

Abstract
Promyelocytic leukemia protein (PML) is an essential organizer of PML nuclear bodies (NBs), which carry out a variety of activities, including antiviral functions. Herpesviruses from all subfamilies encode proteins that counteract PML NB-mediated antiviral defenses by multiple mechanisms. However, because of the species specificity of herpesviruses, only a limited number of in vivo studies have been undertaken to investigate the effect of PML or PML NBs on herpesvirus infection. To address this central issue in herpesvirus biology, we studied the course of infection in wild-type and PML−/− mice using murine gammaherpesvirus 68 (MHV68), which encodes a tegument protein that induces PML degradation. While acute infection in PML−/− mice progressed similarly to that in wild-type mice, the lytic reactivation frequency was higher in peritoneal exudate cells, due to both an increase of MHV68 genome-positive cells and greater reactivation efficiency. We also detected a higher frequency of persistent infection in PML−/− peritoneal cells. These findings suggest that the PML protein can repress the establishment or maintenance of gammaherpesvirus latency in vivo. Further use of the PML−/− mouse model should aid in dissecting the molecular mechanisms that underlie the role of PML in gammaherpesvirus latency and may yield clues for how PML modulates herpesvirus latency in general.
INTRODUCTION
After primary infection, herpesviruses establish long-term chronic infections in their hosts (1–4). Chronic infections are maintained through three mechanisms: (i) alternating cycles of latency and reactivation, (ii) continuous replication, or (iii) invasion of the host genome, which permits vertical spread. Chronic infections caused by herpesviruses are typically associated with the first mechanism. To effectively establish long-term chronic infection, herpesviruses must navigate several host barriers that include intrinsic, innate, and adaptive immune responses (2, 5). A large body of evidence has implicated promyelocytic leukemia nuclear bodies (PML NBs) as an important mediator of intrinsic resistance to herpesviruses. These structures are organized primarily by the PML protein itself and contain several other proteins (6), such as speckled protein 100 (Sp100), Daxx, and the small ubiquitin-like modifier (SUMO) (7), which also contribute to intrinsic antiviral resistance. Consistent with its role as an antiviral factor, PML is an interferon-inducible protein (8) whose absence in cells and mice increases susceptibility to viral infections (9–11). Many RNA and DNA viruses, including all three (alpha-, beta-, and gamma-) herpesvirus subfamilies, have evolved different mechanisms to disrupt PML NBs after infection (12). Examples of human-herpesvirus regulatory proteins that induce PML degradation or PML-NB disruption are herpes simplex virus 1 (HSV-1) ICP0 (13); varicella-zoster virus (VZV) ORF61 (14); human cytomegalovirus (CMV) IE1 (15); Epstein-Barr virus (EBV) proteins EBNA1, Zta, and EBNA-LP (16–18); and Kaposi's sarcoma-associated herpesvirus (KSHV) LANA2 (19). The disruption of PML NBs correlates with an increase of viral gene expression and plaque formation during herpesvirus infection in cell culture systems (20–22), but due to the species specificity of these viruses, only a limited number of in vivo studies (14, 23) have been undertaken to investigate the effect of PML or PML NBs on the establishment, maintenance, and reactivation of latent herpesvirus infection.
Murine gammaherpesvirus 68 (MHV68) is closely related to the human gammaherpesviruses EBV and KSHV (24). It naturally infects inbred laboratory mice and can cause lymphoproliferative disease similar to that induced by human gammaherpesviruses (24, 25). Following intranasal infection, MHV68 transiently replicates in the respiratory tract and spleen and is cleared by 2 weeks postinfection. Latent infection is established in the spleen (B cells and dendritic cells) and peritoneum (macrophages). At 42 days postinfection, splenocytes and peritoneal exudate cells (PECs) harbor latent virus, which can spontaneously reactivate in cell culture (ex vivo reactivation assay). Typically, ongoing virus replication, which we refer to as persistent infection, is not detected or is very low at this time point. Persistent infection can be due to ongoing replication in cells following infection or from reactivation of latent virus that was occurring at the time when cells were explanted from the animal. We have shown that MHV68 induces the proteasome-dependent degradation of PML within 3 h postinfection in murine fibroblast cells and replicates more efficiently in PML−/− fibroblasts than in wild-type fibroblasts. Furthermore, we have determined that the viral tegument protein ORF75c, a homolog of the cellular formylglycinamide ribonucleotide amidotransferase (FGARAT) gene, potentially mediates MHV68-induced PML degradation by an intrinsic ubiquitin E3 ligase activity that specifically targets PML for polyubiquitination (9, 26).
These basic observations led us to use MHV68 as a model system to interrogate how PML modulates gammaherpesvirus infections in vivo. Surprisingly, viral replication in lungs or spleens during acute infection was not different in PML−/− mice relative to wild-type mice. Likewise, the development of splenomegaly and the proportions of lymphoid and mononuclear cells in the spleen were similar between PML−/− and wild-type infected mice. In contrast, MHV68 genome-positive peritoneal cells were more abundant in infected PML−/− mice than in wild-type mice and could be reactivated more efficiently. Our results indicate that PML can modulate establishment and reactivation of latent gammaherpesvirus infection.
MATERIALS AND METHODS
Cells.
