

Abstract
The genus Alphavirus consists of a group of enveloped, single-stranded RNA viruses, many of which are transmitted by arthropods to a wide range of vertebrate host species. Here we report that Sindbis virus (SINV) produced from a representative mammalian cell line consists of at least two unique particle subpopulations, separable on the basis of virion density. In contrast, mosquito-derived SINV consists of a homogeneous population of particles. Our findings indicate that the denser particle subpopulation, SINVHeavy, is more infectious on a per-particle basis than SINVLight. SINV produced in mosquito cell lines (SINVC6/36) exhibited particle-to-PFU ratios similar to those observed for SINVHeavy. In mammalian cells, viral RNA was synthesized and accumulated more rapidly following infection with SINVHeavy or SINVC6/36 than following infection with SINVLight, due partly to enhanced translation of viral genomic RNA early in infection. Analysis of the individual particle subpopulations indicated that SINVHeavy and SINVC6/36 contain host-derived factors whose presence correlates with the enhanced translation, RNA synthesis, and infectivity observed for these particles.
INTRODUCTION
Members of the genus Alphavirus, of the family Togaviridae, are a group of enveloped positive-sense RNA viruses with a wide host range. For the mosquito-borne species, the virus is maintained in the enzootic cycle through transmission between a sylvatic reservoir and the mosquito host (1). The maintenance of this cycle directly affects the genetic fitness of the mosquito-borne alphaviruses. Prolonged disruption of this cycle leads to deleterious effects on viral transmission as the virus becomes adapted to a single host (2–5). Spillover from the enzootic cycle often results in the tangential infection of both humans and equines, which can result in significant outbreaks of disease. The outcome of alphaviral infection is dependent on the host system (6–12). Infection of mosquito cells does not result in the shutoff of host macromolecular synthesis and often culminates in persistent infection for the majority of mosquito cell lines (12–15). Nevertheless, cell death as a result of infection has been reported for several members of the genus in whole mosquitoes (16–21). In contrast, infection of mammalian cells induces the shutoff of host macromolecular synthesis, resulting in a predominantly cytolytic infection. In vertebrates, the immune response to infection generally results in virus clearance. This is initiated by the recognition of viral double-stranded RNA and a rapid type I interferon (IFN-α/β) response (22–25).
Previously, we reported that the infectivity of Sindbis virus (SINV), as measured by the ratio of particles to infectious units, depends on the host cell line from which it is derived (26). SINV derived from mammalian cell lines exhibited a higher particle-to-PFU ratio, on average, than SINV generated from mosquito cell lines. This was due largely to differences in the quantity of total virus particles produced, since the overall numbers of infectious units in infected mosquito and mammalian cell lines were equivalent. These findings indicated that many mammalian cell lines produce a population of virions that are noninfectious, while mosquito cells generally do not.
Several studies have examined the structure and composition of Alphavirus particles derived from both mammalian and mosquito hosts. To date, characterizations of alphaviral particles have not identified any overt differences in morphology between particles derived from the two hosts (27–29). Differences in particle composition between alphaviral particles generated in mammalian and mosquito hosts have been described. Specifically, the glycans linked to the E1 and E2 glycoproteins and the lipid species in the viral envelopes differ due to differences in glycosylation and membrane composition between mammalian and mosquito cells. Nevertheless, the effects of these differences, if any, on viral infectivity are unclear (30–33).
In the present study, we isolated SINV particles from a representative mammalian cell line (BHK-21) that produces SINV with a high particle-to-PFU ratio and from a mosquito cell line (C6/36) that produces SINV with a low particle-to-PFU ratio in order to determine the underlying qualities that modulate particle infectivity. Our findings indicate that the virus derived from BHK-21 cells consists of at least two unique subpopulations, SINVHeavy and SINVLight, whereas the virus produced in C6/36 cells exists as a homogeneous population. The individual subpopulations of BHK-21 cell-derived SINV displayed different particle-to-PFU ratios; the SINVHeavy subpopulation exhibited greater infectivity. SINVC6/36 particles exhibited particle-to-PFU ratios similar to those of the mammalian-cell-derived SINVHeavy particles. The mammalian-cell-derived SINVHeavy and mosquito-derived SINVC6/36 populations were both found to undergo enhanced translation and viral RNA synthesis relative to those of SINVLight immediately following entry. Enhanced translation associated with these particles correlates with the encapsidation of host-derived ribosomal components. Furthermore, infections with SINVHeavy or SINVC6/36 produced significantly less type I IFN than SINVLight infections in a tissue culture model, suggesting an effect on viral pathogenesis. These data potentially explain the differences in alphaviral infectivity between mammalian and mosquito cell lines reported previously (26).
MATERIALS AND METHODS
Cells.
BHK-21, C6/36, 293HEK, and L929 cells were maintained in minimal essential medium (MEM) (Cellgro) supplemented with 10% fetal bovine serum (FBS) (Atlanta Biologicals), 1× antibiotic/antimycotic solution (Cellgro), 1× nonessential amino acid (NEAA) solution (Cellgro), and l-glutamine (Cellgro). Unless otherwise indicated, the mammalian cell lines used in this study were cultured at 37°C in the presence of 5% CO2. Aedes albopictus C6/36 tissue culture cells were cultured at 28°C in the presence of 5% CO2.
Preparation and purification of SINV.
Stocks of SINV TE12, SINV/Fluc (a Toto1101 derivative containing the minimal firefly luciferase coding sequence), and SINVAR86 were prepared by electroporation of infectious RNA as described previously (26). Briefly, a total of 10 μg of full-length RNA was electroporated into BHK-21 cells using a single pulse from a Gene Pulser Xcell system (Bio-Rad) under the following conditions: 1.5 kV, 25 mA, 200 Ω. After a 24-h incubation, the supernatants were clarified via centrifugation at 1,000 × g for 5 min. Zero passage (P0) viral stocks were aliquoted and were stored at −80°C.
Large-scale preparations of SINV were made as follows. A minimum of 2 × 108 tissue culture cells were infected with SINV at a multiplicity of infection (MOI) of 3 PFU/cell. Whole medium was added after aspiration of the initial inoculum, and the monolayers were allowed to incubate under normal conditions for 18 h. After harvesting, the virus-containing supernatant was clarified via centrifugation at 9,000 × g for 10 min. The virus was then concentrated by pelleting through a 27% (mass/vol) sucrose cushion in HNE buffer (10 mM HEPES [pH 7.4]–150 mM NaCl–0.5 mM EDTA) via centrifugation for 1.5 h at 185,000 × g in a 60 Ti rotor. The pelleted virions were resuspended in 500 μl of HNE buffer supplemented with additional EDTA to a final concentration of 40 mM and were incubated for 15 min at 25°C prior to ultracentrifugation over a linear sucrose gradient.
Linear sucrose gradients were prepared over a range of 15 to 45% (mass/vol, in HNE buffer) using a Gradient Master apparatus (BioComp Instruments, Fredericton, NB, Canada). The viral particles were banded over these gradients via centrifugation at 250,000 × g in a SW41 rotor for 2.5 h. The individual populations were removed either via needle aspiration or via gradient fractionation. Particle density was calculated as a function of solution density. Viral titers were determined via standard plaque assay using BHK-21 cells. All purified virions were stored either at 4°C for short-term experimentation or at −80°C for long-term storage in small-volume aliquots.
Particle-to-PFU ratios.
Viral particles were quantitatively examined as described previously by Sokoloski et al. (26). Prior to analysis, the purified viral samples were treated with 100 μg of RNase A and 0.5 U of RNase V1 for 1 h at 25°C and were then extracted with TRIzol reagent. Treatment of the samples with RNase prior to analysis ensures that the RNA species detected via quantitative reverse transcription-PCR (qRT-PCR) are intravirion RNA species and are protected from RNase-mediated degradation. cDNAs specific to the positive-sense viral RNAs were synthesized as described previously from the TRIzol-extracted RNAs by using target-specific primer cocktails (SINV nsP1, 5′-AACATGAACTGGGTGGTG-3′; SINV E1, 5′-ATTGACCTTCGCGGTCGGATTCAT-3′; BHK18S, 5′-AGTCGGCATCGTTTATGGTC-3′; Aedes18S, 5′-ACGACGGTCTACGAATTTCACCTC-3′) (13). The resulting cDNAs were then analyzed by qRT-PCR as described previously using the appropriate primer sets (SINV nsP1F, 5′-AAGGATCTCCGGACCGTA-3′; SINV nsP1R, 5′-AACATGAACTGGGTGGTGTCGAAG-3′; SINV E1F, 5′-TCAGATGCACCACTGGTCTCAACA-3′; SINV E1R, 5′-ATTGACCTTCGCGGTCGGATTCAT-3′; BHK18SF, 5′-CGCGGTTCTATTTTGTTGGT-3′; BHK18SR, 5′-AGTCGGCATCGTTTATGGTC-3′; Aedes18SF, 5′-AGCCCAGCTGCTATTACCTTGAAC-3′; Aedes18SR, 5′-ACGACGGTCTACGAATTTCACCTC-3′) and were compared to standard curves for accurate quantification of nucleic acid content.
cDNAs specific to the SINV minus-strand RNA were synthesized and quantified by using the SINV nsP1 primer sets (26) described above. For these reactions, the primer cocktails used to generate the cDNAs consisted solely of SINV nsP1F and BHK18S to enable detection of the negative-sense viral transcripts.
