Scholar]
- 4.Pecaric-Petkovic T, Didichenko SA, Kaempfer S, Spiegl N, Dahinden CA. Human basophils and eosinophils are the direct target leukocytes of the novel IL-1 family member IL-33. Blood. 2009;113:1526–1534. doi: 10.1182/blood-2008-05-157818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kurowska-Stolarska M, Stolarski B, Kewin P, Murphy G, Corrigan CJ, Ying S, Pitman N, Mirchandani A, Rana B, van Rooijen N, Shepherd M, McSharry C, McInnes IB, Xu D, Liew FY. IL-33 amplifies the polarization of alternatively activated macrophages that contribute to airway inflammation. J Immunol. 2009;183:6469–6477. doi: 10.4049/jimmunol.0901575. [DOI] [PubMed] [Google Scholar]
- 6.Liew FY, Pitman NI, McInnes IB. Disease-associated functions of IL-33: the new kid in the IL-1 family. Nat Rev Immunol. 2010;10:103–110. doi: 10.1038/nri2692. [DOI] [PubMed] [Google Scholar]
- 7.Prefontaine D, Lajoie-Kadoch S, Foley S, Audusseau S, Olivenstein R, Halayko AJ, Lemiere C, Martin JG, Hamid Q. Increased expression of IL-33 in severe asthma: evidence of expression by airway smooth muscle cells. J Immunol. 2009;183:5094–5103. doi: 10.4049/jimmunol.0802387. [DOI] [PubMed] [Google Scholar]
- 8.Prefontaine D, Nadigel J, Chouiali F, Audusseau S, Semlali A, Chakir J, Martin JG, Hamid Q. Increased IL-33 expression by epithelial cells in bronchial asthma. J Allergy Clin Immunol. 2010;125:752–754. doi: 10.1016/j.jaci.2009.12.935. [DOI] [PubMed] [Google Scholar]
- 9.Stolarski B, Kurowska-Stolarska M, Kewin P, Xu D, Liew FY. IL-33 exacerbates eosinophil-mediated airway inflammation. J Immunol. 2010;185:3472–3480. doi: 10.4049/jimmunol.1000730. [DOI] [PubMed] [Google Scholar]
- 10.Rothenberg ME, Hogan SP. The eosinophil. Annu Rev Immunol. 2006;24:147–174. doi: 10.1146/annurev.immunol.24.021605.090720. [DOI] [PubMed] [Google Scholar]
- 11.Hoshino M, Nakamura Y, Sim J, Shimojo J, Isogai S. Bronchial subepithelial fibrosis and expression of matrix metalloproteinase-9 in asthmatic airway inflammation. J Allergy Clin Immunol. 1998;102:783–788. doi: 10.1016/s0091-6749(98)70018-1. [DOI] [PubMed] [Google Scholar]
- 12.Hoshino M, Takahashi M, Aoike N. Expression of vascular endothelial growth factor, basic fibroblast growth factor, and angiogenin immunoreactivity in asthmatic airways and its relationship to angiogenesis. J Allergy Clin Immunol. 2001;107:295–301. doi: 10.1067/mai.2001.111928. [DOI] [PubMed] [Google Scholar]
- 13.Levi-Schaffer F, Garbuzenko E, Rubin A, Reich R, Pickholz D, Gillery P, Emonard H, Nagler A, Maquart FA. Human eosinophils regulate human lung-and skin-derived fibroblast properties in vitro: a role for transforming growth factor beta (TGF-beta) Proc Natl Acad Sci U S A. 1999;96:9660–9665. doi: 10.1073/pnas.96.17.9660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kita H, Ohnishi T, Okubo Y, Weiler D, Abrams JS, Gleich GJ. Granulocyte/macrophage colony-stimulating factor and interleukin 3 release from human peripheral blood eosinophils and neutrophils. J Exp Med. 1991;174:745–748. doi: 10.1084/jem.174.3.745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lopez AF, Begley CG, Williamson DJ, Warren DJ, Vadas MA, Sanderson CJ. Murine eosinophil differentiation factor. An eosinophil-specific colony-stimulating factor with activity for human cells. J Exp Med. 1986;163:1085–1099. doi: 10.1084/jem.163.5.1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shinkai K, Mohrs M, Locksley RM. Helper T cells regulate type-2 innate immunity in vivo. Nature. 2002;420:825–829. doi: 10.1038/nature01202. [DOI] [PubMed] [Google Scholar]
