

Abstract
Arboviruses such as yellow fever virus (YFV) are transmitted between arthropod vectors and vertebrate hosts. While barriers limiting arbovirus population diversity have been observed in mosquitoes, whether barriers exist in vertebrate hosts is unclear. To investigate whether arboviruses encounter bottlenecks during dissemination in the vertebrate host, we infected immunocompetent mice and immune-deficient mice lacking alpha/beta interferon (IFN-α/β) receptors (IFNAR−/− mice) with a pool of genetically marked viruses to evaluate dissemination and host barriers. We used the live attenuated vaccine strain YFV-17D, which contains many mutations compared with virulent YFV. We found that intramuscularly injected immunocompetent mice did not develop disease and that viral dissemination was restricted. Conversely, 32% of intramuscularly injected IFNAR−/− mice developed disease. By following the genetically marked viruses over time, we found broad dissemination in IFNAR−/− mice followed by clearance. The patterns of viral dissemination were similar in mice that developed disease and mice that did not develop disease. Unlike our previous results with poliovirus, these results suggest that YFV-17D encounters no major barriers during dissemination within a vertebrate host in the absence of the type I IFN response.
INTRODUCTION
RNA virus replication is notoriously error prone, giving rise to genetically diverse viral populations. An appropriate amount of viral-population diversity is thought to aid viral adaptation in changing host environments and is required for full virulence in vivo (1–6). While disseminating within a host, a viral population can encounter host barriers that bottleneck, or severely reduce, the population diversity. Bottlenecks are stochastic reductions in the genetic diversity of a viral population and can be caused by physical and other barriers. While bottlenecks can shape viral populations, often through random events, purifying selection remains operative and is one of the most important selective forces acting on arbovirus populations. We previously identified multiple host barriers encountered by poliovirus, a picornavirus, during dissemination in vivo (7–9). These barriers to poliovirus dissemination include inefficient viral transport in neurons and the type I interferon response. Here, we were interested in determining whether other RNA viruses encounter host barriers that limit viral-population diversity.
Arthropod-borne viruses (arboviruses) are transmitted horizontally between arthropod vectors and vertebrate hosts. Interestingly, arboviruses have been shown to undergo low rates of evolution in nature, acquiring substantially fewer mutations per genome per year than other RNA viruses (10–12). This genetic stability is generally attributed to the alternating host cycle, which could constrain evolution because mutations increasing fitness in one host may decrease fitness in the subsequent host (13–16). However, additional constraints on arboviral genetic diversity likely occur during intrahost spread. Arboviruses have been shown to encounter severe bottlenecks in the mosquito vector (17, 18); however, bottlenecks encountered during dissemination in a vertebrate host have not been evaluated.
Yellow fever virus (YFV) is a mosquito-borne flavivirus that causes human disease, with ∼200,000 new cases each year resulting in ∼30,000 deaths (19). YF disease in humans can range from asymptomatic to severe hemorrhagic fever. YF disease has been eliminated in some areas by vaccination with the live attenuated vaccine strain YFV-17D and mosquito control programs. To investigate potential arbovirus bottlenecks in a vertebrate host and to investigate a virus unrelated to our previous poliovirus model, we chose to use YFV-17D as a model system.
Laboratory rodents have various degrees of susceptibility to YFV infection, with disease outcome dependent on age, viral strain, immune status of the host, inoculation route, and viral-inoculum titer (20–26). Immunocompetent infant mice are susceptible to fatal infection with intravenously or intraperitoneally injected virulent strains of YFV, including YFV-Asibi, as well as attenuated strains of YFV such as YFV-17D (23, 24, 26). However, immunocompetent adult mice are not susceptible to fatal infection with virulent or attenuated YFV delivered by intraperitoneal injection. In contrast, immunocompetent infant and adult mice are susceptible to virulent or attenuated YFV strains following olfactory bulb inoculation (27) or direct delivery to the central nervous system (CNS) by intracerebral injection (25, 26, 28–30). YFV-17D can cause encephalitis after peripheral injection of adult mice lacking both alpha/beta interferon (IFN-α/β) receptors and IFN-γ receptors (20–22).
To investigate whether arboviruses encounter bottlenecks during dissemination in the vertebrate host, we infected immunocompetent C57BL/6 mice and immune-deficient C57BL/6 mice lacking IFN-α/β receptors (IFNAR−/− mice) intramuscularly (i.m.) with genetically marked YFV-17D viruses to evaluate dissemination and potential host barriers. We found that while YFV-17D was severely attenuated in immunocompetent mice, YFV-17D rapidly disseminated in IFNAR−/− mice, causing disease in 32% of mice. Surprisingly, viral dissemination was extremely efficient regardless of disease outcome. Our results suggest that the type I IFN response is a major barrier to YFV-17D dissemination and pathogenesis in mice.
MATERIALS AND METHODS
Plasmid construction.
Six viral plasmids were made using site-directed mutagenesis of the YFV-17D viral-cDNA clone (pACNR/FLYF17D) beginning at nucleotide 3185 and ending at nucleotide 3205 (Fig. 1A). Unique restriction sites, MluI at nucleotide 2947 and KpnI at nucleotide 3266, were used for cloning. Each PCR-generated region was confirmed by sequencing (Sequencing Core, UT Southwestern Medical Center, Dallas, TX).
