

Abstract
Crescents consisting of a single lipoprotein membrane with an external protein scaffold comprise the initial structural elements of poxvirus morphogenesis. Crescents enlarge to form spherical immature virions, which enclose viroplasm consisting of proteins destined to form the cores of mature virions. Previous studies suggest that the L2 protein participates in the recruitment of endoplasmic reticulum (ER)-derived membranes to form immature virions within assembly sites of cytoplasmic factories. Here we show that L2 interacts with the previously uncharacterized 42-amino-acid A30.5 protein. An open reading frame similar in size to the one encoding A30.5 is at the same genome location in representatives of all chordopoxvirus genera. A30.5 has a putative transmembrane domain and colocalized with markers of the endoplasmic reticulum and with L2. By constructing a complementing cell line expressing A30.5, we isolated a deletion mutant virus that exhibits a defect in morphogenesis in normal cells. Large electron-dense cytoplasmic inclusions and clusters of scaffold protein-coated membranes that resemble crescents and immature virions devoid of viroplasm were seen in place of normal structures. Crescent-shaped membranes were continuous with the endoplasmic reticulum membrane and oriented with the convex scaffold protein-coated side facing the lumen, while clusters of completed spherical immature-virion-like forms were trapped within the expanded lumen. Immature-virion-like structures were more abundant in infected RK-13 cells than in BS-C-1 or HeLa cells, in which cytoplasmic inclusions were decorated with scaffold protein-coated membrane arcs. We suggest that the outer surface of the poxvirus virion is derived from the luminal side of the ER membrane.
INTRODUCTION
Poxviruses comprise a large group of DNA viruses that infect vertebrates and invertebrates, are responsible for diseases of medical and veterinary importance, and are used as vectors to develop vaccines against infectious diseases and cancer (1). The transcending feature of poxvirus biology is the ability of these viruses to replicate entirely within the cytoplasm, a capability enabled by the encoding of proteins for transcription and replication of the DNA genome, a unique redox system, and assembly of a novel viral membrane. While there is now considerable understanding of many aspects of poxvirus replication, particularly for vaccinia virus (VACV), least is known about the initial steps of morphogenesis. The first recognizable structures are crescents comprised of a single lipoprotein membrane bilayer with an external honeycomb lattice composed of trimers of the D13 protein (2–7). Because the viral membrane displays no obvious continuity with a cellular organelle, a de novo origin was suggested (8). Subsequently, the intermediate compartment between the endoplasmic reticulum (ER) and Golgi membrane was considered the source of the crescent membrane based on the localization of certain viral proteins (9–12). Further studies, however, showed that proteins could traffic from the ER to the crescent membrane and that blockade of the secretory pathway from the ER to the Golgi apparatus did not perturb either crescent formation or their progression to immature virions (IVs) and mature virions (MVs) (13–15). The findings that the VACV L2 membrane protein is required for IV formation and that it is located at the edges of the crescents and the ER further support an ER origin of the viral membrane (16, 17). When expression of the open reading frame (ORF) encoding L2 was repressed or the ORF was deleted, morphogenesis was blocked and large dense inclusions, some of which had short crescent membranes apposed to the surface, formed. In addition, there were “empty IV-like” structures associated and in continuity with smooth ER membranes (17). These studies led us to suggest that L2 participates in the recruitment of ER-derived membranes to virus assembly sites for IV formation. Proteins that appear to be involved in the same or a similar step in morphogenesis as L2, based on the phenotypes of null mutants, will be referred to as viral-membrane assembly proteins (VMAPs). The VMAPS include L2 (16–18), A11 (19–21), H7 (22, 23), A6 (24, 25), and, as will be shown here, A30.5.
The present study arose during further characterization of the L2 protein. We discovered that a previously uncharacterized protein of 42 amino acids encoded by the A30.5L ORF of VACV copurified with L2. (Note that VACV ORFs are designated by a capital letter followed by a number and an “L” or “R” reflecting the direction of transcription; the “L” or “R” is omitted from the name of the corresponding protein.) Although not annotated in the genome sequences of many poxviruses because of their small size, ORFs at the same location as A30.5L were found in representatives of all chordopoxviruses. Here we provide the first characterization of the A30.5 protein and demonstrate that it interacts with L2, associates with the ER, participates in biogenesis of the IV, and is essential for replication, making it a bona fide VMAP. By interrupting IV formation, we demonstrate the direct formation of crescent membranes from the ER and locate the D13 scaffold on the luminal side. These data suggest that the outer surface of the MV is derived from the luminal side of the ER membrane.
MATERIALS AND METHODS
Cells and virus.
BS-C-1, HeLa, RK-13, and RK-A30.5-V5 cells were grown in minimum essential medium with Earle's salt (E-MEM) or Dulbecco's minimum essential medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 100 units of penicillin, and 100 μg of streptomycin per ml (Quality Biologicals, Gaithersburg, MD). The medium of RK-A30.5-V5 cells was supplemented with 300 μg/ml of Zeocin. The VACV Western Reserve (WR) strain and recombinant viruses vL2-HA (16), vA30.5-V5, and vΔL2R (17) were propagated as previously described (26, 27). The A30.5 deletion mutant vΔA30.5 was propagated in RK-A30.5-V5 cells. MVs were purified by sedimentation through a 36%-sucrose cushion followed by a 25- to 40%-sucrose gradient as described previously (26) and a CsCl gradient (28).
Antibodies.
The following antibodies were used: anti-HA.11 mouse monoclonal antibody (MAb) (Covance, Denver, PA) and V5 mouse MAb (Pierce Thermo Scientific); rabbit antisera to A17-N (29), A11 (19), A3 (unpublished data), D13 (B1) (30), L2 (16), A21 (31), L5 (32), H2 (33, 34), A27 (35), and L1 (36); and AB1.1 anti-D8 mouse MAb (37), anti-E3 mouse MAb (38), and mouse MAbs against A13 and A14 (39). Cellular proteins were detected with anti-protein disulfide isomerase (PDI) goat polyclonal IgG (Santa Cruz Biotechnology), anti-calnexin rabbit antibody (Covance, Emeryville, CA), and anti-glyceraldehyde-3-phosphate dehydrogenase (GAPDH) MAb (Covance, Emeryville, CA).
Chemical cross-linking.
HeLa cells were infected with 10 PFU/cell of either VACV strain WR, vL2-HA, or vA30.5-V5. At 10 h postinfection, the cells were washed twice with phosphate-buffered saline (PBS) and incubated with 1% paraformaldehyde for 30 min at room temperature (RT) and quenched with 5 mM glycine for 15 min.
Immunoprecipitation and mass spectrometry.
Cross-linked or non-cross-linked cells were lysed in IP buffer consisting of 1% Nonidet P-40, 50 mM Tris-HCl (pH 7.4), 150 mM NaCl, 1 mM EDTA, and protease inhibitor cocktail tablets (Roche Molecular Biochemicals) for 30 min on ice. The lysates were clarified by centrifugation at 16,000 × g for 15 min at 4°C. The lysates were precleared by incubation with protein G Dynabeads (Invitrogen Dynal A, Oslo, Norway) for 1 h at 4°C and incubated with anti-HA.11 MAb or anti-V5 MAb. The protein-antibody complexes were captured with protein G Dynabeads at 4°C for 1 h. Beads were washed three times with IP buffer and two times with PBS. Bound proteins were eluted with 2× NuPAGE lithium dodecyl sulfate sample loading buffer (Invitrogen) and heated to 70°C for 10 min. The proteins were resolved by electrophoresis in 4 to 12% NuPAGE Novex acrylamide gels (Invitrogen).
For mass spectrometry, proteins in gel slices were reduced, alkylated, and trypsin digested according to standard protocols. The supernatant and two washes (5% formic acid in 50% acetonitrile) of the gel digests were pooled and concentrated with a SpeedVac (Labconco, Kansas, MO) to dryness in 200-μl polypropylene autosampler vials (Sun Sri, Rockwood, TN). The recovered peptides were resuspended in 5 μl of solvent A (0.1% formic acid, 2% acetonitrile, and 97.9% water) and chromatographed without trap cleanup. The bound peptides were separated at 500 nl/min, generating 80 to 120 bars of pressure using AQ C18 reverse-phase media (3-μm particle size and 200-μm pore size) packed in a pulled-tip nanochromatography column (0.100-mm internal diameter [ID] by 150-mm length) from Precision Capillary Columns, San Clemente, CA. Chromatography was carried out in-line with an LTQ-Velos Orbitrap mass spectrometer (Thermo Fisher Scientific, West Palm Beach, FL). The mobile phase consisted of a linear gradient prepared from solvent A and solvent B (0.1% formic acid, 2% water, and 97.9% acetonitrile) at RT. Nanoscale liquid chromatography-tandem mass spectrometry (NanoLC-MS/MS) was performed with a Proxeon Easy-nLC II multidimensional liquid chromatograph and temperature-controlled Ion Max nanospray source (Thermo Fisher Scientific) in-line with the LTQ-Velos Orbitrap mass spectrometer.
