

Abstract
The aim of this study was to gain insights into the tempo and mode of the evolutionary processes that sustain genetic diversity in coxsackievirus B5 (CVB5) and into the interplay with virus transmission. We estimated phylodynamic patterns with a large sample of virus strains collected in Europe by Bayesian statistical methods, reconstructed the ancestral states of genealogical nodes, and tested for selection. The genealogies estimated with the structural one-dimensional gene encoding the VP1 protein and nonstructural 3CD locus allowed the precise description of lineages over time and cocirculating virus populations within the two CVB5 clades, genogroups A and B. Strong negative selection shaped the evolution of both loci, but compelling phylogenetic data suggested that immune selection pressure resulted in the emergence of the two genogroups with opposed evolutionary pathways. The genogroups also differed in the temporal occurrence of the amino acid changes. The virus strains of genogroup A were characterized by sequential acquisition of nonsynonymous changes in residues exposed at the virus 5-fold axis. The genogroup B viruses were marked by selection of three changes in a different domain (VP1 C terminus) during its early emergence. These external changes resulted in a selective sweep, which was followed by an evolutionary stasis that is still ongoing after 50 years. The inferred population history of CVB5 showed an alternation of the prevailing genogroup during meningitis epidemics across Europe and is interpreted to be a consequence of partial cross-immunity.
INTRODUCTION
The enteroviruses (EVs; family Picornaviridae) are the leading cause of acute meningitis (1), hand, foot, and mouth disease (2), and upper respiratory infections (3). Although most EV infections are self-limiting, they represent a burden to human health (4). Thus, they warrant investigation because of their high worldwide prevalence and because they give rise to severe clinical manifestations, particularly in newborns and young children during yearly epidemics of otherwise common illnesses.
Epidemics of human EV infections display a seasonal pattern, occurring in the summer and early autumn in geographical regions with a temperate climate. The basic determinants of the molecular and evolutionary epidemiology of human EVs and their long-term persistence in the general population are not well defined. This is a particularly important issue for devising intervention strategies and developing therapeutics and vaccines against those EV infections that are associated with high morbidity and occasional mortality during extensive outbreaks (5).
The EV particle is made up of 60 copies of four proteins (VP1 to VP4) and has an icosahedra-like three-dimensional (3D) structure (6). Only the VP1 to VP3 proteins have structures (amino acid loops) exposed on the capsid surface. A number of these protruding loops (e.g., those connecting β sheets B and C) usually carry the antigenic properties of each type and are thought to be involved in the neutralization of infectious particles during infections (7), which is consistent with the observation that the humoral immune response is a major factor in the eradication of EV infections (8). The viral genome (7.5 kb of single-stranded, positive-sense RNA) encodes the four structural proteins (gene sequences 1A to 1D, genomic region P1) and at least seven nonstructural proteins, 2A to 2C (region P2) and 3A to 3D (region P3).
On the basis of shared antigenic and genetic similarities, human EVs (HEVs) are grouped into distinct types (or serotypes), which are themselves included within phylogenetically defined viral species named enterovirus A to D (9). Circulating strains of each type are highly heterogeneous and have been divided into different genogroups and subgenogroups on the basis of phylogenetic patterns of structural genes, in particular, the one-dimensional (1D) gene encoding the VP1 protein (10, 11), here designated 1DVP1. Different sentinel systems implemented in Asian countries for the surveillance of hand, foot, and mouth disease (12) and the continuous surveillance in some European countries and the United States have greatly contributed to a better knowledge of the genetic diversity of EVs involved in outbreaks and larger epidemics (13, 14).
A number of picornaviruses have been the subject of comprehensive molecular evolutionary studies investigating the questions of epidemic history and molecular adaptation (15, 16). Phylogenies estimated with the 1DVP1 sequences showed a pattern of continuous lineage turnover for echovirus 30 (enterovirus B species) (17) and enterovirus 71 (enterovirus A species) (18). This indicates that virus variants are continuously emerging and are replacing the earlier prevailing viruses, a pattern similar to that of influenza A virus (19). Studies have suggested the involvement of positive selection at defined sites in the VP1 capsid protein of the virus of foot and mouth disease of cloven-hoofed livestock and of two human EVs (17, 20, 21). However, investigations of human rhinoviruses were unable to detect such patterns (22, 23).