Fig 1.
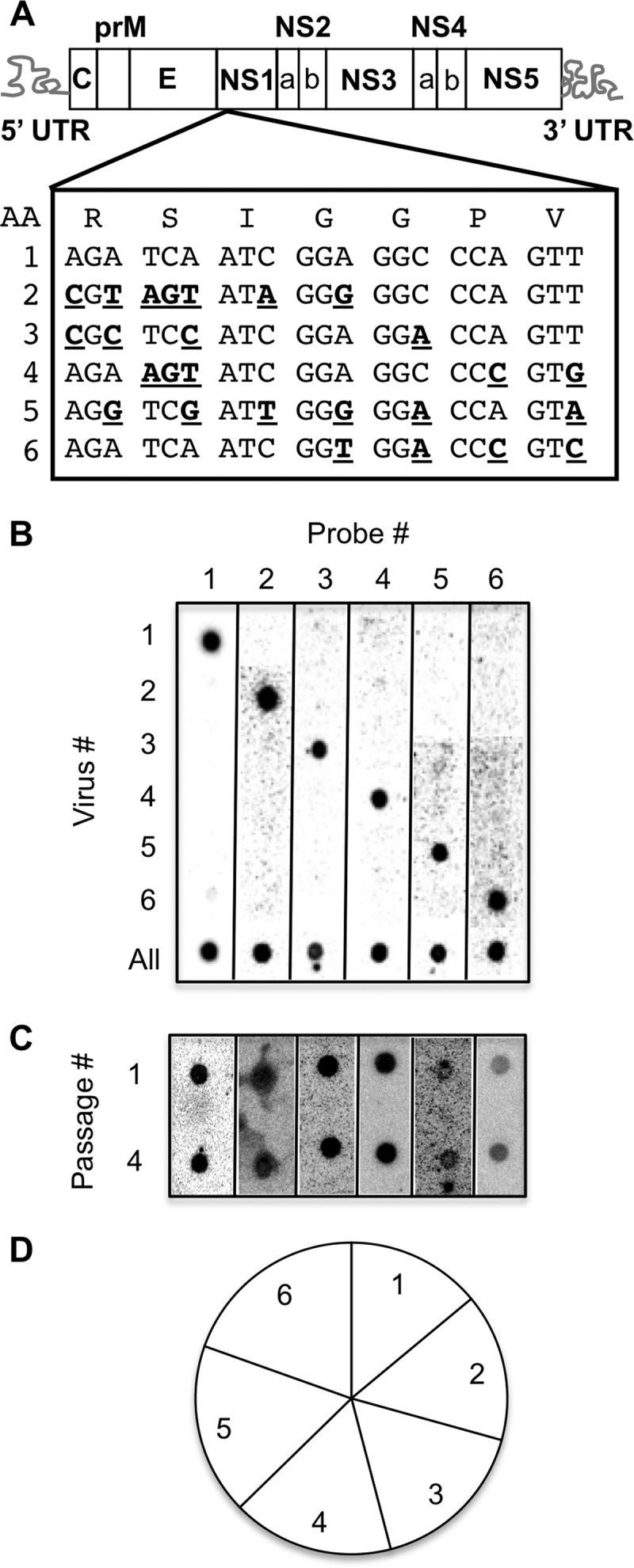
Development of a hybridization-based YFV-17D viral population diversity assay. (A) YFV-17D genome highlighting the NS1 region where groups of silent point mutations (bold, underlined) were incorporated by site-directed mutagenesis. The amino acid sequence is shown at the top. Virus no. 1 was wild-type YFV-17D. (B) Blot showing specificity of each probe for its cognate viral RT-PCR product. The sample marked “All” contained all six viruses mixed in equal ratios. (C) Serial-passage competition experiment. BHK-J cells were infected with equal amounts of each of the six viruses, and amplified virus was harvested after 48 h and used to infect fresh cells. After four passages, the ratios of the six viruses in passage 1 and passage 4 were compared to rule out fitness differences. (D) Relative proportions of the six marked viruses in mouse tissues. BL6-IFNAR−/− mice were inoculated i.m. with 6 × 103 PFU of the YFV-17D viral-pool mixture, tissues (muscle, sciatic nerve, blood, liver, spleen, and brain) were harvested at 7 to 8 days postinfection, and the prevalences of the six viruses were compared using the hybridization assay to ensure equivalent representation. Data were derived from various tissues from 33 mice, consisting of 788 individual hybridization signals (“dots”).
Viruses and titer assay.
Viruses were produced by electroporation of YFV-17D RNAs into BHK-J cells. Briefly, YFV-17D RNA was transcribed in vitro using an mMessage mMachine SP6 kit (Ambion, Grand Island, NY) according to the manufacturer's instructions, and 1 to 5 μl of this in vitro transcription reaction mixture was electroporated into ∼107 BHK-J cells. The Gene Pulser Xcell electroporation system (Bio-Rad, Hercules, CA) was used with settings of 850 V, 5-ms pulse length, 2 pulses, 5-s pulse interval, and 4-mm cuvette. BHK-J cells were grown in Dulbecco's modified Eagle's medium (DMEM) with 2% fetal bovine serum (FBS), plated on 10-cm dishes, and 24 to 48 h later split onto three 15-cm plates. The supernatants were harvested from 15-cm plates when a cytopathic effect was detected. Viral-stock titers were determined by adding serial dilutions of supernatant in serum-free medium to 6-well plates containing ∼106 BHK-J cells for 1 h. The supernatants were removed, cells were washed, and 5 ml of agarose overlay containing 0.5% SeaKem LE agarose (Lonza) in DMEM containing 2% FBS was added. After 5 days, agarose overlays were removed and cells were stained with an alcoholic crystal violet solution for plaque analysis. For the viral serial-passage experiment (Fig. 1C), cells were infected at a multiplicity of infection (MOI) of 0.1 with equivalent PFUs of each virus, and 48 h later the culture supernatant was harvested, used to infect fresh cells, and passaged this way four times. Viruses were detected in cell lysates by performing a viral-population diversity assay (see below).
