

Abstract
Misfolding and aggregation of proteins are common pathogenic mechanisms of a group of diseases called proteinopathies. The formation and spread of proteinaceous lesions within and between individuals were first described in prion diseases and proposed as the basis of their infectious nature. Recently, a similar “prion-like” mechanism of transmission has been proposed in other neurodegenerative diseases such as Alzheimer's disease. We investigated if misfolding and aggregation of corrupted prion protein (PrPTSE) are always associated with horizontal transmission of disease. Knock-in transgenic mice (101LL) expressing mutant PrP (PrP-101L) that are susceptible to disease but do not develop any spontaneous neurological phenotype were inoculated with (i) brain extracts containing PrPTSE from healthy 101LL mice with PrP plaques in the corpus callosum or (ii) brain extracts from mice overexpressing PrP-101L with neurological disease, severe spongiform encephalopathy, and formation of proteinase K-resistant PrPTSE. In all instances, 101LL mice developed PrP plaques in the area of inoculation and vicinity in the absence of clinical disease or spongiform degeneration of the brain. Importantly, 101LL mice did not transmit disease on serial passage, ruling out the presence of subclinical infection. Thus, in both experimental models the formation of PrPTSE is not infectious. These results have implications for the interpretation of tests based on the detection of protein aggregates and suggest that de novo formation of PrPTSE in the host does not always result in a transmissible prion disease. In addition, these results question the validity of assuming that all diseases due to protein misfolding can be transmitted between individuals.
INTRODUCTION
Several studies suggest that neurodegenerative diseases due to protein misfolding (proteinopathies) share a pathogenesis with seeded aggregation of native proteins and the potential for horizontal transmission between individuals. Prion diseases serve as a paradigm for proteinopathies. The pathological hallmark of prion diseases is widespread spongiform encephalopathy (SE) with deposition of disease-associated misfolded prion protein (PrPTSE), which results from the posttranslational conversion of a normal host-encoded cellular isoform of the prion protein (PrPC) (1). Numerous investigators have concluded that PrPTSE is the sole component of the infectious agent (“prion”) causing disease in the complete absence of nucleic acid. Studies using 263K hamster scrapie have shown a strong correlation between PrPTSE and infectivity (1–3). However, prion diseases can develop with high levels of infectivity and very low levels of PrP deposition and, conversely, PrPTSE accumulates in the brain in some situations unaccompanied by the other typical neuropathologic changes or any clinical signs of disease (4–10). Thus, both the molecular basis of prion propagation between individuals and the mechanism by which PrPC converts into PrPTSE and spreads from cell to cell in the affected host remain unclear.
Misfolding and aggregation of other host proteins similar to what is seen in prion diseases are the molecular hallmark of several neurodegenerative diseases (e.g., β-amyloid in Alzheimer's disease [AD] and alpha-synuclein in Parkinson's disease [PD]). It has been reported that in all proteinopathies, misfolded proteins can elicit seeded aggregation of native proteins and spread through the brain in a “prion-like” manner (11). This possibility raises the potential for diseases due to protein misfolding to be transmissible between individuals. However, there may be significant differences between the mechanism by which misfolded protein spreads from cell to cell and that in which PrPTSE acquires infectious properties resulting in horizontal transmission of disease.
Based on the hypothesis that PrPTSE is infectious, conversion of PrPC into PrPTSE has been analyzed as a model of replication of prion infectivity using several methodologies, including in vitro amplification of PrPTSE (12–14). Recent studies concluded that the ratio of the infectivity titer to the amount of PrPTSE (specific infectivity) is much lower when the PrPTSE is generated by amplification in vitro than in infected brain-derived samples (15). Thus, while misfolded protein is readily generated in these assays, it is clear that not all the misfolded protein is infectious. Similarly, attempts to produce de novo infectivity from refolding or fibrillization of recombinant PrP into PrPTSE have resulted in poor transmission rates probably due to low levels of infectivity, and often passage through transgenic mice or hamsters is required before disease can be induced in recipient animals (16, 17). However, others reported that recombinant PrP in the presence of lipids and RNA can efficiently transmit prion disease to mice, suggesting that cofactors are important for prion infectivity (18).
PrPTSE in the form of PrP amyloid is consistently deposited in multiple areas of the cerebrum and cerebellum of patients with Gerstmann-Sträussler-Scheinker (GSS) disease, a prion disease that is most commonly present in individuals who have a proline-to-leucine (P-to-L) substitution at codon 102 (GSS P102L) of the prion protein-encoding gene (PRNP). In most patients with GSS P102L (“typical” phenotype), brain lesions include spongiform degeneration and widespread amyloid deposits. However, other patients present with an “atypical” GSS P102L phenotype, with large amounts of PrP amyloid accumulating in multiple areas of the cerebrum and cerebellum in the absence of spongiform encephalopathy (19–21). By inoculation of brain extracts from patients with typical and atypical GSS P102L into gene-targeted transgenic mice expressing the equivalent mutation in murine PrP (101LL mice) (22), we have demonstrated differences in the ability of misfolded PrP from these two forms of GSS to generate disease in these mice, in which typical GSS produces high transmission rates but atypical GSS transmits poorly (7). Unexpectedly, several asymptomatic mice (101LL-8a) that received an atypical GSS P102L inoculum were found on subsequent histological analysis to have large PrP amyloid plaques only in the corpus callosum (7). However, low transmission rates and long incubation periods are often observed when attempting to transmit prion disease to a new species, in what is known as “species barrier” (e.g., from human to mouse), or in models of synthetic prion transmission, raising the possibility that the apparent absence of transmission is due to low-level agent replication and subclinical infection. This can be addressed by subsequent subpassage in the same mouse line, wherein replication of infectivity and disease is observed following adaptation of the agent to the new species.