In this study, we focused on the evolution of human coxsackievirus B5 (CVB5) by analyzing molecular patterns in serially sampled genes with the aim of clarifying the interplay of selection and transmission. The earliest coxsackievirus B strains were isolated in the 1940s and 1950s in children with symptoms of acute meningitis or paralytic poliomyelitis (24) and were classified into six types, CVB1 to CVB6, as members of the enterovirus B species (9). Coxsackievirus B infections are associated with severe neurological manifestations and are involved in acute myocarditis and fatal neonatal infections (24, 25). CVB5 is frequent in virus isolation records, as it consistently appears in the top five commonly identified EV types in the United States and France (13, 14) and has exhibited the highest annual prevalence in a number of countries (26, 27). Acute meningitis is the typical clinical presentation in CVB5 outbreaks, but the virus was recently involved in an outbreak of neurological hand, foot, and mouth disease in China (28). Previous studies have provided some insight into the genetic diversity within CVB5 (29, 30), and a phylogenetic analysis of the 1DVP1 genes sampled in 41 clinical isolates showed that CVB5 had an estimated origin in 1854 (31). We explored the evolutionary process that shapes the genetic diversity of CVB5 with a larger virus sample and identified variations in the genetic diversity and phylodynamic patterns of two virus lineages, genogroups A and B, involved in seasonal epidemics in Europe.
MATERIALS AND METHODS
Virus strains.
CVB5 strains were collected from seven European countries between the years 1977 and 2011 (see Table S1 in the supplemental material). Virus isolates and sequences are designated throughout the report as follows: a city code including two to four letters plus the isolate number, three letters of the international country code, and the last two digits of the year of collection. All the clinical specimens—cerebrospinal fluid (CSF), feces, throat or nasal swabs, bronchoalveolar lavage fluid, or cell culture supernatants—came from a preexisting collection.
Gene amplification and sequencing.
Viral RNA was extracted with a NucliSENS easyMag automated system (bioMérieux, France). The complete 1DVP1 gene and the 3CD gene fragment were amplified and sequenced using enterovirus B-specific primers described elsewhere (32). The nucleotide sequences of both strands of the PCR products were determined and analyzed with a 3500 Dx genetic analyzer (Applied Biosystems). The CVB5 consensus 1DVP1 sequence (length, 843 nucleotides [nt]; genome positions 2447 to 3295 in prototype strain Faulkner [accession no AF114383]) and the 3CD sequences (length, 1,035 nt; positions 5509 to 6544) were compiled with the BioEdit (v7.0.9.0) program.
Nucleotide sequence data sets.
Two different data sets were generated for comparative analyses. All the complete 1DVP1 CVB5 sequences published with known geographic locations and sampling dates (n = 77) were retrieved from GenBank (as of June 2012; see Table S2 in the supplemental material) and compiled with the 218 sequences determined, generating a data set of 295 taxa. The 1DVP1 sequence data set was split into two subsets for separate investigation of the two identified CVB5 genogroups. Five 3CD sequences were obtained from published CVB5 complete genomes and aligned with the 3CD sequences determined in this study.
Phylodynamic analyses.
To reconstruct the CVB5 evolutionary history, we used a Bayesian statistical approach implemented in the BEAST (v1.5.4) program (33, 34). The evolutionary rate, divergence time, and demographic histories of viral populations were estimated from the whole 1DVP1 and 3CD sequence data sets and the individual 1DVP1 subsets corresponding to the two CVB5 genogroups, A and B.
