

Abstract
Influenza virus is a global health concern due to its unpredictable pandemic potential. This potential threat was realized in 2009 when an H1N1 virus emerged that resembled the 1918 virus in antigenicity but fortunately was not nearly as deadly. 5J8 is a human antibody that potently neutralizes a broad spectrum of H1N1 viruses, including the 1918 and 2009 pandemic viruses. Here, we present the crystal structure of 5J8 Fab in complex with a bacterially expressed and refolded globular head domain from the hemagglutinin (HA) of the A/California/07/2009 (H1N1) pandemic virus. 5J8 recognizes a conserved epitope in and around the receptor binding site (RBS), and its HCDR3 closely mimics interactions of the sialic acid receptor. Electron microscopy (EM) reconstructions of 5J8 Fab in complex with an HA trimer from a 1986 H1 strain and with an engineered stabilized HA trimer from the 2009 H1 pandemic virus showed a similar mode of binding. As for other characterized RBS-targeted antibodies, 5J8 uses avidity to extend its breadth and affinity against divergent H1 strains. 5J8 selectively interacts with HA insertion residue 133a, which is conserved in pandemic H1 strains and has precluded binding of other RBS-targeted antibodies. Thus, the RBS of divergent HAs is targeted by 5J8 and adds to the growing arsenal of common recognition motifs for design of therapeutics and vaccines. Moreover, consistent with previous studies, the bacterially expressed H1 HA properly refolds, retaining its antigenic structure, and presents a low-cost and rapid alternative for engineering and manufacturing candidate flu vaccines.
INTRODUCTION
Influenza virus is the cause of seasonal epidemic and sporadic pandemic flu outbreaks. The hemagglutinin (HA) surface glycoprotein mediates viral recognition of host cells through its interaction with sialic acid receptors (1, 2). The globular “head” domain of HA is immunodominant, likely due to its accessibility on the surface of viruses, and, consequently, antibodies are rapidly generated against it. However, the HA head undergoes continual antigenic drift, which results in escape from the host immune response through amino acid changes on its surface or by masking neutralizing epitopes with glycans. Antibodies generated against HA are typically strain specific, which necessitates nearly annual vaccine strain reformulations. In contrast, the residues that form the receptor binding site (RBS) are functionally constrained for receptor binding and, thus, have restricted mutational freedom. As such, the RBS is a prime target for virus neutralization by broadly neutralizing antibodies that prevent viral-host interactions (3). However, the footprint of the sialoglycan receptor on the RBS is much smaller than that of an antibody. Hence, most antibodies that block the RBS also contact the hypervariable regions surrounding it, which leads to strain-specific binding. Nevertheless, a few antibodies that target the RBS display a broader spectrum of reactivity than those that target HA elsewhere on the head (4–10). S139/1 reaches into the RBS and has heterosubtypic neutralizing activity (7). C05, which also neutralizes highly divergent viruses, similarly enters the RBS and remarkably accomplishes this interaction using essentially a single antibody loop (6). CH65 and CH67 are broadly neutralizing H1-specific antibodies and, unlike S139/1 and C05, make use of receptor mimicry (5, 9); however, CH65 does not neutralize 1918 or 2009 H1 pandemic strains that are now the current seasonal H1 epidemic strains (5).
We have previously reported the identification and characterization of a human monoclonal antibody, 5J8, that possesses neutralization activity and therapeutic efficacy against H1 viruses spanning decades, including the 1918 and 2009 pandemic viruses (11). Notably, the pandemic strains contain a basic amino acid insertion at residue 133a (between residues 133 and 134) that has been proposed to sterically clash with other RBS-targeted antibodies (6, 7). Here, we present the crystal structure of the bacterially expressed HA1 globular head domain from the A/California/07/2009 (H1N1) (Cali07/2009-H1) virus in complex with 5J8 Fab. The complex structure reveals that Lys133a, which is conserved in pandemic H1N1 strains, makes favorable electrostatic interactions with an acidic patch on the antibody. Similar to other RBS-targeted antibodies (6, 7), avidity through a bivalent IgG extends the antibody's breadth of neutralization and allows it to bind divergent HA strains within the H1 subtype. Most strikingly, 5J8 reaches into the RBS and utilizes receptor mimicry, similar to that of CH65 and CH67 (5, 9). That these three antibodies all use receptor mimicry and, hence, display a common theme for receptor site recognition, as well as provide complementary coverage of H1 viruses spanning the past four decades, reinforces the RBS as a promising site of vulnerability on the HA.
MATERIALS AND METHODS
Fab and IgG cloning, expression, and purification.
5J8 and CH65 Fab were cloned in a pFastBac dual vector (Invitrogen) with N-terminal gp67 and honeybee melittin secretion signal peptides fused to the heavy and light chains, respectively, and a C-terminal His6 tag fused to the heavy chain. Recombinant bacmid DNA and baculovirus were generated as previously described (7). The Fabs were purified by Ni-nitrilotriacetic acid (NTA) (Qiagen) and Mono S (GE Healthcare) chromatography. The purified Fabs were then dialyzed into 20 mM HEPES (pH 7.4) and 150 mM NaCl, flash frozen with liquid nitrogen, and stored at −80°C.
The heavy and light chains of the 5J8 and CH65 IgGs were cloned separately in a phCMV3 vector (Genlantis) with an N-terminal Ig kappa secretion signal peptide and a C-terminal His6 tag fused to the heavy chain. The heavy and light chain plasmids were transiently transfected at a 2:1 ratio into HEK293F suspension cells and incubated for six days. The IgGs were purified by Ni-NTA (Qiagen) and protein A (GE Healthcare) chromatography and then buffer exchanged into 20 mM Tris (pH 8) and 150 mM NaCl.
HA expression and purification.
HA was prepared for binding studies and crystallization as previously described (7, 12). Briefly, each HA was fused with an N-terminal gp67 signal peptide and a C-terminal BirA biotinylation site, a thrombin cleavage site, a trimerization domain, and a His6 tag. The HAs were expressed as described above for the Fabs and purified by Ni-NTA. The HAs were either matured by trypsin (New England BioLabs) for crystallization or biotinylated with BirA (12) for binding studies.
Flagellin-HA1 preparation.
The HA1 protein was derived from a recombinant fusion protein, STF2.HA1, that contains N-terminal Salmonella enterica serovar Typhimurium flagellin residues 1 to 505 linked to the N terminus of A/California/07/2009 (H1N1) (Cali07/2009-H1) residues 55 to 271 (H3 numbering), which was expressed in batch bioreactor cultures as previously described (13).
Crystallization.
Apo 5J8 Fab crystals were grown by sitting drop vapor diffusion at 4°C by mixing 0.5 μl of protein (6.3 mg/ml) with 0.5 μl of reservoir solution (0.2 M calcium acetate, 11% [wt/vol] polyethylene glycol 3350 [PEG 3350]). Crystals were cryoprotected in mother liquor supplement with 20% (vol/vol) ethylene glycol, flash cooled, and stored in liquid nitrogen until data collection.
