

Abstract
Borna disease virus (BDV) is a nonsegmented, negative-stranded RNA virus characterized by noncytolytic persistent infection and replication in the nuclei of infected cells. To gain further insight on the intracellular trafficking of BDV components during infection, we sought to generate recombinant BDV (rBDV) encoding fluorescent fusion viral proteins. We successfully rescued a virus bearing a tetracysteine tag fused to BDV-P protein, which allowed assessment of the intracellular distribution and dynamics of BDV using real-time live imaging. In persistently infected cells, viral nuclear inclusions, representing viral factories tethered to chromatin, appeared to be extremely static and stable, contrasting with a very rapid and active trafficking of BDV components in the cytoplasm. Photobleaching (fluorescence recovery after photobleaching [FRAP] and fluorescence loss in photobleaching [FLIP]) imaging approaches revealed that BDV components were permanently and actively exchanged between cellular compartments, including within viral inclusions, albeit with a fraction of BDV-P protein not mobile in these structures, presumably due to its association with viral and/or cellular proteins. We also obtained evidence for transfer of viral material between persistently infected cells, with routing of the transferred components toward the cell nucleus. Finally, coculture experiments with noninfected cells allowed visualization of cell-to-cell BDV transmission and movement of the incoming viral material toward the nucleus. Our data demonstrate the potential of tetracysteine-tagged recombinant BDV for virus tracking during infection, which may provide novel information on the BDV life cycle and on the modalities of its interaction with the nuclear environment during viral persistence.
INTRODUCTION
Borna disease virus (BDV) is an enveloped virus with a nonsegmented, negative-strand RNA genome (1, 2), which represents the prototypic member of the family Bornaviridae. In contrast to other Mononegavirales members, BDV replicates in the nuclei of infected cells (3) and uses the host cell splicing machinery for maturation of viral transcripts (4, 5). The BDV compact genome encodes six proteins, namely, the nucleoprotein (N), phosphoprotein (P), protein X, matrix protein (M), glycoprotein (G), and polymerase (L). Whereas M and G are involved in particle formation, P, N, and L are components of the ribonucleoprotein complex (RNP). The small X protein has been shown to contribute to apoptosis resistance during infection (6) and may also have interferon antagonist activities (7). BDV persistently infects a wide range of mammalian and avian species (8–11), resulting in a large spectrum of neurological disorders ranging from immune-mediated diseases to behavioral syndromes without inflammation (8, 12, 13). BDV is highly neurotropic but can also replicate in other cells of the central nervous system (CNS), as well as in many established cell lines in vitro (9).
To date, the BDV cell cycle remains poorly characterized. It is known that BDV enters cells by receptor-mediated endocytosis and that pH-dependent fusion is required to liberate the RNP into the cell environment (14–16). The postentry events are, however, still ill defined. In particular, the mechanisms of BDV movement to the nucleus are totally unknown. Once inside the nucleus, BDV replicates without any associated cytopathic effect and establishes a long-lasting, persistent infection. It was shown recently that BDV assembles viral factories closely associated with chromatin within the nucleus, which are essential to achieve intranuclear persistence (17). A key issue, however, is to obtain more information on the relationship between viral nuclear inclusions and the other cellular compartments. A better understanding of the dynamics of BDV intracellular movements in infected cells would undoubtedly provide novel information on the life cycle of this unique noncytolytic, nuclear replicating RNA virus.
In the past few years, the use of fluorescently labeled viruses and high-resolution microscopy has proven its great utility for unraveling the dynamic events of virus infection in living cells. Many diverse aspects of virus biology and virus-host cell interplay have been examined, including viral entry or intracellular trafficking of viral ribonucleocapsids. Classically, fluorescent Mononegavirales have been generated by fusing green fluorescent protein (GFP) (or any related variant) in frame with a structural protein in a genomic plasmid. Upon rescue of recombinant virus, the resultant virus is fluorescent, due to incorporation of the GFP fusion protein, thereby allowing live tracking as described for vesicular stomatitis virus (VSV) or rabies virus (18, 19). More recently, many groups have sought to use alternatives to GFP tagging, because the relatively large size of GFP may potentially alter the normal trafficking of viral components. In particular, many have used the tetracysteine technology, initially described by Griffin et al. (20), which consists in adding a small peptide sequence containing two pairs of cysteines (-CCXXCC-) to the protein of interest. Subsequent fluorescent labeling of a recombinant virus results from the capacity of this tetracysteine tag (TCT) to bind to fluorescent biarsenical derivatives (e.g., FlAsH). Because of its small size, TCT appears as the ideal label in virus studies and has been applied to a wide spectrum of viral systems (21).
In this work, our objective was to generate a recombinant BDV (rBDV) suitable for live-imaging studies and to use this virus to gain further insight on the BDV life cycle. Reverse-genetics approaches applied to BDV have demonstrated that the GFP gene can readily be inserted in the viral genome as an additional gene, thereby allowing the visualization of BDV dissemination in the CNS (22, 23). Here, we aimed at inserting a fluorescent label in the context of a fusion construct with a structural protein. In particular, we tested insertions within N, P, and L, the components of BDV RNP, using GFP and TC fluorescent tags. Among all recombinant viruses tested, we succeeded in the rescue of a virus encoding TC-tagged P. Upon characterization, this recombinant virus allowed real-time visualization of BDV dynamics in infected cells.
MATERIALS AND METHODS
Cells.
Vero and HEK293 cells (obtained from the American Type Culture Collection), as well as Vero cells persistently infected with recombinant viruses (rBDV-Pwt, rBDV-P-Flag, and rBDV-P-Flag-TCT), were cultured in Dulbecco's modified Eagle's medium (DMEM)-high glucose supplemented with 10% fetal calf serum (FCS).
Plasmid construction.