Computer-controlled data-dependent automated switching to MS/MS by Xcalibur 2.1 software was used for data acquisition and provided the peptide sequence information. Databank analysis was performed with Mascot software (Matrix Science, Beachwood, OH). The data were searched against a sequence file containing VACV proteins from the UniprotKB TrEMBL database. The data were searched with one missed cleavage allowed and mass tolerances of 10 ppm and 0.8 Da for the precursor and fragment ions, respectively. Carbamidomethylation of cysteine was set as a fixed modification, while oxidation of methionine, deamidation of asparagine and glutamine, and protein N-terminal acetylation were searched as dynamic modifications. The resulting search files were reclustered against the same sequence database for further analysis using ProteoIQ software (Premier Biosoft, Palo Alto, CA). Assignments were filtered using the Protein Prophet algorithm as implemented within ProteoIQ, with cutoff filters set to 95% for peptides and 99% for proteins. Protein assignments were considered only if they met both the probability thresholds and 2-spectra-per-peptide sequence/2-peptides-per-protein minimums.
Plaque assay, virus yield determination, and single-cycle growth experiments.
Plaque assays and virus yield determinations were performed as previously described (17, 18). HeLa cells were infected with 3 PFU per cell of VACV WR, vA30.5-V5, or vΔA30.5. At various times after infection, cells were harvested and crude lysates were prepared by three freeze-thaw cycles and sonication. Cell-associated virus yields were determined by plaque formation on BS-C-1 or RK-A30.5-V5 cell monolayers.
Plasmid and recombinant VACV construction.
The GFP-A30.5-V5 plasmid was constructed using primers to amplify the A30.5 ORF tagged with the V5 coding sequence at the C terminus and a separate enhanced green fluorescent protein (GFP) ORF flanked by VACV DNA sequences to allow homologous recombination (Fig. 1B). The PCR product was cloned using a Zero Blunt TOPO PCR cloning kit (Invitrogen). The endogenous A30.5L ORF of VACV WR was replaced with a V5 epitope-tagged A30.5L ORF and GFP marker gene by transfecting the GFP-A30.5-V5 plasmid using Lipofectamine 2000 (Invitrogen). The recombinant virus vA30.5-V5 was clonally purified by successive picking of fluorescent plaques.
Fig 1.
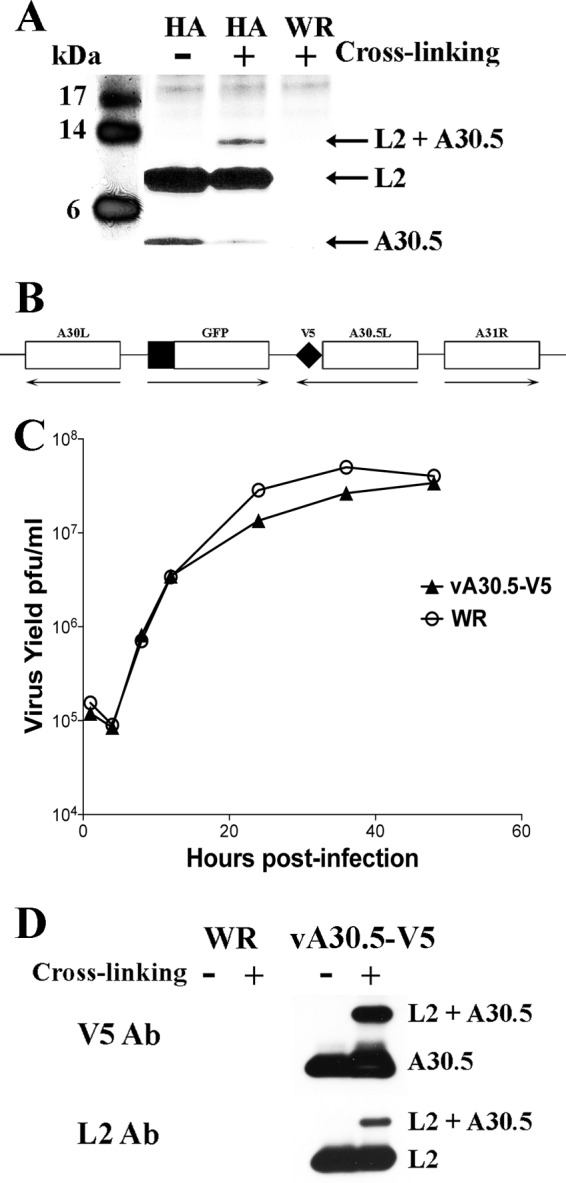
L2 interacts with A30.5. (A) Identification by mass spectrometry of proteins that associate with affinity-purified L2-HA. HeLa cells were infected with vL2-HA (HA) or VACV strain WR (WR) for 10 h. The cells were untreated (−) or cross-linked (+), and clarified cell lysates were incubated with anti-HA MAb. After binding to protein G Dynabeads, the samples were washed, and the bound proteins were eluted, resolved by SDS-PAGE, and stained with Coomassie blue. Each lane of the gel was excised in 12 equally sized pieces, trypsinized, and analyzed by mass spectrometry. The positions and masses of marker proteins are on the left; the positions of A30.5, L2, and the complex of A30.5 plus L2 are on the right. Only the lower part of the gel is shown. (B) Schematic representation of part of the genome of vA30.5-V5. The V5 epitope tag sequence was attached to the C terminus of the A30.5 ORF, while the original A30.5 promoter was retained. The GFP ORF regulated by the late P11 promoter was inserted between the A30L and A30.5L ORFs. Unmodified ORFs encoding A30L and A31R are also shown. Arrows point in the direction of transcription. (C) One-step growth curve of vA30.5-V5. HeLa cells were infected with 3 PFU per cell of wild-type VACV WR or vA30.5-V5. At the indicated hours, the infected cells were harvested and the virus titers were determined in duplicate by plaque assay. Data from two independent experiments were averaged. (D) Confirmation of L2 association with A30.5. HeLa cells were infected with vA30.5-V5 or VACV WR, and after 10 h, the cells were untreated (−) or cross-linked (+). The cells were lysed, and proteins were captured with V5 MAb beads. The proteins were eluted and resolved by SDS-PAGE. Western blotting was performed using L2 polyclonal antibody and V5 MAb. The bands are identified on the right.
Western blotting.
Cells were lysed with lithium dodecyl sulfate and reducing agent. The proteins were resolved by electrophoresis on 4 to 12% NuPAGE Novex acrylamide gels and transferred to nitrocellulose membranes using mini-iBlot gel transfer stacks (Invitrogen). Membranes were blocked for 1 h at RT with 5% nonfat dried milk in PBS containing 0.05% Tween 20 and incubated with the primary antibody for 1 h at RT. The membranes were washed four times with PBS containing 0.05% Tween 20 and then incubated with the secondary antibody for 1 h at RT. Species-specific horseradish peroxidase-conjugated secondary antibodies were used (Pierce, Rockford, IL). The membranes were washed and developed using Dura or Femto chemiluminescent substrate (Pierce).
Detergent extraction of purified virions.
Purified virus particles were incubated in 50 mM Tris-HCl (pH 7.5) containing 150 mM NaCl and 1% Nonidet P-40 detergent in the presence of 50 mM dithiothreitol for 1 h at 37°C. Soluble and insoluble materials were separated by centrifugation at 20,000 × g for 30 min at 4°C. Proteins in the pellet and the supernatant were analyzed by electrophoresis on 4 to 12% NuPAGE Novex acrylamide gels in the absence of reducing agent followed by Western blotting.
Confocal microscopy.
HeLa and RK-13 cells on coverslips were infected or infected and transfected. The cells were subsequently fixed with 4% paraformaldehyde in PBS for 15 min at RT and then washed four times with PBS. The cells were permeabilized with 0.1% Triton X-100 in PBS for 15 min at RT and washed four times. The cells were blocked with 10% FBS, heat inactivated for 30 min, and then incubated with the primary antibody in PBS containing 10% FBS for 1 h at RT. Cells were washed and incubated with the secondary antibody conjugated dye (Molecular Probes, Eugene, OR) for 1 h. The coverslips were incubated for 20 min with 300 nM 4′,6-diamidino-2-phenylindole (DAPI; Invitrogen Life Technologies) in PBS, washed, and mounted on a glass slide by using ProLong Gold (Invitrogen Life Technologies). Micrographs were acquired with a Leica TCS SP5 confocal inverted-base microscope with a 63× oil objective.
Transfection studies.