Mouse experiments.
All animals were handled in strict accordance with good animal practice as defined by the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health (31). All mouse studies were performed at the University of Texas Southwestern Medical Center (Animal Welfare Assurance no. A3472-01) using protocols approved by the UT Southwestern Institutional Animal Care and Use Committee (IACUC). All studies were performed in a manner designed to minimize pain and suffering, and any animals that exhibited severe disease signs were euthanized immediately in accordance with IACUC-approved endpoints. For this study, the wild-type mice were immunocompetent C57BL/6 PVR-Tg21 mice (BL6-IFNAR+/+) expressing the human poliovirus receptor and immune-deficient mice were C57BL/6 PVR-Tg21 mice deficient in the IFN-α/β receptor (BL6-IFNAR−/−) expressing the human poliovirus receptor (provided by S. Koike, Tokyo, Japan) (32). Three- to four-week-old mice were injected intramuscularly in the left quadriceps with 35 μl of viral supernatant containing 6 × 103 PFU comprised of 1 × 103 of each of the genetically marked YFV-17D viruses based on BHK-J titers. Mice were monitored daily for signs of disease as described in the Results section and euthanized upon disease onset (7 to 8 days postinfection). Because mice with severe disease do not recover from infection, we euthanized moribund mice, and data are presented as percentages of mice without disease. For viral-titer and population diversity analyses, tissues were harvested from perfused or nonperfused mice, depending on the time point. Since viral titers in blood were relatively high on day 3 postinfection, mice sacrificed at 1 to 3 days postinfection were perfused with 20 ml of ice-cold phosphate-buffered saline (PBS) prior to tissue harvest to limit blood contamination of tissues. However, viral titers in the blood at 5 to 8 days postinfection were undetectable or at least 10-fold lower than in other tissues. Therefore, mice with disease onset were not perfused and there was no difference in viral tissue titers between perfused and nonperfused mice at disease onset (data not shown).
Tissue processing and viral-population diversity assay.
Tissues (muscle from the inoculated quadricep, whole sciatic nerve from the inoculated leg, brain, liver, spleen, and blood) were weighed and resuspended in 3 volumes of PBS and homogenized with a Bullet Blender tissue homogenizer (Next Advance, Inc., Averill Park, NY) per the manufacturer's instructions. Tissue homogenates were then centrifuged at 13,000 rpm for 1 min and the supernatant virus stocks were used for viral-titer (see above) and diversity assays. For tissues containing low viral titers (e.g., blood), viral amplification in cell culture prior to the viral-population diversity assay was necessary to generate enough viral RNA for the reverse transcription (RT)-PCR-based hybridization assay. The cell culture virus amplification step did not alter the number of pool members observed in tissues for two reasons. First, direct RT-PCR products could be used for tissues containing high viral titers (e.g., brain at disease onset), and in these high-titer tissues, viral amplification in BHK cells did not affect viral-diversity results (data not shown). Second, all viral-pool members were observed in many tissues (see Fig. 3, 4, and 5), indicating that pool members were not lost during viral amplification in BHK cells. For amplification, 50 μl of the tissue homogenate was added to 250 μl of serum-free medium and plated on confluent BHK-J cells for 1 h, virus was removed, cells were washed, and 2 ml of DMEM-2%FBS was added to the cells for 48 h. After 48 h, the supernatant was removed, cells were washed with PBS, 1 ml of TRIzol (Invitrogen, Carlsbad, CA) was added to the plate for 5 min, and RNA extraction and RT-PCR were performed as previously described (7, 8). Primers used for cDNA synthesis and PCR were antisense primer 5′-CATCAATGATCACGCTAG-3′ and sense primer 5′-TCTGGAATTCTTTCCAGA-3′. Fifty to one hundred nanograms of PCR product from each sample was then used for the hybridization-based population diversity assay as previously described (7, 8). Briefly, RT-PCR products were blotted on nylon membranes and hybridized with 32P-labeled primer probes specific for each marked viral sequence. Normalization was performed to eliminate cross-reactivity of probes by using mismatched products (all PCR products except for the one specific for the probe), which were loaded on each membrane as controls.
Fig 3.