Full-length BDV antigenomes harboring Flag tag or TCT were generated by overlapping PCR amplification of the BDV antigenome plasmid (pBRCMV-H1BDVc) (24). The procedures for inserting the different tags are as follows. (i) For insertion of tags in BDV-P, in a first PCR, we amplified part of the P open reading frame (ORF) with a forward primer (prP/F) complementary to the BDV-P sequence upstream of the unique restriction site BbvCI and a reverse primer (prP-tag/R) containing the tag sequence. A second PCR was performed with a forward primer (prP-tag/F) containing the tag sequence and a reverse primer (prP/R) complementary downstream of the restriction site PmeI. These two PCR products then served as templates for a third PCR to amplify a fragment containing the Flag-tagged ORF using the forward primer prP/F and reverse primer prP/R. Finally, the BbvCI/PmeI-digested PCR product was ligated into a BbvCI/PmeI-digested full-length BDV antigenome vector. (ii) For insertion of tags in BDV-N, a first PCR product was amplified with a forward primer prN/F upstream of the unique restriction site SpeI and a reverse primer (prtag-N/R) containing the tag sequence and the 3′ end of the N ORF. A second PCR was performed with a forward primer (prtag-N/R) and a reverse primer (prN/R) downstream of the restriction site MluI. These two PCR products served as templates for a third PCR, amplified with primers prN/F and prN/R. Finally, the SpeI/MluI-digested PCR product was ligated into a SpeI/MluI-digested full-length BDV antigenome vector. (iii) For insertion of tags in BDV-L, a first PCR product was amplified with a forward primer prL/F upstream of the unique restriction site BstXI and a reverse primer (prL-tag/R) containing the 5′ end of the L ORF and the tag sequence. A second PCR was performed with a forward primer (prL-tag/R) and a reverse primer (prL/R) downstream of the restriction site NotI. These two PCR products served as templates for a third PCR, amplified with primers prL/F and prL/R. Finally, the BstXI/NotI-digested PCR product was ligated into a BstXI/NotI-digested full-length BDV antigenome vector. (iv) For insertion of GFP tags, the GFP ORF was amplified by PCR from pCA-Flag-GFP plasmid with a forward and reverse primer consisting of the corresponding nucleotide sequences (∼40 nucleotides [nt]) at the GFP N- and C-terminal fusion sites (prP-GFP/F and prP-GFP-BDV/R, respectively). A second PCR was done using the BDV plasmid (pBRCMV-H1BDVc) as a template and the forward primer (prP/F) and a reverse primer (prP-GFP/R) complementary to prP-GFP/F. The BDV plasmid served as the template for a third PCR that was performed with a forward primer (prP-GFP-BDV/F) complementary to prP-GFP-BDV/R and the reverse primer (prP/R). To incorporate these three PCR products together, two other successive PCRs were performed. One was amplified with prP/F and prP-GFP-BDV/R as primers and GFP and P ORF PCR products as templates. This PCR product and the product from the third PCR served as templates for the final PCR with the primers prP/F and prP/R. Lastly, the BbvCI/PmeI-digested final PCR product was ligated into a BdvCI/PmeI-digested full-length BDV antigenome vector. The same strategy was applied to produce a full-length BDV antigenome coding for GFP-N. The forward (prBDV-GFP-N/F) and reverse (prGFP-N/R) primers used to amplify GFP contained nucleotide sequences at the GFP-N fusion sites. A second PCR was performed with the forward primer prN/F and the reverse primer prBDV-GFP-N/R. A third PCR was performed with the forward primer prGFP-N/F and the reverse primer prN/R. The first two products served as templates for a fourth PCR, amplified by primers prN/F and prGFP-N/R. This PCR product and the third PCR product served as templates for the final PCR amplified by primers prN/F and prN/R. Lastly, the SpeI/MluI-digested final PCR product was ligated into a SpeI/MluI-digested full-length BDV antigenome vector. To generate a full-length BDV antigenome expression plasmid coding for L-GFP, the GFP ORF was amplified with primers prL-GFP/F and prL-GFP-BDV/R. Then, a second PCR was performed with the forward primer prL/F and the reverse primer prL-GFP/R. A third PCR was performed with the forward primer prL-GFP-BDV/F and the reverse primer prL/R. The first two PCR products served as templates for a fourth PCR amplified by primers prL/F and prL-GFP-BDV/R. This PCR product and the third PCR product served as templates for the final PCR amplified by primers prL/F and prL/R. Lastly, the BstXI/NotI-digested final PCR fragment was ligated into a BstXI/NotI-digested full-length BDV antigenome vector.
BDV rescue.
HEK 293T cells grown in 35-mm dishes were transfected with 4 μg of plasmids encoding the full-length BDV antigenome (pBRCMV-H1BDVc) or the BDV antigenome with desired insertions, together with 0.5 μg of pCA-N, 0.1 μg of pCA-L, and 0.05 μg of pCA-P. Three days after transfection, cells were trypsinized and seeded onto T75 flasks together with 106 Vero cells. The cocultures were kept in DMEM supplemented with 4% FCS and were split twice per week. The rescue rate was determined by staining against BDV-N every week as described previously (24). When the infection of Vero cells reached 80% or more, a first virus stock was made. To avoid contamination with input plasmids, the first virus stock was used to infect Vero cells and a second virus stock was made when the infection rate reached 100%. The second virus stock was then used for further experiments.
Preparation of BDV virus stocks.
BDV was isolated from persistently infected Vero cells as described previously (24). Briefly, confluent cells grown in 94-mm dishes were rinsed with 5 ml of 20 mM HEPES (pH 7.4) and incubated for 2 h at 37°C in a 5% CO2-humidified atmosphere with 10 ml of 20 mM HEPES (pH 7.4), 250 mM MgCl2, and 1% FCS. Cell supernatant was centrifuged twice at 2,500 rpm for 5 min to remove cell debris, followed by ultracentrifugation above a 20% sucrose cushion at 24,000 rpm at 8°C for 1 h. Viral pellets were resuspended in 400 μl of phosphate-buffered saline (PBS) without Ca2+ or Mg2+. The virus stock was then dialyzed in PBS without Ca2+ or Mg2+ before usage. Viral titers were determined on Vero cells as described previously (24).
Viral growth curves.
Vero cells (105 cells/well in a 24-well dish) were infected with rBDV-Pwt, rBDV-P-Flag, and rBDV-P-Flag-TCT with a multiplicity of infection (MOI) of 0.01. Three days later, infected cells were trypsinized and seeded into 6-well plates. The cell cultures were split twice per week, and at each passage, the rate of infected cells was monitored by immunofluorescence analysis using a polyclonal BDV-N-specific antibody. Viral growth kinetics was determined 3 times independently.
Western blot analysis.
rBDV-infected and uninfected Vero cells grown in 35-mm dishes were harvested in 200 μl of Laemmli buffer, followed by ultrasonication. Protein extracts were size fractionated by 13% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and blotted onto polyvinylidene difluoride (PVDF) membranes. Membranes were blocked in blocking solution and incubated with primary antibodies (mouse anti-β-tubulin, 1:1,000, Sigma; mouse anti-Flag M2, 1:200, Sigma F3165; rabbit polyclonal anti-BDV-N, 1:1,000; and rabbit polyclonal anti BDV-P, 1:1,000) for 1 h at room temperature. After being washed with PBS–0.1% Tween 20, the membrane was incubated with secondary antibodies (alkaline phosphatase-conjugated goat anti-rabbit IgG, 1:2,000, Jackson ImmunoResearch, and alkaline phosphatase-conjugated goat anti-mouse IgG, 1:2,000, Jackson ImmunoResearch) for 1 h at room temperature. The membrane was washed in PBS with 0.1% Tween 20 and incubated with Tris–Tween-buffered saline and citric acid buffer (15 min each). Finally, protein levels were visualized with the developing solution containing 5 mM MgCl2, 5-bromo-4-chloro-3-indolyl phosphate p-toluidine salt (BCIP; Carl Roth GmbH), and methylthiazolyldiphenyl-tetrazolium bromide (MTT; Sigma-Aldrich) in citrate-buffered saline (CBS).
RNA preparation and Northern blot analysis.
Total RNA was prepared from BDV-infected or uninfected Vero cells in 35-mm dishes with peqGOLD TriFast reagent (PeqLab). For Northern blot analysis, 5 μg of total RNA from each sample was size fractionated on a 1.2% agarose gel containing 3.7% formaldehyde and blotted overnight onto a nylon membrane. The RNA was cross-linked to the membrane by baking at 80°C for 2 h. For prehybridization, the membrane was first treated in 0.2× SSC–0.5% SDS (1× SSC is 0.15 M NaCl plus 0.015 M sodium citrate) at 68°C for 10 min, followed by 2 to 3 h at 42°C in hybridization buffer (5× PIPES, 1× Denhardt's buffer, 50% formamide, 0.2% SDS, and 200 μg/ml of single-stranded herring sperm DNA) under continuous rotation. DNA probes against BDV-N, -P, and -X (nucleotides 976 to 1749) were prepared by PCR, and the PCR products were radiolabeled with 32P according to the manufacturer's protocol (DecaLabel DNA labeling kit; Fermentas). After prehybridization, radiolabeled DNA probes were added into the hybridization buffer and the membrane was incubated overnight at 42°C. The blot was rinsed once with 2× SSC–0.5% SDS at room temperature. The blot was then washed again with 2× SSC–0.5% SDS at 65°C and exposed at −80°C to Biomax MR film (Kodak).