HeLa cells were grown on coverslips in 24-well plates. After reaching 80% confluence, the cells were infected with either vA30.5-V5 or vΔL2R and after 1 h were transfected with a plasmid carrying either the L2 ORF with a hemagglutinin (HA) epitope tag or A30.5 with a V5 epitope tag, respectively. The expression of both plasmids was regulated under their natural promoters. Lipofectamine 2000 (Invitrogen Life Sciences) was used for transfection according to the instructions of the manufacturer. The cells received fresh medium at 6 h after transfection. At 16 h after infection, the cells were fixed and prepared for confocal microscopy.
Construction of the RK-A30.5-V5 cell line.
The methods for construction and isolation of the RK-A30.5-V5 cell line were similar to those previously described for an L2-expressing cell line (17). Briefly, the A30.5L ORF with a V5 tag at the 3′ end was cloned in a pcDNA 3.1/Zeo(+) plasmid (Invitrogen Life Technologies). The plasmid was transfected into RK-13 cells with Lipofectamine 2000 (Invitrogen Life Technologies) according to the manufacturer's instructions. After 48 h, the transfected cells were distributed to new dishes at approximately 25% confluence with fresh medium containing 750 μg/ml Zeocin. The cells were fed with selective medium every 3 days until cell foci were identified on day 10. Individual colonies were isolated with cloning discs (Sigma-Aldrich), transferred to 48-well plates, and screened for V5 epitope expression by Western blotting. The positive colonies were selected, and the recombinant RK-A30.5-V5 cell line was grown as described above and supplemented with 300 μg/ml Zeocin to maintain selection pressure.
Construction of vΔA30.5.
Overlapping PCR was used to assemble DNA containing the GFP ORF regulated by the VACV P11 promoter inserted between ∼500-bp sequences flanking the A30.5L ORF. HeLa cells were infected with VACV WR and transfected with the PCR product using Lipofectamine 2000 (Invitrogen Life Technologies). After 24 h, the lysate was diluted and incubated with RK-A30.5-V5 cells. Plaques that fluoresced green were picked and plaque-purified five times in succession. The mutant virus vΔA30.5 was grown in RK-A30.5-V5 cells and purified by sucrose gradient centrifugation. Deletion of A30.5 was verified by PCR and sequencing.
Transmission electron microscopy.
BS-C-1, RK-13, and HeLa cells were infected with 5 PFU of vΔA30.5-V5 for 20 h. The cells were fixed with 2% glutaraldehyde and embedded in Embed 812 resin (Electron Microscopy Sciences, Hatfield, PA) as described previously (18). Specimens were viewed with a FEI Tecnai Spirit transmission electron microscope (FEI, Hillsboro, OR).
RESULTS
Association of the L2 and A30.5 proteins.
Affinity purification and cross-linking experiments were carried out to identify proteins that associate with L2. We employed vL2-HA, a recombinant VACV with an HA tag at the N terminus of L2 (16), and the parental wild-type (WT) VACV WR as a negative control. At 10 h after infection, the cells were either cross-linked with paraformaldehyde and then lysed with buffer containing NP-40 or lysed directly. After incubation with an anti-HA MAb, the proteins were captured with protein G Dynabeads, resolved by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), and stained with Coomassie blue. The ∼10-kDa band in Fig. 1A corresponds to L2-HA; an additional protein migrating faster than L2 was detected in the non-cross-linked sample, whereas a protein migrating slower than L2 was detected in the cross-linked sample. Neither L2 nor the additional protein bands were detected in cells infected with the control VACV WR. Each lane of the gel was cut into slices, and the proteins were digested with trypsin and analyzed by mass spectrometry. The identity of the 10-kDa protein was confirmed as L2 and the protein migrating faster was identified as A30.5. A30.5 was also present in the cross-linked band migrating more slowly than L2.
To confirm the interaction between L2 and A30.5, we constructed a recombinant VACV called vA30.5-V5 with a V5 epitope tag at the C terminus of the A30.5L ORF. The GFP ORF regulated by the P11 late promoter was inserted between A30.5L and A30L in order to facilitate isolation of the recombinant virus (Fig. 1B). Neither the tag nor the GFP ORF affected plaque formation (not shown) or replication kinetics (Fig. 1C), encouraging the use of the virus for further studies. We then infected cells with vA30.5-V5 and carried out paraformaldehyde cross-linking and immunopurification using an anti-V5 MAb and protein G Dynabeads. Western blotting performed with antibodies to V5 and L2 confirmed the complex of L2 and A30.5 (Fig. 1D).
ORFs corresponding to the location of A30.5L are present in all chordopoxviruses.
ORFs corresponding to the VACV A30.5L ORF had been annotated in some but not all chordopoxviruses. By searching genome databases, we found a short ORF in the same direction and just to the right of the conserved A30L ORF homolog in representatives of all chordopoxvirus genera. The sequences of the short ORFs, however, were widely divergent (Fig. 2A). In contrast, the amino acid identities between orthopoxvirus A30.5 homologs were 88 to 100% (Fig. 2B).
Fig 2.

Computational analysis of VACV A30.5 and corresponding ORFs in other chordopoxvirus genomes. (A) CLUSTAL 2.1 alignment of A30.5L and ORFs at corresponding locations from representative species of the Chordopoxvirinae subfamily. VACV, vaccinia virus strain Western Reserve; SWPV, swinepox virus strain Nebraska; DVP, deerpox virus strain W-848-83; LSDV, lumpy skin disease virus strain Neethling 2490; YMTV, yaba monkey tumor virus strain Amano; RFV, rabbit fibroma virus strain Kasza; ORFV, orf virus strain OV-SA00; FPV, fowlpox virus strain NVSL; COPX, Nile crocodilepox virus strain Zimbabwe/Ume/2001; MOCV, molluscum contagiosum virus subtype 1. (B) Alignment of A30.5 homologs of orthopoxviruses. VARV, variola major virus strain Bangladesh 1975; VACV, vaccinia virus strain Western Reserve; VACV-Acam, vaccinia virus strain Acambis 3000 modified vaccinia virus Ankara; MPXV, monkeypox virus strain Zaire; CMLV, camelpox virus strain CMS; ECTV, ectromelia virus strain Moscow; CPXV, cowpox virus strain Brighton red. Amino acids conserved in all species are marked by asterisks. (C) Kyte-Doolittle hydrophilicity plot of A30.5L amino acid sequence analyzed using MacVector version 11.1.2 software. The predicted amino acid sequence is at the bottom.
Twenty of the 42 amino acids in the A30.5L ORF had hydrophobic side chains with a putative transmembrane domain between amino acids 15 to 33 (Fig. 2C). The A30.5 protein has no recognizable signal peptide or coiled-coil segment; however, there is a potential N-glycosylation site near the C terminus.
Expression of the A30.5 protein.
Transfection studies indicated that expression of the A30.5L ORF is regulated by a postreplicative intermediate promoter (40). In order to determine the kinetics of A30.5 protein synthesis, we infected BS-C-1 cells with vA30.5-V5 and harvested them at various times. A30.5-V5 was detected by Western blotting as a 5-kDa band at 4 h and increased in amount over the next several hours (Fig. 3A). The inability to detect A30.5-V5 in the presence of AraC, an inhibitor of DNA replication, confirmed its classification as a postreplicative protein (Fig. 3A). Known early and late proteins were included in this analysis. E3, an early protein, was also detected at 4 h but increased in the presence of AraC. A3, a late protein, was seen as a faint band immediately after infection due to its presence in the virus core and increased in amount beginning at 6 h (Fig. 3A).
Fig 3.