Viral population diversity in YFV-17D- and poliovirus-infected mice. BL6-IFNAR+/+ and BL6-IFNAR−/− mice were inoculated i.m. with 6 × 103 PFU of the YFV-17D viral-pool mixture or 2 × 107 PFU of 10 genetically marked polioviruses (7, 8), tissues were harvested at disease onset (day 7 to 8 for YFV-17D or day 3 to 8 for poliovirus), and viral population diversity was assessed using the hybridization assay. (A) Representative blot of YFV-17D pool members present, with specific viral probes numbered along the top, in various tissues from a YFV-17D-inoculated BL6-IFNAR−/− mouse. (B) Percentages of input viruses present in muscle (M; black bars) and brain (B; white bars) tissues in poliovirus-infected mice (left) (7, 8) and YFV-17D-infected mice (right) at disease onset or 8 days postinfection for asymptomatic YFV-17D-injected BL6-IFNAR+/+ mice. Means and standard errors of the means (SEMs) compiled from at least 3 mice per condition are shown. Asterisks denote statistically significant differences (P < 0.05, Student's t test), while differences that are not statistically significant are indicated by “n.s.”
Fig 4.
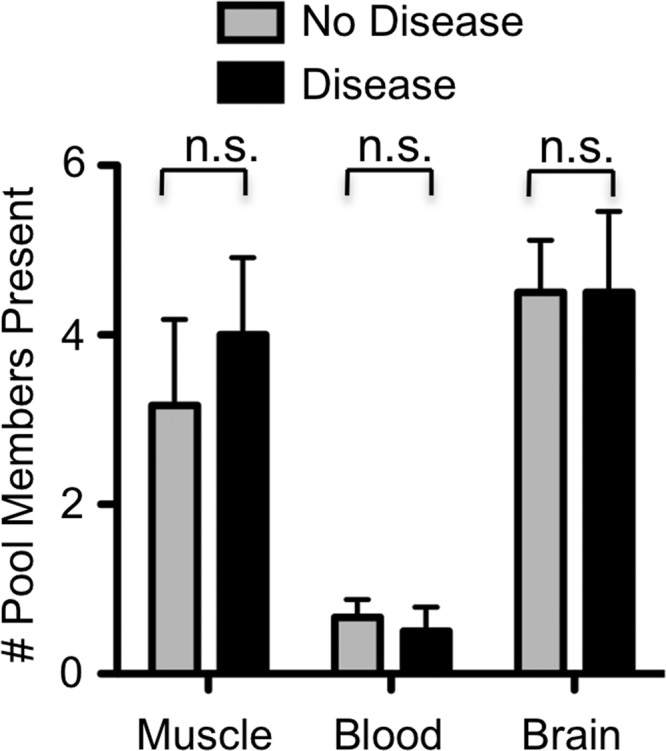
YFV-17D viral population diversity in mice with or without disease signs. BL6-IFNAR−/− mice were inoculated i.m. with 6 × 103 PFU of the YFV-17D viral-pool mixture, tissues were harvested at disease onset (day 7 to 8 postinfection) or day 8 postinfection for mice not showing disease signs, and viral population diversity was assessed using the hybridization assay. Means and SEMs compiled from 4 mice per condition are shown, and differences that are not statistically significant (P > 0.05, Student's t test) are indicated by “n.s.”
Fig 5.
YFV-17D population diversity and titer over time. BL6-IFNAR−/− mice were inoculated i.m. with 6 × 103 PFU of the YFV-17D viral-pool mixture. Tissues were harvested at early (day 1), middle (day 3), or late (days 7 to 8) times postinfection and used for both titer and viral population diversity assays. Titers are represented by black squares (left axis) and the number of pool members present is represented by open circles (right axis), reflecting viral population diversity. Data are shown as PFU/tissue, except for blood titers, which are shown as PFU/ml. Means and SEMs for 3 to 48 mice per time point are shown.
RESULTS
Development of a YFV-17D population diversity assay.
Previously, our lab developed a hybridization-based population diversity assay to identify host barriers that limit poliovirus trafficking in mice (7, 8); herein, we generated a similar assay to study YFV-17D dissemination and trafficking barriers (Fig. 1). We created 6 marked YFV-17D viruses by incorporating silent mutations into the nonstructural 1 (NS1) region of the YFV-17D genome (Fig. 1A). We designed oligonucleotide probes to specifically recognize these tagged regions within the viral genome. Specificity of the YFV-17D population diversity assay was determined by infecting BHK-J cells with individual viruses or a mixture of all 6 viruses, isolating RNA, and blotting RT-PCR products on a nylon membrane, followed by hybridization with each individual 32P-labeled oligonucleotide probe. We found that each probe was specific for its cognate virus (Fig. 1B). To ensure that none of the viral variants had major growth defects, we performed a serial-passage competition experiment. Since subtle growth defects can be masked in single-cycle growth curve experiments and our viruses would be undergoing several rounds of replication in infected mice, we determined whether each of the marked viruses was maintained over four cycles of replication in a mixed infection of cultured cells. For this competition experiment, we infected BHK-J cells with a mixture of all 6 viruses, passaged the virus-containing supernatants to fresh cells, and evaluated the cellular extracts for the presence of each virus. We found that all 6 viruses were present after four serial passages, demonstrating the absence of severe growth defects in vitro (Fig. 1C). To evaluate growth defects in vivo, we compiled 788 hybridization signals from numerous tissues from 33 mice at 7 to 8 days postinfection with our 6 marked YFV-17D viruses (see below) and found that all 6 viruses were represented approximately equally (Fig. 1D). Therefore, none of the 6 viruses demonstrated a growth advantage or disadvantage in vivo.
Survival of YFV-17D-infected mice.