We hypothesize that there is a difference between cell-to-cell spread of misfolded proteins (described in AD, PD, and other diseases) within a host and transmission of an infectious agent between individuals. Infection is defined as invasion and multiplication of a microorganism in body tissues. Disease can arise if the host immune response fails to eradicate the pathogen and damage is inflicted on the host. However, for such organisms to survive and repeat the infectious cycle in other hosts, they must be able to leave the host and be transmitted to a new susceptible individual. In transmissible spongiform encephalopathies (TSEs) such as scrapie in sheep and goats and chronic wasting disease (CWD) in deer, infection appears to be spread by direct contact between infected animals. In the cases of bovine spongiform encephalopathy (BSE) and variant Creutzfeldt-Jakob disease (vCJD), the infection is spread via indirect routes in contaminated feed and blood. However, in diseases such as sporadic CJD (sCJD) and GSS, the misfolding of protein is thought to arise “spontaneously” in the brain in the absence of invasion by an exogenous infectious organism. Spread of these diseases between individuals does not normally occur and happens only in rare occurrences via iatrogenic intervention. To explore the relationship between PrPTSE and infectivity, we performed transmission studies in 101LL mice using as inoculum brain extracts from (i) healthy 101LL mice with PrP plaques (101LL-8a) and (ii) mice overexpressing PrP-101L with neurological disease, spongiform degeneration, and PrPTSE deposits.
MATERIALS AND METHODS
Transgenic mouse lines.
The 101LL mice generated by gene targeting (expressing wild-type levels of mutant PrP (PrP-101L) and the GSS-22 mice generated by microinjection of fertilized eggs overexpressing ∼12-fold levels of mutant PrP-101L were previously described (22, 23). Previous studies have shown that GSS-22 mice develop neurological signs due to overexpression of PrP-101L (23). Conventional wild-type 129/Ola mice served as controls.
Preparation of the inocula and challenge.
For inoculation of 101LL-8a brain extracts, a third serial passage of 101LL-8a was carried out by intracerebral (i.c.) inoculation of 20 μl of 10% brain homogenate prepared from the second passage of atypical GSS P102L in 101LL mice (7) into groups (n = 24) of 101LL and 129/Ola control mice (Fig. 1A). The 101LL mouse used to prepare the homogenate was culled 689 days postinoculation, had no clinical signs or spongiform degeneration, but had large PrP amyloid plaques in the corpus callosum and vicinity.
Fig 1.
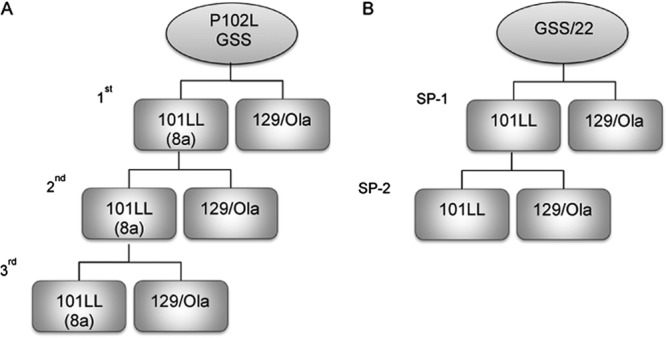
Flowchart outlining the serial passage of brain extract from a patient with “atypical” GSS P102L and uninoculated GSS-22 mice overexpressing PrP-101L into 101LL and 129/Ola mice. (A) Brain extract from a patient with “atypical” GSS P102L was inoculated intracerebrally into 101LL and 129/Ola mice. The absence of prion disease in 101LL-8a and 129/Ola mice was confirmed neuropathologically. Brain extract from selected 101LL-8a mice without disease but with PrP amyloid plaques was serially passaged into 101LL and 129/Ola mice. The first and second passages were described in reference 7. The third passage was performed in the work described here. (B) Brain extract from uninoculated GSS-22 mice overexpressing PrP-101L showing spongiform degeneration and diffuse and amyloid PrP deposits in the brain was serially passaged in 101LL and 129/Ola mice (101LL SP-1). The absence of prion disease was confirmed neuropathologically. Selected 101LL SP-1 tissue without prion disease showing PrP amyloid plaques in the corpus callosum and vicinity was used for serial passage into 101LL and 129/Ola mice (101LL SP-2).
Inoculation of GSS-22 brain extracts.
Twenty-microliter aliquots of 10% homogenates prepared from brains of two terminally ill GSS-22 mice were inoculated i.c. into groups (n = 24) of 101LL and 129/Ola control mice (Fig. 1B). Brain tissues were selected from two recipient 101LL mice culled at the end of their expected normal life span and inoculated into groups of 101LL and 129/Ola mice (Fig. 1B). They showed no evidence of neurological disease or spongiform degeneration but had immunopositive PrP deposits in the brain. Brains were aseptically removed and inoculations performed as described previously (22).
Scoring of clinical TSE.
Clinical signs of TSE were assessed, and incubation times were calculated according to previously described protocols (24). Mice were killed either during terminal disease, at the end of the expected normal life span, or earlier if they developed an intercurrent nonneurological illness. The left half of each brain was fixed in 10% Formol saline. Fixed brain tissue was processed, and tissue sections were prepared as described previously (7). The remaining half-brains were frozen at −70°C for biochemical analysis (22). All mouse experiments were reviewed and approved by the Local Ethical Review Committee and performed under License from the UK Home Office in accordance with the United Kingdom Animal (Scientific Procedures) Act 1986.
Lesion profiles and immunohistochemical analyses.
Tissue sections were assessed for spongiform degeneration following previously described procedures (24). Selected sections were immunostained with monoclonal antibody (MAb) 6H4 (Prionics, Zurich, Switzerland) recognizing residues 143 to 151 of murine PrP (2 μg/ml). Amyloid plaques were visualized with thioflavin S (25). Selected sections were probed with anti-PrP polyclonal antibodies (generously provided by B. Caughey, Rocky Mountain Laboratory, NIAID, NIH, Hamilton, MT); all antibodies were used at a 1:4,000 dilution. The selected antibodies are directed against several regions of PrP: R24 (amino acids 23 to 47), R30 (amino acids 89 to 103), R18 (amino acids 142 to 155), and R20 (amino acids 218 to 232). The presence of gliosis was assessed by incubating brain sections with an antibody to glial fibrillary acidic protein (GFAP; 1.45 μg/ml; Dako UK Ltd.) and anti-Iba1 antibody (0.05 μg/ml−1; Wako Chemicals).