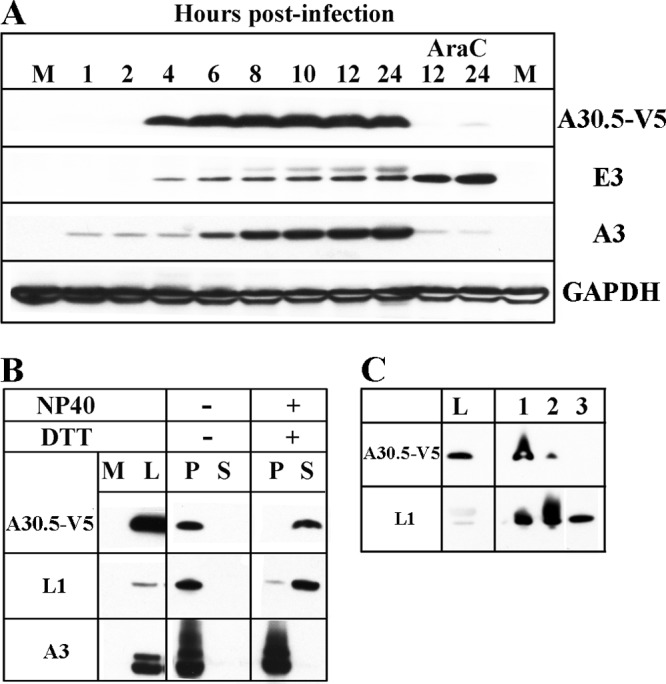
A30.5 expression and analysis of purified virions. (A) Temporal synthesis of A30.5. BS-C-1 cells were mock infected (M) or infected with 10 PFU/cell of vA30.5-V5 in the absence or presence of AraC and harvested at the indicated hours. The proteins from the whole-cell extracts were solubilized with lithium dodecyl sulfate buffer containing reducing agent, resolved by SDS-PAGE, and analyzed by Western blotting with antibodies to the proteins indicated on the right. MAbs were used to detect A30.5-V5 and GAPDH loading control; polyclonal antibodies were used to detect A3 and E3. (B) Extraction of A30.5 from sucrose gradient-purified virions. MVs, purified by sucrose gradient sedimentation from HeLa cells infected with vA30.5-V5, were incubated at 37°C in Tris buffer with (+) or without (−) 1% NP-40 and 50 mM dithiothreitol (DTT) and centrifuged to separate the soluble (S) and insoluble (P) fractions. The insoluble fraction was resuspended in a volume equal to that of the soluble fraction, and aliquots of each were resolved by SDS-PAGE. The proteins were analyzed by Western blotting using antibodies to the viral proteins indicated on the left. M, mock-infected; L, total cell lysate. (C) Further purification of MVs. MVs were purified from infected HeLa cells successively through a sucrose cushion (lane 1), a sucrose gradient (lane 2), and a CsCl gradient (lane 3). The same relative proportions of cell extract (L) and purified fractions were resolved by SDS-PAGE and analyzed by Western blotting with MAb to the V5 epitope tag and polyclonal antibody to L1 as a positive control. Proteins were detected by chemiluminescence.
A30.5-V5, detected by Western blotting of virions purified by sedimentation through one sucrose cushion and a sucrose gradient, was extracted with NP-40 and dithiothreitol, similar to the L1 membrane protein and different from the A3 core protein (Fig. 3B). However, whereas L1 was enriched during virion purification, the amount of A30.5-V5 progressively decreased and was not detected after the final CsCl gradient (Fig. 3C). We concluded that A30.5 is at most a minor component of MVs.
Intracellular localization of A30.5.
L2 was previously shown to localize with the ER throughout the cell. In order to compare the intracellular location of A30.5 with that of L2, we infected HeLa cells with vA30.5-V5 and transfected them with a plasmid expressing L2-HA under the natural promoter. The cells were fixed and stained with antibodies to the V5 and HA epitope tags as well as the membrane and luminal ER markers calnexin and PDI, respectively. In addition, DAPI was used to stain DNA in the nucleus and virus factory. A30.5-V5 was in the cytoplasm mostly outside the virus factory and in a rim around the nucleus (Fig. 4). There was extensive colocalization with the ER markers calnexin and PDI as well as with the viral L2-HA protein (Fig. 4). We also examined the distribution of A30.5-V5 expressed by transfection from its natural promoter in cells infected with an L2 deletion mutant. A30.5-V5 still appeared to have an ER distribution, indicating that this localization was not dependent on L2 (not shown).
Fig 4.

Intracellular localization of A30.5. HeLa cells were infected with vA30.5-V5 and transfected with plasmid encoding L2-HA regulated by the natural promoter. After 16 h, the infected cells were fixed, permeabilized, and stained with anti-calnexin or anti-PDI polyclonal antibody to locate ER and with mouse anti-V5 MAb to detect A30.5 or mouse anti-HA antibody to detect L2, followed by goat anti-rabbit IgG and goat anti-mouse IgG coupled to Alexa Fluor 594 and Alexa 647 and DAPI. The cells were visualized by confocal microscopy. Three-color merges are on the right.
Construction of a stable cell line expressing A30.5.
The conservation of A30.5-like ORFs among chordopoxviruses suggested an important function in replication. This expectation was supported by our inability to construct a viable mutant virus in which the A30.5L ORF was replaced by GFP. An inability to delete the L2R ORF was overcome by making a cell line that expressed L2 and complemented the replication of a mutant virus (17). A similar procedure was undertaken for A30.5L. A mammalian codon-optimized copy of the A30.5L ORF with a C-terminal V5 epitope tag was chemically synthesized and placed under the control of the cytomegalovirus (CMV) promoter and cloned into the pcDNA 3.1/Zeo(+) plasmid. RK-13 cells were transfected with the plasmid and selected with Zeocin. Individual clones were expanded and screened for A30.5-V5 expression by Western blotting. A30.5-V5 was detected in RK-A30.5-V5 cells, but the amount was low compared with that in RK-13 cells infected with 1 to 10 PFU of the recombinant vA30.5-V5 (Fig. 5A).
Fig 5.

Expression of A30.5 in RK-A30.5-V5 cells. (A) Western blotting. Uninfected RK-A30.5-V5 and RK-13 cells infected with 1 to 10 PFU/cell of vA30.5-V5 or 10 PFU/cell of VACV WR for 24 h were lysed. Proteins were resolved by SDS-PAGE and analyzed by Western blotting with MAbs to V5 for the detection of A30.5-V5 and GAPDH as a loading control. (B) Confocal microscopy of the RK-A30.5-V5 cells. Confluent cells were fixed, permeabilized, and stained with the mouse anti-V5 MAb and polyclonal rabbit antibody to calnexin, followed by goat anti-mouse and goat anti-rabbit IgG coupled to Alexa Fluor 488 and 594, respectively. DNA was detected by staining with DAPI. Two (upper)- or three (lower)-color merges are on the right.
The intracellular location and the uniformity of A30.5-V5 expression in the RK-A30.5-V5 cell population were determined by confocal microscopy. RK-A30.5-V5 cells were fixed and costained with anti-V5 MAb to detect A30.5-V5 and antibody against calnexin to show ER. A30.5-V5 colocalized with the ER (Fig. 5B), although the intensity of the stain varied in individual cells. Thus, no additional viral proteins were required for ER localization of A30.5.
Construction of a VACV A30.5L deletion mutant.
Despite the relatively low expression level compared to that in infected cells, the ER localization of A30.5-V5 provided encouragement that it might complement an A30.5-null mutant. The mutant virus was constructed by transfecting DNA, containing the GFP ORF under the P11 late promoter flanked by sequences preceding and following the A30.5L ORF to allow homologous recombination, into HeLa cells that had been infected with VACV (Fig. 6A). The lysate was diluted and virus was adsorbed to a monolayer of RK-A30.5-V5 cells. Green fluorescent plaques were picked and clonally purified by repeated plaque isolations in RK-A30.5-V5 cells. Replacement of the A30.5L ORF by GFP was confirmed by DNA sequencing, and the deletion mutant was named vΔA30.5.
Fig 6.