Immunoprecipitation.
A total of 107 cells grown in 100-mm dishes were harvested in 500 μl of lysis buffer (50 mM Tris-HCl [pH 7.5], 150 mM NaCl, 1% NP-40, 0.5% sodium deoxycholate, 0.1% SDS, and a tablet of protease inhibitor cocktail [Complete; Roche]). Two hundred microliters of lysate was incubated with 0.5 μl of rabbit polyclonal anti-BDV-P antibody and 30 μl of protein G plus agarose beads (Santa Cruz Biotechnology) overnight at 4°C. Immunoprecipitates were centrifuged, and pellets were washed three times in the same buffer and resuspended in Laemmli electrophoresis loading buffer. Total extracts and immunoprecipitated fractions were then subjected to Western blot analysis using anti-BDV-N or anti-BDV-P antibodies.
Plasmid transfections.
Vero cell lines seeded in 12-well plates and grown to 80% confluency were transfected with Fugene 6 (Roche) according to the manufacturer's recommendations. Cells were used 48 to 72 h after transfection.
FlAsH labeling and live-cell imaging.
Cells were seeded in Lab-Tek chambers 8 h before labeling and imaging. Cells were washed once with PBS and once with Opti-MEM prior to FlAsH labeling (Life Technologies). The protocol recommended by the manufacturer was followed precisely, and in another set of experiments, FlAsH labeling was optimized as described below. In this case, FlAsH reagent was diluted in Opti-MEM (2.5 μM final concentration), added to cells, and incubated for 15 min at room temperature (RT) in the dark. Cells were rapidly rinsed twice with Opti-MEM, then once in 5× BAL buffer (2,3-dimercapto-1-propanol) diluted in Opti-MEM for 10 min, and finally once in 1× BAL buffer for 10 min. All washes were performed in the dark at RT. Cells were left in 1× BAL buffer for imaging. For real-time imaging, cells were maintained at 37°C under 5% CO2 in an enclosed chamber and were observed with a confocal microscope (Carl Zeiss LSM 710 equipped with a 63× objective; numerical aperture [NA], 1.4; laser power set at 0.4%). Images were acquired in z stacks every 4 min. Images were processed with Zen software (Carl Zeiss).
Combined FlAsH labeling and immunofluorescence staining.
Cells seeded in Lab-Tek chambers were first labeled with FlAsH as described above. Cells were then fixed with 4% paraformaldehyde–PBS for 20 min, permeabilized with 0.1% Triton X-100–PBS for 4 min, blocked in 5% normal goat serum (NGS), and incubated with anti-BDV-P or -N antibody (each diluted 1:1,000 in PBS-NGS) for 1 h at room temperature, followed by incubation with Alexa Fluor 594-conjugated goat anti-rabbit IgG (Life Technologies). Images were acquired using a confocal microscope (Carl Zeiss LSM 710).
FRAP.
Cells were visualized with a LSM 710 confocal microscope equipped with a heating chamber, using the 488-nm laser line and a 490- to 590-nm emission filter. Disk photobleaching was performed using circular regions of interest (ROI) of 1.5 μm in diameter (ω), placed either on the cytoplasm or on the viral nuclear dots (vSPOTs). The region was photobleached with the 488-nm laser line to obtain 35 to 80% photobleaching compared to the prebleach image. The pinhole was kept fully open to ensure maximum photon collection during recovery. Unbleached control cells were monitored in parallel for photobleaching due to image acquisition after background subtraction. Data were collected every 0.23 s for an average of 1 min. All measurements were carried out at 37°C and 5% CO2. Correction for photobleaching due to acquisition was done for each time point after background correction. For each condition, fluorescence recovery after photobleaching (FRAP) curves were averaged using Excel software and fluorescence intensities were normalized by setting the initial fluorescence to a value of 1. Recovery curves were calculated using a nonlinear least-squares fit equation as described by Soumpasis (25), which was implemented in an Excel spreadsheet. This formula is adapted for the analysis of fluorescence recovery curves measured in fluorescence photobleaching recovery experiments employing uniform circular laser beams, as done here. The characteristic diffusion time τD was obtained from the fitted curve
where I0 and I1 are modified Bessel functions, D is the diffusion coefficient, t is the time in seconds, F∞ is the fluorescence intensity after full recovery, and F0 is the fluorescence intensity immediately after photobleaching. Data fitting was performed using the Excel solver tool with two iterations, which calculates for each time point the square of the difference between the theoretical model and the observed fluorescence (chi-square). The diffusion coefficient D was calculated with the formula D = ω2/(4 τD), with ω being the radius of the ROI set and τD obtained from the fit of the recovery curve. The fit also yielded the viral mobile fraction M, with the formula M = (F∞ − Fprebleach)/(F0 − Fpostbleach).
Results are reported as experimental and fitting FRAP curves, associated with residuals values calculated for each time point (chi-square).
TPEM FLIP.
For two-photon excitation microscopy (TPEM) fluorescence loss in photobleaching (FLIP) experiments in nuclear vSPOTs, a single spot with a diameter of 2 μm within the nucleus was repeatedly photobleached with a femtosecond Chameleon Ultra II (Coherent S.A.) laser line set at 940 nm (iterations at 26% power), using a Carl Zeiss LSM 7MP with a 40× PlanApo lens (NA, 1.3). The measured diameter of a single z-axis plane was 950 nm. Two images were collected after each bleach pulse, with 2.26 s between bleach pulses. After collection of 300 images, two separate measurements on the viral nuclear dots were taken to analyze fluorescence loss. Unbleached control cells were monitored for photobleaching due to image acquisition after background subtraction. The rate of loss in fluorescence on the neighbor viral nuclear point was calculated from fluorescence intensity measurements after a background subtraction using the Carl Zeiss Zen software. The fluorescence intensities were normalized by setting the initial fluorescence to 100%. Plots were drawn for normalized fluorescence over time.
Confocal microscopy FLIP.
A single spot with a diameter of 2 μm within the cytoplasm was repeatedly photobleached with a 488-nm laser line (iterations at 100% power), using a Carl Zeiss LSM 710 with a 63× PlanApo lens (NA, 1.4). The measured diameter of a single z-axis plane was 400 nm. Two images were collected after each bleach pulse, with 3.2 s between bleach pulses. After collection of 200 images, two separate measurements on the cytoplasmic regions were taken to analyze the fluorescence loss. Unbleached control cells were monitored for photobleaching due to image acquisition after a background subtraction. The rate of loss in fluorescence on the neighbor cytoplasmic region or the nuclear viral spot was calculated from fluorescence intensity measurements after a background subtraction using Zen software (Carl Zeiss). Fluorescence intensities were normalized by setting the initial fluorescence to 100%. Plots were drawn for normalized fluorescence over time.
RESULTS
Generation and characterization of tagged BDV.