- 17.Voehringer D, Shinkai K, Locksley RM. Type 2 immunity reflects orchestrated recruitment of cells committed to IL-4 production. Immunity. 2004;20:267–277. doi: 10.1016/s1074-7613(04)00026-3. [DOI] [PubMed] [Google Scholar]
- 18.Wu D, Molofsky AB, Liang HE, Ricardo-Gonzalez RR, Jouihan HA, Bando JK, Chawla A, Locksley RM. Eosinophils sustain adipose alternatively activated macrophages associated with glucose homeostasis. Science. 2011;332:243–247. doi: 10.1126/science.1201475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wills-Karp M, Luyimbazi J, Xu X, Schofield B, Neben TY, Karp CL, Donaldson DD. Interleukin-13: central mediator of allergic asthma. Science. 1998;282:2258–2261. doi: 10.1126/science.282.5397.2258. [DOI] [PubMed] [Google Scholar]
- 20.Li L, Xia Y, Nguyen A, Feng L, Lo D. Th2-induced eotaxin expression and eosinophilia coexist with Th1 responses at the effector stage of lung inflammation. J Immunol. 1998;161:3128–3135. [PubMed] [Google Scholar]
- 21.Chatila TA. Interleukin-4 receptor signaling pathways in asthma pathogenesis. Trends Mol Med. 2004;10:493–499. doi: 10.1016/j.molmed.2004.08.004. [DOI] [PubMed] [Google Scholar]
- 22.Andrews AL, Holloway JW, Holgate ST, Davies DE. IL-4 receptor alpha is an important modulator of IL-4 and IL-13 receptor binding: implications for the development of therapeutic targets. J Immunol. 2006;176:7456–7461. doi: 10.4049/jimmunol.176.12.7456. [DOI] [PubMed] [Google Scholar]
- 23.Kaplan MH, Schindler U, Smiley ST, Grusby MJ. Stat6 is required for mediating responses to IL-4 and for development of Th2 cells. Immunity. 1996;4:313–319. doi: 10.1016/s1074-7613(00)80439-2. [DOI] [PubMed] [Google Scholar]
- 24.Perkins C, Wills-Karp M, Finkelman FD. IL-4 induces IL-13-independent allergic airway inflammation. J Allergy Clin Immunol. 2006;118:410–419. doi: 10.1016/j.jaci.2006.06.004. [DOI] [PubMed] [Google Scholar]
- 25.Munitz A, Brandt EB, Mingler M, Finkelman FD, Rothenberg ME. Distinct roles for IL-13 and IL-4 via IL-13 receptor alpha1 and the type II IL-4 receptor in asthma pathogenesis. Proc Natl Acad Sci U S A. 2008;105:7240–7245. doi: 10.1073/pnas.0802465105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dyer KD, Moser JM, Czapiga M, Siegel SJ, Percopo CM, Rosenberg HF. Functionally competent eosinophils differentiated ex vivo in high purity from normal mouse bone marrow. J Immunol. 2008;181:4004–4009. doi: 10.4049/jimmunol.181.6.4004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, Pimentel H, Salzberg SL, Rinn JL, Pachter L. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc. 2012;7:562–578. doi: 10.1038/nprot.2012.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.de Hoon MJ, Imoto S, Nolan J, Miyano S. Open source clustering software. Bioinformatics. 2004;20:1453–1454. doi: 10.1093/bioinformatics/bth078. [DOI] [PubMed] [Google Scholar]
- 29.Saldanha AJ. Java Treeview--extensible visualization of microarray data. Bioinformatics. 2004;20:3246–3248. doi: 10.1093/bioinformatics/bth349. [DOI] [PubMed] [Google Scholar]
- 30.Matsuba-Kitamura S, Yoshimoto T, Yasuda K, Futatsugi-Yumikura S, Taki Y, Muto T, Ikeda T, Mimura O, Nakanishi K. Contribution of IL-33 to induction and augmentation of experimental allergic conjunctivitis. Int Immunol. 2010;22:479–489. doi: 10.1093/intimm/dxq035. [DOI] [PubMed] [Google Scholar]
- 31.Suzukawa M, Koketsu R, Iikura M, Nakae S, Matsumoto K, Nagase H, Saito H, Matsushima K, Ohta K, Yamamoto K, Yamaguchi M. Interleukin-33 enhances adhesion, CD11b expression and survival in human eosinophils. Lab Invest. 2008;88:1245–1253. doi: 10.1038/labinvest.2008.82. [DOI] [PubMed] [Google Scholar]