Electron microscopy (EM).
Purified SINVLight and SINVHeavy were applied to a Formvar- and carbon-coated 400-mesh copper grid and were stained with 1% uranyl acetate. The stained grids were imaged using a JEOL 1010 transmission electron microscope operating at 80 kV. Images were recorded on a Gatan Ultrascan 4000 charge-coupled-device camera.
One-step growth curves.
BHK-21 cells were infected with the purified SINV subpopulations (diluted in 1× PBS–1% FBS) at an MOI of 10 PFU/cell at 4°C for 1 h. Viral adsorption was conducted at 4°C to allow for attachment, but not entry, of the viral particles (34). Prior to the addition of prewarmed medium and incubation, the cell monolayers were washed twice with excess cold 1× PBS to remove any unbound viral particles. Aliquots were removed as indicated within the figures, and titers were determined using BHK-21 cells.
Viral RNA accumulation.
As described above, confluent monolayers of BHK-21 cells were infected with the individual SINV subpopulations at an MOI of 10 PFU/cell at 4°C. Prior to the addition of prewarmed medium and incubation, the cell monolayers were washed twice with excess cold 1× PBS to remove any contaminating viral particles. At the indicated time points, total cellular RNA was harvested using TRIzol and was used to synthesize cDNA as described previously (26).
The absolute quantities of the viral RNAs were determined via qRT-PCR by comparison to a linear standard as described previously (35). Total cellular RNA (0.5 μg) was used to generate cDNA with the primer sets described above. All quantities were normalized to the presence of cellular 18S rRNA, with threshold values and baseline subtraction as determined by the StepOne analysis software (Agilent). The total sum of the positive-sense viral RNAs, consisting of the genomic and subgenomic (SG) RNAs, was determined via amplification with the E1 primer set. The viral genomic RNA was quantified via qRT-PCR using the nsP1 primer set. The two resulting quantities of viral RNA, as determined via comparison to standard curves of known concentrations, were used to calculate the quantity of SG RNA (total − genomic). The minus-strand RNA was examined using the nsP1 primer set from cDNA generated with the appropriate nsP1 primer.
Attachment and entry assay.
Monolayers of BHK-21 cells were infected with equal numbers of viral particles of the individual SINV subpopulations at 4°C for 1 h. Prior to the addition of prewarmed medium, the monolayers were washed twice with excess cold 1× PBS to remove any unattached/extracellular viral particles. Prewarmed medium was added to the cells, and they were allowed to incubate for 15 min at room temperature under gentle agitation. Following incubation, the medium was removed, and the monolayers were washed twice more with excess 1× PBS. The total cellular RNA was harvested and the number of viral genomes determined as described above.
Viral translational rates.
Subconfluent monolayers of BHK-21 or C6/36 cells were infected with SINV/LucLight, SINV/LucHeavy, or SINV/LucC6/36 at an MOI of 5 PFU/cell. Following the addition of SINV, the cells were incubated at 4°C to allow for adsorption, but not entry, of the viral particles. The cells were washed twice with cold 1× PBS to remove unbound virus prior to the addition of prewarmed whole medium to release the block to viral entry. At the indicated times postrelease, the cells were lysed via the addition of 1× Passive Cell Lysis Buffer (Promega) and were frozen until the completion of the sampling period.
To examine the rate of translation of the genome itself, TRIzol-extracted RNAs from equal numbers of viral particles were transfected into BHK-21 cells by using Lipofectamine LTX as per the manufacturer's instructions, with two notable exceptions. First, the cells were exposed to the transfection reagent for only 30 min. Second, after the 30-min incubation, the cells were washed twice with 1× PBS to remove contaminating RNA complexes. At the indicated times posttransfection, the cells were processed as described above.
SINV nucleocapsid cores were purified as described previously (36). Briefly, purified SINV was treated with 2% Triton X-100 in HNE buffer for 30 min at room temperature. Following detergent treatment, the nucleocapsid cores were concentrated through a 30% sucrose cushion. The resulting nucleocapsid cores were resuspended in HNE buffer. Cells were transfected with the purified nucleocapsids and were processed as described above.
All samples were clarified via centrifugation at 16,000 × g for 2 min after thawing on ice. The samples were then mixed with luciferase reagent (Promega), and luminescence was recorded using a Bioanalyzer microplate reader. In all cases, the luciferase readings were normalized to the levels of SINV genomic RNA present in the assayed sample by using the qRT-PCR methods mentioned above coupled with ΔΔCT calculation of transcript levels.
Measurement of type I IFN.
The synthesis of type I IFN was examined quantitatively as described previously (37, 38). Mouse L929 cells were infected with SINVAR86 at an MOI of 5 PFU/cell for 1 h prior to washing with 1× PBS and the addition of whole medium. Twenty-four hours postinfection, the cell supernatants were harvested and were clarified via centrifugation. To inactivate any SINV present, the samples were acidified to pH 2.0, neutralized to pH 7.4, and UV inactivated for 10 min prior to 1:10 dilution with whole medium. The inactivation of the samples was confirmed via standard plaque assay. The samples were then serially diluted 2-fold in 96-well plates seeded with L929 cells. Twenty-four hours later, each well was challenged with IFN-sensitive Ross River virus with a green fluorescent protein expression cassette (RRV-GFP) at an MOI of 10 PFU/cell. The expression of GFP was detected via a Typhoon 9200 phosphorimager and was analyzed via densitometry using ImageJ 24 h postinfection. Seventy-two hours postinfection, the cells were fixed with 10% formaldehyde and were examined for cytopathic effect (CPE); cell death was found to be highly consistent with the levels of GFP expression observed. For accurate quantification, each 96-well plate contained an IFN standard (catalog no. IF011; Chemicon) to determine the amount (expressed in international units) of type I IFN per sample.
RNA levels of IFN-β were determined by qRT-PCR as described above using the following primer sets as described above for the quantification of the viral RNA species: Murine IFNβ F, 5′-AAGAGTTACACTGCCTTTGCCATC-3′; Murine IFNβ R, 5′-CACTGTCTGCTGGTGGAGTTCATC-3′; Murine 18S F, 5′-CCATTCGAACGTCTGCCCTAT-3′; Murine 18S R, 5′-GTCACCCGTGGTCACCATG-3′). Relative fold changes were determined using standard ΔΔCT analysis.
Mass spectrometry.
The proteins from equal numbers of particles of SINV derived from either BHK-21 or C6/36 cells were precipitated using TRIzol according to the manufacturer's instructions. The samples were resuspended in 100 mM ammonium bicarbonate and were reduced via the addition of dithiothreitol (DTT) to a final concentration of 10 mM. After 1 h of incubation under reducing conditions, the samples were alkylated via the addition of iodoacetamide to a final concentration of 20 mM and incubation for 1 h at room temperature. The samples were digested with trypsin (diluted in HCl) at a trypsin/sample mass ratio of 1:30. The samples were concentrated via Zip-Tip and were diluted in liquid chromatography (LC) solvent A (2% acetonitrile–0.1% formic acid). Each sample was analyzed by nanoscale LC-tandem mass spectrometry (nanoLC–MS-MS) via an Eksigent NanoLC-2D system coupled to a Thermo Scientific LTQ-Orbitrap XL mass spectrometer. Peptide fragments were separated using a 60-min gradient from 95% LC solvent A to 60% LC solvent B (99.9% acetonitrile–0.1% formic acid).
Photoactive residue cross-linking immunoprecipitation (PAR-CLIP).
Monolayers of either BHK-21 or C6/36 cells were infected with SINV at an MOI of 5 PFU/cell. After 16 h of incubation, the virus-containing supernatant was removed, and the infected monolayers were washed twice with 1× PBS to remove residual medium and virus. Fresh medium supplemented with 4 μg/ml actinomycin D and 1 mM 4-thiouridine (4SU) was added to the infected cell monolayers. Two hours later, the supernatant was harvested, and the internally 4-SU labeled SINV particles were collected via sucrose cushion as described above and were resuspended in HNE buffer.
Viral genomes were cross-linked via exposure to 365-nm light prior to the addition of 3 volumes of radioimmunoprecipitation assay (RIPA) buffer (50 mM Tris [pH 7.6]–150 mM NaCl–1% [vol/vol] NP-40–0.5% [wt/vol] sodium deoxycholate–0.1% [wt/vol] sodium dodecyl sulfate) and heating to 80°C for 30 s. The disrupted SINV particles were then incubated in the presence of Pansorbin cells supplemented with either nonspecific IgG or an anti-capsid or anti-RPS14 antibody for 30 min at 4°C with gentle agitation. Unbound RNAs and protein were washed 5 times from the immunoprecipitated materials using RIPA buffer (50 mM Tris [pH 7.6]–150 mM NaCl–1.0% NP-40–0.1% SDS–0.5% sodium deoxycholate). Specificity was ensured by further washing the immunoprecipitated materials with RIPA buffer supplemented with 1 M urea.