PrP immunoblotting.
Brain tissues from terminally ill GSS-22 and asymptomatic 101LL mice (used for subpassage) were digested with proteinase K (PK) (20 μg/ml), incubated at 37°C for 1 h, and prepared for immunoblotting as described previously (26). Brain tissues from 129/Ola mice inoculated with strain ME7 of mouse-adapted TSE (scrapie) and from uninoculated 101LL mice were processed in the same way as controls. In brief, samples were electrophoresed on 12% Tris–glycine gels or 4 to 20% Tris–glycine gels and immunoblotted, and filters were probed with a series of anti-PrP monoclonal and polyclonal antibodies directed against various PrP epitopes. The following antibodies were used: 7A12 (1 mg/ml; monoclonal antibody recognizing amino acids 90 to 145; 1:20,000), 8H4 (1.5 mg/ml; monoclonal antibody recognizing amino acids 145 to 180; Sigma; 1:200), R30 (polyclonal antibody recognizing amino acids 89 to 103; 1:20,000), Mab6664 (polyclonal antibody recognizing amino acids 79 to 97; Abcam; 1:2,000), 1E4 (0.5 mg/ml; monoclonal antibody recognizing amino acids 108 to 119; Abcam; 1:500), 3C10 (1 mg/ml; monoclonal antibody recognizing amino acids 97 to 102; Jena-Bioscience; 1:5,000); and 6G3 (25 μg/ml; monoclonal antibody recognizing amino acids 130 to 150; Santa Cruz; 1:200). Bands were visualized using a secondary antibody labeled with horseradish peroxidase (HRP) (Jackson Immuno Research Laboratories, United Kingdom) and a chemiluminescent substrate (Roche).
Identification of PrPTSE using ligand-coated PrP antigen capture plates.
Brain and spleen extracts from terminally ill GSS-22 and from asymptomatic 101LL mice inoculated with brain extracts from terminally ill GSS-22 mice were screened by the Idexx HerdChek assay following the manufacturer's guidelines with minor modifications to optimize the test for mouse brain and spleen extracts (Idexx, West Yorkshire, United Kingdom). Buffer volumes were adjusted whenever possible to obtain 30% (wt/vol) homogenates. Selected brain and spleen homogenates from all test groups were assayed in duplicate whenever possible both before and after protease digestion using PK (20 μg/ml). Tissues were also assayed from well-characterized mouse-adapted scrapie agents: ME7, 79A, and 139A. A 101LL uninfected brain extract was used as a negative control.
RESULTS
Prion infectivity does not replicate in 101LL-8a mice.
Inefficient agent replication in a new host can produce a subclinical infection, where no clinical signs of disease are observed but tissues are able to transmit disease to recipient animals on subpassage. Therefore, to rule out subclinical infection in the 101LL-8a mice described previously (7), we obtained brains from 101LL-8a mice with no spongiform degeneration but with large PrP amyloid plaques in the corpus callosum following a second passage of atypical GSS P102L in 101LL mice (7) and performed serial passage into 101LL mice and 129/Ola controls (Fig. 1). Except for animals sacrificed due to intercurrent illness (n = 3), all mice remained asymptomatic, and the experiment was terminated 602 days postinoculation. Despite the absence of clinical signs, spongiform degeneration, or widespread gliosis (reactive astrocytes were seen in the periphery of amyloid plaques), all 101LL mice available for examination showed PrP amyloid plaques in the corpus callosum and vicinity (Table 1). To the best of our knowledge, 101LL-8a mice represent the first experimental model in which PrPC is consistently converted (over 3 passages) into PrP amyloid in restricted areas of the brain in the absence of any other signs of prion disease. Collectively, the results show that formation of PrP amyloid can be dissociated from replication of infectivity, given the lack of clinical disease or spongiform degeneration of the brain following 3 passages in mice. Importantly, 129/Ola mice used as controls did not develop any prion disease and did not form PrPTSE (Table 1).
Table 1.
PrP amyloid formation in 101LL mice inoculated with brain extracts from 101LL-8a and overexpressing GSS-22 mice
| Inoculum source | Recipient mice (n) | Avg survival ± SEM (days) | No. of mice affected/total no. of mice |
Output tissue IDa | ||
|---|---|---|---|---|---|---|
| Clinical prion disease | Spongiform degeneration | PrP plaques | ||||
| 101LL-8a | 101LL (16) | 602b | 0/16 | 0/16 | 16/16 | 101LL-8a |
| 101LL-8a | 129/Ola (22) | 602b | 0/22 | 0/22 | 0/22 | 129Ola-8a |
| NA | GSS-22 (9) | 181 ± 8 | 0/9 | 9/9 | 8/9c | GSS-22 |
| GSS-22 | 101LL (37) | 528 ± 23 | 0/37 | 0/37 | 29/37 | 101LL-SP1 |
| GSS-22 | 129/Ola (37) | 526 ± 24 | 0/37 | 0/37 | 0/37 | 129Ola-SP1 |
| 101LL-SP1 | 101LL (47) | 529 ± 14 | 0/47 | 0/47 | 40/47 | 101LL-SP2 |
| 101LL-SP1 | 129/Ola (46) | 558 ± 13 | 0/46 | 0/46 | 0/46 | 129Ola-SP2 |
Identifying tag given to tissues produced in each transmission.
Experiment terminated.
Widespread accumulation of PrP-positive plaques and diffuse deposits in the brain.
Transgenic mice overexpressing PrP-101L (GSS-22) develop the hallmarks of prion disease.