Vero-CRE cells, 3T12 cells (ATCC; catalog no. CCL-164), and mouse embryonic fibroblasts (MEFs) (ATCC; catalog no. SCRC-1008) were grown in Dulbecco's modified Eagle's medium (DMEM)-high glucose (HyClone) with 10% fetal bovine serum (FBS) (Gibco) and 1× antibiotic-antimycotic (Gibco), which we refer to as complete MEM (cMEM). All cells were grown in tissue culture incubators at 37°C and 5% CO2.
Virus and mouse infections.
MHV68 stocks were generated by transfecting the MHV68-bacterial artificial chromosome (BAC) DNA into Vero-CRE cells using jetPRIME (Polyplus transfection) (26). Viruses were harvested as P0 stock when about 50% of transfected cells showed cytopathic effects (CPE). To make a large P1 stock, three 15-cm dishes of 3 × 106 3T12 cells were infected with 50 μl P0 stock per dish and the virus was harvested 5 days postinfection. The titer of P1 virus stocks was determined as described previously (9, 26). Wild-type 129S1/SVImJ mice were purchased from the Jackson Laboratory (Bar Harbor, ME). PML-null (129/SV-Pmltm1Ppp) mice (27, 28) were obtained from the NCI-Charles River Laboratory (strain 01XF8). Each mouse was confirmed to be PML−/− via PCR and free of mouse norovirus (MNV). Vetted mice were bred at Baylor College of Medicine in accordance with all federal and university guidelines. Female mice between 6 and 8 weeks of age were anesthetized for a short period of time and inoculated intranasally with 100 PFU of MHV68 diluted in 20 μl cold cMEM, except for the 50% infective dose (ID50) experiment in which 1, 10, and 100 PFU of MHV68 were used (Table 1). Mice were euthanized at different time points by cervical disarticulation under isoflurane anesthesia and weighed by a portable balance (Sartorius M-power; no. TE313S), and organs were then harvested and weighed. For determining viral titers, lungs and spleens from each mouse were placed in microcentrifuge tubes with silicone O-rings (VWR; catalog no. 89004-308) that contained 0.2 ml of 1-mm-diameter zirconia-silica beads (BioSpec) and 1 ml of cold cMEM. Organs were homogenized with a mini-BeadBeater (BioSpec) 4 times for 1 min each time before plating the homogenates in plaque assays. The animal research protocol was approved by the Institutional Animal Care and Use Committees at Baylor College of Medicine, Houston, TX.
Table 1.
Calculation of ID50 for MHV68 intranasal inoculation of 129S1/SVImJ miceb
| Virus dilution (PFU) | No. of infected test units (mice)a | Cumulative infected (A) | Cumulative uninfected (B) | Ratio of A/(A + B) | % infected |
|---|---|---|---|---|---|
| 100 | 10/10 | 17 | 0 | 17/17 | 100.00 |
| 10 | 6/10 | 7 | 4 | 7/11 | 63.64 |
| 1 | 1/10 | 1 | 13 | 1/14 | 7.14 |
Infection was determined by plaque assay of the lungs harvested at 8 days after inoculation.
Proportionate distance = [(% positive above 50%) − 50%]/[(% positive above 50%) − (% positive below 50%)]; proportionate distance = (63.64 − 50)/(63.64 − 7.14) = 0.24. Log ID50 = (log dilution above 50%) + (proportionate distance × log dilution factor) = 1 + (0.24 × −1) = 0.76; ID50 = 100.76 = 5.75 PFU.
Flow cytometry analysis.
Each spleen was collected in cold cMEM and mashed against a 100 μm nylon cell strainer (BD Falcon). The cells were spun down, and 1 ml of red blood cell lysing buffer (Sigma) was added for 3 min before being neutralized with 11 ml of cold cMEM. The cells were spun down again and resuspended in fluorescence-activated cell sorting (FACS) buffer (phosphate-buffered saline [PBS] with 1% FBS). Then, the cells were filtered again through a 100 μm nylon cell strainer before being counted and stained with antibodies diluted in 0.1 ml FACS buffer. Phycoerythrin (PE) rat anti-mouse CD3 (BD Pharmingen; no. 555275), allophycocyanin (APC) rat anti-mouse CD4 (BD Pharmingen; no. 553051), fluorescein isothiocyanate (FITC) rat anti-mouse CD8a (BD Pharmingen; no. 553031), FITC rat anti-mouse CD19 (BD Pharmingen; no. 557398), and APC-eFluor780 rat anti-mouse F4/80 (eBioscience; no. 47-4801) were used to stain for subpopulations of spleen cells. After antibody staining, the cells were washed with 1 ml FACS buffer, fixed with 0.5 ml 4% paraformaldehyde in PBS for 10 min, and resuspended in 0.5 ml PBS. Data were collected with a BD LSRII cell analyzer and analyzed by FlowJo software at the Cytometry and Cell Sorting Core Facility, Baylor College of Medicine.
Ex vivo reactivation assay.