Retention of the SINV genome was analyzed by RT-PCR using the nsP1 primer set and protocol described above. DNA products were analyzed on a 2% agarose gel and were visualized with ethidium bromide.
Statistical analysis.
Where indicated, the values presented are the mean values of a minimum of three independent biological replicates, with error bars indicating the standard deviations of the means. Where appropriate, statistical analysis of ratios was performed using variable bootstrapping as described previously (26). P values were determined using Student t tests as appropriate.
RESULTS
SINV infection of mammalian cell lines produces at least two infectious-particle subpopulations.
We and others have reported previously that SINV particles derived from mammalian or mosquito tissue culture cells exhibit different infectivities as measured by their particle-to-PFU ratios (1, 26, 39, 40). Quantitative examination of SINV infectivity revealed that SINV particles derived from BHK-21 cells exhibit particle-to-PFU ratios of ∼100:1, whereas the infectivity of SINV derived from C6/36 cells is ∼1:1. To determine if infectious and noninfectious SINV particles could be separated from one another on the basis of particle density, we utilized linear sucrose gradients to examine SINV particles produced in the mammalian BHK-21 and Aedes albopictus C6/36 tissue culture cell lines. Purification of SINV over linear 15-to-60% (wt/vol) sucrose gradients produced a prominent band, with a minor band present at a slightly heavier density. Refinement of the linear sucrose gradients to 15 to 45% (wt/vol) enhanced the resolution of the individual particle species into two distinguishable subpopulations. As indicated in Fig. 1A, BHK-21-cell-derived SINV existed as two particle subpopulations, SINVLight and SINVHeavy, with average particle densities of ∼1.167 g/cm3 and ∼1.184 g/cm3, respectively. Similar results were obtained when SINV was cultured in 293HEK cells, as shown in Fig. 1B, indicating that this phenomenon is not restricted to BHK-21 cells. Moreover, the appearance of two particle subpopulations was not limited to sucrose gradients; similar results were observed using OptiPrep gradients (data not shown). In contrast, SINV derived from the invertebrate C6/36 cell line produced a single particle species (SINVC6/36) with a mean particle density of ∼1.177 g/cm3 (Fig. 1C).
Fig 1.

Mammalian-cell-derived SINV consists of two subpopulations. (A) SINV derived from BHK-21 cells was purified over a 15-to-45% linear sucrose gradient. The gradient was fractionated (left to right corresponds to top to bottom) and was assayed for the presence of infectious units via a plaque assay in BHK-21 cells. Filled circles represent percentages of total infectious viral particles per fraction (values along the left-hand y axis); the dashed line corresponds to densities, in g/cm3 (values along the right-hand y axis). The data are representative of multiple independent biological replicates and purifications. (B) SINV derived from 293HEK cells shows a heavy and a light population. Purification and analysis were performed as for panel A. (C) SINV produced in the Aedes albopictus C6/36 cell line, showing a single peak of infectivity. Purification and analysis were performed as for panels A and B. (D) The entire SINVLight, SINVHeavy (cultured at both 37°C and 30°C, as indicated), and SINVC6/36 subpopulations were analyzed for titers by a standard plaque assay and for particle numbers by qRT-PCR quantification of genome numbers. The values reported are the mean particle-to-PFU ratios for three independent replicates. P values were calculated as described in Materials and Methods. (E) Samples were taken from the margins of each peak to minimize any contamination between the SINVLight and SINVHeavy subpopulations and were analyzed as described above for panel D. The values reported are the particle-to-PFU ratios for a representative purification.
The differences in particle density described above were not due to a heritable genetic trait, as evidenced by the fact that subsequent infection of BHK-21 cells by either mammalian SINV subpopulation or by SINVC6/36 resulted in the production of both SINVLight and SINVHeavy particles as progeny virions. Likewise, infection of C6/36 cells by any of the SINV subpopulations resulted in a single homogeneous population of progeny virions (data not shown).
Further characterization of SINVLight and SINVHeavy indicated that each population contained infectious particles but that their infectivities differed, as determined by the ratio of total particles to infectious units. As shown in Fig. 1D, SINVLight and SINVHeavy exhibited particle-to-PFU ratios of ∼60:1 and ∼20:1, respectively. When only the outside margins of the individual subpopulations were taken from the sucrose gradients and analyzed (thus minimizing cross-contamination of the two subpopulations), SINVLight and SINVHeavy had particle-to-PFU ratios of ∼80:1 and ∼5:1, respectively (Fig. 1E). The observed differences in infectivity between SINVLight and SINVHeavy were not due to thermal inactivation of viral particles, since the particle-to-PFU ratios of SINVLight and SINVHeavy cultured at 37°C were comparable to those for culture at 30°C. SINVC6/36 exhibited a particle-to-PFU ratio of 3:1, approximating that of SINVHeavy.
SINVLight and SINVHeavy are morphologically indistinguishable.
Because density is a function of mass per unit volume, differential migration of a SINV particle could be due to differences in either particle mass or morphology. To determine if SINVLight and SINVHeavy were morphologically different, we examined the purified particle subpopulations by negative staining and transmission electron microscopy. As shown in Fig. 2, both SINVLight and SINVHeavy populations consisted of homogeneous spherical particles ∼70 nm in diameter. The lack of morphological difference between SINVLight and SINVHeavy suggests that the difference in particle density is due to a change in mass.
Fig 2.

SINVLight and SINVHeavy are morphologically similar. Samples of purified SINVLight (A) and SINVHeavy (B) were negatively stained and were examined using transmission electron microscopy. (Left) Representative field of view showing particles from one preparation (magnification, ×25,000). (Right) Particles from three independent preparations (magnification, ×40,000).
SINVHeavy and SINVC6/36 produce infectious virus earlier in infection than SINVLight.
To determine if SINVHeavy and SINVC6/36 particles had a biological or functional advantage over SINVLight particles, one-step growth curves were performed for the individual SINV subpopulations to examine viral growth kinetics. Subconfluent monolayers of BHK-21 cells were infected with SINVLight, SINVHeavy, or SINVC6/36 at an MOI of 10 PFU/cell at 4°C. Infections conducted at this temperature have been shown to allow for receptor binding but not for entry, effectively synchronizing viral infection (34). Following extensive washing with PBS, entry was initiated by the addition of warm medium. At regular times postinfection, aliquots were removed and were assayed for viral titers.
As shown in Fig. 3A, SINVHeavy and SINVC6/36 infections demonstrated modest increases in titer at early times postinfection. On average, SINVHeavy and SINVC6/36 exhibited a log increase in viral titer 1 to 1.5 h earlier than did SINVLight (Fig. 3B). Increases in observed titers due to particle desorption would be expected early in infection but were not observed at the 0.5-h time point, indicating that desorption of bound SINV particles from the cell surface likely contributes little to the increases in titer. All SINV particle subpopulations attained similar titers at 24 h postinfection (data not shown), indicating that the observed enhancement in viral titers was confined to early times postinfection and did not persist throughout the infection.
Fig 3.

Viral growth kinetics and RNA synthesis and accumulation are enhanced during infection with SINVHeavy or SINVC6/36. (A) Monolayers of BHK-21 cells were infected with the indicated SINV subpopulations at an MOI of 10 PFU/cell as described in Materials and Methods. At the indicated times postinfection, cell supernatants were collected, and titers were determined. (B) The average fold change in titer observed for each of the individual SINV subpopulations during infection of BHK-21 cells (from panel A) is plotted against time. Data are mean values for at least two independent biological replicates. Error bars represent standard deviations. (C) Monolayers of BHK-21 cells were infected with the indicated SINV subpopulations at an MOI of 10 PFU/cell as described in Materials and Methods. At the indicated time points postinfection, total cellular RNA was extracted and was used to determine the amount of viral RNA by qRT-PCR. The absolute quantities of the viral RNA species were calculated using standard curves. The absence of a value means that the signal was either below the threshold of detection or below the background of the sample. Data shown are representative of three independent biological replicates. (D) BHK-21 cells were infected with equal numbers of SINVLight, SINVHeavy, or SINVC6/36 particles (in contrast to equal numbers of infectious particles, as for panels A to C). After extensive washing, the viral RNA content was quantified as for panel A. Data are mean values for two independent biological replicates. Error bars represent standard deviations.
Viral RNA synthesis and accumulation occur earlier during SINVHeavy and SINVC6/36 infections than during SINVLight infection.