A comprehensive comparison of molecular clock and demographic models was performed for all sequence data sets, as described earlier (32). The best molecular clock and demographic models were determined by calculating the marginal likelihoods of the data (35) on the basis of all the evolutionary and demographic model parameters. We analyzed all possible combinations between three molecular clock models (strict model and relaxed models with either uncorrelated exponential or uncorrelated lognormal prior distributions of substitution rates among lineages) and four models of demographic history (constant population size, exponential or expansion population growth, and Bayesian skyline reconstruction). In these investigations, two nucleotide substitution models, GTR+I+Γ4 and SRD06 (36), were also compared. Markov chain Monte Carlo (MCMC) analyses were run for 200 million generations, with a tree sampled every 5,000 steps, and the first 10% was discarded as burn-in. MCMC convergence and effective sample sizes (ESS) were checked using the Tracer (v1.5.0) program (34). A Bayes factor (BF) was calculated for each pair of models with Tracer, and a log10 BF of >5 was considered substantial evidence in support of model combination 1 over model combination 2 (37). The overall analyses indicated that the best-fit model combination for both the 1DVP1 (log10 BF = 114) and 3CD (log10 BF = 9) sequence data sets was a relaxed clock model with an uncorrelated exponential distribution of evolution rates estimated with an SRD06 substitution model and a Bayesian skyline population model as a population model (see Table S3 in the supplemental material). Using the new version of BEAST, v1.7.5, we also tested four demographic models, Bayesian skyline, Gaussian Markov random fields Bayesian skyride, birth-death serially sampled, and birth-death basic reproductive number (38, 39). Model selection was performed with the path sampling (PS) and stepping-stone sampling (SS) methods to calculate the marginal likelihoods (40).
Maximum clade credibility (MCC) trees were estimated with the TreeAnnotator program (http://beast.bio.ed.ac.uk/TreeAnnotator) and produced with the FigTree program (http://tree.bio.ed.ac.uk/software/figtree). Statistical support for the tree nodes was assessed by a posterior probability (pp) value. The time to the most recent common ancestor (tMRCA) at each node in the phylogenies was calculated from the height value in the MCC tree. Statistical uncertainty in the tMRCA calculations was estimated as 95% highest probability density (HPD) intervals. The reproducibility of all phylogenetic trees was tested by multiple BEAST runs.
The variations in the relative CVB5 genetic diversity, a measure of the effective number of infections over time, were estimated with a Bayesian skyline plot (BSP) model (41) implemented in BEAST. Investigations were performed with the 1DVP1 data set and subsets. A piecewise-constant population size model with 20 groups was used for BSP calculation.
Site-specific selection pressure.
Site-specific selection pressure was measured as the relative number of nonsynonymous (dN) and synonymous (dS) nucleotide substitutions per site for the CVB5 VP1 protein. Estimates of the dN-to-dS ratio per site were calculated on phylogenetic trees inferred with the neighbor-joining method incorporating the general time reversible nucleotide substitution model. The dN/dS ratio was estimated on entire phylogenetic trees with methods implemented in the HyPhy package available at the Datamonkey web facility (http://www.hyphy.org/), the single-likelihood ancestor counting (SLAC) and the two-rate fixed-effects likelihood (FEL) methods (42). The FEL and the fast, unconstrained Bayesian approximation (FUBAR) (43) methods were also applied on internal branches of phylogenetic trees (internal FEL [IFEL] method). P values less than 0.1 were used as thresholds for strong evidence of selection. A Shannon entropy score was calculated at each position in the VP1 protein alignment to estimate the amino acid variability of the capsid protein in CVB5 genogroups A and B (http://www.hiv.lanl.gov/). The CVB5 ancestral nucleotide and amino acid sequences of all internal nodes were reconstructed using the probabilistic ancestral state reconstruction (ASR) method (42) implemented in the HyPhy software package. The ancestral reconstruction analyses included joint (44), marginal (45), and sampled (46) methods. Amino acid changes between lineages were determined and mapped along phylogenetic trees inferred for the 1DVP1 gene.
Nucleotide sequence accession numbers.