The 5J8 Fab-STF2.HA1 complex was prepared by mixing individually prepared proteins in a 1.1:1 molar ratio and then purified by gel filtration. Fractions corresponding to the complex were pooled and concentrated to 16 mg/ml for crystallization screening. However, STF2.HA1 degraded over time, likely at or around the linker connecting flagellin and HA1, as only HA1 in complex with 5J8 Fab crystallized. Crystals grew at 23°C by sitting drop vapor diffusion by mixing 0.6 μl of protein solution (16 mg/ml) with 0.5 μl of reservoir solution (0.1 M Tris [pH 8.5], 23% [wt/vol] PEG 8000, 0.2 M magnesium chloride). Crystals were cryoprotected in mother liquor supplemented with 15% (vol/vol) glycerol, flash cooled, and stored in liquid nitrogen until data collection.
The trimer-stabilized A/California/04/2009 (H1N1) (Cali04/2009-H1 HA2 E47G) HA crystals were grown by sitting drop vapor diffusion. The HA was concentrated to 17 mg/ml in 20 mM Tris (pH 8.0), 100 mM NaCl, and 0.02% (vol/vol) NaN3 and crystallized in 0.1 M Tris (pH 8.8), 25% (wt/vol) methoxypolyethylene glycol 2000 (MPEG 2000) at 23°C. Crystals were flash cooled in mother liquor supplemented with 12% (vol/vol) ethylene glycol and stored in liquid nitrogen until data collection.
X-ray structure determination and refinement.
X-ray diffraction data for the apo 5J8 Fab were collected to a 1.55-Å resolution at the GM-CA CAT 23ID-D beamline at the Advanced Photon Source (APS). The data were processed in space group P212121 using XDS (14). The structure was determined by molecular replacement with Phaser (15) by using the variable and constant domains of the anti-HIV-1 V3 Fab 3074 (Protein Data Bank [PDB] accession no. 3MLY, chains H and L) as search models, and two Fab copies were found in the asymmetric unit. The crystal exhibited pseudotranslational symmetry. The model was iteratively built using Coot (16) and refined in Phenix (17). Refinement parameters included rigid-body refinement (set for each Ig domain), simulated annealing, and restrained refinement, including translation/libration/screw (TLS) refinement (for each Ig domain).
X-ray diffraction data for the 5J8-Cali07/2009-H1 HA1 complex were collected to 2.25 Å at the Canadian Light Source beamline 08B1-1 (CMGF-BM). The data were processed in space group P3121 using XDS (14). The complex was determined by molecular replacement with Phaser (15) by first using one copy of the HA1 from Cali04/2009-H1 (PDB accession no. 3LZG, chain A) residues 55 to 271 (H3 numbering). Next, one copy of the variable and constant domains of the high-resolution 5J8 Fab structure were used as search models after fixing the position and orientation of the HA head. The model was iteratively built using Coot (16) and refined in Phenix (17). Refinement parameters included rigid-body refinement (set for the HA1 and each Ig domain), simulated annealing, and restrained refinement, including TLS refinement (set for the HA1 and each Ig domain).
X-ray diffraction data for the trimer-stabilized HA (Cali04/2009-H1 HA2 E47G) were collected to a 2.20-Å resolution at beamline 12-2 at the Stanford Synchrotron Radiation Lightsource (SSRL). The mutant HA structure was determined by molecular replacement using the program Phaser (15) by using the native Cali04/2009-H1 HA (PDB accession no. 3LZG) as the starting model. The refinement was performed in Refmac5 (18) and Phenix (17), and model building was carried out with Coot (16).
Sample preparation and imaging by electron microscopy.
Copper grids (400 mesh) were coated in nitrocellulose and a thin layer of carbon. The grids received samples shortly (<20 min) after glow discharging. Negative staining was performed through application of 4 μl of sample (∼0.02 mg/ml of Fab-HA complex diluted in Tris-buffered saline [TBS]) to the grid, with blotting to remove excess sample, followed by two cycles of staining with 4 μl of “Nano W” stain (2% methylamine tungstate [Nanoprobes, Yaphank, NY]), 20 s of incubation, and blotting to remove excess stain.
Micrographs were acquired on an FEI Tecnai Spirit transmission electron microscope (TEM) operating at an accelerating voltage of 120 kV. A Tietz charge-coupled-device camera was used to record 2,048- by 2,048-pixel images at a magnification of ×52,000 and a defocus range of 900 to 1,300 nm. The stage was tilted in five-degree increments, from 0° to 55°, to increase the number of observed orientations. The pixel size was previously calibrated to be 2.65 Å using a two-dimensional catalase crystal. The Leginon software package (19, 20) was used to automate some steps of data acquisition.
Image processing, volume map determination, and interpretation.
Particles were automatically selected using a difference of Gaussian algorithm (21) provided in the Appion package (22), and most subsequent processing steps were facilitated using Appion. Particle boxing was performed using Eman1.9 (23) and Spider (24). Xmipp (25) was used to normalize the boxed images. There was no correction applied for the contrast transfer function. Initial classification was performed with the CL2D program (26) provided in the Xmipp package (25). Classification steps were followed by manual analysis in which heterogeneities or low-quality picks were excluded.
Images corresponding to homogeneous complexes were inputted into a projection-matching algorithm implemented in Eman 1.9 (23). An unliganded HA (PDB accession no. 4FQV) was low-pass filtered to 30 Å and used as an initial model for the reconstruction by first refining against class averages. The resulting map was then refined against raw particles (128- by 128-pixel box size) for 80 to 90 cycles. Volumes were visualized and interpreted using UCSF Chimera (27). Fourier shell correlation (FSC) curves were calculated using the eotest protocol in Eman 1.9 (23) and were fit to a tanh-based function to estimate the resolutions at an FSC value of 0.5.
Structural analyses.
Hydrogen bonds and van der Waals contacts were calculated using HBPLUS and CONTACSYM, respectively (28, 29). Surface area upon Fab binding was calculated using MS (30). MacPyMOL (DeLano Scientific) was used to render structure figures. Kabat numbering was applied to the coordinate files using the AbNum server (31). The final coordinates were validated using the JCSG quality control server (version 2.8), which includes MolProbity (32). Structural alignments to calculate root mean square deviation (RMSD) values were performed by iterative fitting on the alpha carbons using the McLachlan algorithm (33) as implemented in the program ProFit (A. C. R. Martin and C. T. Porter, http://www.bioinf.org.uk/software/profit).
Sequence analysis of the antibody epitopes.
The full-length and nonredundant influenza A HA sequences were downloaded from the Influenza Virus Resource at the NCBI database (34). At the time of download (20 March 2013), the data set includes 3,700 human sequences from the H1 subtype. The sequences were aligned using MUSCLE (35) and analyzed using GCG (Accelrys) and custom shell scripts (available from the authors upon request).
Kd determination.