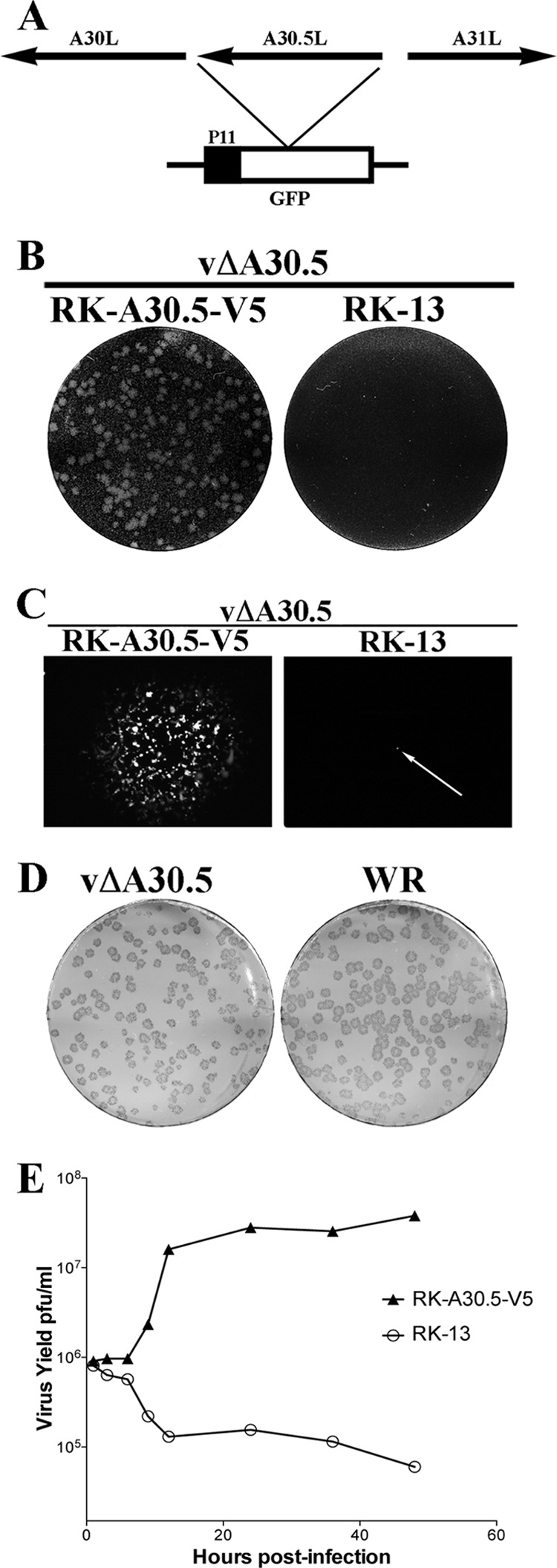
Construction and characterization of vΔA30.5. (A) Schematic representation of the genome structure of vΔA30.5. The A30.5L ORF was replaced with the GFP ORF regulated by the VACV P11 late promoter. Arrows indicate the direction of transcription. (B) Plaque phenotype of vΔA30.5. RK-A30.5-V5 and RK-13 cells were infected with vΔA30.5. After 48 h, the cells were stained with crystal violet. (C) Plaques formed by vΔA30.5 in RK-A30.5-V5 cells and a single infected cell (arrow) in RK-13 cells visualized with a fluorescence microscope. (D) Comparison of plaques formed by vΔA30.5 and wild-type VACV strain WR on RK-A30.5-V5 cells. After 72 h, plaques were visualized by staining with antibody to VACV followed by anti-protein A conjugated to alkaline phosphatase. (E) One-step growth curve of vΔA30.5. RK-A30.5-V5 and RK-13 cells were infected with 3 PFU/cell of vΔA30.5. At the indicated times, the infected cells were harvested and virus titers were determined in duplicate by plaque assay on RK-A30.5-V5 cells. Data from two independent experiments were averaged.
vΔA30.5 formed plaques in RK-A30.5-V5 cells but not in the parental RK-13 cells (Fig. 6B). Individual RK-13 cells infected with vΔA30.5 were visualized by GFP fluorescence, but infection of neighboring cells failed to occur (Fig. 6C), indicating a defect in the formation or spread of infectious progeny. Despite the relatively low level of expression of A30.5 in RK-A30.5-V5 cells, the plaques formed by vΔA30.5 were indistinguishable in size and shape from wild-type VACV WR plaques (Fig. 6D). Furthermore, vΔA30.5 replicated with normal kinetics in RK-A30.5-V5 cells but not in RK-13 cells (Fig. 6E), indicating a defect in the formation of infectious virus. The yields of vΔA30.5 in RK-A30.5-V5 cells were similar to that of WT virus, allowing preparation of large stocks for infection and purification.
Electron-dense cytoplasmic inclusions and IV-like structures associated with ER formed in the absence of A30.5.
The association of A30.5 with L2, as well as its localization with ER and requirement for replication, suggested that it might be involved in an early stage of morphogenesis. To evaluate this possibility, we analyzed cells infected with vΔA30.5 by transmission electron microscopy. Since we recently found differences in the phenotypes of an A11-inducible null mutant in RK-13 and BS-C-1 cells (21), both types of cells as well as HeLa cells were infected for the present study. The three cell types are permissive for WT VACV but not for vΔA30.5. BS-C-1 and HeLa cells infected with vΔA30.5 appeared generally similar. The predominant structures were large electron-dense spherical inclusions (Fig. 7A and C) identical to those shown to contain core proteins with other morphogenesis mutants (19). Sometimes the masses of dense viroplasm were juxtaposed to more lacey structures in BS-C-1 cells (Fig. 7B). In both cell types, membrane arcs decorated the surfaces of many inclusions, although the arcs were on average larger in HeLa cells than in BS-C-1 cells (Fig. 7A and C). Higher magnification of the membrane arcs revealed the typical outer scaffold protein structure comprised of D13 trimers that form a lattice but appear as spicules in cut sections (Fig. 7A and C, insets) like those on the crescents that form during normal VACV infection. Dense inclusions were also seen in RK-13 cells infected with vΔA30.5, although they were not decorated with crescents (Fig. 7D and Table 1).
Fig 7.

Transmission electron microscopy images of dense inclusions in cells infected with vΔA30.5. BS-C-1 (A and B), HeLa (C), and RK-13 (D) cells were infected with 5 PFU/cell of vΔA30.5. After 16 h, the cells were fixed and prepared for transmission electron microscopy. Insets are at higher magnification to reveal spicules on the convex surfaces of the crescents. Arrows in panels point to crescents; arrows in insets point to spicules on the crescents. Scale bars are at the bottoms of panels and insets.
Table 1.
Frequency of virus structures
| Structuresa | BS-C-1 (23 cells) | HeLa (20 cells) | RK-13 (22 cells) |
|---|---|---|---|
| DV without crescents | 0 | 0 | 19 |
| DV with crescents | 22 | 19b | 0 |
| IV-like | 4 | 3 | 14 |
| IV, MV, WV | 0 | 0 | 0 |
DV, dense mass of viroplasm.
Crescents were larger than in BS-C-1 cells.
MVs and typical IVs were not seen in infected BS-C-1, HeLa, or RK-13 cells. However, circular IV-like structures that appeared devoid of dense viroplasm were found in the three cell types (Fig. 8A and B and data not shown), though they were more numerous in the RK-13 cells (Table 1). The IV-like structures that formed in the absence of A30.5 were identical in appearance to those previously described for cells infected with L2 and A11 deletion mutants and shown to contain the A17 and A14 viral membrane proteins and the D13 scaffold protein (17, 21). Higher magnification revealed the scaffold protein spicules on the IV-like membranes (Fig. 8A and B, insets). The IV-like structures appeared packed together in large clusters partially surrounded by a smooth membrane, which we showed contained the ER integral membrane protein calnexin with L2 and A11 mutants (17, 21). These clusters were located in the virus factory but were separated from dense inclusions. Although IV-like structures were abundant and present in most RK-13 cell sections, the number of IV-like structures seemed less than the number of IVs and MVs in a typical WT VACV infection.
Fig 8.

Transmission electron microscopy images of crescent- and IV-like structures associated with ER in cells infected with vΔA30.5. HeLa (A) and RK-13 (B through F) cells were infected and prepared for transmission electron microscopy as described in the legend to Fig. 7. Clusters of IV-like structures surrounded by ER membranes are shown in panels A and B. Insets have higher magnification images of ER membranes next to IV-like structures with spicules. In panel C there is a cluster of IV-like membranes within the expanded lumen of the membrane surrounding the nucleus (Nu). A single IV-like structure within the ER lumen is seen in panel D. In panels E and F, crescent membranes are continuous with the ER membrane and the convex surfaces face the lumen, which contains IV-like structures. Symbols: arrowheads, ER membrane; arrows, spicules; asterisks, crescents.
The IV-like structures were in greatly expanded ER lumens, as is readily seen in Fig. 8C, which shows the separation of the inner and outer nuclear membranes, and in Fig. 8D, which shows a single luminal IV-like structure. Furthermore, the spicule-coated membranes of crescent-like structures are continuous with the smooth ER membrane (Fig. 8E and F). In the majority of images, the convex spicule-coated side of the crescent-like structures face the lumen. It would appear that the IV-like structures pinch off from the ER after the crescents extend to complete spheres and are then trapped in the lumen. Discontinuities appear in the ER membrane surrounding the IV-like clusters, which could account for the entry of the D13 scaffold protein into the lumen where it can interact with the A17 protein in the nascent viral membrane.
Effect of the A30.5 deletion on the localization of other viral proteins.
Previous studies had shown that some viral proteins are abnormally localized when L2 is not expressed, presumably because of the absence of normal IV and MV membranes. Confocal microscopy was used to compare the location of the core protein A3, the MV membrane proteins A13, A14, A17, and D8, and the IV scaffold protein D13 in HeLa cells infected with vA30.5-V5 and vΔA30.5. In the cells expressing A30.5, the A3 protein had a punctate distribution within the cytoplasmic viral factories (Fig. 9, left). In contrast, A3 appeared as rings in the factory regions of cells not expressing A30.5 (Fig. 9, right). The rings outline the dense inclusions of viroplasm seen by electron microscopy and result from the inability of the antibody to penetrate them after fixation. Similarly, A13, A14, and D8 membrane proteins formed rings in the cells infected with vΔA30.5 but not vA30.5-V5, consistent with the presence of crescent membranes around the dense inclusions. The A17 and D13 proteins had a punctate appearance in cells infected with both viruses and in the vΔA30.5-infected cells appeared more widely dispersed than A13, A14, and D8.