Spleens and peritoneal exudate cells (PECs) from four infected mice per group were harvested and pooled in cold cMEM. Spleens were mashed against a 100 μm nylon cell strainer (BD Falcon) and spun down, and 4 ml of red blood cell lysing buffer (Sigma) was added for 3 min before being neutralized with 45 ml of cold cMEM. Splenocytes and PECs were spun down again, resuspended in 15 ml and 5 ml cold cMEM, respectively, and counted (for each cell sample, 2 to 5 vials of 5 × 106 cells were saved and stored at −80°C for limiting-dilution nested-PCR assay). To detect virus reactivation, serial 2-fold dilutions of splenocytes or PECs were added onto monolayers of MEFs that had been plated in 96-well flat-bottom tissue culture plates 2 days previously at a density of 5 × 103 cells/well. Twenty-four wells of the MEF monolayers were used for each dilution of cells, starting from 1 × 105 cells/well to 50 cells/well as described previously (5, 29). Cells were incubated in a 5% CO2 tissue culture incubator at 37°C for 21 days, and wells scoring positive for CPE for each dilution were counted. To determine the amount of preformed infectious virus in cell samples, similar amounts of cells were aliquoted into microcentrifuge tubes with silicone O-rings (VWR) that contained 0.2 ml of 0.5-mm-diameter zirconia-silica beads (BioSpec) and were mechanically disrupted by using a mini-BeadBeater (BioSpec) 4 times for 1 min each before the serial 2-fold dilutions were added onto MEF monolayers, in a manner similar to that for nondisrupted cells.
Limiting-dilution nested-PCR assay.
Genome frequency was determined as described previously (5, 30, 31). Briefly, PECs from infected mice were thawed, washed once with IsoLB buffer (10 mM Tris [pH 7.5], 150 mM KCl, and 1.5 mM MgCl2), and resuspended with 0.2 ml IsoLB buffer. Cells were counted, and five serial 3-fold dilutions were performed, starting at 2 × 106 cells/ml (1 × 104 cells/PCR). For each cell dilution, 5 μl of diluted cells was added into 12 wells of a semiskirted 96-well PCR plate (Eppendorf) that contained 5 μl of 0.2-mg/ml proteinase K (PK) (Ambion) in PK diluent buffer (10 mM Tris [pH 8.5], 1.5 mM MgCl2, 1% NP-40, and 1% Tween 20) and 5 μl of 2 × 106 3T12 cells/ml (1 × 104 cells/PCR) as a background. Two microliters of pBamHI-N plasmid (30, 32), at concentrations of 5, 0.5, and 0.05 copies/μl, was added to the 3T12 cell background (6 wells/each concentration) as positive controls for the MHV68 ORF50 gene. pBamHI-N standards were prepared in water containing 0.1-mg/ml baker's yeast tRNA (Sigma; no. R8508). Another 6 wells containing only 3T12 cell background and PK were included as negative controls. Reaction mixtures were incubated at 56°C for 6 h to lyse the cells, and then PK was inactivated at 95°C for 20 min. Nested PCR was then performed. For the first round of PCR, 10 μl of a mixture containing 20 ng of each primer (5′-AAC TGG AAC TCT TCT GTG GC-3′ and 5′-GGC CGC AGA CAT TTA ATG AC-3′), 0.4 mM deoxynucleoside triphosphates (dNTPs) (New England BioLabs), 1 unit of Taq DNA polymerase (Promega), 10 mM Tris (pH 9.0), 50 mM KCl, and 1.5 mM MgCl2 was added to each reaction mixture. PCR was performed on a Mastercycler Pro (Eppendorf) with 45 cycles of 94°C for 30 s, 60°C for 45 s, and 72°C for 45 s, followed by 72°C for 5 min. The second round of PCR was done by adding another 10-μl mixture, which contained 40 ng of each primer (5′-CCC CAA TGG TTC ATA AGT GG-3′ and 5′-ATC AGC ACG CCA TCA ACA TC-3′), 0.6 mM dNTPs, 1 unit of Taq DNA polymerase, 30 mM Tris (pH 9.0), 150 mM KCl, 4.5 mM MgCl2, and 1.5% Triton X-100. PCR was then performed for another 30 cycles with the same PCR conditions as those used for the first round. The reaction mixtures containing a positive 382-bp PCR product were detected by ethidium bromide staining after electrophoresis on a 2% agarose gel.
Statistical analysis.
Analysis of variance (two-way ANOVA) was used to test the significance of white blood cell populations in spleens from wild-type and PML−/− mice at 19 days postinfection. These analyses were performed with Prism5 (GraphPad Software, Inc.). The generalized linear model for limiting-dilution experiments (33) was performed with R software to calculate the frequency of reactivation, number of MHV68-genome positive cells, and level of persistent infection.
RESULTS
Absence of PML does not alter the course of acute infection.