GSS-22 transgenic mice were previously engineered (23) to express PrP-101L at high levels (∼12-fold). A group of 12 mice were monitored for signs of clinical prion disease as they aged. All mice were culled due to intercurrent illness between 147 and 221 days of age (Table 1). Nine of these mice presented with a neurological phenotype that was neither consistent between animals nor consistent with clinical prion disease. Histopathologic studies of brain from uninoculated GSS-22 mice showed widespread spongiform degeneration (Table 1). The degree of spongiform degeneration in the brain as determined by lesion profile analysis is an important parameter to define prion disease. Comparative analysis of the lesion profiles seen in uninoculated aged GSS-22 mice with those described in terminally ill 101LL mice inoculated with brain homogenates from patients with “typical” GSS P102L (having severe spongiform degeneration of the brain) showed that they were clearly distinct, indicating that the neurological disorder observed in GSS-22 mice is distinct from classical GSS (Fig. 2). Immunohistochemical studies of uninoculated GSS-22 mice showed widespread and severe gliosis; in addition, accumulation of PrPTSE, mostly in the form of coarse and plaque-like deposits, was seen in multiple areas of the cerebrum (e.g., cerebral cortex, caudate nucleus, septum, hippocampus, thalamus, hypothalamus, cerebellum, brain stem, and spinal cord) in all mice (Fig. 3A6H4 and A*6H4). Several PrP-immunopositive plaque-like deposits fluoresced in sections treated with thioflavin S, confirming the presence of amyloid in the brain of these animals (Fig. 3AThioflavin). Similar results were obtained when samples were probed with a panel of antibodies directed against several PrP regions spanning residues 96 to 185 (mouse PrP nomenclature). In contrast to previous studies in mice overexpressing PrP-101L, we observed that brain homogenate from the oldest GSS-22 mouse culled with end stage disease at 221 days showed an electrophoretic pattern of PK-resistant PrP bands identical to that observed in most prion diseases (Fig. 4). However, we were unable to detect similar PK-resistant PrP bands in any of the younger mice analyzed, despite the identification of PrP amyloid plaques by immunohistochemistry. Although prion infectivity can be detected in the presence of low levels of PK-resistant PrP (6, 27), the identification of protease-resistant PrPTSE in the oldest GSS-22 mice indicates that this mouse model reproduces some aspects of prion disease. However, the inconsistency of clinical signs and general lack of PK-resistant PrP confirms that this model may instead develop a disorder caused by overexpression of the mutant PrP and amyloid accumulation and not replication of an infectious, transmissible prion disease.
Fig 2.
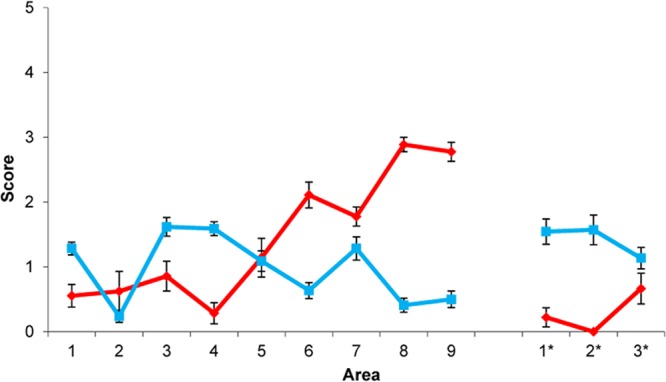
Pattern of vacuolation in 101LL mice with prion disease after inoculation of brain extract from a patient with “typical” GSS P102L and in uninoculated GSS-22 mice overexpressing PrP-101L. Lesion profile comparison of uninoculated mice overexpressing mutant PrP (GSS-22) (red) and 101LL mice with prion disease (blue). Data show mean lesion profiles ± standard errors of the means. Areas 1 to 9, gray matter scoring regions: 1, dorsal medulla; 2, cerebellar cortex; 3, superior colliculus; 4, hypothalamus; 5, thalamus; 6, hippocampus; 7, septum; 8, retrosplenial and adjacent motor cortex; 9, cingulate and adjacent motor cortex. Areas 1* to 3*, white matter scoring regions: 1*, cerebellar white matter; 2*, mesencephalic tegmentum; 3*, pyramidal tract.
Fig 3.

Contrast between abundant PrPTSE accumulation and gliosis in the brain of uninoculated GSS-22 mice and sparse PrP deposition in the brain of 101LL mice after serial subpassage. Shown are abundant PrPTSE accumulations in multiple areas of the cerebral cortex, hippocampus, and thalamus of uninoculated GSS-22 mice with terminal disease (A6H4, A*6H4), PrPTSE accumulation restricted to the corpus callosum and hippocampus in 101LL-SP1 (B6H4, B*6H4), small amounts of PrPTSE in the corpus callosum of 101LL-SP2 (C6H4, C*6H4), PrP fluorescent amyloid deposits in the corpus callosum and hippocampus of GSS-22 (AThioflavin) and 101LL-SP1 mice (BThioflavin), severe microgliosis (AIBA1) and astrogliosis (AGFAP) in the hippocampus of GSS-22 mice, milder microgliosis (BIBA1) and astrogliosis (BGFAP) in the hippocampus and vicinity of 101LL-SP1, and minimal microgliosis (CIBA1) and astrogliosis (CGFAP) in 101LL-SP2 mice and in 101LL aged controls (DIBA1 and DGFAP).
Fig 4.
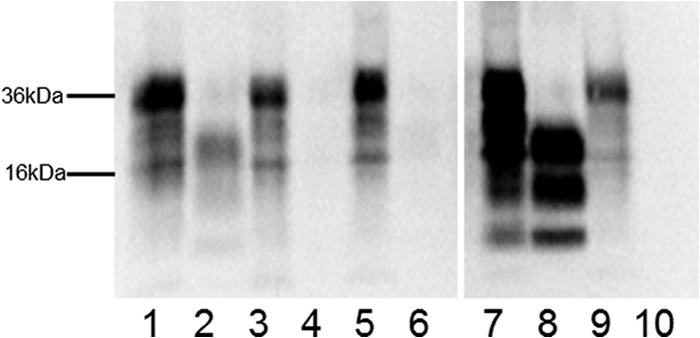
Presence of PK-resistant PrP in uninoculated GSS-22 mice. Western blotting using the 7A12 anti-PrP antibody was carried out in brain homogenates of uninoculated GSS-22 mice (lanes 1 and 2), 101LL-SP1 mice (lanes 3 and 4), and 101LL-SP2 mice (lanes 5 and 6). Positive control, 101LL mice inoculated with scrapie agent ME7 (lanes 7 and 8); negative control, 101LL noninoculated mouse (lanes 9 and 10). PK-resistant PrP is observed in GSS-22 (lane 2) and in 101LL mouse inoculated with scrapie agent ME7 (lane 8). Non-PK-treated samples (lanes 1, 3, 5, 7, and 9); PK-treated samples (lanes 2, 4, 6, 8, and 10). The image was cropped from a single blot to remove irrelevant lanes.