All the CVB5 sequences determined in the present study were deposited in the GenBank sequence database under accession numbers HF948028 to HF948444.
RESULTS
Phylogenetic clustering of CVB5 taxa in the 1DVP1 gene tree.
We analyzed 295 1DVP1 sequences (218 determined in the present study and 77 recovered from GenBank) of CVB5 strains sampled between 1952 and 2011 in 23 countries (see Tables S1 and S2 in the supplemental material). The CVB5 1DVP1 gene trees shown in Fig. 1 and Fig. S1 in the supplemental material were obtained with a Bayesian procedure that infers demographic history as a function of time with the program BEAST (33, 34). On the basis of this expansive phylogeny, the CVB5 strains separated into two consistent and coevolving clades or genogroups (Fig. 1). The clade close to the tree root was given the letter A (genogroup CVB5-A) and corresponds to genogroup II in an earlier study (31). The two clades had different phylogenetic patterns. Genogroup A consisted of multiple lineages arising from the main backbone, a pattern that indicates cocirculation of multiple virus lineages, and included five main subgenogroups (named A0 to A4) with a consistent posterior probability (pp) density of 1. The taxa within genogroup A had 14 geographic origins in the Americas, Europe, and Asia over 6 decades (see Fig. S1A in the supplemental material). In contrast, the phylogenetic pattern of genogroup B had a long backbone with short side branches, due to the combined effects of temporal sampling and rapid coalescence, a shape characteristic of influenza A viruses (47). The topology of genogroup B was consistent with a turnover of virus lineages over time and reflected virus sampling (n = 169/295 taxa) more consistent than that in genogroup A (n = 125/295 taxa, excluding the prototype strain isolated in 1952) over the same time period, 1970 to 2010/2011. The phylogenetic pattern of genogroup B also reflected differences in geographic sampling, as most taxa were collected in Western Europe and only 12 were collected in the Americas and Australia (see Fig. S1B in the supplemental material). The sampling period of taxa within subgenogroups varied from 7 years (subgenogroup B2) to 26 years (subgenogroups A0, B0, and B1), and the various geographical origins indicated the wide dissemination of virus lineages in Europe and throughout the world (i.e., subgenogroup A1). Multiple occurrences of cocirculation of different subgenogroups were observed in individual countries, for instance, in France, Denmark, Cyprus, and Germany (Table 1). The monophyletic clusters in the 1DVP1 tree included isolates from at least three of the four most frequently collected samples, feces, CSF, throat swabs, and nasopharyngeal aspirates (see Fig. S2 in the supplemental material). This pattern suggests that the sample diversity was not a major bias in the phylogenetic reconstruction.
Fig 1.

Evolutionary patterns reconstructed for the VP1 capsid protein gene of 295 CVB5 strains sampled between 1952 and 2011. Each tip branch represents a sampled virus sequence, and the colors show the locations where it was sampled. The topology shown is the MCC tree reconstructed with the 1DVP1 gene sequences (849 nt) and generated by the MCMC procedure implemented in the software program BEAST (34). Samples are explicitly dated on the x axis. The genealogy is represented so that lineages that leave more descendants are placed upward. This sorting places the backbone along a raw diagonal. The lineages determined from the phylogeny inferred with the 3CD locus (Fig. 2) are indicated to show the distribution of subgenogroups over time. Full circles along the numbered backbone branches indicate the location of amino acid changes significant for the evolutionary pathway of CVB5 genogroups A and B (see the caption table [upper left]). For nodes 1 and 2, see Fig. 4. AA, amino acid.
Table 1.