During infection, 49S genomic RNA functions initially as an mRNA to encode the viral replicase and later acts as a template for the synthesis of minus-strand viral RNA (41). The minus-strand RNA serves as a template for the synthesis of nascent 49S genomic RNA and the transcription of 26S SG RNA, which encodes the viral structural proteins. Synthesis and subsequent translation of the viral RNAs are required for particle formation. Since infectious progeny were produced earlier during SINVHeavy and SINVC6/36 infections than during SINVLight infections, we hypothesized that viral transcription and the accumulation of all of the viral RNAs occurred more rapidly during SINVHeavy and SINVC6/36 infections.
To examine the synthesis and accumulation of the viral RNAs, monolayers of BHK-21 cells were infected with SINVLight, SINVHeavy, or SINVC6/36 at an MOI of 10 PFU/cell at 4°C. After extensive washing, prewarmed medium was added to the cells to release the block to viral entry. Following cell lysis and RNA extraction, absolute quantities of the viral RNAs were determined by qRT-PCR at the times postinfection indicated in Fig. 3C.
SINV RNAs accumulated earlier in infections initiated with SINVHeavy or SINVC6/36 than in infections with SINVLight (Fig. 3C). Minus-strand RNA was synthesized and accumulated, on average, 2 h earlier during SINVHeavy or SINVC6/36 infection than during SINVLight infection. Similarly, the 26S SG RNA, which encodes the structural proteins of the SINV virion necessary for particle assembly and release, accumulated earlier during SINVHeavy or SINVC6/36 infections than during SINVLight infections.
The infections performed to obtain the data in Fig. 3C took into account the particle-to-PFU ratio for each virus and used equivalent infectious units of each virus. More genomes are detected early in SINVLight infection than in SINVHeavy or SINVC6/36 infection, suggesting that particles incapable of initiating productive infection are in fact entering the cell. To test this hypothesis, monolayers of BHK-21 cells were infected as described above with equal numbers of SINVLight, SINVHeavy, or SINVC6/36 particles as opposed to infectious units. Prior to the addition of prewarmed medium, the cell monolayers were washed extensively to remove unbound SINV particles. Shortly after the addition of prewarmed medium, and before the extraction of total cellular RNA, the cell monolayers were washed again. Quantitative analysis of the number of SINV genomic RNAs indicated that all of the SINV particle subpopulations were equivalently able to bind to and presumably enter the host cell (Fig. 3D) but differed in their abilities to replicate.
SINVHeavy and SINVC6/36 exhibit vertebrate-cell-specific enhancement of genomic RNA translation relative to that of SINVLight.
The data presented above suggest that SINVHeavy and SINVC6/36 have a biological advantage over SINVLight with regard to viral growth kinetics early in infection as a result of enhanced RNA accumulation. Furthermore, these data indicate that there is no general deficit in particle attachment and entry for any of the SINV subpopulations. This led us to question whether the step between entry and viral RNA synthesis, translation of the incoming genomic RNA, was responsible for the enhanced kinetics of viral RNA accumulation observed for SINVHeavy and SINVC6/36.
Previous studies have utilized a SINV construct carrying the firefly luciferase open reading frame (ORF) sequence in frame with the nsP3 gene (for brevity, this construct is referred to as SINV/Luc) to examine the rate of viral genome translation during infection (25, 42). Monolayers of BHK-21 and C6/36 cells were infected with SINV/LucHeavy, SINV/LucLight, or SINV/LucC6/36 at an MOI of 3 PFU/cell at 4°C to synchronize the onset of viral infection. At regular intervals after the addition of prewarmed medium, samples were harvested and were assayed for luciferase activity and RNA content.
As shown in Fig. 4A, the rate of translation, as indicated by luciferase activity per encoding RNA, was on average 9-fold greater in SINVHeavy or SINVC6/36 infections than in SINVLight infections of BHK-21 cells. Curiously, this effect was specific to BHK-21 cells; the rates of translation observed for the individual SINV subpopulations in C6/36 cells were essentially identical, although SINV was less translationally active than in BHK-21 cells over a similar period (Fig. 4B).
Fig 4.

Rates of translation of SINVHeavy and SINVC6/36 are enhanced over those of SINVLight in mammalian cells. (A and B) BHK-21 cells (A) and C6/36 cells (B) were infected with the indicated SINV/Luc subpopulations at an MOI of 3 PFU/cell as described in Materials and Methods. At the indicated times postinfection, samples were taken and were analyzed for luciferase activity, which was standardized to the quantity of viral genomic RNA present. The values shown along the y axis represent the relative light units detected. The P value given in panel A was determined by Student's t test. (C) Genomic RNAs were extracted from equal numbers of SINVLight and SINVHeavy particles and were transfected into BHK-21 cells. At regular times posttransfection, samples were taken, analyzed for luciferase activity, and reported as for panels A and B. (D) Nucleocapsid cores from SINVLight and SINVHeavy were transfected into BHK-21 cells. As described for panel C, samples were taken and were analyzed for the level of luciferase activity per genomic RNA. For all panels, data are mean values for at least three independent biological replicates, and error bars represent standard deviations.
This result indicates that the enhanced translation of SINV genomic RNA during SINVHeavy and SINVC6/36 infections may be specific to the mammalian host.
The minimal viral component capable of initiating an alphaviral infection is the viral genomic RNA. Hence, we next sought to determine if the genomic RNAs isolated from SINVHeavy and SINVLight exhibited the same translational profiles observed during infection. Genomic RNA extracted from SINVHeavy and SINVLight particles was transfected into BHK-21 cells. As demonstrated in Fig. 4C, the rates of translation for SINVHeavy and SINVLight genomic RNAs were similar. It should be noted that in contrast to the viral infections described above, the introduction of the viral RNA into the cell by transfection could not be synchronized. For this reason, the zero-hour time point could not be analyzed, since the abundance of the viral RNA to which the luciferase activity is normalized would be zero. Hence, a general increase in luciferase expression per viral RNA with time, similar to that observed with genuine viral infection (Fig. 4A), could not be observed for technical reasons.
Since the translation of the viral genome was enhanced for SINVHeavy over that for SINVLight during a genuine infection, but not during transfection of the genome itself, we hypothesized that a component of the nucleocapsid core was responsible for the enhanced translation, replication, and growth kinetics associated with SINVHeavy. SINV nucleocapsid cores from SINVLight and SINVHeavy were prepared by detergent treatment and were recaptured using sucrose step gradients as described previously (36, 43). The number of nucleocapsid cores was verified by qRT-PCR as described above for virus particles. Equal numbers of nucleocapsid cores were transfected into BHK-21 cells, and at regular intervals, samples were harvested and were assayed for luciferase activity and RNA content. Despite the bypass of entry by receptor-mediated endocytosis, the translational activities of the nucleocapsids closely resembled those of their intact virion counterparts during viral infection. As indicated in Fig. 4D, transfection of SINVHeavy nucleocapsids resulted in ∼6-fold-greater translational activity than transfection of SINVLight nucleocapsids. Once again, zero time point values could not be analyzed for the reasons stated above.
Taken together, these data indicate that the translational activities of SINVHeavy and SINVC6/36 are enhanced during the infection of mammalian cells and, furthermore, are dependent on the nucleocapsid core.
Infections with SINVHeavy or SINVC6/36 induce less type I IFN than infections with SINVLight in cultured cells.
SINVHeavy or SINVC6/36 infections exhibit enhanced translation of genomic RNA and increased viral RNA synthesis early in infection. The expression of the SINV nonstructural proteins in mammalian cells is known to inhibit host transcription (7, 44). We next hypothesized that the enhanced translation of the SINVHeavy and SINVC6/36 genomic RNAs in the mammalian host could limit the initiation of an effective host response (22–25, 45, 46). To test this hypothesis, we utilized a tissue culture model to examine the induction of type I IFN production during SINVLight, SINVHeavy, and SINVC6/36 infections.
IFN-competent L929 cells were infected with SINVLight, SINVHeavy, or SINVC6/36 at an MOI of 5 PFU/cell for 1 h. At 24 h postinfection, the cell supernatants were harvested and were clarified by centrifugation; after inactivation, they were assayed for the levels of IFN-α/β. As demonstrated in Fig. 5A, ∼3-fold more type I IFN was produced during SINVLight infections than during SINVHeavy infections. Examination of the abundances of IFN-β mRNA indicated that SINVLight on average resulted in ∼6.5-fold more IFN-β mRNA transcripts than SINVHeavy (Fig. 5B).
Fig 5.