GSS-22 mice do not transmit disease to 101LL mice.
As the disease characterized in the GSS-22 mice did not resemble a classical TSE phenotype, we assayed for the presence of a transmissible prion agent in brain tissue of these mice by serial passage into 101LL mice, a knock-in model of the same PrP mutation. Homogenates were prepared from brains of two GSS-22 mice culled at 197 and 221 days of age, and each homogenate was inoculated i.c. into groups of 101LL (101LL-SP1) and 129/Ola (129/Ola-SP1) mice (Table 1 and Fig. 1B). The GSS-22 mice used to generate each inoculum showed severe spongiform degeneration and high levels of immunopositive PrP in most brain areas. All 101LL-SP1 and 129/Ola-SP1 mice were culled either for intercurrent illness or at the end of their expected normal life span > 520 days postinoculation. None of these animals developed neurological signs or spongiform degeneration of the brain (Table 1). A total of 37 101LL mice (101LL-SP1) and 37 129/Ola mice (129/Ola-SP1) were available from both transmissions for analysis. All animals were examined for PrP deposition by immunohistochemistry using antibody 6H4. We observed that, in contrast to the widespread and severe PrP deposition seen in overexpressing GSS-22 mice, brains of 101LL-SP1 (29/37) mice showed a restricted pattern of PrP accumulation in the form of PrP amyloid plaques mostly limited to the corpus callosum and stratum lacunosum-moleculare of the hippocampus (Fig. 3B6H4 and B*6H4). In a few animals, limited PrP immunopositivity was seen in the subependymal region of the lateral ventricle. Some plaques showed unstained centers, probably due to failure of the antibody to penetrate the amyloid cores. However, the poorly stained plaques fluoresced in sections treated with thioflavin S, indicating that they were amyloid deposits (Fig. 3BThioflavin). In 101LL-SP1 mice, astrocytic gliosis and microgliosis were seen in the vicinity of amyloid plaques but were less severe than the gliosis seen in uninoculated GSS-22 mice (Fig. 3BIBA1 and BGFAP). No PrP deposits were observed in the brains of any 129/Ola-SP1 mice (Table 1). These findings show that expression of mutant PrP in the host is important for amyloidogenesis and that amyloid deposition occurs in the inoculated 101LL-SP1 mice in the absence of clinical disease or spongiform degeneration of the brain.
Are 101LL SP-1 mice asymptomatic carriers?
To investigate the possibility of subclinical infection, we prepared brain homogenates from two 101LL-SP1 mice (776 and 807 days old) for further passage (Fig. 1B). To increase the chances of disease transmission, we prepared brain homogenates from selected mice with large PrP deposits in the corpus callosum and vicinity (the only regions of the brain having moderate to abundant PrP accumulations in this group of animals). Each brain homogenate was inoculated i.c. into 101LL mice (101LL-SP2) and 129/Ola mice (129/Ola-SP2) (Fig. 1B and Table 1). None of those mice developed neurological signs, and they were finally culled due to intercurrent illness or at the end of their expected normal life span at over 540 days postinoculation (Table 1). Neuropathologic examination revealed no spongiform degeneration in any of those mice. No accumulations of PrP were detected in 129/Ola-SP2 mice; however, 101LL-SP2 mice showed small numbers of PrP-positive plaques located mostly in the hippocampus and corpus callosum (Fig. 3C6H4 and C*6H4 and Table 1). Smaller amounts of PrP accumulation were seen in 101LL-SP2 mice than in 101LL-SP1 mice. In a few animals, limited PrP immunopositivity was also seen in the subependymal region of the lateral ventricle. The level of gliosis detected in 101LL-SP2 mice is comparable to that observed in age-matched uninoculated 101LL mice (control) (Fig. 3CIBA1, CGFAP, DIBA1, and DGFAP). In contrast to the immunopositive PrP seen in tissue sections, no protease-resistant PrPTSE was detected in the brains of 101LL mice from subpassages (101LL-SP1 and 101LL-SP2) tested by immunoblotting. Thus, inoculation of PrPTSE from GSS-22 mice into 101LL mice induces the formation of PrP amyloid but no disease on 2 passages.
Some prion strains have been shown to replicate preferentially in lymphoid tissue without generating a fatal neurological disease. Under this scenario, the apparent absence of transmission could represent subclinical infection (28). A ligand-coated PrP antigen capture immunoassay (Idexx HerdCheck) has been widely utilized for the screening of natural prion diseases in animals. Therefore, we used this immunoassay (which has been shown to be highly sensitive for the detection of aggregated PrP) to explore the presence of PrPTSE in the brain and spleen of spontaneously sick GSS-22 and in asymptomatic 101LL-SP1 and 101LL-SP2 mice. Using Idexx HerdCheck, we detected PrPTSE in brains of aged/sick, uninoculated GSS-22 mice, in brains of mice in the first subpassage (101LL-SP1), and in control animals following inoculation with mouse-adapted scrapie agents (ME7 or 79A with large amounts of PrPTSE and infectivity titers). Brains from mice in the second subpassage experiments (101LL-SP2 mice) yielded negative results. In addition, spleens from all experimental groups (GSS22, 101LL-SP1, and 101LL-SP2) were negative for PrPTSE. The results argue for absence of PrPTSE in the periphery of GSS-22, 101LL-SP1, and 101LL-SP2 mice, despite the formation of plaques in the brain.