Temporal distribution of CVB5 lineages over the last 12 years showing instances of transient local lineage coexistencea
| Countryb | Lineage(s) found in the following sampling yr: |
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 2000 | 2001 | 2002 | 2003 | 2004 | 2005 | 2006 | 2007 | 2008 | 2009 | 2010 | 2011 | |
| CYP | B1c | A4a, B2a | B2a | A4a | ||||||||
| DEU | B1c | B1a, B1c | A1, B1a | B1c | B2b | B2a, B2b | B1c | B2b | B2a, B2c, B2e | B1c, B2f | ||
| DNK | B1c | A1, B1c | A1, B1c | B1c | B1c | B1c, B2b | A1, B2b | A4b | B2a, B2c | A4a, B2e | ||
| FRA | A3b, B1c | A3b | A1, A3b, A3c, B1c | A1, A3c | A1, A3c | A3d, A4a | A3e, B2a, B2b, B2c | A3e | B2a, B2c | B2c, B2d | B2c, B2d, B2f | B2c, B2e, B2f |
| CHN | A4 | A4 | A4 | A4 | A4b, A4c | |||||||
The lineages are indicated with the 3CD clade designations, unless the 3CD sequence is unknown. Genogroups A and B are shown in bold and nonbold, respectively, for clarity.
The countries are designated with the international 3-letter code: CYP, Cyprus; DEU, Germany; DNK, Denmark; FRA, France; CHN, China.
Phylogenetic clustering of CVB5 taxa in the 3CD gene tree.
The completely different phylogenetic topology estimated for the nonstructural 3CD locus (Fig. 2) allowed us to differentiate a number of virus lineages (with consistent node support; pp = 1) within all CVB5 subgenogroups except A1. Each lineage was designated by its subgenogroup (allowing connection with the 1DVP1 gene phylogenetic pattern; Fig. 1), followed by a letter specifying the 3CD lineage. The lineages were named consecutively according to the sampling period of the taxa; e.g., two taxa included in the A0 subgenogroup split into the distantly related lineages A0a and A0b. The A2 subgenogroup included viruses for which the 3CD sequences were not available in GenBank. The A3 and A4 subgenogroups split into five and four 3CD lineages, respectively. The B1 subgenogroup split into three 3CD lineages, one of which (B1c) was related to the A3d and A4d lineages in a consistent tree partition (pp = 1). The B2 subgenogroup split into six lineages in the 3CD tree. Three genogroup A strains, the prototype strain Faulkner (A0a), one strain isolated in France in 2005 (A3d), and one strain isolated in China in 2010 (A4d), grouped within the genogroup B in the 3CD tree. B0 and three B2 lineages (B2b, nine taxa; B2d, six taxa; B2f, three taxa) were interspersed among lineages arising from the CVB5-A subgenogroups. The epidemiological interest in identifying lineages in subgenogroups was that virus succession over time (Fig. 1) and the origin of epidemics were defined precisely. For instance, the 2000 epidemic (Denmark, France, and Germany) was caused by subgenogroup B1c, which arose in 1995 (tMRCA 95% HPD interval, 1994 to 1996), and the 2003 upsurge (France) was associated with the A3c subgenogroup, a variant population that arose 9 to 11 years earlier. The latest epidemic occurred in 2006, 7 years after the time origin of subgenogroup B2c (tMRCA = 1999 [95% HPD interval, 1996 to 2001]). We found substantial differences in the mean circulation time of the most recent lineages, in particular, the B2 lineages and the A3 and A4 lineages (see Table S4 in the supplemental material). Sampling bias may have contributed to the high circulation times of the lineages in the most ancient subgenogroups, A1 and B1.
Fig 2.

Phylogenetic tree topology inferred with the CVB5 3CD sequences. The MCC tree (phylogram) was reconstructed with a Bayesian MCMC procedure implemented in the software program BEAST (34) for analyzing the sequences (1,035 nt) of 223 virus strains sampled between 1952 and 2011. The branches of the phylogram were drawn to the indicated scale (number of nucleotide substitutions per site). Posterior probability values of >0.9 are indicated with asterisks at key nodes.
Population dynamics of CVB5 strains.