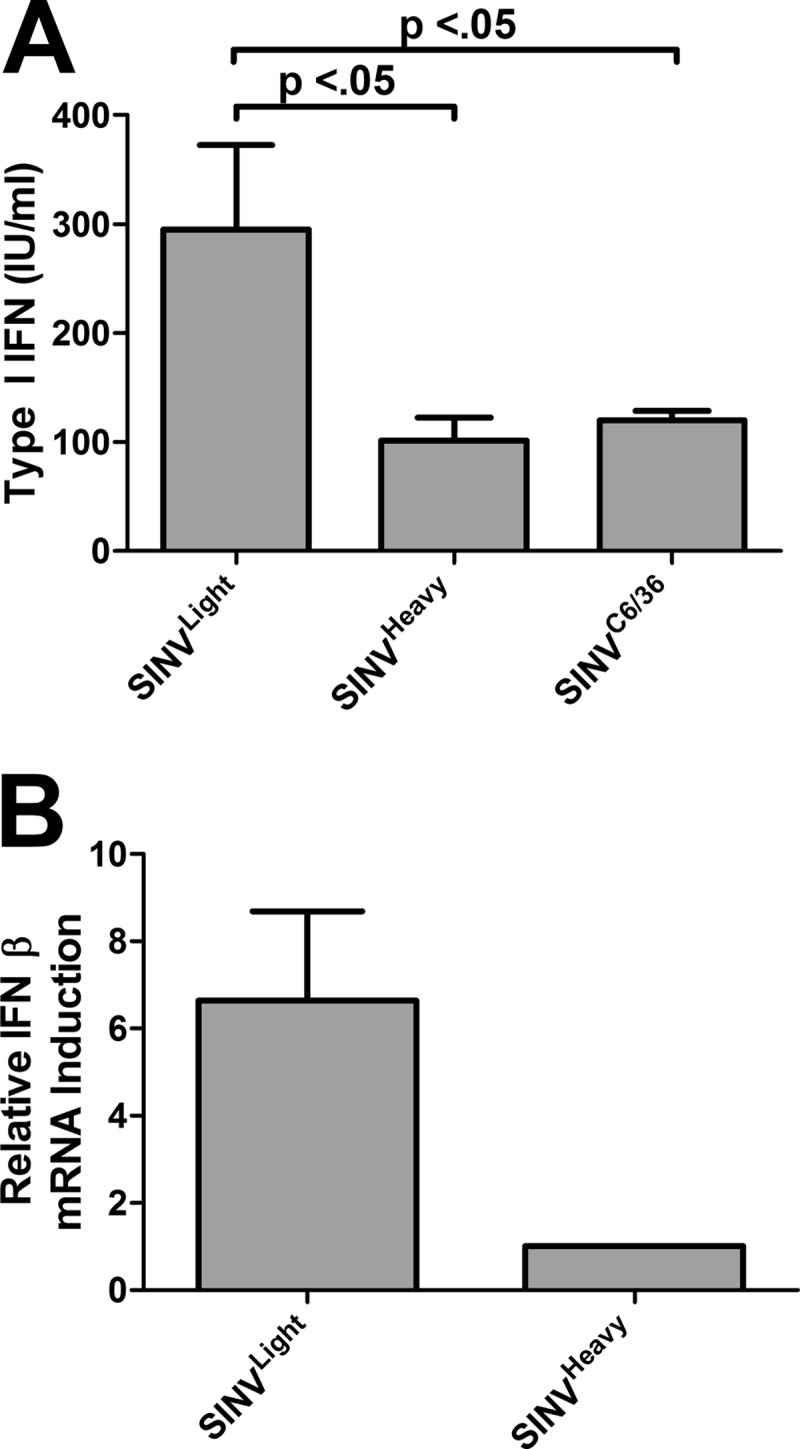
SINVHeavy and SINVC6/36 induce less type I IFN than SINVLight in tissue culture cells. (A) L929 cells were infected with SINVLight, SINVHeavy, or SINVC6/36 at an MOI of 5 PFU/cell. Twenty-four hours later, the supernatant was harvested, inactivated, and assayed for type I IFN as described in Materials and Methods. Data are mean values for three independent biological replicates. Error bars represent standard deviations. (B) Quantitative analysis of IFN-β mRNA levels, normalized to the host 18S rRNA levels, of the SINVLight and SINVHeavy samples for which results are shown in panel A. The mRNA abundances reported are relative to that of SINVHeavy, since the extent of type I IFN induction in mock-treated samples, both at the level of protein production and at the level of mRNA abundance, was below the limits of detection.
Previously, it was demonstrated that mosquito-derived viral particles elicit a lower type I IFN response than mammalian-cell-derived virions (38). Interestingly, SINVHeavy and SINVC6/36 induce similar levels of type I IFN, suggesting that SINVLight (Fig. 5A) may contribute to the activation of type I IFN during infections using mammalian-cell-derived particles.
SINVHeavy and SINVC6/36 contain host-derived ribosomal factors.
Since we had observed a difference in SINV virion density without an alteration of particle morphology, we next sought to determine what accounted for the apparent difference in particle mass. The proteins from equal numbers of SINVLight, SINVHeavy, and SINVC6/36 particles were precipitated, digested with trypsin, and analyzed by mass spectrometry. Mass spectrometric analysis of SINVHeavy and SINVC6/36 indicated the presence of the rRNA binding proteins RPS14 (7.3% sequence coverage), RPS18 (16.4% sequence coverage), and RBM3 (35.3% sequence coverage). The presence of these factors indicated that some portions of the host ribosomal complexes may be associated with SINVHeavy and SINVC6/36.
Using a modified photoactive cross-linking immunoprecipitation (PAR-CLIP) strategy, we sought to determine if the ribosomal components were encapsidated and in contact with the viral genome in purified particles. The PAR-CLIP method has traditionally been used to identify the site(s) of interaction for RNA binding proteins (47–49). A method similar to that described here has been used to identify the positioning of a translating ribosome via the site of interaction between RPS14 and the cognate mRNA (50).
Purified SINV particles with the genomic RNAs internally labeled with 4-thiouridine (4SU) were harvested from BHK-21 cells 18 h postinfection. After purification, the SINV particles were exposed to 365-nm UV light in order to cross-link proteins to genomic RNA. The particles were disrupted, and proteins were immunoprecipitated using a nonspecific or anti-capsid antibody. The presence of SINV genomic RNA with immunoprecipitated proteins was detected by RT-PCR as indicated in Fig. 6A. The lack of a PCR product in the absence of either UV cross-linking or 4SU incorporation indicates that the modified PAR-CLIP method described here is specific. As shown in Fig. 6B, immunoprecipitation of purified SINV particles with an antibody to either SINV capsid or RPS14 revealed an interaction between these proteins and the viral genome in highly purified SINV particles, indicating that host-derived ribosomal components are in direct with the viral genome in the nucleocapsid. Despite a high degree of similarity between the RPS14 proteins of vertebrates and invertebrates, the anti-RPS14 antibody was not cross-reactive with mosquito RPS14 (data not shown).
Fig 6.

RPS14 is in direct contact with the viral genome within SINV nucleocapsids. (A) SINV-infected BHK-21 cells were incubated for 16 h postinfection. Then they were washed, and actinomycin D was added. The photoactive nucleoside analogue 4-thiouridine (4SU) was added as indicated. Virus production was allowed to proceed for 2 h, after which the internally 4SU labeled virus was harvested and was cross-linked by exposure to UV irradiation as indicated. Heat-disrupted particles were immunoprecipitated either with a nonspecific antibody (Ab) or with an antibody against SINV capsid. The retention of SINV genomic RNA only in samples that had been cross-linked and UV irradiated indicated that the modified PAR-CLIP assay was specific. (B) Internally 4SU labeled SINV derived from BHK-21 cells was cross-linked and was immunoprecipitated with either a nonspecific antibody, an anti-SINV capsid antibody, or an anti-RPS14 antibody. The results indicated that SINV genomic RNA was in contact with both SINV capsid and RPS14 in purified SINV particles. In both panels, the lane marked “Load” represents the analysis of the initial materials prior to immunoprecipitation as a reference.
Inadvertent copurification of the host-derived ribosomal components is highly unlikely for numerous reasons. First, during the initial stage of the purification strategy, when SINV particles are purified through a 27% sucrose cushion, the force and time of centrifugation would be insufficient to pellet intact 80S ribosomes, much less their individual subcomponents. While polysomes could copurify with SINV particles during the initial stage of purification, further treatment of the samples prior to the second stage of purification greatly reduces the likelihood of contamination. The requirement of divalent magnesium cations for the assembly, structure, and function of the 80S ribosome is well known. Prior to the second stage of purification, whereby SINV particles are resolved on linear sucrose gradients, the SINV samples are treated with excess EDTA to disrupt any intact polysomes and ribosomes that may be present. This ensures that any ribosomal material in the sample is present as an individual 40S or 60S subunit incapable of copurifying with SINV deep in the linear gradient. Moreover, prior to any analysis of RNA content, as described below, the samples are treated with excess RNase A and RNase V1 to degrade any extravirion RNAs that may be present.
SINVHeavy and SINVC6/36, but not SINVLight, contain host-derived 18S rRNAs.
While the data presented above confirm that there are indeed host-derived factors interior to SINV nucleocapsid cores, the presence of ribosomal proteins would not account for the entire difference in particle density between SINVLight and SINVHeavy. Since several RNA binding proteins that are known to associate with 18S rRNA were detected by mass spectrometry, we next sought to determine if host-derived rRNAs were also present in highly purified SINV particles.