In order to visualize BDV traffic in cells using live imaging, we first generated BDV rescue plasmids encoding GFP-tagged BDV-N, -P, or -L proteins (Fig. 1A). However, we failed to rescue these viruses, suggesting that fusion to GFP is not tolerated in any case. Next, we tried to rescue recombinant BDV (Fig. 1A), encoding either BDV-N, -P, or -L fused to a tetracysteine tag (TCT). Again, rescue of these recombinant viruses (BDV-N-TCT, BDV-P-TCT, or BDV-L-TCT) failed. To test whether any tag could be fused to BDV proteins in the context of an infectious clone, we then tried to rescue BDV harboring a Flag tag at the N terminus of BDV-N or at the C terminus of either BDV-P or -L (Fig. 1A). Although BDV coding for Flag-tagged BDV-N or BDV-L could not be generated, we successfully rescued a virus expressing Flag-tagged BDV-P protein, designated rBDV-P-Flag. Encouraged by these findings, we generated and successfully rescued a virus harboring a TCT in addition to the C-terminal Flag tag of BDV-P, designated rBDV-P-Flag-TCT (Fig. 1A). Sequencing of both rBDV-Flag and rBDV-Flag-TCT viruses revealed no compensatory mutations in the BDV-P ORF (data not shown).
Fig 1.

Rescue attempts and characterization of recombinant BDV encoding a tetracysteine-tagged BDV-P. (A) Diagram of recombinant full-length antigenome constructs depicting the position of BDV open reading frames fused to GFP, TCT, and/or Flag. Note that only viruses highlighted in red could be rescued, with the Flag or Fag-TCT tag fused to the C terminus of BDV-P. (B) Viral growth characteristics of wild-type recombinant BDV (rBDV-Pwt), Flag-tagged rBDV (rBDV-P-Flag), and rBDV dually tagged with Flag and TCT (rBDV-P-Flag-TCT) in Vero cells infected with an initial MOI of 0.01. The infection rate was determined by immunocytochemistry with an N-specific antibody at the indicated time points. Error bars represent standard errors of the means of three independent experiments. (C) Viral transcript levels in Vero cells persistently infected with tagged and untagged rBDV. Northern blot analysis was carried out with 5 μg of total RNA from the uninfected or persistently infected Vero cells. A DNA probe comprising nucleotides 976 to 1749 (BDV-N/P/X) was used to visualize the indicated viral RNA species. 18S RNA confirmed that similar amounts of RNA were loaded in each lane of the gel (bottom). (D) Protein levels in Vero cells persistently infected with tagged and untagged rBDV. Cell extracts of Vero cells infected with the indicated viruses were subjected to 13% SDS-PAGE, and antigens were visualized with antibodies recognizing BDV-N, BDV-P, Flag, and β-tubulin.
To study the growth properties of these recombinant BDV, we infected Vero cells with rBDV-Pwt, rBDV-P-Flag, and rBDV-P-Flag-TCT at an MOI of 0.01 and monitored viral spread by staining for BDV-N. In contrast to findings for rBDV-Pwt, the infection rates of Vero cells were delayed with both rBDV-P-Flag and rBDV-P-Flag-TCT (Fig. 1B). After an initial delay in the early steps of infection, all recombinant viruses, however, achieved 100% infection and established persistence.
Next, we determined steady-state levels of viral transcripts in persistently infected Vero cells by Northern blotting, using a DNA probe comprising nucleotides 976 to 1749 (BDV-N/P/X). As shown in Fig. 1C, levels of the 0.8-kb and 1.2-kb transcripts were comparable between cells infected with recombinant wild-type and mutant viruses. Compared to wild-type-infected cells, we observed in the mutant virus-infected cells a decrease in 1.9-kb transcripts accompanied by an increase of 4.7-kb transcripts. The accumulation of the latter transcripts is most likely due to a read-through at the transcription termination sites T1 and T2 (26). However, Western blot analysis revealed comparable levels of BDV-N and -P in Vero cells persistently infected with rBDV-Pwt, rBDV-P-Flag, and rBDV-P-Flag-TCT (Fig. 1D). In addition, coimmunoprecipitation experiments showed that BDV-P dually tagged with Flag and TCT was still able to interact with BDV-N and hence to presumably be incorporated in the RNP (Fig. 2). Thus, despite the difficulties in rescue of recombinant BDV bearing a component of the RNP fused to a fluorescent tag, we were able to recover a recombinant virus with a TC-tagged BDV-P.
Fig 2.
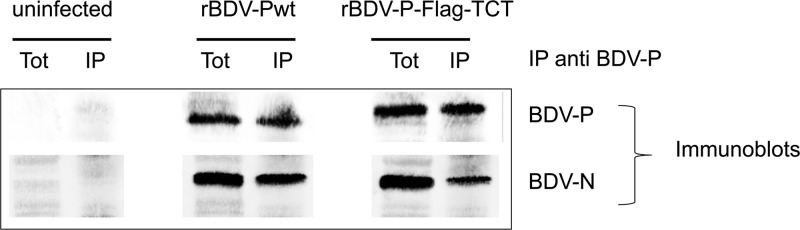
BDV-P-Flag-TCT and BDV-N interact in infected cells. Extracts from control Vero cells or Vero cells infected with either wild-type recombinant BDV (rBDV-Pwt) or rBDV dually tagged with Flag and TCT (rBDV-P-Flag-TCT) were subjected to immunoprecipitation using a rabbit polyclonal anti BDV-P antibody. Western blots of fractions (Tot, whole-cell extracts; IP, immunoprecipitates) were probed with a rabbit polyclonal anti-BDV-P (top) or anti-BDV-N (bottom) antibody.
Characterization of FlAsH-labeled cells.
The TCT added to BDV-P forms a putative hairpin that strongly and specifically reacts with the FlAsH biarsenical reagents to covalently label the protein of interest. In application to persistently infected cells using the staining conditions suggested by the manufacturer, we observed that FlAsH labeling yielded a nonspecific cytoplasmic punctate background (i.e., also observed in noninfected cells) (Fig. 3A). Although staining appeared clearly different in infected cells, the unspecific background under these staining conditions was unacceptable for further subcellular localization studies. We thus sought to optimize FlAsH labeling (see Materials and Methods), in particular by increasing the stringency of washes after staining. This procedure led to much better labeling conditions that revealed a specific staining only in rBDV-P-Flag-TCT-infected cells (Fig. 3A). Staining was reminiscent of that classically observed in BDV-infected cells and was characterized by a diffuse cytoplasmic pattern and a punctate staining in the nucleus (Fig. 3A). To demonstrate that FlAsH-labeled protein reflected the authentic BDV-P protein in BDV-infected cells, we performed a combination of FlAsH labeling and immunofluorescence analysis for BDV-P. As shown in Fig. 3B, antibody staining for BDV-P and FlAsH labeling largely overlapped (Mander's correlation coefficient: 0.79 ± 0.11; n = 10) (27), confirming that FlAsH labeling of the TCT can truly detect BDV-P protein in infected cells. FlAsH labeling was also largely superimposable with immunofluorescence staining for BDV-N (Fig. 3C), suggesting that labeling of this TCT allows detection of BDV RNP complexes, although we cannot exclude the possibility that part of the staining could be due to soluble or complexes of BDV-P not associated with RNP.
Fig 3.

Development and validation of FlAsH-labeling. (A) Biarsenical dye FlAsH labeling of Vero cells persistently infected with rBDV-P-Flag-TCT. Infected or control cells were FlAsH labeled as recommended by the vendor (left column, condition 1) or using optimized conditions, including more stringent washes as described in Materials and Methods (right column, condition 2). (B and C) Dual-labeling experiments. Following FlAsH labeling and washing of the dye, cells were fixed and subjected to immunofluorescence staining for BDV-P (B) or BDV-N (C), followed by anti-rabbit Alexa 594-conjugated secondary antibody. The merged images of the two stainings are shown on the right. In the graphs are plotted the intensity profiles of green and red fluorescent signals along a line drawn across the cell, indicated by the path of a 3-pixel-wide line scan. Bar = 10 μm.