In designing experiments to investigate how PML modulates MHV68 infection in vivo, we were concerned that infection with excessively high viral doses might mask any effects of the protein. We therefore sought to determine the minimal amount of virus needed, following intranasal infection, to ensure that most of the mice became infected. Wild-type 129S1/SVImJ mice were inoculated intranasally with viral doses of 100, 10, and 1 PFU. Successful establishment of infection was determined by plaque assays of lung tissue harvested from mice at 8 days postinoculation. All mice were infected at a dose of 100 PFU, and the ID50 was 5.75 PFU, as calculated by the Reed-Muench method (34) (Table 1). Hence, for this study, the mice were inoculated intranasally with 100 PFU of MHV68. In five different experiments using 100 PFU intranasal inoculation in a total of 103 PML−/− and wild-type mice each, we had a 2% rate of uninfected mice in both groups (data not shown). To begin to define the role of PML during acute infection with MHV68, we studied viral replication in the lungs and spleens as well as the development of splenomegaly (a sign of lymphoproliferative responses during primary infection) (35, 36), in PML−/− mice compared with wild-type mice on days 7 to 22 postinfection. Neither the viral titers (Fig. 1A) nor the development of splenomegaly (Fig. 1B) in these animals differed significantly over the observation period. To determine whether the cellular components contributing to splenomegaly might be different in the absence of PML, we measured the sizes of T, B, and macrophage cell populations from the spleens of infected mice by flow cytometry. The relative percentages of these cell types were similar between uninfected PML−/− and wild-type mice (Fig. 1C). MHV68 infection significantly reduced the proportion of B cells in spleens from infected wild-type and PML−/− mice probably because of significant increases in non-T, non-B, or nonmacrophage cell types, which we classified as “other” (Fig. 1C). We also have completed an analysis of serum cytokine levels (Milliplex CytoChemo 22-plex) during infection between PML−/− mice and wild-type mice and found no statistically significant differences for 22 cytokines, including gamma interferon (IFN-γ), interleukin 2 (IL-2), IL-6, and IL-10, which have been shown previously to be induced during MHV68 infection (37) (data not shown).
Fig 1.

Acute-phase replication in PML−/− and wild-type mice. (A) Age-matched 129/SV-Pmltm1Ppp (PML−/−) and PML+/+ mice were infected with 100 PFU MHV68 intranasally, and spleen and lung were harvested at 7, 10, 13, 16, 19, and 22 days postinoculation. The virus present in each organ was quantitated by plaque assay. Virus titers are shown as mean log titers ± standard deviation per milligram of organ from at least three independent experiments and a total of 6 to 12 mice. (B) The average spleen size from mice in panel A was measured as the percentage of spleen weight to total body weight. Spleen sizes are shown as the means ± standard deviations. (C) Percentages of four types of white blood cells (CD3+/CD4+, CD3+/CD8+, CD19+, and F4/80+ cells) in spleens from mock- and MHV68-infected PML−/− and PML+/+ mice at 19 days postinoculation. Five mice were used for each group. There was a statistically significant difference in the percentages of CD19+ cells in PML+/+ infected mice and in uninfected PML+/+ mice (P < 0.0003) and in uninfected PML−/− and infected PML−/− mice (P < 0.04). Likewise, there was a statistically significant increase in non-B, non-T, or nonmacrophage cells, designated “other,” between uninfected and infected PML+/+ mouse spleens (P < 0.0002) and between uninfected and infected PML−/− mouse spleens (P < 0.004).
PML maintains latency and controls persistent infection by MHV68 in vivo.
We next investigated whether PML might affect chronic infection with MHV68 by comparing the reactivation frequencies, viral genome frequencies, and levels of persistent MHV68 infection in PML−/− and wild-type mice at 42 days after infection, when the mice are presumed to have resolved acute infection (38). Reactivation frequency was determined by ex vivo reactivation assays of splenocytes and peritoneal exudate cells (PECs), which have been identified as reservoirs of MHV68 latency (31, 39, 40). The frequency of reactivation among splenocytes was approximately 1 in 1 × 106 cells in both PML−/− and wild-type mice (Fig. 2A). However, the frequency of reactivation among PECs from PML−/− mice was about 3-fold higher than that in wild-type mice (Fig. 2A and Table 2, P < 0.001). Factors that may contribute to higher reactivation frequency include higher numbers of genome-positive PECs or the continuing presence of replicating virus, which has been defined as persistent replication (2–4). To determine viral genome frequency, PECs from the same cell pools used in the ex vivo reactivation assays were analyzed by limiting-dilution nested-PCR assays. Using this assay, the frequency of genome-positive PECs from PML−/− mice was determined to be about 1.55-fold higher than that in wild-type mice (Table 2, P = 0.03). Despite this increase in genome-positive cells, PML−/− PECs still reactivated at an efficiency nearly 2-fold higher than that seen in wild-type mice when normalized to viral genome frequency (Table 2). To distinguish between preformed infectious virus present in PECs and reactivating virus, cells were also mechanically disrupted before setting up the ex vivo reactivation assays. Although the frequency of reactivation from disrupted PECs was not high enough to significantly impact the frequency derived from nondisrupted cells, it did show that infected PML−/− mice had a frequency of persistent infection approximately 8-fold higher than that seen in wild-type-derived PECs (Fig. 2B and Table 2, P < 0.001). At this point, it is unclear whether preformed virus detected in these assays is due to a failure to maintain latency in infected PECs in vivo (leading to higher levels of reactivation in vivo) or whether increased levels of preexisting infectious virus were due to ongoing replication after infection because PML generally helps promote establishment of the latent state. Together, these results indicate that the presence of PML reduces reactivation efficiency and persistent infection in peritoneal cells, which are a major reservoir for MHV68 latency in vivo.
Fig 2.