Prior to analysis, the highly purified SINV virions were treated with RNase A and RNase V1 to effectively degrade any extraparticle RNA. Assessment of the nucleic acid contents of SINVLight, SINVHeavy, and SINVC6/36 by qRT-PCR confirmed that host-derived 18S rRNAs were present in both SINVHeavy and SINVC6/36. Mock gradients loaded with native cellular RNAs indicated that no contaminating RNA penetrated the gradient under the conditions used in this assay (data not shown). As shown in Fig. 7, the number of 18S rRNA molecules present per infectious unit was ∼1.2 to 1 for both SINVHeavy and SINVC6/36. In contrast, SINVLight exhibited ∼0.2 18S rRNA molecule per infectious unit.
Fig 7.
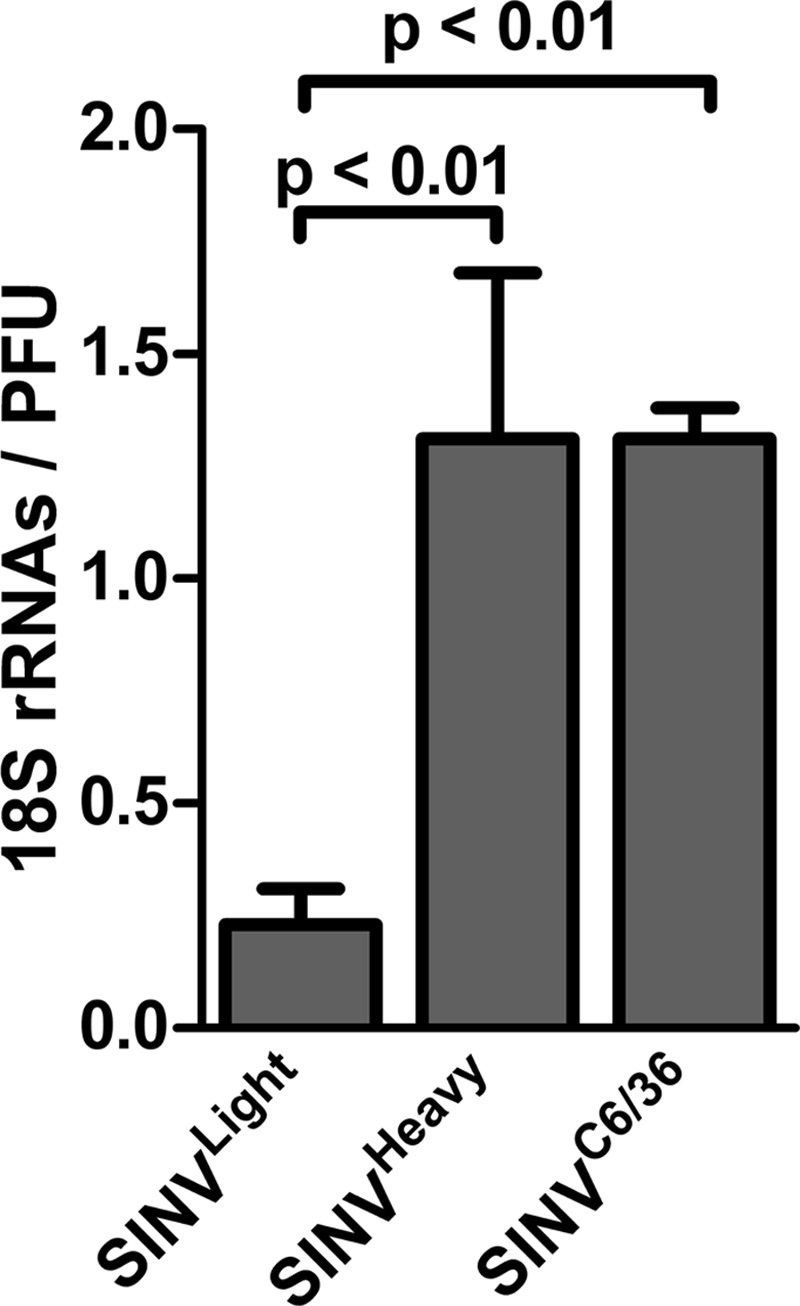
SINVHeavy and SINVC6/36 contain host-derived rRNAs. The 18S rRNA content per infectious particle was quantitatively analyzed for nuclease-treated purified SINVLight, SINVHeavy, and SINVC6/36 particles (see Materials and Methods for details). Titers were determined using BHK-21 cells. rRNA contents were determined by qRT-PCR following treatment of intact particles with RNase A and RNase V1. Data are mean values for a minimum of three independent biological replicates. Error bars represent standard deviations.
DISCUSSION
The data presented above indicate that at least two biologically unique SINV subpopulations are produced during the infection of BHK-21 tissue culture cells, but only one population is produced from C6/36 cells. These BHK-21 cell-derived subpopulations, SINVLight and SINVHeavy, were found to differ on the basis of infectivity, as indicated by their relative particle-to-PFU ratios. Comparative examination of the growth kinetics of the individual SINV subpopulations revealed that progeny SINV virions are produced early during SINVHeavy and SINVC6/36 infections. The enhanced viral growth kinetics observed for SINVHeavy and SINVC6/36 correlated with increased translation of the viral genome early during the infection of BHK-21 cells. Additionally, SINVHeavy and SINVC6/36 induced less type I IFN than SINVLight, indicating that the host cell response may be modulated during SINVHeavy and SINVC6/36 infections. Host-derived factors were detected in both SINVHeavy and SINVC6/36 particles, suggesting that the encapsidation of host factors is responsible for the difference in particle density and may be responsible in part for the enhanced translation early during infection.
Alphaviruses produced from mammalian cells consist of at least two individual subpopulations.
The data presented above indicate that SINV infections of vertebrate cell lines result in the formation of a heterogeneous population of viral particles, whereas infection of insect cells results in a homogeneous population. Prior to this report, mammalian-cell-derived alphaviruses were described as consisting of a single particle species. Prior characterization of purified SINV derived from chicken embryo fibroblasts indicated that two particle species could be distinguished from one another on the basis of particle density (51). Nevertheless, these studies stopped short of determining if the individual particle subpopulations exhibited different infectivities or if they were biologically distinct. Perhaps the simplest explanation of why mammalian-cell-derived alphaviruses are commonly reported as a single particle species has to do with the particle-to-PFU ratios of the two subpopulations. The difference in the particle-to-PFU ratios between the light and heavy subpopulations indicates that the predominant species, by optical density, would be the lighter species, as we observed. As a consequence, when a low concentration of particles is purified, they would appear as a single band. Additionally, technical factors, such as gradient range and length, could also affect the visibility and resolution of the two particle subpopulations.
In contrast to mammalian-cell-derived SINV particles, SINV particles produced in the Aedes albopictus C6/36 cell line were purified as a single population under similar purification conditions. Interestingly, mosquito-derived SINV exhibits a particle-to-PFU ratio similar to that of SINVHeavy, suggesting that perhaps these populations are functionally synonymous.
Examinations of mammalian-cell-derived alphavirus particles by cryo-EM, even those at a resolution higher than 5 Å, were likely performed on mixed virus populations (33). The data presented above indicate that for every SINVHeavy particle, there are at least 5 SINVLight particles within the mixed population. Therefore, differences in electron density within the nucleocapsids of mammalian-cell-derived SINVs are most likely lost during the averaging procedure. Additionally, if the position of the host-derived components, either on the genomic RNA or within the nucleocapsid, was not consistent, then the density of the host-derived components would not be detected by use of icosahedral averaging. Structural studies of the individual SINVLight and SINVHeavy subpopulations are ongoing.
SINVHeavy and SINVC6/36 initiate infection more rapidly than SINVLight.
On average, the burst time for the alphaviruses has been reported to be after 3 h postinfection, consistent with the observed lag prior to the initiation of exponential growth during SINVLight infection. Interestingly, SINVHeavy and SINVC6/36 produce progeny viruses early during viral infection and attain exponential growth more rapidly than SINVLight. Similarly, early synthesis and accumulation of the viral RNAs was observed, confirming that the RNA components necessary for the production of SINV virions were present. The enhanced synthesis and accumulation of viral RNAs observed during SINVHeavy and SINVC6/36 infections correlated with the superior translation of the viral genome during infection of BHK-21 cells. Moreover, the data suggest that the production of progeny virions is not necessarily restricted directly by the overall magnitudes of the viral RNA species but depends on the time by which they were synthesized. For instance, SINVHeavy and SINVC6/36 exhibit exponential growth between ∼3 and 4 h postinfection, whereas SINVLight exhibits delayed growth kinetics, demonstrating exponential growth ∼5 h postinfection. The overall magnitudes of the SINV RNA species for all of the individual SINV subpopulations are similar at 4 h postinfection; however, the accumulation of the RNA species began, on average, 2 h earlier during SINVHeavy and SINVC6/36 infections than during SINVLight infection. Therefore, the onset of exponential production of SINV virions appears to be related to the timing of the synthesis and accumulation of the SG and minus-strand RNAs.