Dissociation constant (Kd) values were determined by biolayer interferometry using an Octet RED instrument (ForteBio, Inc.) as previously described (12). Briefly, biotinylated HAs at ∼10 to 50 μg/ml in 1× kinetics buffer (1× phosphate-buffered saline [PBS; pH 7.4], 0.01% bovine serum albumin [BSA], and 0.002% Tween 20) were immobilized onto streptavidin-coated biosensors and incubated with various concentrations of Fab or IgG of 5J8 or CH65. All binding data were collected at 30°C. The kon and koff values of each Fab or IgG were measured in real time to determine the Kd values for each HA tested. The sequences of the HA proteins used in the binding studies and the experimental binding curves for each Fab or IgG for fitting kon and koff are reported in the supplemental material.
Protein structure accession numbers.
The atomic coordinates and structure factors reported in this paper are deposited in the Protein Data Bank (www.pdb.org; PDB accession no. 4M5Y, 4M5Z, and 4M4Y). The reconstruction data reported in this paper are deposited in the Electron Microscopy Data Bank (www.emdatabank.org; EMDB accession no. EMDB-5731 and EMDB-5733).
RESULTS
Crystal structure of 5J8 Fab in complex with bacterially expressed 2009 H1 HA1.
To understand the mechanism that antibody 5J8 uses to neutralize a large number of H1 viruses, we determined the crystal structure of its Fab in complex with the globular HA1 head domain of Cali07/2009-H1 (HA1 residues 55 to 271 based on H3 numbering) at a 2.25-Å resolution (Table 1). In addition, the crystal structure of the 5J8 Fab alone was determined at a 1.55-Å resolution and served as a high-resolution starting model for refinement of the complex (Table 1). The asymmetric unit of the complex contains one copy of the 5J8 Fab bound to the monomeric HA1 (Fig. 1). Interestingly, the HA1 was generated from a flagellin-HA1 recombinant fusion protein in Escherichia coli and is a vaccine candidate termed STF2.HA1 (13). However, STF2.HA1 was cleaved over time in our storage buffer and crystallization conditions (Fig. 2), which differ from the vaccine formulation buffer where the vaccine candidates are stable, and only the 5J8-HA1 complex crystallized. In spite of being produced in bacteria and undergoing refolding procedures, the HA1 does indeed properly fold and aligns well with a similar bacterially expressed and refolded HA1 (36) as well as full-length HAs produced from insect cells (4) (Fig. 3A). The lack of carbohydrates on the bacterially expressed HA1 does not affect the folding and antigenic integrity; almost all of the conventional HA antigenic sites, Sa, Sb, Ca1, Ca2, and Cb, which have been classically characterized using polyclonal mouse antisera (37, 38), have good structural integrity and align well with the bacterially expressed and refolded HA1 and full-length HAs from the H1 subtype (4, 36) (Fig. 3A). However, residues in and around the Cb site as well as the eight C-terminal residues, which are located at the opposite end of the RBS, have weak electron density and could not be completely modeled. This conformational heterogeneity is likely attributable to the absence of normal stabilizing secondary structural elements in the minimal construct used. A longer construct may provide additional secondary structural elements that stabilize this region of the structure, as observed in other antibody complexes with HA1 fragments (6, 39) (Fig. 3B).
Table 1.
X-ray data collection and refinement statistics
| Data collection | Resulta |
||
|---|---|---|---|
| 5J8 Fab | 5J8-Cali07/2009-H1 HA1 | Cali04/2009-H1 HA2 E47G | |
| Beamline | APS 23ID-D | CLS 08ID-1 | SSRL 12-2 |
| Wavelength (Å) | 1.0332 | 0.97949 | 0.97950 |
| Space group | P212121 | P3121 | P212121 |
| Unit cell parameters (Å, °) | a = 90.6, b = 100.0, c = 144.3; α = β = γ = 90.0 | a = b = 67.5, c = 259.6; α = β = 90.0, γ = 120.0 | a = 71.4, b = 132.2, c = 201.8; α = β = γ = 90.0 |
| Resolution (Å) | 50–1.55 (1.57–1.55) | 50–2.25 (2.32–2.25) | 50–2.20 (2.24–2.20) |
| No. of observations | 1,340,148 | 219,878 | 321,115 |
| No. of unique reflections | 184,085 (6,687) | 33,082 (2,526) | 95,786 (4,847) |
| Rmerge (%)b | 4.6 (91.0) | 6.0 (75.5) | 13.5 (71.1) |
| Rpim (%)b | 1.8 (41.2) | 2.4 (37.6) | 8.0 (42.0) |
| I/sigma | 21.9 (1.8) | 14.9 (1.9) | 11.2 (1.5) |
| Completeness (%) | 96.9 (72.5) | 97.7 (83.1) | 96.3 (98.9) |
| Multiplicity | 7.3 (5.6) | 6.6 (4.4) | 3.4 (3.2) |
| Refinement statistics | |||
| Resolution (Å) | 49.2–1.55 | 48.5–2.25 | 48.5–2.20 |
| No. of reflections (total) | 183,666 | 32,999 | 95,720 |
| No. of reflections (test) | 9,196 | 1,667 | 4,796 |
| Rcryst (%)c | 17.2 | 19.1 | 17.9 |
| Rfree (%)d | 18.7 | 23.8 | 23.2 |
| No. of protein atoms | 6,607 | 4,928 | 11,752 |
| No. of carbohydrate atoms | 200 | ||
| No. of waters | 866 | 90 | 940 |
| Other | 97 | 0 | 0 |
| Average B value (Å2) | |||
| Overall | 27.9 | 64.1 | 36.1 |
| HA | 74.1 | 35.3 | |
| Fab | 26.6 | 59.2 | |
| Wilson | 20.1 | 54.2 | 41.7 |
| RMSD from ideal geometry | |||
| Bond length (Å) | 0.009 | 0.010 | 0.008 |
| Bond angles (°) | 1.26 | 1.55 | 1.13 |
| Ramachandran statistics (%)e | |||
| Favored | 97.8 | 96.0 | 97.2 |
| Outliers | 0 | 0.3 | 0.1 |
| PDB accession no. | 4M5Y | 4M5Z | 4M4Y |
Numbers in parenthesis refer to the highest resolution shell.
Rmerge = Σhkl Σi | Ihkl,i − <Ihkl> |/Σhkl Σi Ihkl,i and Rpim = Σhkl[1/(N − 1)]1/2Σi | Ihkl,i − <Ihkl>|/ΣhklΣi Ihkl,i, where Ihkl,i is the scaled intensity of the ith measurement of reflection h, k, or l, <Ihkl> is the average intensity for that reflection, and N is the redundancy.
Rcryst = Σ | Fo − Fc |/Σ | Fo | × 100, where Fo and Fc are the observed and calculated structure factors, respectively.
Rfree was calculated as described for Rcryst, but on a random test set comprising 5% of the data excluded from refinement.
Calculated using MolProbity (32).
Fig 1.

Human monoclonal antibody 5J8 recognizes the HA receptor binding site. (A) Crystal structure of 5J8 Fab in complex with the bacterially expressed Cali07/2009-H1 HA1. The HA1 is colored yellow, the Fab heavy chain is blue, the Fab light chain is light blue, and the HCDR3 is red. 5J8 inserts its HCDR3 into the HA receptor binding site (B) and overlaps with the human α2,6 sialoglycan receptor (PDB accession no. 3UBE) (C).