- 32.Schleimer RP, Sterbinsky SA, Kaiser J, Bickel CA, Klunk DA, Tomioka K, Newman W, Luscinskas FW, Gimbrone MA, Jr, McIntyre BW, et al. IL-4 induces adherence of human eosinophils and basophils but not neutrophils to endothelium. Association with expression of VCAM-1. J Immunol. 1992;148:1086–1092. [PubMed] [Google Scholar]
- 33.Heller NM, Gwinn WM, Donnelly RP, Constant SL, Keegan AD. IL-4 engagement of the type I IL-4 receptor complex enhances mouse eosinophil migration to eotaxin-1 in vitro. PLoS One. 2012;7:e39673. doi: 10.1371/journal.pone.0039673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rehli M, Sulzbacher S, Pape S, Ravasi T, Wells CA, Heinz S, Sollner L, El Chartouni C, Krause SW, Steingrimsson E, Hume DA, Andreesen R. Transcription factor Tfec contributes to the IL-4-inducible expression of a small group of genes in mouse macrophages including the granulocyte colony-stimulating factor receptor. J Immunol. 2005;174:7111–7122. doi: 10.4049/jimmunol.174.11.7111. [DOI] [PubMed] [Google Scholar]
- 35.Doherty TA, Khorram N, Sugimoto K, Sheppard D, Rosenthal P, Cho JY, Pham A, Miller M, Croft M, Broide DH. Alternaria induces STAT6-dependent acute airway eosinophilia and epithelial FIZZ1 expression that promotes airway fibrosis and epithelial thickness. J Immunol. 2012;188:2622–2629. doi: 10.4049/jimmunol.1101632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wen T, Mingler MK, Blanchard C, Wahl B, Pabst O, Rothenberg ME. The pan-B cell marker CD22 is expressed on gastrointestinal eosinophils and negatively regulates tissue eosinophilia. J Immunol. 2012;188:1075–1082. doi: 10.4049/jimmunol.1102222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Piehler D, Stenzel W, Grahnert A, Held J, Richter L, Kohler G, Richter T, Eschke M, Alber G, Muller U. Eosinophils contribute to IL-4 production and shape the T-helper cytokine profile and inflammatory response in pulmonary cryptococcosis. Am J Pathol. 2011;179:733–744. doi: 10.1016/j.ajpath.2011.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Munitz A, Waddell A, Seidu L, Cole ET, Ahrens R, Hogan SP, Rothenberg ME. Resistin-like molecule alpha enhances myeloid cell activation and promotes colitis. J Allergy Clin Immunol. 2008;122:1200–1207. e1201. doi: 10.1016/j.jaci.2008.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Melo RC, Spencer LA, Perez SA, Ghiran I, Dvorak AM, Weller PF. Human eosinophils secrete preformed, granule-stored interleukin-4 through distinct vesicular compartments. Traffic. 2005;6:1047–1057. doi: 10.1111/j.1600-0854.2005.00344.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Spencer LA, Melo RC, Perez SA, Bafford SP, Dvorak AM, Weller PF. Cytokine receptor-mediated trafficking of preformed IL-4 in eosinophils identifies an innate immune mechanism of cytokine secretion. Proc Natl Acad Sci U S A. 2006;103:3333–3338. doi: 10.1073/pnas.0508946103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Suzukawa M, Iikura M, Koketsu R, Nagase H, Tamura C, Komiya A, Nakae S, Matsushima K, Ohta K, Yamamoto K, Yamaguchi M. An IL-1 cytokine member, IL-33, induces human basophil activation via its ST2 receptor. J Immunol. 2008;181:5981–5989. doi: 10.4049/jimmunol.181.9.5981. [DOI] [PubMed] [Google Scholar]