Visualization of BDV-P kinetics in infected cells.
Having shown that FlAsH labeling of TC-tagged BDV-P allowed study of the behavior of BDV in live infected cells, we analyzed BDV kinetics by confocal microscopy, upon FlAsH labeling of Vero cells persistently infected with rBDV-P-Flag-TCT. In the nuclei of infected cells, the punctate staining appeared extremely static, and we did not detect any change in their number or distribution throughout the observation period (up to 12 h) (Fig. 4A; see also Video S1 in the supplemental material; other data not shown). The labeling characteristics were strikingly different in the cytoplasm of infected cells, with very dynamic movements of viral material, which was even sometimes exchanged between adjacent cells (Fig. 4A; see also Video S1).
Fig 4.

Dynamics of BDV-P in persistently infected Vero cells. (A) Real-time visualization of BDV-P dynamics in persistently infected cells. BDV-infected Vero cells were FlAsH labeled and observed by confocal microscopy. Images were acquired every 4 min for over 3 h. Note the static behavior of BDV-P within nuclear dots. (B and C) FRAP analysis to evaluate BDV-P dynamics in the nucleus (B) or in the cytoplasm (C). The pre- and postbleached images are shown, sampled until 70 s postbleach. The white circles indicate photobleached spots, and the arrow indicates the moment when photobleaching was performed. Images were collected at 0.23-s intervals, and correction for photobleaching due to acquisition was done for each time point after background correction. The left side shows both experimental and fitted fluorescence recovery curves of FRAP experiments. FRAP recovery curves were averaged and fitted with a diffusion model, as described in Materials and Methods. Each fit allowed the calculation of D (diffusion coefficient), M (percentage of mobile fraction), and chi-square (on the right of each curve), which confirms that experimental values differ from the estimated values by less than 8% in all cases. Data were generated from 3 independent experiments, for a total of 33 viral nuclear (B) dots and 17 cytoplasmic (C) FRAP measurements. Bar = 5 μm.
Recently, Matsumoto et al. demonstrated that the viral inclusions revealed by the punctate nuclear staining for BDV, designated vSPOTs (viral speckles of transcripts), represent viral factories using the cellular chromatin as a scaffold (17). Since viral factories are sites of ongoing viral replication, this finding implies that the dynamics of viral components in vSPOTs should include active exchanges with other viral components such as BDV-P. However, this appears at first sight in contradiction with our real-time imaging findings, which revealed that vSPOTs were very stable, although rather slow exchange processes might not be easily visualized by real-time imaging. To analyze further the relative nuclear and cytoplasmic mobility of BDV-P, we used fluorescence recovery after photobleaching (FRAP) to photobleach a circular defined region of the nucleus or the cytoplasm, and the time taken to refill the photobleached area was measured. When applied to a nuclear vSPOT (Fig. 4B), this analysis revealed a progressive but incomplete recovery of the photobleached area, contrasting with the very rapid and complete recovery when FRAP was applied to a cytoplasmic area (Fig. 4C). To compare the relative mobility of viral material, we analyzed the fluorescence recovery curves in both cases. Since we used a uniform disk photobleaching for FRAP experiments and because of the extension of the photobleaching in the z axis due to confocal microscopy, diffusion along the z axis can be neglected. We thus used a two-dimensional (2D) fitting model to analyze the fluorescence recovery curves (25). We first calculated the mobile fraction (M), obtained as a result of the curve fit and which is defined as the maximum fractional recovery of the fluorescent protein in the region of interest. In vSPOTs, the mobile fraction accounted for 86% (i.e., showing that 14% of BDV-P is immobile within vSPOTs, the rest being mobile), contrasting with the 100% rate of the mobile fraction in the cytoplasm (Fig. 4B and C, center). We also measured the diffusion coefficient values (D), representing the rate at which a protein repopulates a photobleached area (Fig. 4B and C, center). These measures confirmed that BDV-P had a lower mobility in vSPOTs (D = 0.190 μm2/s [Fig. 4B]). Importantly, the D value for BDV-P in vSPOTs is comparable to those previously reported for nucleosomal binding proteins, such as HMG-17 (28). Moreover, this value is much lower than values reported for free solutes in the nucleus (29), suggesting that it is due to interaction with nuclear components. In contrast, the D value obtained for the cytoplasm (D = 1.335 μm2/s [Fig. 4C]) confirmed the virtual absence of immobile molecules. Taken together, these findings suggest that a fraction of BDV-P in the nucleus is associated or interacts with nuclear proteins, thereby resulting in significant immobility.
To determine the relative trafficking of BDV-P between different vSPOTs or between the different cellular compartments, we used the approach of fluorescence loss induced by photobleaching (FLIP), in which a defined area is continuously photobleached using high laser power. The loss of fluorescence in a defined area outside the bleached region is measured over time and provides information on the rate of dissociation of the protein from this particular compartment. To perform FLIP on vSPOTs, we wanted to ensure that continuous photobleaching that characterizes this technique would be restricted to this focal area in the nucleus. To this end, we developed a 2-photon-based procedure, in which photobleaching is indeed restricted to this nuclear area, due to the confined excitation at the focal plane (Fig. 5A). By repeatedly photobleaching a vSPOT area, more than 60% fluorescence was lost from a neighboring vSPOT (Fig. 5B), showing that BDV-P is continuously exchanged between vSPOTs. Moreover, FLIP revealed that continuous photobleaching of the nucleoplasm (Fig. 5C) led to a fast decay of fluorescence in a neighboring vSPOT, suggesting that BDV-P continuously dissociates from this compartment and that its movements are not restricted to any particular nuclear domain. Finally, we also applied confocal FLIP to a defined area of the cytoplasm and monitored trafficking in three regions: another area of the cytoplasm, the nucleoplasm, and a vSPOT (Fig. 5D). This analysis revealed a rapid trafficking of BDV-P in the cytoplasm, but also trafficking between the cytoplasm and the nucleus. The trafficking between these two compartments was, however, much slower, in particular for vSPOTs, consistent with the low diffusion coefficient value of BDV-P in this area as determined by FRAP analyses.
Fig 5.

FLIP experiments after photobleaching of cellular compartments. (A) Rationale for the development of a 2-photon-based FLIP approach to study vSPOTs. A confocal microscope will excite fluorescence throughout the specimen volume (i.e., above and below the focal point centered in the region of interest; see left side) and will contribute to the photobleached path. In contrast, with a 2-photon microscope, excitation occurs only at the focal plane (see right side), and photobleaching is therefore confined to the vSPOT. (B and C) 2-photon-based FLIP analysis of BDV-P trafficking between vSPOTs (B) and between one vSPOT and the nucleoplasm (C). The arrow points to the vSPOT that was continuously photobleached, and the circles represent the areas of fluorescence intensity measurements. Images were collected at 1.13-s intervals, and images are shown at different time points during FLIP to illustrate the fluorescence decay in the regions of interest. The fluorescent intensities areas were measured and are shown on the right side, the dark curve corresponding to the permanently photobleached area and the gray curve to the other one. Correction for photobleaching due to acquisition was done for each time point after background correction. For each condition, FLIP recovery curves were averaged. The figure represents the sum of 3 independent experiments, for a total of 10 vSPOTs (B) and 11 nucleoplasm (C) FLIP measurements. (D) Confocal FLIP analysis of BDV-P trafficking between the different cellular compartments. The arrow points to the cytoplasmic area that was photobleached. Fluorescence intensity measurements were performed in separate areas represented by circles, which were numbered as follows: 1 for cytoplasm, 2 for nucleoplasm, and 3 for a nuclear vSPOT. FLIP recovery curves are shown on the right side and were numbered accordingly. Images were collected at 3.2-s intervals. The figure represents the sum of 3 independent experiments for a total of 15 cytoplasm FLIP measurements. Correction for photobleaching due to acquisition was done for each time point after background correction. For each condition, FLIP recovery curves were averaged. Bars = 10 μm (B and D) and 50 μm (C).