Curiously, these effects were absent in C6/36 cells, indicating possible host specificity. The directionality of this phenomenon—the fact that mosquito particles exhibit enhanced mammalian infection, but not vice versa—indicates possible enhancement of transmission of the alphaviruses to a mammalian host.
The early replication and release of progeny virions could benefit the establishment of viral infection in the vertebrate host. Frolov et al. have demonstrated that events early in infection can significantly influence the IFN response and the outcome of alphavirus infection (45). As shown above, SINVHeavy infections induce less type I IFN, both at the level of protein production and at the level of transcription, than SINVLight, which correlates with the enhanced translation exhibited by SINVHeavy. It is not clear if the reduced induction of type I IFN is due to either inhibition of the host or evasion of the recognition required for the induction of the host response. Nonetheless, a potential consequence of enhanced translation would be modulation of the host response. Rapid expression of the viral nonstructural proteins would enable SINV to shut off host transcription and translation quickly following infection, presumably reducing the ability of the host to mount an effective response (22, 46, 52). This, in turn, would have far-reaching consequences for ongoing viral replication and gene expression, well beyond the initial round of infection. Additionally, the release of infectious particles earlier during infection likely serves to increase the dissemination of the viral infection locally, potentially limiting the efficacy of the vertebrate innate immune response following transmission.
The mechanism by which SINVHeavy and SINVC6/36 promote the translation of their viral genomes is currently unknown. The data presented above indicate that the enhanced translation of SINVHeavy and SINVC6/36 genomic RNAs was contextually dependent on the nucleocapsid core. The requirement for the nucleocapsid core strongly suggested that a component within the viral particles themselves is responsible for enhancing the translation of the viral genome. SINVHeavy and SINVC6/36 particles were found to encapsidate host-derived factors, as indicated by the encapsidation of 18S rRNA, correlating with the enhanced translation described above. Whether the 18S rRNA is the sole host-derived factor present is unclear, since the particle-to-PFU and 18S rRNA-to-PFU ratios indicate that there may be SINVHeavy particles lacking the 18S rRNA subunit. This discrepancy could be due simply to cross-contamination of the individual subpopulations during purification.
We propose a model in which the inclusion of these host-derived factors improves the assembly of the functional messenger ribonucleoprotein complex necessary for efficient translation following release of the genomic RNA. The enhanced translation of the genomic RNA serves both to mute the host response and to accelerate the timing of viral RNA synthesis, leading to a more efficient SINV infection.
Host cell shutoff may play a role in the formation of multiple alphaviral subpopulations.
Precisely why mammalian-cell-derived alphaviruses exist as heterogeneous populations whereas mosquito-derived alphaviruses exist as a homogeneous population is not clear. Examination of SINV particles produced in mammalian and mosquito cells indicated that host-derived factors copurified with SINVHeavy and SINVC6/36 particles. The formation of a single host-factor-containing SINV particle species upon infection of mosquito cells indicates that incorporation of host factors is favored during particle assembly in the mosquito host. The outcome of alphaviral infection is known to depend on the host. SINV infection of mammalian cell lines results in the inhibition of cellular transcription and translation, a feature largely absent from infection of invertebrate cells (8, 44). Perhaps the inhibition of host translation results in the formation of viral particles lacking the host-derived ribosomal components. Alternatively, the affinity with which the host factors interact with the SINV genome may differ between the vertebrate and invertebrate hosts, resulting in the assembly of different particle species. Studies focusing on identifying and characterizing these mechanisms are ongoing.
Prior to this report, the incorporation of host factors into alphaviral particles has been reported only for mutant strains that exhibit budding defects (53). Furthermore, the inclusion of any component of the host ribosomal machinery has, to date, been described for only one other virus family. Members of the Arenaviridae have been shown to incorporate 80S ribosomes into mature infectious virions (54, 55). The packaged ribosomes observed in Pichinde virus were found to be translationally competent but unnecessary for viral infection (54). Similarly, as reported here, inclusion of the host factors was not absolutely necessary for SINV infection, since SINVLight particles are clearly infectious, as is purified genomic RNA; however, inclusion of the host factors correlated strongly with the enhanced translation and viral RNA synthesis observed for SINVHeavy and SINVC6/36.
ACKNOWLEDGMENTS
We thank the members of the Danthi, Hardy, and Mukhopadhyay labs for reading and editing of the manuscript. We also thank Mark Heise and Kristin Long (UNC—Chapel Hill) for assistance with the interferon assays.
The research reported in this work was supported by grant R01 AI090077 from the NIAID/NIH to R.W.H., by MCB-1157716 from the NSF to S.M., and by grant F32AI104217 from the NIAID/NIH to K.J.S. A.J.S. was supported by Indiana University Genetics, Cellular and Molecular Sciences training grant T32-GM007757.
Footnotes
Published ahead of print 4 September 2013
REFERENCES
- 1.Stollar V, Stollar BD, Koo R, Harrap KA, Schlesinger RW. 1976. Sialic acid contents of Sindbis virus from vertebrate and mosquito cells. Equivalence of biological and immunological viral properties. Virology 69:104–115 [DOI] [PubMed] [Google Scholar]
- 2.Coffey LL, Vasilakis N, Brault AC, Powers AM, Tripet F, Weaver SC. 2008. Arbovirus evolution in vivo is constrained by host alternation. Proc. Natl. Acad. Sci. U. S. A. 105:6970–6975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Coffey LL, Vignuzzi M. 2011. Host alternation of Chikungunya virus increases fitness while restricting population diversity and adaptability to novel selective pressures. J. Virol. 85:1025–1035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Greene IP, Wang E, Deardorff ER, Milleron R, Domingo E, Weaver SC. 2005. Effect of alternating passage on adaptation of Sindbis virus to vertebrate and invertebrate cells. J. Virol. 79:14253–14260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Weaver SC, Brault AC, Kang W, Holland JJ. 1999. Genetic and fitness changes accompanying adaptation of an arbovirus to vertebrate and invertebrate cells. J. Virol. 73:4316–4326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Garmashova N, Atasheva S, Kang W, Weaver SC, Frolova E, Frolov I. 2007. Analysis of Venezuelan equine encephalitis virus capsid protein function in the inhibition of cellular transcription. J. Virol. 81:13552–13565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Garmashova N, Gorchakov R, Volkova E, Paessler S, Frolova E, Frolov I. 2007. The Old World and New World alphaviruses use different virus-specific proteins for induction of transcriptional shutoff. J. Virol. 81:2472–2484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gorchakov R, Frolova E, Frolov I. 2005. Inhibition of transcription and translation in Sindbis virus-infected cells. J. Virol. 79:9397–9409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Karpf AR, Blake JM, Brown DT. 1997. Characterization of the infection of Aedes albopictus cell clones by Sindbis virus. Virus Res. 50:1–13 [DOI] [PubMed] [Google Scholar]
- 10.Stollar V, Shenk TE, Koo R, Igarashi A, Schlesinger RW. 1975. Observations of Aedes albopictus cell cultures persistently infected with Sindbis virus. Ann. N. Y. Acad. Sci. 266:214–231 [DOI] [PubMed] [Google Scholar]
- 11.Stollar V, Thomas VL. 1975. An agent in the Aedes aegypti cell line (Peleg) which causes fusion of Aedes albopictus cells. Virology 64:367–377 [DOI] [PubMed] [Google Scholar]
- 12.Tooker P, Kennedy SI. 1981. Semliki Forest virus multiplication in clones of Aedes albopictus cells. J. Virol. 37:589–600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mims CA, Day MF, Marshall ID. 1966. Cytopathic effect of Semliki Forest virus in the mosquito Aedes aegypti. Am. J. Trop. Med. Hyg. 15:775–784 [DOI] [PubMed] [Google Scholar]
- 14.Raghow RS, Davey MW, Dalgarno L. 1973. The growth of Semliki Forest virus in cultured mosquito cells: ultrastructural observations. Arch. Gesamte Virusforsch. 43:165–168 [DOI] [PubMed] [Google Scholar]
- 15.Raghow RS, Grace TD, Filshie BK, Bartley W, Dalgarno L. 1973. Ross River virus replication in cultured mosquito and mammalian cells: virus growth and correlated ultrastructural changes. J. Gen. Virol. 21:109–122 [DOI] [PubMed] [Google Scholar]
- 16.Bowers DF, Abell BA, Brown DT. 1995. Replication and tissue tropism of the alphavirus Sindbis in the mosquito Aedes albopictus. Virology 212:1–12 [DOI] [PubMed] [Google Scholar]