In order to evaluate pathogenesis of the genetically marked pool of YFV-17D viruses, we inoculated 3- to 4-week-old BL6-IFNAR+/+ or BL6-IFNAR−/− mice intramuscularly (i.m.) with 6 × 103 PFU of the YFV-17D viral-pool mixture and monitored animals daily for signs of disease. We chose a relatively low concentration of virus since host barriers can be overcome by high virus concentrations in certain circumstances (9). We found that BL6-IFNAR+/+ mice did not exhibit signs of disease up to 30 days postinfection (Fig. 2 and data not shown). In contrast, YFV-17D i.m.-inoculated BL6-IFNAR−/− mice displayed various degrees of illness. At 5 days postinfection, all i.m.-inoculated mice were limping on the injected leg; however, by 6 days postinfection the mice were walking normally. At 7 to 8 days postinfection, 32% of the i.m.-inoculated BL6-IFNAR−/− mice presented with one of two distinct disease phenotypes, neurotropic or viscerotropic disease. Neurotropic disease manifested as paralysis ranging from one paralyzed leg to severe dual-leg paralysis, with occasional lethargy, walking in circles, and eye discharge. Viscerotropic disease manifested as lethargy, hunched posture, piloerection, and rapid breathing quickly leading to immobility and death. We found that all mice that were asymptomatic at 8 days postinfection remained asymptomatic through 40 days postinfection.
Fig 2.
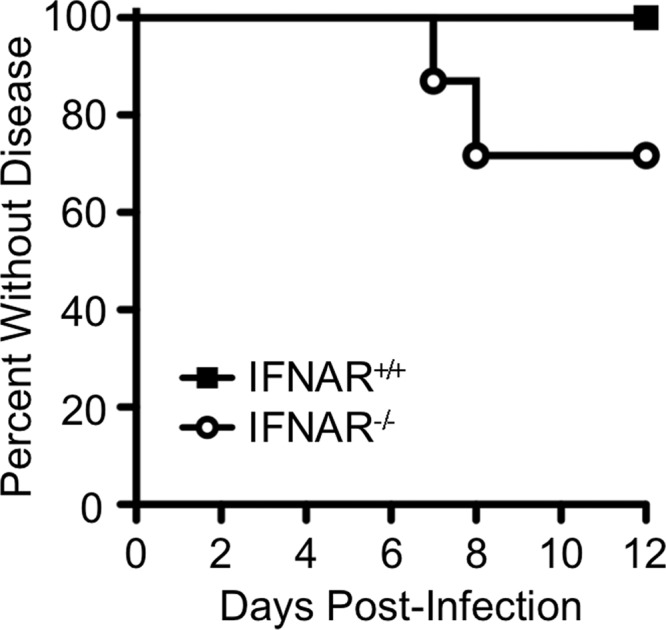
Survival of YFV-17D-infected mice. Three- to four-week-old mice were injected i.m. with 6 × 103 PFU of the YFV-17D viral-pool mixture and disease onset was monitored. Since moribund mice do not recover from infection, they were euthanized upon severe disease onset; therefore, data are shown as percentages without disease. Data are from 48 BL6-IFNAR−/− mice and 8 BL6-IFNAR+/+ mice. Statistically significant differences were observed (P < 0.005, Mantel-Cox test).
Viral-population diversity in BL6-IFNAR+/+ and BL6-IFNAR−/− mice.
While arbovirus population diversity and trafficking are limited by host barriers in mosquitoes (17, 18), whether arboviruses encounter barriers during dissemination in vertebrate hosts was unknown. Previously our lab found that poliovirus, a neurotropic picornavirus, encounters multiple host barriers that limit viral-population diversity, trafficking to the CNS, and virulence in mice (7, 8). We hypothesized that, like that of poliovirus, YFV-17D population diversity would be reduced by host barriers encountered during trafficking from the inoculation site (muscle) to peripheral tissues and the brain. To determine whether YFV-17D encounters major host barriers that limit viral diversity, we infected BL6-IFNAR+/+ or BL6-IFNAR−/− mice i.m. with 6 × 103 PFU of the YFV-17D viral-pool mixture. At 8 days postinfection, viral-population diversity was analyzed in various tissues, including blood, brain, spleen, liver, sciatic nerve, and muscle. A representative blot from an infected BL6-IFNAR−/− mouse is shown in Fig. 3A and demonstrates that all 6 viruses were detected in the majority of tissues at disease onset. Our previous work, summarized on the left side of Fig. 3B, demonstrated that poliovirus encountered a major bottleneck between the inoculation site (muscle) and the brain in BL6-IFNAR+/+ mice, with 100% of viral-pool members present in muscle but only 10% of viral-pool members present in the brain at disease onset (Fig. 3B) (7, 8). This poliovirus bottleneck was diminished but not abolished in BL6-IFNAR−/− mice, with ∼60% of viral-pool members present in the brain. We determined that inefficient virion retrograde axonal transport in neurons constitutes much of the remaining barrier to poliovirus CNS invasion in BL6-IFNAR−/− mice (8). In contrast, dissemination of YFV-17D in BL6-IFNAR+/+ mice was severely restricted, with asymptomatic infection and only 30% of viral-pool members present in the inoculation tissue (muscle) and 10% of viral-pool members present in the brain at 8 days postinfection. Strikingly, the barrier to YFV-17D dissemination was almost completely overcome in BL6-IFNAR−/− mice, where 60 to 100% of viral-pool members were present in the muscle and brain tissues at 8 days postinfection (Fig. 3B). In fact, there were more marked viruses in the brain than in muscle of YFV-17D-infected BL6-IFNAR−/− mice. These results suggest that, unlike for poliovirus, which is limited by multiple host barriers (7, 8), the type I IFN response is the main barrier limiting YFV-17D dissemination in mice.