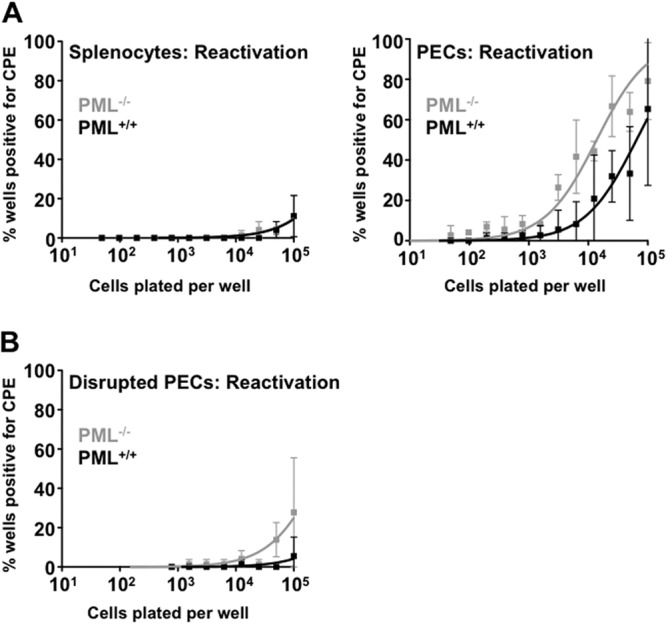
PML represses MHV68 reactivation and persistent infection. (A) Ex vivo reactivation from latency. Limiting-dilution ex vivo reactivation of virus in intact splenocytes and peritoneal exudate cells (PECs) from PML−/− and PML+/+ mice at 42 days postinoculation. Each sample contained pooled cells from four infected mice. (B) Parallel samples from PECs in panel A were mechanically disrupted prior to culture to detect preformed virus. In both panels, the data are presented as means ± standard deviations of three independent experiments.
Table 2.
Increased frequencies of reactivation, MHV68-genome-positive cells, persistent infection, and reactivation efficiency in PECs from PML−/− mice at 42 days postinfection
| Mouse strain | Frequency of reactivation | Frequency of genome-positive cells | Reactivation efficiency (%)b | Frequency of persistent infection |
|---|---|---|---|---|
| PML−/− | 1/29,849 (1/25,319, 1/34,380)a | 1/848 (1/619, 1/1,078)a | 2.84% | 1/351,445 (1/238,334, 1/464,556)a |
| PML+/+ | 1/84,050 (1/68,665, 1/99,435)a | 1/1,316 (1/958, 1/1,673)a | 1.57% | 1/2,827,263 (1/352,524, 1/5,302,002)a |
| Fold increase | 2.8 (P < 0.001) | 1.55 (P = 0.03) | 1.8 | 8 (P < 0.001) |
Values in parentheses are upper and lower limits (95% confidence intervals).
Reactivation efficiency (%) = (frequency of reactivation/frequency of genome-positive cells) × 100.
DISCUSSION
Here, we demonstrate that PML can exert a repressive effect on establishment of the latent state and reactivation of the MHV68 strain of gammaherpesvirus (summarized in Fig. 3). This conclusion is based on comparisons of these endpoints in peritoneal cells from PML−/− mice with those in wild-type mice (Fig. 2 and Table 2). Surprisingly, we did not observe any significant differences in the progression of acute infection between PML−/− and wild-type mice (Fig. 1). Our findings provide the first direct evidence for the regulation of herpesvirus latency by PML within a tractable animal model system that overcomes the species specificity limits of pathogenesis and immunity studies with human herpesviruses.
Fig 3.
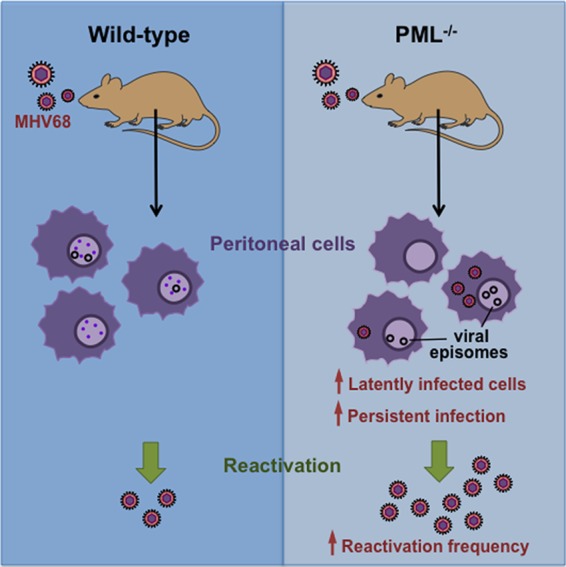
Graphic summary of the differences in establishment and reactivation from latency after MHV68 infection in wild-type and PML-null mice.
Several cell culture systems have implicated PML as a repressor of primary replication and reactivation of HSV from latency (41, 42). Fewer studies have extended these observations into animal model systems. Studies with HSVs containing a deletions or mutations of ICP0, whose product mediates PML degradation, indicated that such viruses are defective for both primary infection in mice and latency reactivation from mouse neurons (43–45). However, ICP0 acts on multiple cellular pathways in addition to PML (e.g., RNF8 and IRF3) (46, 47), making it difficult to interpret the contributions of PML to the observed phenotypes of infection in vivo. In one study, however, PML−/− mice apparently repressed replication of an ICP0− HSV with the same kinetics as those in wild-type mice, which would suggest that PML might not exert a significant effect on HSV primary infection in vivo (23). Limitations of this earlier study are that it did not compare the mutant virus with wild-type virus infection, nor did it compare different viral doses or routes of infection. After corneal inoculation with HSV in mice, a second study showed that PML appeared to limit the number of viral genomes that establish latency in trigeminal ganglion (48), which is consistent with observations made in this investigation. A human skin xenograft-SCID mouse model was used to investigate the effects of PML on VZV infection (14). ORF61, the VZV homolog to HSV ICP0, is known to disrupt PML NBs (14). Mutation of the ORF61 SUMO-interacting motifs (SIMs) resulted in viruses unable to disrupt PML NBs, and viral replication was severely impaired in this infection model system. However, to our knowledge, no in vivo studies have been undertaken to directly assess the contribution of PML to herpesvirus establishment and reactivation from latency.