Fig 2.
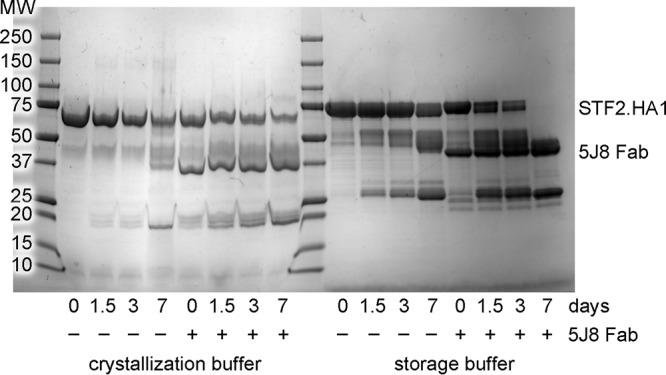
Degradation of unliganded STF2.HA1 and 5J8 Fab-STF2.HA1 complex. Unliganded STF2.HA1 (∼77 kDa) and 5J8 Fab (∼45 kDa) in complex with STF2.HA1 were incubated at room temperature in the crystallization buffer or the storage buffer (150 mM NaCl, 20 mM Tris [pH 8]). A total of 10 μg of each reaction was quenched at each time point by the addition of nonreducing SDS buffer and was boiled for ∼2 min. Samples were analyzed by SDS-PAGE. Over time, a band at ∼25 kDa appears, which is consistent with the mass of HA1 in the 5J8 Fab-Cali07/2009-H1 complex crystal.
Fig 3.

Structural relationships between the bacterially expressed Cali07/2009-H1 HA1 subunit to other HAs. (A) Comparison of the structure of the antigenic sites between the bacterially expressed HA1 (colored yellow) and the full-length baculovirus-expressed Cali04/2009-H1 HA (colored green; PDB accession no. 3LZG) with RMSD values for each site labeled. The Cb antigenic site in our structure is disordered and is not fully modeled. The global-fit RMSD between these HAs is 0.64 Å. A previously solved bacterial HA1 (PDB accession no. 3MLH) is shown in pink and has a global-fit RMSD of 0.6 Å to our structure. (B) Alignment of the HA1 subunits between Cali07/2009-H1 and A/Hong Kong/1/1968 (H3N2) (PDB accession no. 4FP8), colored yellow and gray, respectively. The longer H3 HA1 construct contains additional secondary structural elements of the vestigial esterase domain that extend away from the RBS toward the stem domain as well as two additional disulfide bridges, which are circled and depicted as green sticks. The 5J8 Fab is colored in light and dark blue (heavy and light chains), and its HCDR3 is in red.
Consistent with previous epitope mapping and hemagglutination inhibition studies (11), 5J8 recognizes the area in and around the RBS and contacts the Sb and Ca2 antigenic sites, a region of HA that has not previously been observed to be involved in crystal structures of other HA complexes with antibodies (Fig. 4). In particular, 5J8 contacts conserved residues in the structural elements that form the RBS: the 130 loop, 190 helix, and 220 loop. Additional contacts are made outside the RBS at the 140 loop. The antibody recognizes HA using all three complementarity determining region loops (CDRs) of both the heavy and light chains as well as the light chain framework 3 region. A total of 1,298 Å2 is buried upon binding (657 Å2 on HA and 641 Å2 on the Fab). The heavy chain and light chain contribute 56% and 44% of the buried surface area, respectively, and the light chain contributes a larger proportion of the buried surface area than seen for some other Fab-HA complexes that target the RBS (5–7, 9, 10, 39).
Fig 4.

Interaction of 5J8 with Cali07/2009-H1 illustrating the neutralizing epitope. (A) Sequence conservation of the 5J8 epitope across human H1 strains. HA residues contacted by 5J8 are represented as yellow sticks. The percent conservation for the most common residue at each position is shown, which is identical to the residues of Cali07/2009-H1. (B) HA is illustrated as a surface in the same orientation as panel A, with the HA contact residues on the surface colored by sequence conservation according to the inserted scale. 5J8 contact residues are labeled and shown as sticks, with the heavy chain in dark blue and the light chain in light blue.
5J8 binds HA using receptor mimicry.
The HCDR3 of 5J8 reaches into the HA RBS, makes up 47% of the buried surface area contributed by the antibody, and contacts highly conserved residues that participate in sialic acid receptor binding (Fig. 4). Most notably, the carboxylate moiety of AspH100b overlaps closely with the sialic acid carboxylate and utilizes the same network of hydrogen bonding interactions with conserved RBS residues (Fig. 5). In addition, ProH100a inserts into a hydrophobic pocket formed by the highly conserved Trp153 and Leu194, which are conserved in 100% and 95.8% of human H1 strains, respectively, in 3,700 sequences from the Influenza Virus Resource at the NCBI database (34). Other RBS-targeted antibodies have also been observed to insert a hydrophobic residue into this RBS pocket, indicating a common recurring recognition motif at this site (5–7, 9, 10).
Fig 5.

5J8 binds to the HA receptor binding site using receptor mimicry. The carboxylate of AspH100b (A) overlaps with the carboxylate of the α2,6 sialoglycan (PDB accession no. 3UBE) (B) and uses identical hydrogen bonding interactions, which are shown as black dashed lines.
In addition to the RBS contacts, the 140 loop of HA is buried by the antibody and comprises 22% of the buried surface on HA. Although the 140 loop is established as part of the Ca2 antigenic site, no other antibody complex structure has exhibited such extensive contacts with this site. Most contacts between 5J8 and the 140 loop are van der Waals interactions. Additionally, a salt bridge is formed between Lys145 and AspL51, as well as a main-chain–main-chain hydrogen bond between Lys145 and GlyL29 that stabilizes the interaction (Fig. 6A). Three other electrostatic and seven other hydrogen bonding interactions stabilize the interface between 5J8 and Cali07/2009-H1.
Fig 6.

Electrostatic and hydrogen bonding interactions between 5J8 Fab and Cali07/2009-H1 HA1, with hydrogen bonds depicted by dashed lines. The HA is colored yellow, the Fab is colored blue, and the HCDR3 is colored red.
Electron microscopy (EM) reconstructions of 5J8 in complex with full-length stabilized HA trimers.
Due to the weak association of the HA ectodomain protomers in the wild-type 2009 H1 protein that causes it to have a high propensity to be purified as a monomer (40–42), we investigated mutations that would stabilize the HA stem trimer interface. A mutation from HA2 Glu47 (H3 numbering), which interacts with a γ-turn at HA1 position 30 from a neighboring protomer, to Gly47 was found to stabilize the trimer (Fig. 7). We then determined the stabilized-trimer 2009 H1 crystal structure at a 2.20-Å resolution (Table 1) and observed that the HA2 Glu47Gly mutation appears to relieve steric restraints between HA protomers. We previously reported use of a similar HA2 Ala47Gly mutation to stabilize the bat H17 HA to determine its structure (43). The crystal structures of the native and mutant HA are nearly identical, with Cα root mean square deviation (RMSD) values of 0.5 Å (HA1), 0.4 Å (HA2), and 0.6 Å (HA protomer). We are currently investigating whether this mutation is also beneficial for increased stability of other HA trimers that have weak association between protomers.