Since we previously found that high poliovirus population diversity correlated with a high level of pathogenesis (7), we quantified the number of YFV-17D pool members present in tissues of BL6-IFNAR−/− mice with or without disease signs at day 7 to 8 postinfection. We found that, in contrast to that of the poliovirus system, YFV-17D population diversity, as measured by the presence of our six marked viruses, did not correlate with disease state (Fig. 4). For example, a majority of the six marked viruses were present in muscle and brain of BL6-IFNAR−/− mice regardless of whether the mice showed signs of disease.
In order to determine whether YFV-17D encounters barriers early after infection, we inoculated BL6-IFNAR+/+ or BL6-IFNAR−/− mice i.m. with 6 × 103 PFU of the YFV-17D viral-pool mixture and compared viral titers and viral-population diversity at early (1 day postinfection), middle (3 days postinfection), and late (7 to 8 days postinfection) time points in various tissues. Shortly after inoculation, 1 day postinfection, 100% of the viral-pool members were present in the inoculation tissue (muscle) with ∼104 PFU of virus, likely reflecting inoculum virus (Fig. 5). Similarly, at 1 day postinfection, 100% of the viral-pool members were present in the sciatic nerve of the injected leg. These results suggest that similar to poliovirus, YFV-17D is able to enter neurons and may be able to traffic to the central nervous system via peripheral neurons such as the sciatic nerve (8, 33). At 1 day postinfection, we detected virus only in the spleen, where approximately 50% of the viral-pool members were present. However, at 3 days postinfection we found high viral-population diversity in all tissues, corresponding with high viral titers in all tissues. These results indicate that the viruses did not encounter a bottleneck during trafficking from the inoculation site to other tissues. Moreover, these results confirm that none of the marked viruses has a growth advantage or disadvantage in vivo and that our sample processing did not alter the abundance of individual viruses. At 7 to 8 days postinfection/disease onset, viral-population diversity and titers were decreased in muscle, sciatic nerve, and spleen and were undetectable in the blood and liver, suggesting viral clearance in the periphery. However, viral titers in the brain were highest at disease onset, although viral-population diversity was reduced, suggesting a dynamic interplay between viral replication and clearance in the brain (Fig. 5). In contrast to our results for poliovirus infection of BL6-IFNAR−/− mice (7, 8), in YFV-17D infection the number of YFV-17D pool members present and YFV-17D titer were linked throughout infection in all tissues examined, where high-titer sites contained a high number of pool members and low-titer sites contained a low number of pool members (Fig. 5). Overall our data suggest that although YFV-17D infection of BL6-IFNAR−/− mice does not always induce disease, the virus population is able to efficiently replicate to high titers and easily disseminate to peripheral and CNS tissues without encountering major bottlenecks.
DISCUSSION
Previous studies have found that arboviruses encounter population bottlenecks during dissemination in the mosquito vector (17, 18); however, potential bottlenecks encountered during dissemination in the vertebrate host have not been evaluated. Using a pool of 6 genetically tagged viruses, we found that dissemination of the YFV-17D population was severely diminished in BL6-IFNAR+/+ mice such that viruses were restricted to the inoculation tissue (muscle) and very rarely disseminated to the brain (Fig. 3B). These results are consistent with previous findings that the mouse type I IFN response is extremely effective at blocking YFV-17D and virulent YFV-Asibi replication in mice (21) as well as West Nile virus (WNV) infection (5, 6, 34). Additionally it was found that while virulent strains of YFV caused disease in subcutaneously inoculated 129Sv[ev]-IFNAR−/− mice, YFV-17D was severely attenuated (21), suggesting that additional factors such as inoculation route limit YFV-17D infection. However, we found that i.m. inoculation of YFV-17D caused disease in BL6-IFNAR−/− mice and the viral-pool members did not encounter any major barriers during dissemination (Fig. 3B).
In nature, vertebrates are infected with YFV by subcutaneous mosquito bite. However, in this study we used intramuscular injection of mice, partly to compare YFV-17D and poliovirus dissemination and bottlenecks. During natural infection, the YFV population may encounter different environments and may be influenced by specific cell subsets in the skin and other factors such as mosquito saliva. These factors likely influence initial replication and dissemination. Indeed, our preliminary experiments suggest that the six marked YFV-17D viruses have limited dissemination in subcutaneously inoculated BL6-IFNAR−/− mice.