An unexpected finding was that acute infection in PML−/− mice progressed similarly to that in wild-type mice. One possible explanation is that ORF75c induces complete degradation of PML within 3 h postinfection, essentially converting infected PML-positive cells to PML-null cells (9, 26). Hence, the absence of PML in the PML knockout mice does little to enhance virus infection. One strategy to explore this idea would be to generate MHV68 viruses that express a mutant form of ORF75 that is unable to degrade PML and then compare the pathogenesis of this virus in PML−/− and wild-type mice. We have successfully generated a mutant virus that possesses these characteristics and have used it to show that MHV68 encodes at least one other protein that can disrupt (but not degrade) PML NBs, an action that is normally masked because ORF75c generally degrades the bulk of PML within a cell (unpublished results). Whether this protein acts during reactivation from latency or under other circumstances when wild-type ORF75c is not usually expressed remains unknown. Nonetheless, further studies are needed to generate MHV68 recombinant viruses with mutations in a second viral protein, leaving PML NBs intact so that the role of PML can be further assessed during acute infection in this model system.
Another unusual finding is that the effects of PML on latency and reactivation appeared to be confined to peritoneal cells and did not extend to splenic cells. Previous studies have shown that at 42 days postinfection, the majority of infected splenocytes are latently infected B cells, and by 3 months postinfection, these latently infected cells are confined to memory B cells (49, 50). The peritoneum is the other major latency site that has been extensively characterized, where macrophages are the major cell type harboring latent virus (31). It is noteworthy that B cells are not required for the establishment of long-term chronic infection in mice, suggesting that macrophages serve as a critical site for maintaining chronic long-term infection. Given the central importance of macrophages for long-term chronic infection, it is perhaps not too surprising that the absence of PML had the strongest observable effect on latency in this cell type.
The antiviral role of PML during herpesvirus infection in vivo has been relatively unexplored. This investigation in a well-established mouse model system demonstrates that PML modulates gammaherpesvirus reactivation and persistent infection in a natural host infection model. Because reactivation and persistent infection are common features of the human oncogenic gammaherpesviruses EBV and KSHV, further studies to determine the molecular mechanism(s) by which PML limits reactivation efficiency and the number of latently infected cells might provide new strategies to prevent gammaherpesvirus-associated cancers in humans and may provide conceptual benefits for the treatment of herpesvirus-associated disease in general.
ACKNOWLEDGMENTS
We thank Kathleen S. Gray and Samuel H. Speck for training and advice with mouse infections and limiting-dilution assays.
This work was supported by a scholarship from the Faculty of Medicine Siriraj Hospital, Mahidol University, Bangkok, Thailand, and a Baylor College of Medicine Comprehensive Cancer Training Program grant RP101499 from the Cancer Prevention and Research Institute of Texas (CPRIT) and NIH grant 5R01AI080681 (P.D.L.).
Footnotes
Published ahead of print 28 August 2013
REFERENCES
- 1.Pellett PE, Roizman B. 2007. The family Herpesviridae: a brief introduction, p 2479–2499 In Knipe DM, Howley PM, Griffin DE, Lamb RA, Martin MA, Roizman B, Straus SE. (ed), Fields virology, 5th ed. Lippincott Williams & Wilkins, Philadelphia, PA [Google Scholar]
- 2.Speck SH, Ganem D. 2010. Viral latency and its regulation: lessons from the gamma-herpesviruses. Cell Host Microbe 8:100–115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Virgin HW, Wherry EJ, Ahmed R. 2009. Redefining chronic viral infection. Cell 138:30–50 [DOI] [PubMed] [Google Scholar]
- 4.Jung JU, Speck SH. 2013. Insights into chronic gamma-herpesvirus infections. Curr. Opin. Virol. 3:225–226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tibbetts SA, van Dyk LF, Speck SH, Virgin HW. 2002. Immune control of the number and reactivation phenotype of cells latently infected with a gammaherpesvirus. J. Virol. 76:7125–7132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lallemand-Breitenbach V, de Thé H. 2010. PML nuclear bodies. Cold Spring Harbor Perspect. Biol. 2:a000661. 10.1101/cshperspect.a000661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Negorev D, Maul GG. 2001. Cellular proteins localized at and interacting within ND10/PML nuclear bodies/PODs suggest functions of a nuclear depot. Oncogene 20:7234–7242 [DOI] [PubMed] [Google Scholar]