- 42.Pushparaj PN, Li D, Komai-Koma M, Guabiraba R, Alexander J, McSharry C, Xu D. Interleukin-33 exacerbates acute colitis via Interleukin- 4 in mice. Immunology. 2013 doi: 10.1111/imm.12111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Komai-Koma M, Brombacher F, Pushparaj PN, Arendse B, McSharry C, Alexander J, Chaudhuri R, Thomson NC, McKenzie AN, McInnes I, Liew FY, Xu D. Interleukin-33 amplifies IgE synthesis and triggers mast cell degranulation via interleukin-4 in naive mice. Allergy. 2012;67:1118–1126. doi: 10.1111/j.1398-9995.2012.02859.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Humphreys NE, Xu D, Hepworth MR, Liew FY, Grencis RK. IL-33, a potent inducer of adaptive immunity to intestinal nematodes. J Immunol. 2008;180:2443–2449. doi: 10.4049/jimmunol.180.4.2443. [DOI] [PubMed] [Google Scholar]
- 45.Goh YP, Henderson NC, Heredia JE, Red Eagle A, Odegaard JI, Lehwald N, Nguyen KD, Sheppard D, Mukundan L, Locksley RM, Chawla A. Eosinophils secrete IL-4 to facilitate liver regeneration. Proc Natl Acad Sci U S A. 2013;110:9914–9919. doi: 10.1073/pnas.1304046110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Heredia JE, Mukundan L, Chen FM, Mueller AA, Deo RC, Locksley RM, Rando TA, Chawla A. Type 2 innate signals stimulate fibro/adipogenic progenitors to facilitate muscle regeneration. Cell. 2013;153:376–388. doi: 10.1016/j.cell.2013.02.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Miller AM, Asquith DL, Hueber AJ, Anderson LA, Holmes WM, McKenzie AN, Xu D, Sattar N, McInnes IB, Liew FY. Interleukin-33 induces protective effects in adipose tissue inflammation during obesity in mice. Circ Res. 2010;107:650–658. doi: 10.1161/CIRCRESAHA.110.218867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wang HB, Weller PF. Pivotal advance: eosinophils mediate early alum adjuvant-elicited B cell priming and IgM production. J Leukoc Biol. 2008;83:817–821. doi: 10.1189/jlb.0607392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.McKee AS, Munks MW, MacLeod MK, Fleenor CJ, Van Rooijen N, Kappler JW, Marrack P. Alum induces innate immune responses through macrophage and mast cell sensors, but these sensors are not required for alum to act as an adjuvant for specific immunity. J Immunol. 2009;183:4403–4414. doi: 10.4049/jimmunol.0900164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Voehringer D, Reese TA, Huang X, Shinkai K, Locksley RM. Type 2 immunity is controlled by IL-4/IL-13 expression in hematopoietic non-eosinophil cells of the innate immune system. J Exp Med. 2006;203:1435–1446. doi: 10.1084/jem.20052448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dong L, Wang SJ, Camoretti-Mercado B, Li HJ, Chen M, Bi WX. FIZZ1 plays a crucial role in early stage airway remodeling of OVA-induced asthma. J Asthma. 2008;45:648–653. doi: 10.1080/02770900802126941. [DOI] [PubMed] [Google Scholar]
- 52.Liu T, Dhanasekaran SM, Jin H, Hu B, Tomlins SA, Chinnaiyan AM, Phan SH. FIZZ1 stimulation of myofibroblast differentiation. Am J Pathol. 2004;164:1315–1326. doi: 10.1016/S0002-9440(10)63218-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Munitz A, Seidu L, Cole ET, Ahrens R, Hogan SP, Rothenberg ME. Resistin-like molecule alpha decreases glucose tolerance during intestinal inflammation. J Immunol. 2009;182:2357–2363. doi: 10.4049/jimmunol.0803130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cherry WB, Yoon J, Bartemes KR, Iijima K, Kita H. A novel IL-1 family cytokine, IL-33, potently activates human eosinophils. J Allergy Clin Immunol. 2008;121:1484–1490. doi: 10.1016/j.jaci.2008.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chow JY, Wong CK, Cheung PF, Lam CW. Intracellular signaling mechanisms regulating the activation of human eosinophils by the novel Th2 cytokine IL-33: implications for allergic inflammation. Cell Mol Immunol. 2010;7:26–34. doi: 10.1038/cmi.2009.106. [DOI] [PMC free article] [PubMed] [Google Scholar]