Thus, despite an apparent static behavior of BDV-P as initially suggested by real-time imaging, our FRAP and FLIP data indicate a permanent trafficking of this protein between the different cellular compartments, albeit with a lower mobility of a fraction of BDV-P in vSPOTs, presumably resulting from its association with viral and/or cellular proteins.
Analysis of BDV cell-to-cell transfer.
In order to test whether rBDV-P-Flag-TCT would allow imaging BDV transfer between cells, we analyzed FlAsH-labeled infected Vero cells for longer periods using a temperature-controlled confocal microscope setting. Since we had observed very dynamic movements of fluorescent material in the cytoplasm of persistently infected Vero cells (Fig. 4A), we first sought to investigate this finding further. We observed that viral transfer between adjacent infected cells actually occurred, preferentially through cytoplasmic projections connecting two cells. The transferred viral material was thereafter progressively routed toward the nuclei of the target cells (Fig. 6A to C; see also Video S2 in the supplemental material). Interestingly, viral transfer was still observed in cells treated with cytochalasin D, despite efficient disruption of the actin network, suggesting that cell-to-cell BDV transfer is not absolutely dependent on cytoskeleton rearrangements (Fig. 7).
Fig 6.

Real-time imaging of BDV intracellular traffic in Vero cells. (A to C) Visualization of viral transfer between two infected Vero cells. Persistently infected Vero cells were FlAsH labeled and imaged for 10 h by confocal microscopy. Images were acquired every 4 min. FlAsH-labeled viral material (arrow) was transmitted through a cytoplasmic extension of a neighboring cell and was progressively routed toward the nucleus of the cell shown. (D to F) Visualization of BDV traffic during early stages of infection. Noninfected Vero cells transfected with an mRFP-Cherry expression plasmid (red) were cocultivated with FlAsH-labeled BDV-infected Vero cells (green). Thereafter, this coculture was imaged by confocal microscopy for 6 h. Images were acquired every 4 min. The arrow points to viral material that was transmitted to the noninfected cell and that was progressively routed toward its nucleus throughout the observation period. Bar = 10 μM.
Fig 7.
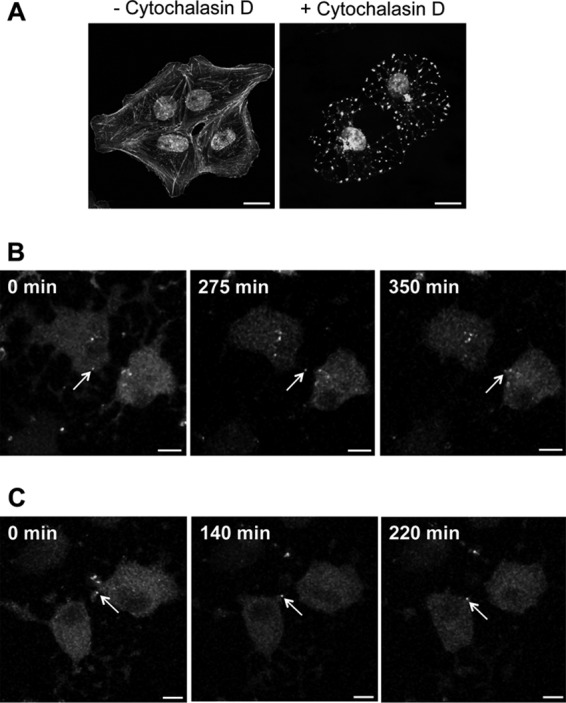
Cytoskeleton rearrangement is not essential for BDV cell-to-cell spread. Vero cells persistently infected by rBDV-P-Flag-TCT were labeled with FlAsH and treated with 5 μM cytochalasin D throughout live-cell imaging. (A) Validation of cytochalasin D impact on the cytoskeleton network. Cells were treated for different times (1 to 6 h, results being identical in all cases) and labeled with Alexa 488-conjugated phalloidin. Bar = 20 μm. (B and C) Two representative examples of cell-to-cell BDV spread using cytochalasin D-treated, FlAsH-labeled BDV-infected Vero cells. These cultures were imaged by confocal microscopy for 6 h. Images were acquired every 4 min. The arrows point to viral material that was transmitted between cells and that was progressively routed toward the nucleus throughout the observation period. Bar = 10 μm.
We then sought to analyze the early steps of BDV infection. The low yield of cell-free viral particles that characterizes BDV infection (30) was very limiting for performing this analysis with FlAsH-labeled, cell-free virus. Rather, we transiently transfected noninfected Vero cells with an mRFP-Cherry-expressing plasmid and cocultivated these cells with FlAsH-labeled infected Vero cells. Viral transfer between infected and noninfected cells was evidenced by the appearance of green fluorescent material in the red cells. Real-time imaging revealed that viral transfer first occurred at the contact with the infected cells and was followed by a progressive movement of viral complexes toward the nuclear area (Fig. 6D to F; see also Video S3 in the supplemental material).
DISCUSSION
In this work, we have sought to generate infectious rBDV bearing a fluorescent tag fused to one of the components of the RNP. We succeeded in generating a recombinant virus bearing a TCT on BDV-P, which was stable and readily established a persistent infection of susceptible cells, albeit with a delayed infection rate compared to that of recombinant wild-type virus. Upon optimized biarsenical labeling, we were able to perform real-time fluorescence imaging of BDV-P kinetics in living cells. Notably, we measured the kinetics of BDV-P in the nuclei and cytoplasm of infected cells using photobleaching techniques (FRAP and FLIP), including a new 2-photon-based FLIP approach suited to imaging BDV-P dynamics in viral nuclear inclusions (vSPOTs). The novelty of our study lies in the first description of BDV dynamics in the context of virus infection and on the visualization of BDV trafficking in live, infected cells.
Initially, we wished to generate a recombinant BDV bearing a GFP fused in frame with either BDV-N, -P, or -L, but none of these recombinant viruses could be rescued, presumably due to the compact nature of the BDV genome and its complex transcription map. Even addition of a smaller tag such as TCT, which supposedly can be fused to the protein of interest with minimal disruption of protein function, proved to be intrinsically difficult for BDV. Indeed, insertion of TCT yielded an infectious recombinant virus only when it was inserted together with a Flag tag. The reason for this is unclear, and it may be the result of a spacer function of the Flag tag. Alternatively, the addition of the highly charged and hydrophilic Flag tag may have compensated the charge of the tetracysteine tag, thereby leading a successful rescue. The recent description of a new recombinant BDV bearing an additional transcription cassette in the intercistronic noncoding region between the BDV-P and -M genes (23) may provide new leads to insert fluorescent fusion proteins, since this vector appears to be more stable than the previously reported backbones used for BDV rescue.