The absence of a barrier limiting YFV-17D dissemination in intramuscularly inoculated BL6-IFNAR−/− mice was surprising considering our previous results with poliovirus. Poliovirus encountered a bottleneck during trafficking from muscle to the brain in BL6-IFNAR−/− mice due to inefficient retrograde axonal transport in neurons (8). For poliovirus, titer and viral diversity were not linked, such that some high-titer sites (brain) had very low viral-population diversity, indicative of viral replication after a major bottleneck (7, 8). However, for YFV-17D, we found that viral titer and diversity were linked and that by 3 days postinfection all six marked viruses could be found in most tissues, demonstrating that the YFV-17D population did not encounter a major bottleneck during dissemination (Fig. 5). While we did not sequence the YFV-17D population to examine population diversity directly, the presence of all six marked viruses in several tissues indicates that no major barrier limited trafficking of these marked pool members in intramuscularly inoculated BL6-IFNAR−/− mice. Therefore, there was a difference in poliovirus and YFV-17D population dissemination from the intramuscular injection site to other tissues. It is possible that YFV-17D spreads through blood to the CNS and other tissues while poliovirus spreads through less efficient neural routes.
A model based on our data depicting temporal YFV-17D population movement in i.m.-inoculated BL6-IFNAR−/− mice is shown in Fig. 6. At day 1 postinfection, the viral population was restricted to the inoculation area (muscle, sciatic nerve) with low titers and high population diversity, likely reflecting the inoculum. At days 3 to 5 postinfection, viral dissemination was widespread, with high viral titers and a high number of viral-pool members present in many tissues, including the brain. At days 7 to 8 postinfection, the time of disease onset, viral titers and the numbers of pool members were limited in most tissues except for brain. This profile suggests widespread dissemination in the middle of the infection course followed by viral clearance. By using genetically marked viruses to monitor viral movement, we found that, overall, YFV-17D dissemination in BL6-IFNAR−/− mice is a dynamic process.
Fig 6.
Model of YFV-17D population movement in i.m.-inoculated BL6-IFNAR−/− mice. Viral population diversity is indicated by the six different colors of circles (representing the six marked viruses), and total titers are indicated by the relative numbers of circles. Early after injection, most of the six marked viruses were present in the inoculated tissue (leg muscle, sciatic nerve) and viral titers were relatively low and likely reflect the inoculum. Elsewhere, viral titers and the numbers of pool members present were low. In the middle of the infection course, viral titers and the numbers of pool members present were high in nearly all tissues, reflecting efficient replication and dissemination. Late in infection, viral titers were highest in the brain, with lower viral titers and numbers of pool members present in other tissues, reflecting clearance in the periphery.
ACKNOWLEDGMENTS
We thank Charlie Rice for the YFV-17D infectious clone, Satoshi Koike for the mice used in this study, and Chris Etheredge for assistance in generating the genetically tagged YFV clones. We also thank Nick Conrad, Chris Robinson, and John Schoggins for helpful comments on the manuscript.
This work was supported by NIAID/National Institutes of Health grant AI-74668, a Pew Scholar award, and a Burroughs Wellcome Foundation Investigators in the Pathogenesis of Infectious Diseases award (to J.K.P.).
The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Footnotes
Published ahead of print 11 September 2013
REFERENCES
- 1.Pfeiffer JK, Kirkegaard K. 2005. Increased fidelity reduces poliovirus fitness and virulence under selective pressure in mice. PLoS Pathog. 1:e11. 10.1371/journal.ppat.0010011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Vignuzzi M, Stone JK, Arnold JJ, Cameron CE, Andino R. 2006. Quasispecies diversity determines pathogenesis through cooperative interactions in a viral population. Nature 439:344–348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Coffey LL, Beeharry Y, Borderia AV, Blanc H, Vignuzzi M. 2011. Arbovirus high fidelity variant loses fitness in mosquitoes and mice. Proc. Natl. Acad. Sci. U. S. A. 108:16038–16043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Domingo E, Holland JJ. 1997. RNA virus mutations and fitness for survival. Annu. Rev. Microbiol. 51:151–178 [DOI] [PubMed] [Google Scholar]
- 5.Gnädig NF, Beaucourt S, Campagnola G, Borderia AV, Sanz-Ramos M, Gong P, Blanc H, Peersen OB, Vignuzzi M. 2012. Coxsackievirus B3 mutator strains are attenuated in vivo. Proc. Natl. Acad. Sci. U. S. A. 109:E2294–E2303. 10.1073/pnas.1204022109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Graham RL, Becker MM, Eckerle LD, Bolles M, Denison MR, Baric RS. 2012. A live, impaired-fidelity coronavirus vaccine protects in an aged, immunocompromised mouse model of lethal disease. Nat. Med. 18:1820–1826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kuss SK, Etheredge CA, Pfeiffer JK. 2008. Multiple host barriers restrict poliovirus trafficking in mice. PLoS Pathog. 4:e1000082. 10.1371/journal.ppat.1000082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lancaster KZ, Pfeiffer JK. 2010. Limited trafficking of a neurotropic virus through inefficient retrograde axonal transport and the type I interferon response. PLoS Pathog. 6:e1000791. 10.1371/journal.ppat.1000791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pfeiffer JK, Kirkegaard K. 2006. Bottleneck-mediated quasispecies restriction during spread of an RNA virus from inoculation site to brain. Proc. Natl. Acad. Sci. U. S. A. 103:5520–5525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Xie H, Cass AR, Barrett AD. 1998. Yellow fever 17D vaccine virus isolated from healthy vaccinees accumulates very few mutations. Virus Res. 55:93–99 [DOI] [PubMed] [Google Scholar]