- 17.Bowers DF, Coleman CG, Brown DT. 2003. Sindbis virus-associated pathology in Aedes albopictus (Diptera: Culicidae). J. Med. Entomol. 40:698–705 [DOI] [PubMed] [Google Scholar]
- 18.Kelly EM, Moon DC, Bowers DF. 2012. Apoptosis in mosquito salivary glands: Sindbis virus-associated and tissue homeostasis. J. Gen. Virol. 93:2419–2424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang H, Gort T, Boyle DL, Clem RJ. 2012. Effects of manipulating apoptosis on Sindbis virus infection of Aedes aegypti mosquitoes. J. Virol. 86:6546–6554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Weaver SC, Lorenz LH, Scott TW. 1992. Pathologic changes in the midgut of Culex tarsalis following infection with Western equine encephalomyelitis virus. Am. J. Trop. Med. Hyg. 47:691–701 [DOI] [PubMed] [Google Scholar]
- 21.Weaver SC, Scott TW, Lorenz LH, Lerdthusnee K, Romoser WS. 1988. Togavirus-associated pathologic changes in the midgut of a natural mosquito vector. J. Virol. 62:2083–2090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fragkoudis R, Breakwell L, McKimmie C, Boyd A, Barry G, Kohl A, Merits A, Fazakerley JK. 2007. The type I interferon system protects mice from Semliki Forest virus by preventing widespread virus dissemination in extraneural tissues, but does not mediate the restricted replication of avirulent virus in central nervous system neurons. J. Gen. Virol. 88:3373–3384 [DOI] [PubMed] [Google Scholar]
- 23.Klimstra WB, Ryman KD, Bernard KA, Nguyen KB, Biron CA, Johnston RE. 1999. Infection of neonatal mice with Sindbis virus results in a systemic inflammatory response syndrome. J. Virol. 73:10387–10398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ryman KD, Klimstra WB, Nguyen KB, Biron CA, Johnston RE. 2000. Alpha/beta interferon protects adult mice from fatal Sindbis virus infection and is an important determinant of cell and tissue tropism. J. Virol. 74:3366–3378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ryman KD, Meier KC, Nangle EM, Ragsdale SL, Korneeva NL, Rhoads RE, MacDonald MR, Klimstra WB. 2005. Sindbis virus translation is inhibited by a PKR/RNase L-independent effector induced by alpha/beta interferon priming of dendritic cells. J. Virol. 79:1487–1499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sokoloski KJ, Hayes CA, Dunn MP, Balke JL, Hardy RW, Mukhopadhyay S. 2012. Sindbis virus infectivity improves during the course of infection in both mammalian and mosquito cells. Virus Res. 167:26–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.He L, Piper A, Meilleur F, Myles DA, Hernandez R, Brown DT, Heller WT. 2010. The structure of Sindbis virus produced from vertebrate and invertebrate hosts as determined by small-angle neutron scattering. J. Virol. 84:5270–5276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kostyuchenko VA, Jakana J, Liu X, Haddow AD, Aung M, Weaver SC, Chiu W, Lok SM. 2011. The structure of Barmah Forest virus as revealed by cryo-electron microscopy at a 6-angstrom resolution has detailed transmembrane protein architecture and interactions. J. Virol. 85:9327–9333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mukhopadhyay S, Zhang W, Gabler S, Chipman PR, Strauss EG, Strauss JH, Baker TS, Kuhn RJ, Rossmann MG. 2006. Mapping the structure and function of the E1 and E2 glycoproteins in alphaviruses. Structure 14:63–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Boehme KW, Williams JC, Johnston RE, Heidner HW. 2000. Linkage of an alphavirus host-range restriction to the carbohydrate-processing phenotypes of the host cell. J. Gen. Virol. 81:161–170 [DOI] [PubMed] [Google Scholar]
- 31.Burke DJ, Keegstra K. 1976. Purification and composition of the proteins from Sindbis virus grown in chick and BHK cells. J. Virol. 20:676–686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hafer A, Whittlesey R, Brown DT, Hernandez R. 2009. Differential incorporation of cholesterol by Sindbis virus grown in mammalian or insect cells. J. Virol. 83:9113–9121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang R, Hryc CF, Cong Y, Liu X, Jakana J, Gorchakov R, Baker ML, Weaver SC, Chiu W. 2011. 4.4 Å cryo-EM structure of an enveloped alphavirus Venezuelan equine encephalitis virus. EMBO J. 30:3854–3863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang G, Hernandez R, Weninger K, Brown DT. 2007. Infection of cells by Sindbis virus at low temperature. Virology 362:461–467 [DOI] [PubMed] [Google Scholar]
- 35.Garneau NL, Sokoloski KJ, Opyrchal M, Neff CP, Wilusz CJ, Wilusz J. 2008. The 3′ untranslated region of Sindbis virus represses deadenylation of viral transcripts in mosquito and mammalian cells. J. Virol. 82:880–892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Paredes A, Alwell-Warda K, Weaver SC, Chiu W, Watowich SJ. 2003. Structure of isolated nucleocapsids from Venezuelan equine encephalitis virus and implications for assembly and disassembly of enveloped virus. J. Virol. 77:659–664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cruz CC, Suthar MS, Montgomery SA, Shabman R, Simmons J, Johnston RE, Morrison TE, Heise MT. 2010. Modulation of type I IFN induction by a virulence determinant within the alphavirus nsP1 protein. Virology 399:1–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shabman RS, Morrison TE, Moore C, White L, Suthar MS, Hueston L, Rulli N, Lidbury B, Ting JP, Mahalingam S, Heise MT. 2007. Differential induction of type I interferon responses in myeloid dendritic cells by mosquito and mammalian-cell-derived alphaviruses. J. Virol. 81:237–247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Flynn DC, Olmsted RA, Mackenzie JM, Jr, Johnston RE. 1988. Antibody-mediated activation of Sindbis virus. Virology 166:82–90 [DOI] [PubMed] [Google Scholar]
- 40.Hernandez R, Sinodis C, Horton M, Ferreira D, Yang C, Brown DT. 2003. Deletions in the transmembrane domain of a Sindbis virus glycoprotein alter virus infectivity, stability, and host range. J. Virol. 77:12710–12719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Strauss JH, Strauss EG. 1994. The alphaviruses: gene expression, replication, and evolution. Microbiol. Rev. 58:491–562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bick MJ, Carroll JW, Gao G, Goff SP, Rice CM, MacDonald MR. 2003. Expression of the zinc-finger antiviral protein inhibits alphavirus replication. J. Virol. 77:11555–11562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Coombs K, Brown DT. 1987. Topological organization of Sindbis virus capsid protein in isolated nucleocapsids. Virus Res. 7:131–149 [DOI] [PubMed] [Google Scholar]
- 44.Garmashova N, Gorchakov R, Frolova E, Frolov I. 2006. Sindbis virus nonstructural protein nsP2 is cytotoxic and inhibits cellular transcription. J. Virol. 80:5686–5696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Frolov I, Akhrymuk M, Akhrymuk I, Atasheva S, Frolova EI. 2012. Early events in alphavirus replication determine the outcome of infection. J. Virol. 86:5055–5066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Frolova EI, Fayzulin RZ, Cook SH, Griffin DE, Rice CM, Frolov I. 2002. Roles of nonstructural protein nsP2 and alpha/beta interferons in determining the outcome of Sindbis virus infection. J. Virol. 76:11254–11264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jungkamp AC, Stoeckius M, Mecenas D, Grun D, Mastrobuoni G, Kempa S, Rajewsky N. 2011. In vivo and transcriptome-wide identification of RNA binding protein target sites. Mol. Cell 44:828–840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Juzumiene D, Shapkina T, Kirillov S, Wollenzien P. 2001. Short-range RNA-RNA crosslinking methods to determine rRNA structure and interactions. Methods 25:333–343 [DOI] [PubMed] [Google Scholar]
- 49.Kishore S, Jaskiewicz L, Burger L, Hausser J, Khorshid M, Zavolan M. 2011. A quantitative analysis of CLIP methods for identifying binding sites of RNA-binding proteins. Nat. Methods 8:559–564 [DOI] [PubMed] [Google Scholar]
- 50.Pisarev AV, Kolupaeva VG, Yusupov MM, Hellen CU, Pestova TV. 2008. Ribosomal position and contacts of mRNA in eukaryotic translation initiation complexes. EMBO J. 27:1609–1621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mussgay M. 1964. Studies on the structure of a hemagglutinating component of a group A arbo virus (Sindbis). Virology 23:573–581 [DOI] [PubMed] [Google Scholar]
- 52.Breakwell L, Dosenovic P, Karlsson Hedestam GB, D'Amato M, Liljestrom P, Fazakerley J, McInerney GM. 2007. Semliki Forest virus nonstructural protein 2 is involved in suppression of the type I interferon response. J. Virol. 81:8677–8684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Snyder AJ, Sokoloski KJ, Mukhopadhyay S. 2012. Mutating conserved cysteines in the alphavirus E2 glycoprotein causes virus-specific assembly defects. J. Virol. 86:3100–3111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Leung WC, Rawls WE. 1977. Virion-associated ribosomes are not required for the replication of Pichinde virus. Virology 81:174–176 [DOI] [PubMed] [Google Scholar]
- 55.Pedersen IR, Konigshofer EP. 1976. Characterization of ribonucleoproteins and ribosomes isolated from lymphocytic choriomeningitis virus. J. Virol. 20:14–21 [DOI] [PMC free article] [PubMed] [Google Scholar]