DISCUSSION
The original concept that prion diseases constitute a special group among proteinopathies, because the seeded polymerization of PrPTSE replicates its misfolded conformation indefinitely and provides the molecular basis for spread within defined anatomical pathways and transmission between individuals, has more recently been expanded to include other neurodegenerative diseases (29, 30). The mechanism of transport of misfolded protein aggregates from one cell to another remains to be determined but could include endocytosis, tunneling nanotubes, or a transynaptic mechanism. The last was proposed for the cell-to-cell spread of hyperphosphorylated microtubule-associated protein (tau) in patients with AD (31). However, our results suggest that there is a difference between cell-to-cell spread of a misfolded protein and the transfer of infectivity from one organism to another, a process that clearly requires a different kind of interaction between the transmissible agent and its host.
101LL-8a mice do not have subclinical infection.
In our original study (7), we found no evidence of replication of infectivity in 101LL-8a mice with PrP amyloid plaques following inoculation with PrPTSE purified from a patient with atypical GSS P102L. Absence of cross-species transmission to host species that are susceptible to many prion agents has been observed in few instances. Importantly, analysis following serial subpassage of hamster prion agent into mice has demonstrated that in some instances subclinical infections have two distinct phases, a persistent phase followed by a replicative phase (32). This issue has practical consequences because in wildlife and agricultural settings prion agents might both persist and adapt over long periods of time (years). Here, we demonstrate that even following subpassages the formation of mouse PrP amyloid plaques triggered by the inoculation of PrPTSE obtained from a patient with atypical GSS P102L is dissociated from disease. In fact, the only form of GSS shown to be transmissible to experimental animals is typical GSS P102L.
GSS-22 mice overexpressing PrP 101L develop a nontransmissible prion disease.
The dramatic differences in the amounts and distribution of PrPTSE seen in 101LL-SP1 and 101LL-SP2 compared with GSS-22 mice can be interpreted in several ways. PrPTSE in the GSS-22-derived inoculum might be unstable and readily degraded, preventing the misfolding and accumulation of endogenous PrP that would cause disease. However, the presence of amyloid plaques in the corpus callosum and vicinity in many animals strongly suggest that PrPTSE in the inoculum is stable and remains capable of seeding further amyloid formation. Another possibility is that low levels of infectivity in the GSS-22 inoculum caused subclinical infection in 101LL animals. Other investigators reported a long incubation time and slow progression of clinical disease in mice and hamsters inoculated with synthetic prions (32) and lack of clinical disease in animals expressing anchorless PrP inoculated with mouse-adapted scrapie agent (33) or in transmission experiments across species (34, 35). Interestingly, in all these models disease was consistently detected after serial passage in wild-type animals, demonstrating efficient adaptation and spread of the infectious agent. To rule out the occurrence of subclinical infection in 101LL mice after the initial injection with GSS-22 brain suspension, we performed subpassage experiments. We observed that although 101LL recipient animals remained asymptomatic and without any spongiform encephalopathy, they again accumulated PrPTSE but only in restricted areas of the brain. These results differ substantially from those obtained by others, whereby generation of prion infectivity and disease transmission was evident following subpassage (32–35).
The data presented in this work could be interpreted to show an infection, where introduction of the amyloid seed from brains of patients with atypical GSS P102L or GSS-22 mice leads to the propagation of further amyloid in the brains of recipient mice. However, this “transmission” is iatrogenic, and we have yet to determine whether such seeding can be initiated by following “natural” routes such as oral exposure. Such infection would also appear to be nonproductive, as plaques are restricted to a limited brain area and, following up to 3 subpassages, no disease is observed in 101LL mice. Limited spread is also seen within the brain, and no spread to the periphery is apparent. We also failed to induce the plaque-forming phenotype in wild-type mice. In addition, previous ultrastructural analysis showed that cell membrane alterations consistently seen in murine scrapie and other infectious prion diseases were not present in 101LL-8a mice with amyloid plaques, suggesting differences in the pathogenesis of these conditions (9). Therefore, our model would appear to represent a different mechanism from what is generally understood as infection.
Relevance to natural protein misfolding diseases.
Most proteinopathies (e.g., AD and PD) have previously not been considered to be infectious diseases, and indeed there is no epidemiological evidence to suggest that they are. It would seem important therefore to try to understand when a misfolded protein has the potential to be transmitted between individuals, which should be considered a very different scenario from a cell-to-cell spread within an individual, the former having a consequence for a population and the latter for an individual. If even within prion diseases some affect the individual only, then this would provide precedence for other protein misfolding diseases to be specific to individuals rather than having consequences for populations. We believe that the experiments outlined in this paper start to address this important issue, namely, that protein seeding within an individual and transmission of disease between individuals represent different processes.
In our experiments, we consider that the 101LL mouse model would, with an extended life span, overcome the long lag phase required to form endogenous PrP amyloid plaques naturally via a nucleated polymerization mechanism. This would reflect what is seen in atypical GSS P102L, where PrP plaques are seen in the absence of spongiform change in the brains of older patients. By introducing the PrP-8a or GSS-22 amyloid seed into 101LL mice, the lag phase is reduced, accelerating plaque formation in restricted areas of the brain. GSS-22 mice ubiquitously overexpress PrP-101L and therefore are likely to shift the balance of seed nucleation due to unregulated overproduction of the mutated protein. These data mirror what has been described in overexpressing transgenic models of human amyloid precursor protein (APP), seeded with AD brain (30).
In vitro studies suggested that infectious and noninfectious aggregates of PrP might be structurally similar but that cofactors such as lipids and nucleic acids might also be critical in determining the infectious characteristics of PrPTSE (36). Another possibility for the lower specific infectivity associated with PrP amyloid is that extracts with smaller PrPTSE aggregates might be more infectious than the larger ones (37). While disease-associated PrP (i.e., PrPTSE) was originally described as insoluble in detergents and PK resistant, it is increasingly recognized that multiple distinct disease-related PrP isoforms may be important in the pathogenesis of prion diseases (5, 23, 38–40). Our data suggest that the formation of PrPTSE does not necessarily correlate with the replication of prion infectivity. The results from bioassays reported here have implications for the interpretation of tests based on the detection of protein aggregates. Thus, determining the difference between PrPTSE aggregates associated with infectivity and those aggregates that are not infectious will be essential to determine which diseases associated with protein misfolding are threats to public health (transmissible between individuals).