- 8.Heuser M, van der Kuip H, Falini B, Peschel C, Huber C, Fischer T. 1998. Induction of the pro-myelocytic leukaemia gene by type I and type II interferons. Mediators Inflamm. 7:319–325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ling PD, Tan J, Sewatanon J, Peng R. 2008. Murine gammaherpesvirus 68 open reading frame 75c tegument protein induces the degradation of PML and is essential for production of infectious virus. J. Virol. 82:8000–8012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bonilla WV, Pinschewer DD, Klenerman P, Rousson V, Gaboli M, Pandolfi PP, Zinkernagel RM, Salvato MS, Hengartner H. 2002. Effects of promyelocytic leukemia protein on virus-host balance. J. Virol. 76:3810–3818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chee AV, Lopez P, Pandolfi PP, Roizman B. 2003. Promyelocytic leukemia protein mediates interferon-based anti-herpes simplex virus 1 effects. J. Virol. 77:7101–7105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Everett RD, Chelbi-Alix MK. 2007. PML and PML nuclear bodies: implications in antiviral defence. Biochimie 89:819–830 [DOI] [PubMed] [Google Scholar]
- 13.Everett RD, Freemont P, Saitoh H, Dasso M, Orr A, Kathoria M, Parkinson J. 1998. The disruption of ND10 during herpes simplex virus infection correlates with the Vmw110 and proteasome-dependent loss of several PML isoforms. J. Virol. 72:6581–6591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang L, Oliver SL, Sommer M, Rajamani J, Reichelt M, Arvin AM. 2011. Disruption of PML nuclear bodies is mediated by ORF61 SUMO-interacting motifs and required for varicella-zoster virus pathogenesis in skin. PLoS Pathog. 7:e1002157. 10.1371/journal.ppat.1002157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ahn JH, Hayward GS. 1997. The major immediate-early proteins IE1 and IE2 of human cytomegalovirus colocalize with and disrupt PML-associated nuclear bodies at very early times in infected permissive cells. J. Virol. 71:4599–4613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sivachandran N, Sarkari F, Frappier L. 2008. Epstein-Barr nuclear antigen 1 contributes to nasopharyngeal carcinoma through disruption of PML nuclear bodies. PLoS Pathog. 4:e1000170. 10.1371/journal.ppat.1000170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ling PD, Peng RS, Nakajima A, Yu JH, Tan J, Moses SM, Yang W-H, Zhao B, Kieff E, Bloch KD, Bloch DB. 2005. Mediation of Epstein-Barr virus EBNA-LP transcriptional coactivation by Sp100. EMBO J. 24:3565–3575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Adamson AL, Kenney S. 2001. Epstein-Barr virus immediate-early protein BZLF1 is SUMO-1 modified and disrupts promyelocytic leukemia bodies. J. Virol. 75:2388–2399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Marcos-Villar L, Lopitz-Otsoa F, Gallego P, Muñoz-Fontela C, González-Santamaría J, Campagna M, Shou-Jiang G, Rodriguez MS, Rivas C. 2009. Kaposi's sarcoma-associated herpesvirus protein LANA2 disrupts PML oncogenic domains and inhibits PML-mediated transcriptional repression of the survivin gene. J. Virol. 83:8849–8858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Everett RD, Rechter S, Papior P, Tavalai N, Stamminger T, Orr A. 2006. PML contributes to a cellular mechanism of repression of herpes simplex virus type 1 infection that is inactivated by ICP0. J. Virol. 80:7995–8005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang S, Long J, Zheng C-F. 2012. The potential link between PML NBs and ICP0 in regulating lytic and latent infection of HSV-1. Protein Cell 3:372–382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tavalai N, Papior P, Rechter S, Leis M, Stamminger T. 2006. Evidence for a role of the cellular ND10 protein PML in mediating intrinsic immunity against human cytomegalovirus infections. J. Virol. 80:8006–8018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Halford WP, Weisend C, Grace J, Soboleski M, Carr DJJ, Balliet JW, Imai Y, Margolis TP, Gebhardt BM. 2006. ICP0 antagonizes Stat 1-dependent repression of herpes simplex virus: implications for the regulation of viral latency. Virol. J. 3:44. 10.1186/1743-422X-3-44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Simas JP, Efstathiou S. 1998. Murine gammaherpesvirus 68: a model for the study of gammaherpesvirus pathogenesis. Trends Microbiol. 6:276–282 [DOI] [PubMed] [Google Scholar]
- 25.Nash AA, Dutia BM, Stewart JP, Davison AJ. 2001. Natural history of murine gamma-herpesvirus infection. Philos. Trans. R. Soc. Lond. B Biol. Sci. 356:569–579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sewatanon J, Ling PD. 2013. Murine gammaherpesvirus 68 ORF75c contains ubiquitin E3 ligase activity and requires PML SUMOylation but not other known cellular PML regulators, CK2 and E6AP, to mediate PML degradation. Virology 440:140–149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang ZG, Delva L, Gaboli M, Rivi R, Giorgio M, Cordon-Cardo C, Grosveld F, Pandolfi PP. 1998. Role of PML in cell growth and the retinoic acid pathway. Science 279:1547–1551 [DOI] [PubMed] [Google Scholar]
- 28.Wang ZG, Ruggero D, Ronchetti S, Zhong S, Gaboli M, Rivi R, Pandolfi PP. 1998. PML is essential for multiple apoptotic pathways. Nat. Genet. 20:266–272 [DOI] [PubMed] [Google Scholar]