- 11.Pisano MR, Nicoli J, Tolou H. 1997. Homogeneity of yellow fever virus strains isolated during an epidemic and a post-epidemic period in West Africa. Virus Genes 14:225–234 [DOI] [PubMed] [Google Scholar]
- 12.Jenkins GM, Rambaut A, Pybus OG, Holmes EC. 2002. Rates of molecular evolution in RNA viruses: a quantitative phylogenetic analysis. J. Mol. Evol. 54:156–165 [DOI] [PubMed] [Google Scholar]
- 13.Coffey LL, Vasilakis N, Brault AC, Powers AM, Tripet F, Weaver SC. 2008. Arbovirus evolution in vivo is constrained by host alternation. Proc. Natl. Acad. Sci. U. S. A. 105:6970–6975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Weaver SC. 2006. Evolutionary influences in arboviral disease. Curr. Top. Microbiol. Immunol. 299:285–314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Holmes EC, Twiddy SS. 2003. The origin, emergence and evolutionary genetics of dengue virus. Infect. Genet. Evol. 3:19–28 [DOI] [PubMed] [Google Scholar]
- 16.Jerzak G, Bernard KA, Kramer LD, Ebel GD. 2005. Genetic variation in West Nile virus from naturally infected mosquitoes and birds suggests quasispecies structure and strong purifying selection. J. Gen. Virol. 86:2175–2183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Forrester NL, Guerbois M, Seymour RL, Spratt H, Weaver SC. 2012. Vector-borne transmission imposes a severe bottleneck on an RNA virus population. PLoS Pathog. 8:e1002897. 10.1371/journal.ppat.1002897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ciota AT, Ehrbar DJ, Van Slyke GA, Willsey GG, Kramer LD. 2012. Cooperative interactions in the West Nile virus mutant swarm. BMC Evol. Biol. 12:58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Robertson SE, Hull BP, Tomori O, Bele O, LeDuc JW, Esteves K. 1996. Yellow fever: a decade of reemergence. JAMA 276:1157–1162 [PubMed] [Google Scholar]
- 20.Lee E, Lobigs M. 2008. E protein domain III determinants of yellow fever virus 17D vaccine strain enhance binding to glycosaminoglycans, impede virus spread, and attenuate virulence. J. Virol. 82:6024–6033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Meier KC, Gardner CL, Khoretonenko MV, Klimstra WB, Ryman KD. 2009. A mouse model for studying viscerotropic disease caused by yellow fever virus infection. PLoS Pathog. 5:e1000614. 10.1371/journal.ppat.1000614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Thibodeaux BA, Garbino NC, Liss NM, Piper J, Blair CD, Roehrig JT. 2012. A small animal peripheral challenge model of yellow fever using interferon-receptor deficient mice and the 17D-204 vaccine strain. Vaccine 30:3180–3187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zisman B, Wheelock EF, Allison AC. 1971. Role of macrophages and antibody in resistance of mice against yellow fever virus. J. Immunol. 107:236–243 [PubMed] [Google Scholar]
- 24.Horsfall FL, Tamm I. (ed). 1965. Viral and rickettsial infections of man, 4th ed. J. P. Lippincott Co., Philadelphia, PA [Google Scholar]
- 25.Theiler M. 1930. Susceptibility of white mice to the virus of yellow fever. Science 71:367. [DOI] [PubMed] [Google Scholar]
- 26.Fitzgeorge R, Bradish CJ. 1980. The in vivo differentiation of strains of yellow fever virus in mice. J. Gen. Virol. 46:1–13 [DOI] [PubMed] [Google Scholar]
- 27.Schlesinger JJ, Chapman S, Nestorowicz A, Rice CM, Ginocchio TE, Chambers TJ. 1996. Replication of yellow fever virus in the mouse central nervous system: comparison of neuroadapted and non-neuroadapted virus and partial sequence analysis of the neuroadapted strain. J. Gen. Virol. 77:1277–1285 [DOI] [PubMed] [Google Scholar]
- 28.Fox JP. 1943. Immunity to yellow fever encephalitis of monkeys and mice immunized by neural and extraneural routes. J. Exp. Med. 77:487–506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sellards AW. 1931. The behavior of the virus of yellow fever in monkeys and mice. Proc. Natl. Acad. Sci. U. S. A. 17:339–343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Goodman GT, Koprowski H. 1962. Macrophages as a cellular expression of inherited natural resistance. Proc. Natl. Acad. Sci. U. S. A. 48:160–165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.National Research Council 2011. Guide for the care and use of laboratory animals, 8th ed. National Academies Press, Washington, DC [Google Scholar]
- 32.Ida-Hosonuma M, Iwasaki T, Yoshikawa T, Nagata N, Sato Y, Sata T, Yoneyama M, Fujita T, Taya C, Yonekawa H, Koike S. 2005. The alpha/beta interferon response controls tissue tropism and pathogenicity of poliovirus. J. Virol. 79:4460–4469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ohka S, Yang WX, Terada E, Iwasaki K, Nomoto A. 1998. Retrograde transport of intact poliovirus through the axon via the fast transport system. Virology 250:67–75 [DOI] [PubMed] [Google Scholar]
- 34.Freudenberg MA, Meier-Dieter U, Staehelin T, Galanos C. 1991. Analysis of LPS released from Salmonella abortus equi in human serum. Microb. Pathog. 10:93–104 [DOI] [PubMed] [Google Scholar]