ACKNOWLEDGMENTS
We thank David M. Asher (Laboratory of Bacterial and TSE Agents, U.S. FDA) for critical reading of the manuscript, V. Thomson, S. Cumming, K. Hogan, S. Carpenter, and R. Greenan for care and scoring of the animals, and A. Coghill, A. Boyle, S. Mack, and G. McGregor for tissue processing and lesion profile analysis.
These studies were partially funded by the Biotechnology and Biological Sciences Research Council (BBSRC) Institute Strategic Grant BB/J004332/1, NIH-NIAID Agreement no. Y1-AI-4893-02, FDA Agreement no. 224-05-1307, and a Public Health Service Cooperative Research and Development Agreement between U.S. FDA and The Roslin Institute, University of Edinburgh, United Kingdom.
The findings and conclusions in this article have not been formally disseminated by the Food and Drug Administration and should not be construed to represent any Agency determination of policy.
Footnotes
Published ahead of print 11 September 2013
REFERENCES
- 1.Prusiner S. 1982. Novel proteinaceous infectious particles cause scrapie. Science 216:136–144 [DOI] [PubMed] [Google Scholar]
- 2.McKinley MP, Bolton DC, Prusiner SB. 1983. A protease-resistant protein is a structural component of the scrapie prion. Cell 35:57–62 [DOI] [PubMed] [Google Scholar]
- 3.Beekes M, Baldauf E, Diringer H. 1996. Sequential appearance and accumulation of pathognomonic markers in the central-nervous-system of hamsters orally infected with scrapie. J. Gen. Virol. 77:1925–1934 [DOI] [PubMed] [Google Scholar]
- 4.Lasmezas CI, Deslys J, Robain O, Jaegly A, Beringue V, Peyrin J, Fournier J, Hauw J, Rossier J, Dormont D. 1997. Transmission of the BSE agent to mice in the absence of detectable abnormal prion protein. Science 275:402–405 [DOI] [PubMed] [Google Scholar]
- 5.Chiesa R, Piccardo P, Quaglio E, Drisaldi B, Si-Hoe SL, Takao M, Ghetti B, Harris DA. 2003. Molecular distinction between pathogenic and infectious properties of the prion protein. J. Virol. 77:7611–7622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Barron RM, Campbell SL, King D, Bellon A, Chapman KE, Williamson RA, Manson JC. 2007. High titres of TSE infectivity associated with extremely low levels of PrPSc in vivo. J. Biol. Chem. 282:35878–35886 [DOI] [PubMed] [Google Scholar]
- 7.Piccardo P, Manson JC, King D, Ghetti B, Barron RM. 2007. Accumulation of prion protein in the brain that is not associated with transmissible disease. Proc. Natl. Acad. Sci. U. S. A. 104:4712–4717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Balkema-Buschmann A, Eiden M, Hoffmann C, Kaatz M, Ziegler U, Keller M, Groschup MH. 2011. BSE infectivity in the absence of detectable PrPSc accumulation in the tongue and nasal mucosa of terminally diseased cattle. J. Gen. Virol. 92:467–476 [DOI] [PubMed] [Google Scholar]
- 9.Jeffrey M, McGovern G, Chambers EV, King D, González L, Manson JC, Ghetti B, Piccardo P, Barron RM. 2012. Mechanism of PrP-amyloid formation in mice without transmissible spongiform encephalopathy. Brain Pathol. 22:58–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Miyazawa K, Emmerling K, Manuelidis L. 2011. High CJD infectivity remains after prion protein is destroyed. J. Cell Biochem. 112:3630–3637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Frost B, Diamond MI. 2010. Prion-like mechanisms in neurodegenerative diseases. Nat. Rev. Neurosci. 11:155–159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Saborio GP, Permanne B, Soto C. 2001. Sensitive detection of pathological prion protein by cyclic amplification of protein misfolding. Nature 411:810–813 [DOI] [PubMed] [Google Scholar]
- 13.Colby DW, Zhang Q, Wang S, Groth D, Legname G, Riesner D, Prusiner SB. 2007. Prion detection by an amyloid seeding assay. Proc. Natl. Acad. Sci. U. S. A. 104:20914–20919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Atarashi R, Wilham JM, Christensen L, Hughson AG, Moore RA, Johnson LM, Onwubiko HA, Priola SA, Caughey B. 2008. Simplified ultrasensitive prion detection by recombinant PrP conversion with shaking. Nat. Methods 5:211–212 [DOI] [PubMed] [Google Scholar]
- 15.Klingeborn M, Race B, Meade-White KD, Chesebro B. 2011. Lower specific infectivity of protease-resistant prion protein generated in cell-free reactions. Proc. Natl. Acad. Sci. U. S. A. 108:E1244–E1253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Makarava N, Kovacs GG, Bocharova O, Savtchenko R, Alexeeva I, Budka H, Rohwer RG, Baskakov IV. 2010. Recombinant prion protein induces a new transmissible prion disease in wild-type animals. Acta Neuropathol. 119:177–187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Raymond GJ, Race B, Hollister JR, Offerdahl DK, Moore RA, Kodali R, Raymond LD, Hughson AG, Rosenke R, Long D, Dorward DW, Baron GS. 2012. Isolation of novel synthetic prion strains by amplification in transgenic mice coexpressing wild-type and anchorless prion proteins. J. Virol. 86:11763–11778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang F, Wang X, Yuan C-G, Ma J. 2010. Generating a prion with bacterially expressed recombinant prion protein. Science 327:1132–1135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ghetti B, Tagliavini F, Kovacs GG, Piccardo P. 2011. Gerstmann-Straussler-Sheinker disease. In Dickinson DW, Weller RO. (ed), Neurodegeneration: the molecular pathology of dementia and movement disorders, p 364–377. Blackwell Publishing Ltd., Oxford, United Kingdom [Google Scholar]