- 29.Weck KE, Barkon ML, Yoo LI, Speck SH, Virgin HW., IV 1996. Mature B cells are required for acute splenic infection, but not for establishment of latency, by murine gammaherpesvirus 68. J. Virol. 70:6775–6780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Weck KE, Kim SS, Virgin HW, IV, Speck SH. 1999. B cells regulate murine gammaherpesvirus 68 latency. J. Virol. 73:4651–4661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Weck KE, Kim SS, Virgin HW, IV, Speck SH. 1999. Macrophages are the major reservoir of latent murine gammaherpesvirus 68 in peritoneal cells. J. Virol. 73:3273–3283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Efstathiou S, Ho YM, Minson AC. 1990. Cloning and molecular characterization of the murine herpesvirus 68 genome. J. Gen. Virol. 71:1355–1364 [DOI] [PubMed] [Google Scholar]
- 33.Bonnefoix T, Bonnefoix P, Callanan M, Verdiel P, Sotto JJ. 2001. Graphical representation of a generalized linear model-based statistical test estimating the fit of the single-hit Poisson model to limiting dilution assays. J. Immunol. 167:5725–5730 [DOI] [PubMed] [Google Scholar]
- 34.Condit RC. 2007. Principles of virology, p 39–40 In Knipe DM, Howley PM, Griffin DE, Lamb RA, Martin MA, Roizman B, Straus SE. (ed), Fields virology, 5th ed. Lippincott Williams & Wilkins, Philadelphia, PA [Google Scholar]
- 35.Sunil-Chandra NP, Efstathiou S, Arno J, Nash AA. 1992. Virological and pathological features of mice infected with murine gamma-herpesvirus 68. J. Gen. Virol. 73:2347–2356 [DOI] [PubMed] [Google Scholar]
- 36.Usherwood EJ, Ross AJ, Allen DJ, Nash AA. 1996. Murine gammaherpesvirus-induced splenomegaly: a critical role for CD4 T cells. J. Gen. Virol. 77:627–630 [DOI] [PubMed] [Google Scholar]
- 37.Sarawar SR, Cardin RD, Brooks JW, Mehrpooya M, Tripp RA, Doherty PC. 1996. Cytokine production in the immune response to murine gammaherpesvirus 68. J. Virol. 70:3264–3268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tibbetts SA, Loh J, Van Berkel V, McClellan JS, Jacoby MA, Kapadia SB, Speck SH, Virgin HW. 2003. Establishment and maintenance of gammaherpesvirus latency are independent of infective dose and route of infection. J. Virol. 77:7696–7701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sunil-Chandra NP, Efstathiou S, Nash AA. 1992. Murine gammaherpesvirus 68 establishes a latent infection in mouse B lymphocytes in vivo. J. Gen. Virol. 73:3275–3279 [DOI] [PubMed] [Google Scholar]
- 40.Flano E, Husain SM, Sample JT, Woodland DL, Blackman MA. 2000. Latent murine gamma-herpesvirus infection is established in activated B cells, dendritic cells, and macrophages. J. Immunol. 165:1074–1081 [DOI] [PubMed] [Google Scholar]
- 41.Efstathiou S, Preston CM. 2005. Towards an understanding of the molecular basis of herpes simplex virus latency. Virus Res. 111:108–119 [DOI] [PubMed] [Google Scholar]
- 42.Boutell C, Everett RD. 2013. Regulation of alphaherpesvirus infections by the ICP0 family of proteins. J. Gen. Virol. 94:465–481 [DOI] [PubMed] [Google Scholar]
- 43.Cai W, Astor TL, Liptak LM, Cho C, Coen DM, Schaffer PA. 1993. The herpes simplex virus type 1 regulatory protein ICP0 enhances virus replication during acute infection and reactivation from latency. J. Virol. 67:7501–7512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Leib DA, Coen DM, Bogard CL, Hicks KA, Yager DR, Knipe DM, Tyler KL, Schaffer PA. 1989. Immediate-early regulatory gene mutants define different stages in the establishment and reactivation of herpes simplex virus latency. J. Virol. 63:759–768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Halford WP, Schaffer PA. 2001. ICP0 is required for efficient reactivation of herpes simplex virus type 1 from neuronal latency. J. Virol. 75:3240–3249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lilley CE, Chaurushiya MS, Boutell C, Landry S, Suh J, Panier S, Everett RD, Stewart GS, Durocher D, Weitzman MD. 2010. A viral E3 ligase targets RNF8 and RNF168 to control histone ubiquitination and DNA damage responses. EMBO J. 29:943–955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lin R, Noyce RS, Collins SE, Everett RD, Mossman KL. 2004. The herpes simplex virus ICP0 RING finger domain inhibits IRF3- and IRF7-mediated activation of interferon-stimulated genes. J. Virol. 78:1675–1684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Catez F, Picard C, Held K, Gross S, Rousseau A, Theil D, Sawtell N, Labetoulle M, Lomonte P. 2012. HSV-1 genome subnuclear positioning and associations with host-cell PML-NBs and centromeres regulate LAT locus transcription during latency in neurons. PLoS Pathog. 8:e1002852. 10.1371/journal.ppat.1002852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Flano E, Kim I-J, Woodland DL, Blackman MA. 2002. Gamma-herpesvirus latency is preferentially maintained in splenic germinal center and memory B cells. J. Exp. Med. 196:1363–1372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Willer DO, Speck SH. 2003. Long-term latent murine gammaherpesvirus 68 infection is preferentially found within the surface immunoglobulin D-negative subset of splenic B cells in vivo. J. Virol. 77:8310–8321 [DOI] [PMC free article] [PubMed] [Google Scholar]