Fig 7.

Engineered trimer-stabilized Cali04/2009-H1 HA2 Glu47Gly used for the EM reconstructions. (A) Overview of the HA structure with the HA2 Glu47Gly mutation circled in red. (B) Zoomed-in view of the HA trimer interface. The wild-type HA is colored gray, with Glu47 and the HA1 γ-turn residues (Leu30′ and Glu31′) from the neighboring HA protomer shown as sticks. The trimer-stabilized chains are differentially colored.
Extensive crystallization trials were performed with 5J8 Fab in complex with full-length HAs, and although we were able to grow crystals of the Fab in complex with full-length HAs from 1918, 1986, and 2009 viruses, these crystals diffracted only to ∼8 Å and quickly decayed from radiation damage. Instead, we turned to EM and generated reconstructions of 5J8 Fab in complex with full-length A/Singapore/6/1986 (H1N1) and trimer-stabilized A/California/04/2009 (H1N1) HAs at resolutions of 22 Å and 23 Å, respectively (Fig. 8). These EM reconstructions confirm the binding data and show that 5J8 does indeed target the apex of HA at and around the RBS in the HA trimer, occupying all three potential binding sites.
Fig 8.
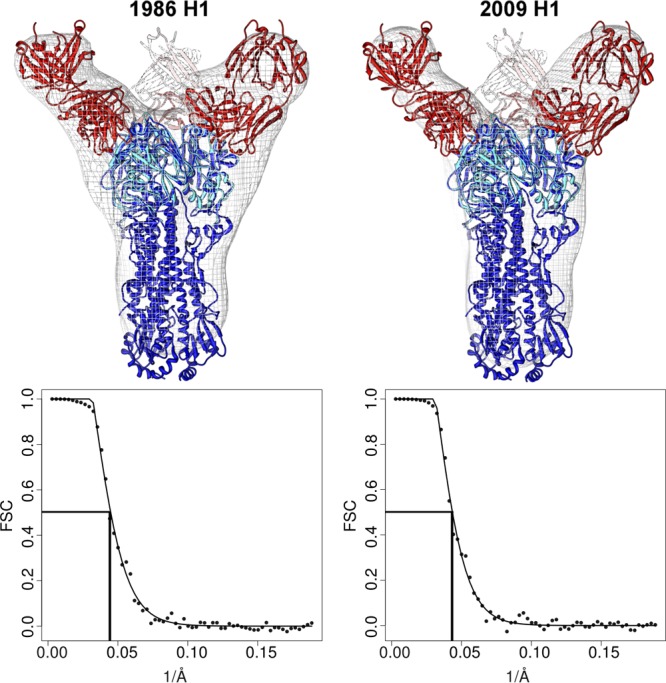
EM reconstructions of 5J8 in complex with HA. Negative-stain EM was used to determine volume maps of 5J8 Fab (red) in complex with the full-length HAs (blue) from the A/Singapore/6/1986 (H1N1) and trimer-stabilized A/California/04/2009 (H1N1) strains. The reconstructions show that the HA-antibody interactions described in the 5J8-Cali07/2009-H1 HA1 crystal structure (red and cyan) are recapitulated with the HA trimers. The HA from PDB accession no. 3LZG (blue) was used as a reference structure. Fourier shell correlation (FSC) curves indicate that the resolution for the 5J8-A/Singapore/6/1986 map is 22 Å and for the 5J8-Cali04/2009-H1 map is 23 Å.
Sequence analysis of the 5J8 epitope.
To further probe the binding specificity and breadth of 5J8, binding studies of Fab and IgG were performed by biolayer interferometry against a panel of 12 H1 strains that spanned from 1918 to 2009. Nine strains were bound by 5J8 ranging from very weak (Kd > 1 μM) to very strong (Kd < 1 nM) affinity (Table 2). Remarkably, 5J8 extends its binding breadth using avidity, as the bivalent binding of IgG has substantially higher apparent affinity to HA than the monovalent Fab. Moreover, the IgG recognizes divergent H1 strains where no detectable binding was observed for the Fab, as for A/AA/Marton/1943, A/Beijing/262/1995, and A/New Caledonia/20/1999. However, these strains are bound by the IgG with a Kd around or greater than 250 nM, which we have previously shown is not sufficient for neutralization, as for antibody S139/1 (7). These binding data are in good agreement with the published neutralization data, which show that 5J8 binds HAs that span decades, including the pandemic strains that circulated in 1918, a sporadic case in 1977, and the most recent 2009 outbreak (11).
Table 2.
Binding of the influenza HA strains by 5J8 and CH65 Fab and IgG
| H1N1 strain |
Kda |
|||
|---|---|---|---|---|
| 5J8 Fab | 5J8 IgG | CH65 Fab | CH65 IgG | |
| A/South Carolina/1/1918 | ++ | ++++ | − | − |
| A/WSN/1933 | − | − | − | − |
| A/Puerto Rico/8/1934 | − | − | − | − |
| A/AA/Marton/1943 | − | ++ | − | + |
| A/duck/Alberta/345/1976 | − | − | − | − |
| A/USSR/90/1977 | + | ++++ | − | ++ |
| A/Singapore/6/1986 | +++ | ++++ | +++ | ++++ |
| A/Texas/36/1991 | ++ | ++++ | ++++ | ++++ |
| A/Beijing/262/1995 | − | + | ++++ | ++++ |
| A/New Caledonia/20/1999 | − | ++ | ++ | ++++ |
| A/Solomon Islands/3/2006 | ++ | +++ | +++ | ++++ |
| A/California/04/2009 | +++ | ++++ | − | − |
−, no detectable binding of >5 μM; +, 500 to 5,000 nM; ++, 50 to 500 nM; +++, 5 to 50 nM; ++++, <5 nM.
Among the pandemic HA strains bound by 5J8, the single-residue insertion at position 133a is in common. This insertion is present in 71.7% of H1 isolates (Fig. 4). The 133a residue is typically lysine (in 97.5% of the human H1 strains that possess the 133a insertion) but can also be a similarly positively charged arginine. The Lys133a amine forms an electrostatic interaction with the AspL53 carboxylate and appears to be a common determinant for recognition by 5J8 (Fig. 6B). The importance of this residue has been previously determined from escape mutants and mutagenesis studies, as deletion of the 133a residue, or mutation to Gln or Ile, eliminates binding by 5J8 (11). Another important electrostatic interaction is formed between Lys222 (conserved in 99.7% of human H1 strains) and AspL95a, which are both buried in the interface (Fig. 6C). The Lys222Gln escape mutant (11) would disrupt favorable electrostatic interactions and bury a charged residue in the interface and, hence, eliminate recognition by 5J8. In addition, A/duck/Alberta/345/1976 HA possesses Glu222, which would likely form a destabilizing interaction with AspL95a. The strains that are not bound by 5J8 contain Glu190 versus Asp190 for the strains that are bound by 5J8. Although Asp190 and Glu190 are both acidic, it is probable that the longer Glu side chain would clash with the antibody (Fig. 6D). Interestingly, Asp190 mediates favorable interactions with α2,6-linked glycans (42) and is highly conserved in 90.7% of human H1 strains.