Although FlAsH/TCT-based labeling of recombinant viruses has proven its considerable utility for tracking viral entry and trafficking, it often requires that the TC-tagged virus be grown at high titers and purified after labeling prior to analysis (21). The paucity of cell-free virus associated with BDV infection and the inability to grow cell-free infectious BDV to high titers do not allow adoption of such a strategy. Thus, FlAsH labeling and imaging of TCT-tagged BDV need to be performed on persistently infected cells. In this case, the intracellular background staining due to binding of the FlAsH reagent to off-target cysteine-rich proteins represents a considerable issue, as revealed by our initial attempts using the labeling conditions recommended in the commercial kit. This issue has also been encountered by others, for example, when trying to label amyloid precursor or prion proteins, or to track hepatitis C virus (HCV) core or HIV Gag proteins, and these groups have developed modified and optimized staining conditions or new imaging techniques to circumvent this problem (31–33). To achieve a satisfying signal-to-noise ratio, we also developed our own optimized FlAsH labeling protocol that satisfactorily allowed live BDV imaging in persistently infected Vero cells. To date, it remains, however, to be determined whether such optimized staining conditions will also be applicable to any type of cells or to other infection models. Alternatively, it may be interesting in the future to test other approaches to generate fluorescent BDV. One attractive alternative may be the split-GFP technology, as elegantly described recently for influenza virus and its cotransportation with the cellular protein Rab11 (34).
We were particularly interested in obtaining further insight on the dynamics of BDV components in the nuclear vSPOTs, because it was recently shown that they represent viral nuclear factories associated with the host cell chromatin (17). FRAP analysis revealed that BDV-P is mobile but that part of this protein remains tightly associated with vSPOTs. Interestingly, the diffusion coefficient measured for BDV-P was in the same range as those reported for nucleosomal binding proteins (28). This finding is consistent with previous results showing that BDV interacts with high-mobility-group box protein 1 (HMGB-1), a nonhistone DNA architectural protein (17, 35, 36), and further suggests that shuttling of BDV RNP involves dynamic interaction with this cellular protein and/or perhaps with other cellular factors yet to be identified. Moreover, FLIP analysis demonstrated that a permanent shuttling of BDV-P between vSPOTs occurs, as well as with the other cellular compartments.
Long-term live imaging of persistently infected cells revealed very dynamic movements of viral material occurring at cell contacts. Surprisingly, we observed that viral components, presumably RNP, could be exchanged between cells, and video capture of these events suggested that viral exchange might result from microfusion events occurring at contact sites between cells. The determinants of these events, notably the potential contribution of BDV surface glycoprotein (G), remain to be fully deciphered and could be addressed using BDV-G neutralizing antibodies (37). Even more surprising was the fact that the transferred material was routed toward the nucleus of the neighboring cell, because it was shown previously that BDV-infected cells are resistant to superinfection (38, 39). It is likely, however, that infection will not proceed further because of unbalanced intracellular levels of viral nucleocapsid components.
It has long been postulated that BDV could spread in the form of bare RNP (40, 41). In agreement with this hypothesis, our coculture experiments using infected and noninfected cells revealed a sizeable cell-to-cell transfer of viral material, although it remains to be shown whether this material represents viral RNPs. This process was, however, not seen in all noninfected neighboring cells. At present, it is difficult to assess whether the relative inefficiency of this cell-to-cell BDV transfer actually reflects the situation of natural infection or if it is due to the particular phenotype of our recombinant virus. In any event, and similar to what was observed in persistently infected cells, this viral material was thereafter routed toward the nucleus, which is in agreement with a nuclear localization of BDV replication and transcription. Thus, this recombinant virus clearly allows the visualization of BDV cell-to-cell transfer and its progressive transport to the nucleus. Recently, a small interfering RNA (siRNA)-based functional genetic screen allowed the identification of host cell factors involved in BDV entry (42). It may be interesting in the future to probe the identified target genes by live imaging using our TC-tagged virus. This will allow assessing whether such genes play a role in cell-to-cell transfer or intracellular or even intranuclear dynamics of BDV RNP. In conclusion, our fluorescent BDV allowed us to image some characteristics of BDV life cycle and has provided the first description of BDV dynamics in persistently infected cells. We are currently examining whether this TC-tagged virus could be used to track BDV infection in neurons, the main target for BDV in the CNS.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported financially by grants from INSERM, the ANR (Agence Nationale de la Recherche, project ANR-10-Blanc-1322), the Fondation Recherche Médicale (“Equipe FRM 2009”) to D.G.-D., the CNRS (to D.G.-D.), and the DFG (to M.S). C.M.C. was the recipient of a doctoral fellowship from the French Ministry of Research. Y.-J.W. was supported by the Spemann Graduate School of Biology and Medicine, University of Freiburg, Germany.
We thank M. Fallet (CIML imaging facility, Marseille, France) for assistance in analysis of fluorescence recovery curves, D. Sapède (Carl Zeiss SAS, France) for expert assistance during the initial steps of imaging studies, and M. Szelechowski, R. Liblau, and A. Saoudi for critical reading of the manuscript.
Footnotes
Published ahead of print 11 September 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JVI.01127-13.
REFERENCES
- 1.de la Torre JC. 1994. Molecular biology of Borna disease virus: prototype of a new group of animal viruses. J. Virol. 68:7669–7675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schneemann A, Schneider PA, Lamb RA, Lipkin WI. 1995. The remarkable coding strategy of Borna disease virus: a new member of the nonsegmented negative strand RNA viruses. Virology 210:1–8 [DOI] [PubMed] [Google Scholar]
- 3.Briese T, Schneemann A, Lewis AJ, Park YS, Kim S, Ludwig H, Lipkin WI. 1994. Genomic organization of Borna disease virus. Proc. Natl. Acad. Sci. U. S. A. 91:4362–4366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cubitt B, Oldstone C, Valcarcel J, de la Torre JC. 1994. RNA splicing contributes to the generation of mature mRNAs of Borna disease virus, a non-segmented negative strand RNA virus. Virus Res. 34:69–79 [DOI] [PubMed] [Google Scholar]
- 5.Schneider PA, Schneemann A, Lipkin WI. 1994. RNA splicing in Borna disease virus, a nonsegmented, negative-strand RNA virus. J. Virol. 68:5007–5012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Poenisch M, Burger N, Staeheli P, Bauer G, Schneider U. 2009. Protein X of Borna disease virus inhibits apoptosis and promotes viral persistence in the central nervous systems of newborn-infected rats. J. Virol. 83:4297–4307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wensman JJ, Munir M, Thaduri S, Hornaeus K, Rizwan M, Blomstrom AL, Briese T, Lipkin WI, Berg M. 2013. The X proteins of Bornaviruses interfere with type I interferon signalling. J. Gen. Virol. 94:263–269 [DOI] [PubMed] [Google Scholar]
- 8.Ludwig H, Bode L. 2000. Borna disease virus: new aspects on infection, disease, diagnosis and epidemiology. Rev. Sci. Tech. 19:259–288 [DOI] [PubMed] [Google Scholar]
- 9.Staeheli P, Sauder C, Hausmann J, Ehrensperger F, Schwemmle M. 2000. Epidemiology of Borna disease virus. J. Gen. Virol. 81:2123–2135 [DOI] [PubMed] [Google Scholar]