Fig 9.

Confocal microscopy of HeLa cells infected with vA30.5-V5 or vΔA30.5. (A) HeLa cells were infected with 5 PFU/cell of vA30.5-V5 or vΔA30.5. After 10 h, the infected cells were fixed, permeabilized, and stained with primary polyclonal antibodies for A3, A17, D13, and calnexin and the mouse MAbs to A13, A14, D8, and V5 followed by goat anti-rabbit IgG and goat anti-mouse coupled to Alexa Fluor 594 and 647 and DAPI. Three-color merges include DAPI.
We also examined the distribution of proteins in RK-13 cells that were infected with vΔA30.5 (Fig. 10). The A3 core protein also appeared as rings around the inclusions, whereas A14 and A13 membrane proteins had faint ring staining and the D8 membrane protein localized mostly with the ER. These differences were consistent with the electron microscope data showing absence of the short crescent membranes around the inclusions in RK-13 cells infected with vΔA30.5.
Fig 10.

Confocal microscopy of RK-13 cells infected with vA30.5-V5 or vΔA30.5. (A) RK-13 cells were infected with 5 PFU/cell of vA30.5-V5 or vΔA30.5. After 10 h, the infected cells were fixed, permeabilized, and stained with primary polyclonal antibodies for A3, D13, A11, and calnexin and the mouse MAbs to A13, A14, and D8 followed by goat anti-rabbit IgG and goat anti-mouse coupled to Alexa Fluor 594 and 647 and DAPI. Three-color merges are shown on the right.
Effect of the A30.5 deletion on the accumulation of other viral proteins.
In our previous studies (16, 17), we found that the absence of L2 results in instability of certain viral membrane proteins, particularly those comprising the entry-fusion complex (EFC). In view of the association of A30.5 with L2 and the similar mutant phenotypes, we suspected that there would be instability of the same proteins in cells infected with vΔA30.5. Western blots of 15 viral proteins from HeLa cells that were infected with ΔA30.5 and with vA30.5-V5 and wild-type VACV WR controls are shown in Fig. 11. In cells infected with vΔA30.5, the following proteins in addition to A30.5 were specifically reduced or not detected: the EFC proteins L1, A21, H2, L5, and G3 and the A11 membrane protein required for IV formation. In addition, the A14 protein had reduced mobility, apparently due to glycosylation, and the A3 and A17 membrane proteins were unprocessed. However, the absence of A30.5 appeared to have only mild effects on the amounts of the D13 scaffold protein, the membrane proteins L2, A13, and D8, and the A27 protein, which associates with the MV membrane proteins via attachment to A17. The reductions in specific proteins in the A30.5 deletion mutant were similar to findings with L2-null mutants (16, 17).
Fig 11.
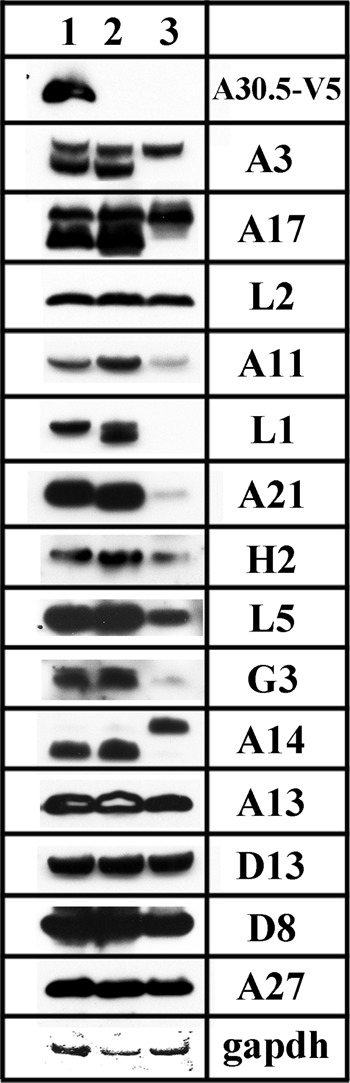
Western blots of proteins from cells infected with vΔA30.5. HeLa cells were infected with 3 PFU/cell of (1) vA30.5-V5, (2) wild-type VACV WR, and (3) vΔA30.5. After 9 h, the cells were lysed with lithium dodecyl sulfate, resolved by SDS-PAGE, and analyzed by Western blotting with antibodies to the viral proteins indicated on the right. MAbs were used to detect A30.5-V5 and GAPDH as a loading control. Proteins were visualized by chemiluminescence.
DISCUSSION
Our previous studies of the L2 and A11 proteins and the corresponding null mutants provided strong support for the origin of viral membranes from the ER (16, 17, 21). Those findings led us to investigate whether other viral proteins with a related function might be associated with L2. We discovered such an association with the previously uncharacterized A30.5 protein by mass spectrometric analysis of proteins that copurified with affinity-tagged L2 and confirmed the association by Western blotting. In the absence of cross-linking, A30.5 migrated as a 5-kDa protein by SDS-PAGE, whereas, after cross-linking, A30.5 was identified in a more slowly migrating complex associated with the 10-kDa L2 protein. The A30.5L ORF had been annotated in VACV and some other poxvirus genomes but not in others, presumably because of its small size. However, by examining sequences of representatives of all chordopoxvirus genera, we detected a short ORF adjacent to and in the same direction as the A30L ORF in each case. Although the amino acid identities of the short ORFs with VACV A30.5 were highly conserved among orthopoxviruses, they were widely divergent among other chordopoxvirus genera. Whether the sequence-divergent ORFs have a similar function remains to be determined.
The small size of the A30.5 protein and the failure to recognize the presence of related ORFs in other chordopoxviruses can explain why the protein was not previously characterized. Inspection of the A30.5L ORF revealed a putative transmembrane domain but no signal peptide or any other conserved motif except for a potential N-glycosylation site near the C terminus. In contrast to L2, A30.5 was synthesized exclusively after DNA replication. However, like L2, A30.5 colocalized with ER throughout the cytoplasm. As predicted from cross-linking data, A30.5 and L2 colocalized with each other as determined by confocal microscopy. However, the association of A30.5 with the ER was not dependent on L2 expression. We have been unable to specifically determine the localization of A30.5 by immunoelectron microscopy, in contrast to our results with confocal microscopy, and other epitope tags may be needed. However, since we have located L2 by electron microscopy, we can presume that A30.5 would have a similar association with viral crescents.
Our inability to produce a viable A30.5L deletion mutant by conventional procedures led us to create a cell line that stably expressed A30.5 and which served as the basis for isolation of an A30.5L deletion mutant. The mutant replicated as well as wild-type virus in the complementing cell line but did not produce infectious virus in normal cells. The A30.5-expressing cell line is only the fifth example of a VACV-complementing cell line; the other cell lines express the D4 replication protein (41), the A8 and A23 transcription factors (42), and the L2 (17) and H7 (23) morphogenesis proteins. While conditional lethal mutants are extremely valuable and usually straightforward to produce, deletion mutants eliminate the possibility of low but significant levels of expression of a functional protein. This appears to be the case for the H7 protein, for which the deletion mutant appeared to be more stringent than an inducible mutant (23).
The most significant findings of the present study relate to the phenotype of the A30.5L deletion mutant, which mimicked that of the L2R- and A11R-null mutants (17, 21), and the implications for IV formation. The absence of A30.5 synthesis resulted in a block in viral early morphogenesis and the absence of MVs or typical IVs. A conspicuous feature was the accumulation in the cytoplasm of large electron-dense inclusions. Such inclusions occur with a variety of mutants that exhibit absent or reduced IV formation. The electron-dense material has the same appearance as the viroplasm within normal IVs and contains the core proteins (19). It is reasonable to infer that the core precursors aggregate when not partitioned by membranes. In infected BS-C-1 and HeLa cells, many of the inclusions had short membrane crescents on their surfaces, whereas the inclusions were naked in RK-13 cells. In addition to the inclusions and separated from them were scaffold-coated membrane structures that resembled crescents and IVs devoid of viroplasm. These structures were present in large clusters in most vΔA30.5-infected RK-13 cell sections but were less frequent in infected BS-C-1 and HeLa cells. A similar disparity between the predominant structures in BS-C-1 and RK-13 cells had previously been noted with an A11R mutant (21). In our previous studies of RK-13 cells infected with L2R and A11R mutants, we demonstrated the presence of A17 and A14 viral membrane proteins and D13 trimers in the IV-like membranes and the protein disulfide isomerase and calnexin ER proteins in the closely associated smooth membranes (17, 21). In the present study, we provided images from cells infected with the A30.5L deletion mutant that showed clusters of spicule-coated IV-like structures in the greatly expanded lumens of partially interrupted ER membranes. The presence of IV-like structures within the lumen derived from the membrane surrounding the nucleus was unambiguous. Additional images showed continuity between the crescent membrane precursors of the IV-like structures and the ER membrane. Remarkably, the convex scaffold-coated side of most crescent-like membranes faced the ER lumen. While this orientation seems counterintuitive because of the cytoplasmic location of the D13 scaffold protein and the N terminus of the A17 protein, which interacts with D13 (43, 44), we suggest that the arrangement ensures that the crescents form only after the ER membrane is partially broken. Moreover, the topology implies a preference for interactions of viral proteins with cellular proteins or lipids on the luminal side of the ER membrane. An important difference between the formation of IV-like structures and that of normal IVs is that the membrane of the enlarging crescents remained continuous with the ER membrane until the spherical IV-like structures pinched off and were trapped in the lumen. The membranes of crescents that form during wild-type VACV infection appear to be connected to short open membrane structures when analyzed by electron tomography (7). Importantly, IV-like structures identical to those seen in cells infected with vΔA30.5 were also seen in cells infected with L2 and A11 mutants. It will be interesting to determine whether an IV-like membrane as well as dense inclusions form in cells infected with the H7 (22, 23)- and A6 (25)-null mutants.