DISCUSSION
Here, we describe the recognition of H1 pandemic viruses by the human monoclonal antibody 5J8. This antibody inserts its HCDR3 into the RBS of HA and thus blocks viral-host interactions. In addition, 5J8 utilizes avidity through bivalency to extend its breadth of recognition and increase its affinity against highly divergent HA strains, as the bivalent IgG is more potent compared to monovalent Fab. Avidity has also been observed in other previously characterized antibodies that target the RBS (6, 7) as well as in CH65, which has been further characterized in this study (Table 2). These data indicate that avidity through bivalency is critical for extending the breadth of neutralization and may be a general mode of antibody recognition against the HA RBS. In addition, a bivalent IgG relaxes the specificity of antibody recognition, allowing it to tolerate some of the hypervariable residues in divergent strains that would have otherwise been moderately or weakly bound by monovalent Fab.
The HCDR3 of 5J8 inserts into the HA RBS and closely mimics the natural sialoglycan receptor. The carboxylate of AspH100b is oriented nearly identically to that of the sialic acid carboxylate and makes similar hydrogen bonding networks to conserved receptor binding residues. This mode of receptor mimicry has also been observed in related broadly neutralizing H1 antibodies CH65 and CH67 (5, 9) (n.b. 5J8 uses D3-3*02 and J4*02, while CH65 and CH67 use D1-1 and J6 germ line genes). In addition, the 5J8 ProH100a inserts into a universally conserved hydrophobic pocket in the HA RBS that would be occupied by the acetamide group of sialic acid, which has also been similarly targeted by other antibodies (5–7, 9, 10). The compounding structural information has revealed a number of common binding modes and recognition hot spots. For instance, these binding details may serve as a template for structure-guided drug discovery by combining common recognition elements for the design of small molecules. The antibody-antigen interactions can also be recapitulated through proteins engineered to target the HA RBS, as has been successfully performed against the HA stem (44, 45). In addition, the structural information can be used to design immunogens that elicit RBS-targeted antibodies, along similar lines as the germ line-targeting engineered outer domain of HIV-1 gp120 (46).
Although 5J8 and other antibodies mimic certain moieties of the receptor, the region of the binding site occupied by the glycerol moiety of sialic acid is not contacted by these antibodies (Fig. 9B). As there is only space for a single antibody loop to enter into the binding groove, the level of receptor mimicry therefore has spatial limitations. Sterics also play a role in antibody recognition, as the 133a insertion present in pandemic H1 strains appears to be an important binding determinant for these H1-specific antibodies. For example, binding by 5J8 depends largely on the presence of the 133a insertion, whereas CH65 appears to favor binding to strains without the insertion (Table 2), although CH67 modestly neutralizes pandemic strains (5). The 133a insertion may thus dictate the specificity of any subsequent design efforts against the RBS of H1 isolates. Obviously, it is overly simplistic to distinguish the antibodies by a single amino acid, considering they have distinct binding footprints on HA and use different angles of approach (Fig. 9). However, it is compelling to note that these antibodies complement each other and jointly recognize all H1 human isolates tested since the H1N1 virus reemerged in humans in 1977 (Table 2). As avidity by bivalent IgG increases the affinity of each antibody to HA, it could be possible to use a bispecific antibody (47), i.e., with one arm as 5J8 and the other as CH65 or CH67, as a potential therapeutic or diagnostic for existing and emerging H1 viruses.
Fig 9.

Comparison of 5J8 and CH65 contacts on HA. (A) Sequence alignment of the H1 strains tested for binding by biolayer interferometry. (B) Residues contacted by 5J8 (blue), CH65 (red), or both antibodies (violet) are mapped onto the alignment and colored on the surface of Cali07/2009-H1. The sialoglycan (PDB accession no. 3UBE) is represented as sticks. The glyph “.” indicates sequence identity with the Cali07/2009-H1 strain.
Our study also highlights the potential for developing an alternative immunogenic and effective vaccination strategy using an E. coli expression system. Eukaryotic expression systems are widely used for the preparation of recombinant HAs, as these proteins have glycans on their N-linked glycosylation sites. However, it has been previously reported that HA1 can be recombinantly produced and refolded from E. coli inclusion bodies with similar biophysical properties to HA produced in insect cells (36). Moreover, E. coli-expressed HA1 has been shown to elicit a protective immune response in ferrets (48), suggesting that a protective antibody response can be generated despite the lack of glycan shielding on the surface of HA, at least in the case of the 2009 H1 pandemic strain. In support of this, E. coli-expressed fusions of flagellin and Cali07/2009-H1 HA1 or A/Solomon Islands/3/1986 HA1 have been shown to elicit protective antibody titers in humans (49, 50). We show that the bacterially expressed HA1, which was originally expressed as a fusion protein with flagellin, is properly refolded and is recognized by broadly neutralizing human antibody 5J8. As such, these results provide further evidence that this strategy of producing recombinant HA in E. coli is suitable for vaccination, as also noted from the ferret experiments (48) and human studies (49, 50). The structural knowledge of how 5J8 functions, in combination with the other RBS-targeted antibodies, will potentially further aid and inform vaccine and therapeutic design against the influenza A H1 subtype.
Supplementary Material
ACKNOWLEDGMENTS
We thank H. Tien of the Robotics Core at the Joint Center for Structural Genomics for automated crystal screening, the staff of the APS GM/CA-CAT 23ID-D, CLS 08ID-1, and SSRL BL12-2 for beamline support, X. Dai and M. Elsliger for crystallographic and computational support, W. Yu, Y. Hua, and A. Arnell for materials and expertise, as well as J.-P. Julien, R. L. Stanfield, and L. Kong for helpful discussions. We thank A. Cheng, T. Nieusma, and J. H. Lee for assistance with EM data collection and interpretation.
The work was funded in part by NIH R56 AI099275 (I.A.W.), by the Skaggs Institute for Chemical Biology, by GM080209 from the NIH Molecular Evolution Training Program (P.S.L.), by a Saper Aude fellowship from the Danish Council for Independent Research, Natural Sciences (N.S.L.), and with federal funds from the Office of the Assistant Secretary for Preparedness and Response, BARDA, HHS, contract no. HHSO100201100011C. Isolation and production of the antibody was supported by NIH grant R01 AI106002 and NIH contract HHSN272200900047C (both to J.E.C.). The electron microscopy data presented here were collected at the National Resource for Automated Molecular Microscopy at TSRI, which is supported by the NIH though the P41 program (RR017573) at the National Center for Research Resources. X-ray diffraction data were collected at the Canadian Light Source, which is supported by the Natural Sciences and Engineering Research Council of Canada, the National Research Council Canada, the Canadian Institutes of Health Research, the Province of Saskatchewan, Western Economic Diversification Canada, and the University of Saskatchewan. The GM/CA CAT has been funded in whole or in part with federal funds from National Cancer Institute (Y1-CO-1020) and National Institute of General Medical Sciences (Y1-GM-1104). Use of the Advanced Photon Source was supported by the U.S. Department of Energy, Basic Energy Sciences, Office of Science, under contract no. DE-AC02-06CH11357.