- 20.Piccardo P, Dlouhy SR, Lievens PMJ, Young K, Thomas DP, Nochlin D, Dickson DW, Vinters HV, Zimmerman TR, Mackenzie IRA, Kish SJ, Ang LC, De Carli C, Pocchiari M, Brown P, Gibbs CJ, Gajdusek DC, Bugiani O, Ironside J, Tagliavini F, Ghetti B. 1998. Phenotypic variability of Gerstmann-Straussler-Scheinker disease is associated with prion protein heterogeneity. J. Neuropathol. Exp. Neurol. 57:979–988 [DOI] [PubMed] [Google Scholar]
- 21.Parchi P, Chen SG, Brown P, Zou WQ, Capellari S, Budka H, Hainfellner J, Reyes PF, Golden GT, Hauw JJ, Gajdusek DC, Gambetti P. 1998. Different patterns of truncated prion protein fragments correlate with distinct phenotypes in P102L Gerstmann-Straussler-Scheinker disease. Proc. Natl. Acad. Sci. U. S. A. 95:8322–8327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Manson JC, Jamieson E, Baybutt H, Tuzi NL, Barron R, McConnell I, Somerville R, Ironside J, Will R, Sy MS, Melton DW, Hope J, Bostock C. 1999. A single amino acid alteration (101L) introduced into murine PrP dramatically alters incubation time of transmissible spongiform encephalopathy. EMBO J. 18:6855–6864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nazor KE, Kuhn F, Seward T, Green M, Zwald D, Pürro M, Schmid J, Biffiger K, Power AM, Oesch B, Raeber A, Telling G. 2005. Immunodetection of disease-associated mutant PrP, which accelerates disease in GSS transgenic mice. EMBO J. 24:2472–2480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fraser H, Dickinson AG. 1967. Distribution of experimentally induced scrapie lesions in the brain. Nature 216:1310–1311 [DOI] [PubMed] [Google Scholar]
- 25.Schmidt ML, Robinson KA, Lee VMY, Trojanowski JQ. 1995. Chemical and immunological heterogeneity of fibrillar amyloid in plaques of Alzheimer's disease and Downs Syndrome brains revealed by confocal microscopy. Am. J. Pathol. 147:503–515 [PMC free article] [PubMed] [Google Scholar]
- 26.Plinston C, Hart P, Chong A, Hunter N, Foster J, Piccardo P, Manson JC, Barron RM. 2011. Increased susceptibility of human-PrP transgenic mice to bovine spongiform encephalopathy infection following passage in sheep. J. Virol. 85:1174–1181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tzaban S, Friedlander G, Schonberger O, Horonchik L, Yedidia Y, Shaked G, Gabizon R, Taraboulos A. 2002. Protease-sensitive scrapie prion protein in aggregates of heterogeneous sizes. Biochemistry 41:12868–12875 [DOI] [PubMed] [Google Scholar]
- 28.Beringue V, Herzog L, Jaumain E, Reine F, Sibille P, Le Dur A, Vilotte JL, Laude H. 2012. Facilitated cross-species transmission of prions in extraneural tissue. Science 335:472–475 [DOI] [PubMed] [Google Scholar]
- 29.Westermark GT, Westermark P. 2010. Prion-like aggregates: infectious agents in human disease. Trends Mol. Med. 16:501–507 [DOI] [PubMed] [Google Scholar]
- 30.Brundin P, Melki R, Kopito R. 2010. Prion-like transmission of protein aggregates in neurodegenerative diseases. Nat. Rev. Mol. Cell Biol. 11:301–307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.de Calignon A, Polydoro M, Suarez-Calvet M, William C, Adamowicz DH, Kopeikina KJ, Pitstick R, Sahara N, Ashe KH, Carlson GA, Spires-Jones TL, Hyman BT. 2012. Propagation of tau pathology in a model of early Alzheimer's disease. Neuron 73:685–697 (Erratum, 76:461.) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Makarava N, Kovacs GG, Savtchenko R, Alexeeva I, Budka H, Rohwer RG, Baskakov IV. 2012. Stabilization of a prion strain of synthetic origin requires multiple serial passages. J. Biol. Chem. 287:30205–30214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chesebro B, Trifilo M, Race R, Meade-White K, Teng C, LaCasse R, Raymond L, Favara C, Baron G, Priola S, Caughey B, Masliah E, Oldstone M. 2005. Anchorless prion protein results in infectious amyloid disease without clinical scrapie. Science 308:1435–1439 [DOI] [PubMed] [Google Scholar]
- 34.Race R, Meade-White K, Raines A, Raymond GJ, Caughey B, Chesebro B. 2002. Subclinical scrapie infection in a resistant species: persistence, replication, and adaptation of infectivity during four passages. J. Infect. Dis. 186:S166–S170 [DOI] [PubMed] [Google Scholar]
- 35.Hill AF, Antoniou M, Collinge J. 1999. Protease-resistant prion protein produced in vitro lacks detectable infectivity. J. Gen. Virol. 80:11–14 [DOI] [PubMed] [Google Scholar]
- 36.Deleault NR, Kascsak R, Geoghegan JC, Supattapone S. 2010. Species-dependent differences in cofactor utilization for formation of the protease-resistant prion protein in vitro. Biochemistry 49:3928–3934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Silveira JR, Raymond GJ, Hughson AG, Race R, Sim VL, Hayes SF, Caughey B. 2005. The most infectious prion protein particles. Nature 437:257–261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Biasini E, Seegulam ME, Patti BN, Solforosi L, Medrano AZ, Christensen HM, Senatore A, Chiesa R, Williamson RA, Harris DA. 2008. Non-infectious aggregates of the prion protein react with several PrPSc-directed antibodies. J. Neurochem. 105:2190–2204 [DOI] [PubMed] [Google Scholar]
- 39.Miller MB, Geoghegan JC, Supattapone S. 2011. Dissociation of infectivity from seeding ability in prions with alternate docking mechanism. PLoS Pathog. 7:e1002128. 10.1371/journal.ppat.1002128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chiesa R, Piccardo P, Biasini E, Ghetti B, Harris DA. 2008. Aggregated, wild-type prion protein causes neurological dysfunction and synaptic abnormalities. J. Neurosci. 28:13258–13267 [DOI] [PMC free article] [PubMed] [Google Scholar]