The failure to produce normal IVs and MVs has a profound effect on the stability of certain membrane-associated proteins. This phenomenon was previously shown for A11 (19)-, L2 (16, 17)-, and A6 (25)-null mutants and was also true for the A30.5 mutant described here. The EFC proteins show the most dramatic decreases, with some not detected at all in the absence of A30.5 or other proteins needed to form IVs. We previously showed that the decrease is due to rapid degradation (16) and suggested that the affected proteins may transit directly to IV or MV membranes during normal infection, which cannot occur when early stages of morphogenesis are interrupted. The failure of A17 processing and increased glycosylation of the A14 protein in cells infected with vΔA30.5 were similar to those found with other mutants that have defects in morphogenesis (17, 19, 25, 45). The glycosylation site on A14 as well as the cysteines that form disulfide-linked dimers are consistent with a luminal location of the C terminus of A14 (44, 45). Based on our present experiments, this would place the N and C termini of A14 on the surfaces of the IV and MV rather than the interior as previously thought. Two- and four-membrane-spanning topological models for A17 have been considered (10, 29, 44, 46). However, both models predict that the N and C termini would be in the cytoplasm, which is consistent with in vitro translation in the presence of microsomes. In addition, the two-membrane-spanning model places the intramolecular disulfide bond in the lumen (46). However, our present electron microscopy images clearly show crescents with D13 on the luminal side of the ER; the N-terminal segment of A17 must also be on the luminal side since it interacts with D13 (43, 44). Further topological studies of A17 are needed to resolve the apparent differences.
A tentative model for viral-membrane formation in mutant and WT virus-infected cells is presented in Fig. 12. We posit that the VMAPs comprising A30.5, L2, A11, H7, and A6 work in concert with unidentified cellular proteins to produce breaks in the ER membrane containing the viral A17 and A14 transmembrane proteins, stabilize the broken membrane ends, and elongate the crescents by membrane fusion. In the absence of A30.5 or other members of this group, these functions are impaired in a cell-type-dependent manner. Breaks in the ER, which are caused or stabilized by the association of VMAPs, occur rarely in RK-13 cells. The breaks in the ER membrane allow the association of D13 with the N terminus of A17, which is on the luminal side of the ER. The curvature imposed by D13 and A17 leads to the formation of an IV-like sphere that pinches off the modified ER into the lumen (Fig. 12). The sequestration of viral membranes in the lumen prevents their interaction with core proteins, which form masses of dense viroplasm unadorned by membranes. For undetermined reasons, more stabilized breaks occur in the ER membrane of BS-C-1 and HeLa cells, leading to the formation of short crescent-like structures that can escape from the lumen and associate with viroplasm. However, the crescent-like structures are unable or have limited ability to elongate by membrane fusion, resulting in their decoration of the large dense occlusions (Fig. 12). By extrapolating from the analysis of the mutants, we propose a model for IV formation by WT virus. Thus, the VMAPS promote ER breakage and stabilize membrane segments containing the A17 and A14 proteins. These segments interact with viroplasm and elongate by fusion with additional ER-derived membranes to form spherical IVs (Fig. 12). A further consequence of our model is that the outer surface of the MV, which is derived from the IV, represents the luminal side of the ER. This topology can explain the exposure of phosphatidylserine, which is more abundant on the luminal side of the ER (47, 48), on the surface of the MV (49).
Fig 12.

Model of viral membrane assembly. Crescent and IV-like structures forming in RK-13 (left) and BS-C-1 or HeLa (center) cells infected with vΔA30.5 are depicted. The right side of the diagram represents crescents and IVs formed by WT VACV. The cytoplasmic and luminal sides of the ER bilayer are represented by purple and blue lines, respectively. The pink balls at the ends of ER fragments represent VMAPs that include L2 and A11 but lack A30.5. Green balls at the ends of ER fragments represent VMAPs that include L2, A11, and A30.5. The spicules attached to the luminal side of the ER membrane represent D13 trimers attached to the N-terminal segment of the A17 protein forming crescents. In RK-13 cells infected by vΔA30.5, three crescents bulging into the ER lumen and one IV-like particle that has pinched off the ER and is in the lumen are shown. The large gray masses depict inclusions of core proteins that are either unadorned by crescent membranes in RK-13 cells infected with vΔA30.5 or decorated with short crescents in HeLa or BS-C-1 cells infected with vΔA30.5. The small gray masses on the right represent viroplasm associated with crescents and IVs during a WT VACV infection.
ACKNOWLEDGMENTS
We thank Catherine Cotter for providing cells; the Protein Chemistry Section, NIAID, for mass spectroscopy studies, especially L. Renee Olano for the computational analysis; Yan Xiang of the University of Texas Health Center, San Antonio, for MAbs; and John Heuser of the Washington University School of Medicine for comments on our previous work with L2.
The research was supported by the Division of Intramural Research, NIAID, NIH.
Footnotes
Published ahead of print 11 September 2013
REFERENCES
- 1.Moss B. 2013. Poxviridae, p 2129–2159 In Knipe DM, Howley PM, Cohen JL, Griffin DE, Lamb RA, Martin MA, Racaniello VR, Roizman B. (ed), Fields virology, 6th ed, vol 2 Lippincott Williams & Wilkins, Philadelphia, PA [Google Scholar]
- 2.Dales S, Siminovitch L. 1961. The development of vaccinia virus in Earle's L strain cells as examined by electron microscopy. J. Biophys. Biochem. Cytol. 10:475–503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Grimley PM, Rosenblum EN, Mims SJ, Moss B. 1970. Interruption by rifampin of an early stage in vaccinia virus morphogenesis: accumulation of membranes which are precursors of virus envelopes. J. Virol. 6:519–533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hollinshead M, Vanderplasschen A, Smith GL, Vaux DJ. 1999. Vaccinia virus intracellular mature virions contain only one lipid membrane. J. Virol. 73:1503–1517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Heuser J. 2005. Deep-etch EM reveals that the early poxvirus envelope is a single membrane bilayer stabilized by a geodetic “honeycomb” surface coat. J. Cell Biol. 169:269–283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Szajner P, Weisberg AS, Lebowitz J, Heuser J, Moss B. 2005. External scaffold of spherical immature poxvirus particles is made of protein trimers, forming a honeycomb lattice. J. Cell Biol. 170:971–981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chlanda P, Carbajal MA, Cyrklaff M, Griffiths G, Krijnse-Locker J. 2009. Membrane rupture generates single open membrane sheets during vaccinia virus assembly. Cell Host Microbe 6:81–90 [DOI] [PubMed] [Google Scholar]
- 8.Dales S, Mosbach EH. 1968. Vaccinia as a model for membrane biogenesis. Virology 35:564–583 [DOI] [PubMed] [Google Scholar]
- 9.Sodeik B, Doms RW, Ericsson M, Hiller G, Machamer CE, van't Hof W, van Meer G, Moss B, Griffiths G. 1993. Assembly of vaccinia virus: role of the intermediate compartment between the endoplasmic reticulum and the Golgi stacks. J. Cell Biol. 121:521–541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Krijnse-Locker J, Schleich S, Rodriguez D, Goud B, Snijder EJ, Griffiths G. 1996. The role of a 21-kDa viral membrane protein in the assembly of vaccinia virus from the intermediate compartment. J. Biol. Chem. 271:14950–14958 [DOI] [PubMed] [Google Scholar]