The content is solely the responsibility of the authors and does not necessarily represent the official views of NIGMS or the NIH. This is The Scripps Research Institute manuscript number 23085.
Vanderbilt University submitted a patent covering the diagnostic and therapeutic use of the antibody described in this paper; J.C.K. and J.E.C. are two of the coinventors on that application.
Footnotes
Published ahead of print 11 September 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JVI.01388-13.
REFERENCES
- 1.Wilson IA, Skehel JJ, Wiley DC. 1981. Structure of the haemagglutinin membrane glycoprotein of influenza virus at 3 Å resolution. Nature 289:366–373 [DOI] [PubMed] [Google Scholar]
- 2.Weis W, Brown JH, Cusack S, Paulson JC, Skehel JJ, Wiley DC. 1988. Structure of the influenza virus haemagglutinin complexed with its receptor, sialic acid. Nature 333:426–431 [DOI] [PubMed] [Google Scholar]
- 3.Julien JP, Lee PS, Wilson IA. 2012. Structural insights into key sites of vulnerability on HIV-1 Env and influenza HA. Immunol. Rev. 250:180–198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Xu R, Ekiert DC, Krause JC, Hai R, Crowe JE, Jr, Wilson IA. 2010. Structural basis of preexisting immunity to the 2009 H1N1 pandemic influenza virus. Science 328:357–360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Whittle JR, Zhang R, Khurana S, King LR, Manischewitz J, Golding H, Dormitzer PR, Haynes BF, Walter EB, Moody MA, Kepler TB, Liao HX, Harrison SC. 2011. Broadly neutralizing human antibody that recognizes the receptor-binding pocket of influenza virus hemagglutinin. Proc. Natl. Acad. Sci. U. S. A. 108:14216–14221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ekiert DC, Kashyap AK, Steel J, Rubrum A, Bhabha G, Khayat R, Lee JH, Dillon MA, O'Neil RE, Faynboym AM, Horowitz M, Horowitz L, Ward AB, Palese P, Webby R, Lerner RA, Bhatt RR, Wilson IA. 2012. Cross-neutralization of influenza A viruses mediated by a single antibody loop. Nature 489:526–532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lee PS, Yoshida R, Ekiert DC, Sakai N, Suzuki Y, Takada A, Wilson IA. 2012. Heterosubtypic antibody recognition of the influenza virus hemagglutinin receptor binding site enhanced by avidity. Proc. Natl. Acad. Sci. U. S. A. 109:17040–17045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tsibane T, Ekiert DC, Krause JC, Martinez O, Crowe JE, Jr, Wilson IA, Basler CF. 2012. Influenza human monoclonal antibody 1F1 interacts with three major antigenic sites and residues mediating human receptor specificity in H1N1 viruses. PLoS Pathog. 8:e1003067. 10.1371/journal.ppat.1003067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schmidt AG, Xu H, Khan AR, O'Donnell T, Khurana S, King LR, Manischewitz J, Golding H, Suphaphiphat P, Carfi A, Settembre EC, Dormitzer PR, Kepler TB, Zhang R, Moody MA, Haynes BF, Liao HX, Shaw DE, Harrison SC. 2013. Preconfiguration of the antigen-binding site during affinity maturation of a broadly neutralizing influenza virus antibody. Proc. Natl. Acad. Sci. U. S. A. 110:264–269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Xu R, Krause JC, McBride R, Paulson JC, Crowe JE, Jr, Wilson IA. 2013. A recurring motif for antibody recognition of the receptor-binding site of influenza hemagglutinin. Nat. Struct. Mol. Biol. 20:363–370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Krause JC, Tsibane T, Tumpey TM, Huffman CJ, Basler CF, Crowe JE., Jr 2011. A broadly neutralizing human monoclonal antibody that recognizes a conserved, novel epitope on the globular head of the influenza H1N1 virus hemagglutinin. J. Virol. 85:10905–10908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ekiert DC, Friesen RH, Bhabha G, Kwaks T, Jongeneelen M, Yu W, Ophorst C, Cox F, Korse HJ, Brandenburg B, Vogels R, Brakenhoff JP, Kompier R, Koldijk MH, Cornelissen LA, Poon LL, Peiris M, Koudstaal W, Wilson IA, Goudsmit J. 2011. A highly conserved neutralizing epitope on group 2 influenza A viruses. Science 333:843–850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu G, Tarbet B, Song L, Reiserova L, Weaver B, Chen Y, Li H, Hou F, Liu X, Parent J, Umlauf S, Shaw A, Tussey L. 2011. Immunogenicity and efficacy of flagellin-fused vaccine candidates targeting 2009 pandemic H1N1 influenza in mice. PLoS One 6:e20928. 10.1371/journal.pone.0020928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kabsch W. 2010. XDS. Acta Crystallogr. D Biol. Crystallogr. 66:125–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. 2007. Phaser crystallographic software. J. Appl. Crystallogr. 40:658–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Emsley P, Lohkamp B, Scott WG, Cowtan K. 2010. Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66:486–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH. 2010. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66:213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Murshudov GN, Vagin AA, Dodson EJ. 1997. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr. 53:240–255 [DOI] [PubMed] [Google Scholar]
- 19.Suloway C, Pulokas J, Fellmann D, Cheng A, Guerra F, Quispe J, Stagg S, Potter CS, Carragher B. 2005. Automated molecular microscopy: the new Leginon system. J. Struct. Biol. 151:41–60 [DOI] [PubMed] [Google Scholar]
- 20.Carragher B, Kisseberth N, Kriegman D, Milligan RA, Potter CS, Pulokas J, Reilein A. 2000. Leginon: an automated system for acquisition of images from vitreous ice specimens. J. Struct. Biol. 132:33–45 [DOI] [PubMed] [Google Scholar]
- 21.Voss NR, Yoshioka CK, Radermacher M, Potter CS, Carragher B. 2009. DoG Picker and TiltPicker: software tools to facilitate particle selection in single particle electron microscopy. J. Struct. Biol. 166:205–213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lander GC, Stagg SM, Voss NR, Cheng A, Fellmann D, Pulokas J, Yoshioka C, Irving C, Mulder A, Lau PW, Lyumkis D, Potter CS, Carragher B. 2009. Appion: an integrated, database-driven pipeline to facilitate EM image processing. J. Struct. Biol. 166:95–102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ludtke SJ, Baldwin PR, Chiu W. 1999. EMAN: semiautomated software for high-resolution single-particle reconstructions. J. Struct. Biol. 128:82–97 [DOI] [PubMed] [Google Scholar]
- 24.Frank J, Radermacher M, Penczek P, Zhu J, Li Y, Ladjadj M, Leith A. 1996. SPIDER and WEB: processing and visualization of images in 3D electron microscopy and related fields. J. Struct. Biol. 116:190–199 [DOI] [PubMed] [Google Scholar]