- 10.Honkavuori KS, Shivaprasad HL, Williams BL, Quan PL, Hornig M, Street C, Palacios G, Hutchison SK, Franca M, Egholm M, Briese T, Lipkin WI. 2008. Novel Borna virus in psittacine birds with proventricular dilatation disease. Emerg. Infect. Dis. 14:1883–1886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kistler AL, Gancz A, Clubb S, Skewes-Cox P, Fischer K, Sorber K, Chiu CY, Lublin A, Mechani S, Farnoushi Y, Greninger A, Wen CC, Karlene SB, Ganem D, DeRisi JL. 2008. Recovery of divergent avian Bornaviruses from cases of proventricular dilatation disease: identification of a candidate etiologic agent. Virology J. 5:88. 10.1186/1743-422X-5-88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pletnikov M, Gonzalez-Dunia D, Stitz L. 2002. Experimental infection: pathogenesis of neurobehavioral disease, p 125–178 In Carbone K. (ed), Borna disease virus and its role in neurobehavioral disease. ASM Press, Washington, DC [Google Scholar]
- 13.Gonzalez-Dunia D, Volmer R, Mayer D, Schwemmle M. 2005. Borna disease virus interference with neuronal plasticity. Virus Res. 111:224–234 [DOI] [PubMed] [Google Scholar]
- 14.Clemente R, de la Torre JC. 2009. Cell entry of Borna disease virus follows a clathrin-mediated endocytosis pathway that requires Rab5 and microtubules. J. Virol. 83:10406–10416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gonzalez-Dunia D, Cubitt B, de la Torre JC. 1998. Mechanism of Borna disease virus entry into cells. J. Virol. 72:783–788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gonzalez-Dunia D, Cubitt B, Grässer FA, de la Torre JC. 1997. Characterization of Borna disease virus p56 protein, a surface glycoprotein involved in virus entry. J. Virol. 71:3208–3218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Matsumoto Y, Hayashi Y, Omori H, Honda T, Daito T, Horie M, Ikuta K, Fujino K, Nakamura S, Schneider U, Chase G, Yoshimori T, Schwemmle M, Tomonaga K. 2012. Bornavirus closely associates and segregates with host chromosomes to ensure persistent intranuclear infection. Cell Host Microbe 11:492–503 [DOI] [PubMed] [Google Scholar]
- 18.Finke S, Brzozka K, Conzelmann KK. 2004. Tracking fluorescence-labeled rabies virus: enhanced green fluorescent protein-tagged phosphoprotein P supports virus gene expression and formation of infectious particles. J. Virol. 78:12333–12343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Das SC, Nayak D, Zhou Y, Pattnaik AK. 2006. Visualization of intracellular transport of vesicular stomatitis virus nucleocapsids in living cells. J. Virol. 80:6368–6377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Griffin BA, Adams SR, Tsien RY. 1998. Specific covalent labeling of recombinant protein molecules inside live cells. Science 281:269–272 [DOI] [PubMed] [Google Scholar]
- 21.Whitt MA, Mire CE. 2011. Utilization of fluorescently-labeled tetracysteine-tagged proteins to study virus entry by live cell microscopy. Methods 55:127–136 [DOI] [PubMed] [Google Scholar]
- 22.Ackermann A, Guelzow T, Staeheli P, Schneider U, Heimrich B. 2010. Visualizing viral dissemination in the mouse nervous system, using a green fluorescent protein-expressing Borna disease virus vector. J. Virol. 84:5438–5442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Daito T, Fujino K, Honda T, Matsumoto Y, Watanabe Y, Tomonaga K. 2011. A novel Borna disease virus vector system that stably expresses foreign proteins from an intercistronic noncoding region. J. Virol. 85:12170–12178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schneider U, Schwemmle M, Staeheli P. 2005. Genome trimming: a unique strategy for replication control employed by Borna disease virus. Proc. Natl. Acad. Sci. U. S. A. 102:3441–3446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Soumpasis DM. 1983. Theoretical analysis of fluorescence photobleaching recovery experiments. Biophys. J. 41:95–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schneider U. 2005. Novel insights into the regulation of the viral polymerase complex of neurotropic Borna disease virus. Virus Res. 111:148–160 [DOI] [PubMed] [Google Scholar]
- 27.Manders EM, Stap J, Brakenhoff GJ, van Driel R, Aten JA. 1992. Dynamics of three-dimensional replication patterns during the S-phase, analysed by double labelling of DNA and confocal microscopy. J. Cell Sci. 103:857–862 [DOI] [PubMed] [Google Scholar]
- 28.Phair RD, Misteli T. 2000. High mobility of proteins in the mammalian cell nucleus. Nature 404:604–609 [DOI] [PubMed] [Google Scholar]
- 29.Seksek O, Biwersi J, Verkman AS. 1997. Translational diffusion of macromolecule-sized solutes in cytoplasm and nucleus. J. Cell Biol. 138:131–142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Carbone KM, Rubin SA, Sierra-Honigmann AM, Lederman HM. 1993. Characterization of a glial cell line persistently infected with Borna disease virus (BDV): influence of neurotrophic factors on BDV protein and RNA expression. J. Virol. 67:1453–1460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Taguchi Y, Shi ZD, Ruddy B, Dorward DW, Greene L, Baron GS. 2009. Specific biarsenical labeling of cell surface proteins allows fluorescent- and biotin-tagging of amyloid precursor protein and prion proteins. Mol. Biol. Cell 20:233–244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rudner L, Nydegger S, Coren LV, Nagashima K, Thali M, Ott DE. 2005. Dynamic fluorescent imaging of human immunodeficiency virus type 1 gag in live cells by biarsenical labeling. J. Virol. 79:4055–4065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Counihan NA, Rawlinson SM, Lindenbach BD. 2011. Trafficking of hepatitis C virus core protein during virus particle assembly. PLoS Pathog. 7:e1002302. 10.1371/journal.ppat.1002302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Avilov SV, Moisy D, Naffakh N, Cusack S. 2012. Influenza A virus progeny vRNP trafficking in live infected cells studied with the virus-encoded fluorescently tagged PB2 protein. Vaccine 30:7411–7417 [DOI] [PubMed] [Google Scholar]
- 35.Kamitani W, Shoya Y, Kobayashi T, Watanabe M, Lee BJ, Zhang G, Tomonaga K, Ikuta K. 2001. Borna disease virus phosphoprotein binds a neurite outgrowth factor, amphoterin/HMG-1. J. Virol. 75:8742–8751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang G, Kobayashi T, Kamitani W, Komoto S, Yamashita M, Baba S, Yanai H, Ikuta K, Tomonaga K. 2003. Borna disease virus phosphoprotein represses p53-mediated transcriptional activity by interference with HMGB1. J. Virol. 77:12243–12251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bajramovic JJ, Mn̈ter S, Syan S, Nerhbass U, Brahic M, Gonzalez-Dunia D. 2003. Borna disease virus glycoprotein is required for viral dissemination in neurons. J. Virol. 77:12222–12231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Formella S, Jehle C, Sauder C, Staeheli P, Schwemmle M. 2000. Sequence variability of Borna disease virus: resistance to superinfection may contribute to high genome stability in persistently infected cells. J. Virol. 74:7878–7883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Geib T, Sauder C, Venturelli S, Hassler C, Staeheli P, Schwemmle M. 2003. Selective virus resistance conferred by expression of Borna disease virus nucleocapsid components. J. Virol. 77:4283–4290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Clemente R, de la Torre JC. 2007. Cell-to-cell spread of Borna disease virus proceeds in the absence of the virus primary receptor and furin-mediated processing of the virus surface glycoprotein. J. Virol. 81:5968–5977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cubitt B, de la Torre JC. 1994. Borna disease virus (BDV), a nonsegmented RNA virus, replicates in the nuclei of infected cells where infectious BDV ribonucleoproteins are present. J. Virol. 68:1371–1381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Clemente R, Sisman E, Aza-Blanc P, de la Torre JC. 2010. Identification of host factors involved in Borna disease virus cell entry through a small interfering RNA functional genetic screen. J. Virol. 84:3562–3575 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.