- 11.Salmons T, Kuhn A, Wylie F, Schleich S, Rodriguez JR, Rodriguez D, Estban M, Griffiths G, Locker JK. 1997. Vaccinia virus membrane proteins p8 and p16 are cotranslationally inserted into the rough endoplasmic reticulum and retained in the intermediate compartment. J. Virol. 71:7404–7420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Risco C, Rodriguez JR, Lopez-Iglesias C, Carrascosa JL, Esteban M, Rodriguez D. 2002. Endoplasmic reticulum-Golgi intermediate compartment membranes and vimentin filaments participate in vaccinia virus assembly. J. Virol. 76:1839–1855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Husain M, Moss B. 2003. Evidence against an essential role of COPII-mediated cargo transport to the endoplasmic reticulum-Golgi intermediate compartment in the formation of the primary membrane of vaccinia virus. J. Virol. 77:11754–11766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Husain M, Weisberg AS, Moss B. 2007. Sequence-independent targeting of transmembrane proteins synthesized within vaccinia virus factories to nascent viral membranes. J. Virol. 81:2646–2655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Husain M, Weisberg AS, Moss B. 2006. Existence of an operative pathway from the endoplasmic reticulum to the immature poxvirus membrane. Proc. Natl. Acad. Sci. U. S. A. 103:19506–19511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Maruri-Avidal L, Weisberg AS, Moss B. 2011. Vaccinia virus L2 protein associates with the endoplasmic reticulum near the growing edge of crescent precursors of immature virions and stabilizes a subset of viral membrane proteins. J. Virol. 85:12431–12441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Maruri-Avidal L, Weisberg AS, Bisht H, Moss B. 2013. Analysis of viral membranes formed in cells infected by a vaccinia virus L2-deletion mutant suggests their origin from the endoplasmic reticulum. J. Virol. 87:1861–1871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Maruri-Avidal L, Domi A, Weisberg AS, Moss B. 2011. Participation of vaccinia virus L2 protein in the formation of crescent membranes and immature virions. J. Virol. 85:2504–2511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Resch W, Weisberg AS, Moss B. 2005. Vaccinia virus nonstructural protein encoded by the A11R gene is required for formation of the virion membrane. J. Virol. 79:6598–6609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wu X, Meng X, Yan B, Rose L, Deng J, Xiang Y. 2012. Vaccinia virus virion membrane biogenesis protein A11 associates with viral membranes in a manner that requires the expression of another membrane biogenesis protein, A6. J. Virol. 86:11276–11286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Maruri-Avidal L, Weisberg AS, Moss B. 2013. Association of the vaccinia virus A11 protein with the endoplasmic reticulum and crescent precursors of immature virions. J. Virol. 87:10195–10206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Satheshkumar PS, Weisberg A, Moss B. 2009. Vaccinia virus H7 protein contributes to the formation of crescent membrane precursors of immature virions. J. Virol. 83:8439–8450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Meng X, Wu X, Yan B, Deng J, Xiang Y. 2013. Analysis of the role of vaccinia virus H7 in virion membrane biogenesis with an H7-deletion mutant. J. Virol. 87:8247–8253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Meng XZ, Embry A, Sochia D, Xiang Y. 2007. Vaccinia virus A6L encodes a virion core protein required for formation of mature virion. J. Virol. 81:1433–1443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Meng X, Embry A, Rose L, Yan B, Xu C, Xiang Y. 2012. Vaccinia virus A6 is essential for virion membrane biogenesis and localization of virion membrane proteins to sites of virion assembly. J. Virol. 86:5603–5613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Earl PL, Moss B, Wyatt LS, Carroll MW. 2001. Generation of recombinant vaccinia viruses. Curr. Protoc. Mol. Biol. 43:16.17.1–16.17.19. 10.1002/0471142727.mb1617s43. [DOI] [PubMed] [Google Scholar]
- 27.Earl PL, Cooper N, Wyatt LS, Moss B, Carroll MW. 2001. Preparation of cell cultures and vaccinia virus stocks. Curr. Protoc. Mol. Biol. 43:16.16.1–16.16.13. 10.1002/0471142727.mb1616s43 [DOI] [PubMed] [Google Scholar]
- 28.Laliberte JP, Moss B. 2009. Appraising the apoptotic mimicry model and the role of phospholipids for poxvirus entry. Proc. Natl. Acad. Sci. U. S. A. 106:17517–17521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Betakova T, Wolffe EJ, Moss B. 1999. Membrane topology of the vaccinia virus A17L envelope protein. Virology 261:347–356 [DOI] [PubMed] [Google Scholar]
- 30.Sodeik B, Griffiths G, Ericsson M, Moss B, Doms RW. 1994. Assembly of vaccinia virus: effects of rifampin on the intracellular distribution of viral protein p65. J. Virol. 68:1103–1114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Townsley A, Senkevich TG, Moss B. 2005. Vaccinia virus A21 virion membrane protein is required for cell entry and fusion. J. Virol. 79:9458–9469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Townsley A, Senkevich TG, Moss B. 2005. The product of the vaccinia virus L5R gene is a fourth membrane protein encoded by all poxviruses that is requried for cell entry and cell-cell fusion. J. Virol. 79:10988–10998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nelson GE, Wagenaar TR, Moss B. 2008. A conserved sequence within the H2 subunit of the vaccinia virus entry/fusion complex is important for interaction with the A28 subunit and infectivity. J. Virol. 82:6244–6250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wagenaar TR, Ojeda S, Moss B. 2008. Vaccinia virus A56/K2 fusion regulatory protein interacts with the A16 and G9 subunits of the entry fusion complex. J. Virol. 82:5153–5160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Howard AR, Senkevich TG, Moss B. 2008. Vaccinia virus A26 and A27 proteins form a stable complex tethered to mature virions by association with the A17 transmembrane protein. J. Virol. 82:12384–12391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lustig S, Fogg C, Whitbeck JC, Eisenberg RJ, Cohen GH, Moss B. 2005. Combinations of polyclonal or monoclonal antibodies to proteins of the outer membranes of the two infectious forms of vaccinia virus protect mice against a lethal respiratory challenge. J. Virol. 79:13454–13462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Parkinson JE, Smith GL. 1994. Vaccinia virus gene A36R encodes a Mr 43-50 K protein on the surface of extracellular enveloped virus. Virology 204:376–390 [DOI] [PubMed] [Google Scholar]
- 38.Yuwen H, Cox JH, Yewdell JW, Bennink JR, Moss B. 1993. Nuclear localization of a double-stranded RNA-binding protein encoded by the vaccinia virus E3l gene. Virology 195:732–744 [DOI] [PubMed] [Google Scholar]
- 39.Meng X, Zhong Y, Embry A, Yan B, Lu S, Zhong G, Xiang Y. 2011. Generation and characterization of a large panel of murine monoclonal antibodies against vaccinia virus. Virology 409:271–279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yang Z, Reynolds SE, Martens CA, Bruno DP, Porcella SF, Moss B. 2011. Expression profiling of the intermediate and late stages of poxvirus replication. J. Virol. 85:9899–9908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Holzer G, Falkner FG. 1997. Construction of a vaccinia virus deficient in the essential DNA repair enzyme uracil DNA glycosylase by a complementing cell line. J. Virol. 71:4997–5002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Warren RD, Cotter C, Moss B. 2012. Reverse genetic analysis of poxvirus intermediate transcription factors. J. Virol. 86:9514–9519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bisht H, Weisberg AS, Szajner P, Moss B. 2009. Assembly and disassembly of the capsid-like external scaffold of immature virions during vaccinia virus morphogenesis. J. Virol. 83:9140–9150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Unger B, Mercer J, Boyle KA, Traktman P. 2013. Biogenesis of the vaccinia virus membrane: genetic and ultrastructural analysis of the contributions of the A14 and A17 proteins. J. Virol. 87:1083–1097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mercer J, Traktman P. 2003. Investigation of structural and functional motifs within the vaccinia virus A14 phosphoprotein, an essential component of the virion membrane. J. Virol. 77:8857–8871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Betakova T, Moss B. 2000. Disulfide bonds and membrane topology of the vaccinia virus A17L envelope protein. J. Virol. 74:2438–2442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dominski J, Binaglia L, Dreyfus H, Massarelli R, Mersel M, Freysz L. 1983. A study on the topological distribution of phospholipids in microsomal membranes of chick brain using phospholipase C and trinitrobenzenesulfonic acid. Biochim. Biophys. Acta 734:257–266 [DOI] [PubMed] [Google Scholar]
- 48.Fairn GD, Schieber NL, Ariotti N, Murphy S, Kuerschner L, Webb RI, Grinstein S, Parton RG. 2011. High-resolution mapping reveals topologically distinct cellular pools of phosphatidylserine. J. Cell Biol. 194:257–275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mercer J, Helenius A. 2008. Vaccinia virus uses macropinocytosis and apoptotic mimicry to enter host cells. Science 320:531–535 [DOI] [PubMed] [Google Scholar]