- 25.Marabini R, Masegosa IM, San Martin MC, Marco S, Fernandez JJ, de la Fraga LG, Vaquerizo C, Carazo JM. 1996. Xmipp: an image processing package for electron microscopy. J. Struct. Biol. 116:237–240 [DOI] [PubMed] [Google Scholar]
- 26.Sorzano CO, Bilbao-Castro JR, Shkolnisky Y, Alcorlo M, Melero R, Caffarena-Fernandez G, Li M, Xu G, Marabini R, Carazo JM. 2010. A clustering approach to multireference alignment of single-particle projections in electron microscopy. J. Struct. Biol. 171:197–206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. 2004. UCSF Chimera—a visualization system for exploratory research and analysis. J. Comput. Chem. 25:1605–1612 [DOI] [PubMed] [Google Scholar]
- 28.McDonald IK, Thornton JM. 1994. Satisfying hydrogen bonding potential in proteins. J. Mol. Biol. 238:777–793 [DOI] [PubMed] [Google Scholar]
- 29.Sheriff S, Hendrickson WA, Smith JL. 1987. Structure of myohemerythrin in the azidomet state at 1.7/1.3 Å resolution. J. Mol. Biol. 197:273–296 [DOI] [PubMed] [Google Scholar]
- 30.Connolly ML. 1983. Analytical molecular surface calculation. J. Appl. Crystallogr. 16:548–558 [Google Scholar]
- 31.Abhinandan KR, Martin AC. 2008. Analysis and improvements to Kabat and structurally correct numbering of antibody variable domains. Mol. Immunol. 45:3832–3839 [DOI] [PubMed] [Google Scholar]
- 32.Chen VB, Arendall WB, III, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, Richardson DC. 2010. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 66:12–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.McLachlan AD. 1982. Rapid comparison of protein structures. Acta Crystallogr. A. 38:871–873 [Google Scholar]
- 34.Bao Y, Bolotov P, Dernovoy D, Kiryutin B, Zaslavsky L, Tatusova T, Ostell J, Lipman D. 2008. The influenza virus resource at the National Center for Biotechnology Information. J. Virol. 82:596–601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Edgar RC. 2004. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32:1792–1797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.DuBois RM, Aguilar-Yanez JM, Mendoza-Ochoa GI, Oropeza-Almazan Y, Schultz-Cherry S, Alvarez MM, White SW, Russell CJ. 2011. The receptor-binding domain of influenza virus hemagglutinin produced in Escherichia coli folds into its native, immunogenic structure. J. Virol. 85:865–872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gerhard W, Yewdell J, Frankel ME, Webster R. 1981. Antigenic structure of influenza virus haemagglutinin defined by hybridoma antibodies. Nature 290:713–717 [DOI] [PubMed] [Google Scholar]
- 38.Caton AJ, Brownlee GG, Yewdell JW, Gerhard W. 1982. The antigenic structure of the influenza virus A/PR/8/34 hemagglutinin (H1 subtype). Cell 31:417–427 [DOI] [PubMed] [Google Scholar]
- 39.Bizebard T, Gigant B, Rigolet P, Rasmussen B, Diat O, Bosecke P, Wharton SA, Skehel JJ, Knossow M. 1995. Structure of influenza virus haemagglutinin complexed with a neutralizing antibody. Nature 376:92–94 [DOI] [PubMed] [Google Scholar]
- 40.Yang H, Carney P, Stevens J. 2010. Structure and receptor binding properties of a pandemic H1N1 virus hemagglutinin. PLoS Curr. 2:RRN1152. 10.1371/currents.RRN1152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhang W, Qi J, Shi Y, Li Q, Gao F, Sun Y, Lu X, Lu Q, Vavricka CJ, Liu D, Yan J, Gao GF. 2010. Crystal structure of the swine-origin A (H1N1)-2009 influenza A virus hemagglutinin (HA) reveals similar antigenicity to that of the 1918 pandemic virus. Protein Cell 1:459–467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Xu R, McBride R, Nycholat CM, Paulson JC, Wilson IA. 2012. Structural characterization of the hemagglutinin receptor specificity from the 2009 H1N1 influenza pandemic. J. Virol. 86:982–990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhu X, Yu W, McBride R, Li Y, Chen LM, Donis RO, Tong S, Paulson JC, Wilson IA. 2013. Hemagglutinin homologue from H17N10 bat influenza virus exhibits divergent receptor-binding and pH-dependent fusion activities. Proc. Natl. Acad. Sci. U. S. A. 110:1458–1463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fleishman SJ, Whitehead TA, Ekiert DC, Dreyfus C, Corn JE, Strauch EM, Wilson IA, Baker D. 2011. Computational design of proteins targeting the conserved stem region of influenza hemagglutinin. Science 332:816–821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Whitehead TA, Chevalier A, Song Y, Dreyfus C, Fleishman SJ, De Mattos C, Myers CA, Kamisetty H, Blair P, Wilson IA, Baker D. 2012. Optimization of affinity, specificity and function of designed influenza inhibitors using deep sequencing. Nat. Biotechnol. 30:543–548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jardine J, Julien JP, Menis S, Ota T, Kalyuzhniy O, McGuire A, Sok D, Huang PS, MacPherson S, Jones M, Nieusma T, Mathison J, Baker D, Ward AB, Burton DR, Stamatatos L, Nemazee D, Wilson IA, Schief WR. 2013. Rational HIV immunogen design to target specific germline B cell receptors. Science 340:711–716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ridgway JB, Presta LG, Carter P. 1996. ‘Knobs-into-holes' engineering of antibody CH3 domains for heavy chain heterodimerization. Protein Eng. 9:617–621 [DOI] [PubMed] [Google Scholar]
- 48.Aguilar-Yanez JM, Portillo-Lara R, Mendoza-Ochoa GI, Garcia-Echauri SA, Lopez-Pacheco F, Bulnes-Abundis D, Salgado-Gallegos J, Lara-Mayorga IM, Webb-Vargas Y, Leon-Angel FO, Rivero-Aranda RE, Oropeza-Almazan Y, Ruiz-Palacios GM, Zertuche-Guerra MI, DuBois RM, White SW, Schultz-Cherry S, Russell CJ, Alvarez MM. 2010. An influenza A/H1N1/2009 hemagglutinin vaccine produced in Escherichia coli. PLoS One 5:e11694. 10.1371/journal.pone.0011694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Treanor JJ, Taylor DN, Tussey L, Hay C, Nolan C, Fitzgerald T, Liu G, Kavita U, Song L, Dark I, Shaw A. 2010. Safety and immunogenicity of a recombinant hemagglutinin influenza-flagellin fusion vaccine (VAX125) in healthy young adults. Vaccine 28:8268–8274 [DOI] [PubMed] [Google Scholar]
- 50.Taylor DN, Treanor JJ, Sheldon EA, Johnson C, Umlauf S, Song L, Kavita U, Liu G, Tussey L, Ozer K, Hofstaetter T, Shaw A. 2012. Development of VAX128, a recombinant hemagglutinin (HA) influenza-flagellin fusion vaccine with improved safety and immune response. Vaccine 30:5761–5769 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.