The tMRCA and mean evolutionary rate values estimated with the Bayesian skyline were different from those estimated with the other demographic models (Table 2), but the phylogenies reconstructed with the different models exhibited overall similar topologies. The Bayesian credibility intervals of the two parameters were estimated with narrow 95% HPD intervals with the skyride and both birth-death models. The marginal likelihood calculated with the PS and SS methods indicated that the Bayesian skyline model fit the data better than the other demographic models. Accordingly, the phylodynamic features of CVB5 described below were determined with this demographic model. The mean rate of nucleotide substitution estimated for both CVB5 subgenogroups was similar since their Bayesian credibility intervals displayed significant overlap. Genogroup A was older than genogroup B (1908 and 1941), but the 95% HPD intervals displayed some overlap (1849 to 1948 and 1912 to 1962, respectively).
Table 2.
Selection of demographic models for the Bayesian phylogenetic analysis of the CVB5 1DVP1 genea
| Parameter | Genogroup A (n = 126) |
Genogroup B (n = 169) |
||||||
|---|---|---|---|---|---|---|---|---|
| Marginal likelihood |
tMRCA (calendar yr) | Mean substitution rate (10−3 substitutions/site/yr) | Marginal likelihood |
tMRCA (calendar yr) | Mean substitution rate (10−3 substitution/site/yr) | |||
| PSb | SSc | PS | SS | |||||
| BSP | −8,627.8152 | −8,628.3069 | 1908 (1849–1948) | 5.449 (3.69–7.367) | −11,049.2201 | −11,050.0717 | 1941 (1912–1962) | 5.613 (4.54–6.615) |
| GMRF/BS | −8,682.8956 | −8,683.7626 | 1949 (1944–1952) | 7.307 (6.097–8.56) | −11,087.8397 | −11,088.8417 | 1963 (1959–1966) | 6.159 (5.383–6.968) |
| BD/SS | −8,690.9063 | −8,691.4030 | 1951 (1950–1952) | 9.054 (7.748–10.428) | −11,087.1253 | −11,087.8962 | 1964 (1962–1966) | 7.106 (6.182–8.024) |
| BD/BRN | −8,697.4840 | −8,697.9942 | 1951 (1950–1952) | 9.059 (7.784–10.438) | −11,091.9033 | −11,092.6482 | 1964.5 (1962–1966) | 7.212 (6.313–8.213) |
The phylogenetic reconstructions were done with the SRD06 substitution model and an uncorrelated relaxed clock model including an underlying exponential distribution with the BEAST (v1.7.5) program. Abbreviations: n, number of sequences; BSP, Bayesian skyline plot; GMRF/BS, Gaussian Markov random fields Bayesian skyride; BD/SS, birth-death serial sampling; BD/BRN, birth-death basic reproduction number. Years in parentheses are 95% HPD intervals.
The log marginal-likelihood estimate was calculated with the path sampling (PS) method.
The log marginal-likelihood estimate was calculated with the stepping-stone (SS) method.
The CVB5 population dynamics were reconstructed from the 1DVP1 sequence data set (Fig. 3). The plot indicated successive upward and downward fluctuations in viral genetic diversity (i.e., rapid variation in coalescence rates) across Europe during the period from 1995 to 2011 (Fig. 3A). The pattern was characterized by three rapid expansions followed by declines. Separate analyses for each genogroup were performed, and the superposition of the two plots (Fig. 3B) depicted similar temporal patterns and provided details of the temporal occurrence of the two genogroups. Genogroup A showed two minor increases in genetic diversity in 2000 and 2006 and a sharp increase in 2003. Genogroup B showed a different pattern, with two major increases in the early 2000s and in 2006, both of which were followed by slow variations over 4 years. The estimated changes in genetic diversity were consistent with epidemiological data recorded by the French network for surveillance of enteroviruses (Fig. 3C). Over the period from 2000 to 2010, three epidemic peaks were observed: in 2000, 2003, and 2006. Moreover, the temporal distribution of the two genogroups, estimated from the sampling of CVB5 French isolates, matched the demographic inference calculated with the BSP model. The isolates recovered during the epidemic in 2003 were exclusively related to genogroup A, while in 2000 and 2006, the two genogroups cocirculated with predominant B isolates.
Fig 3.
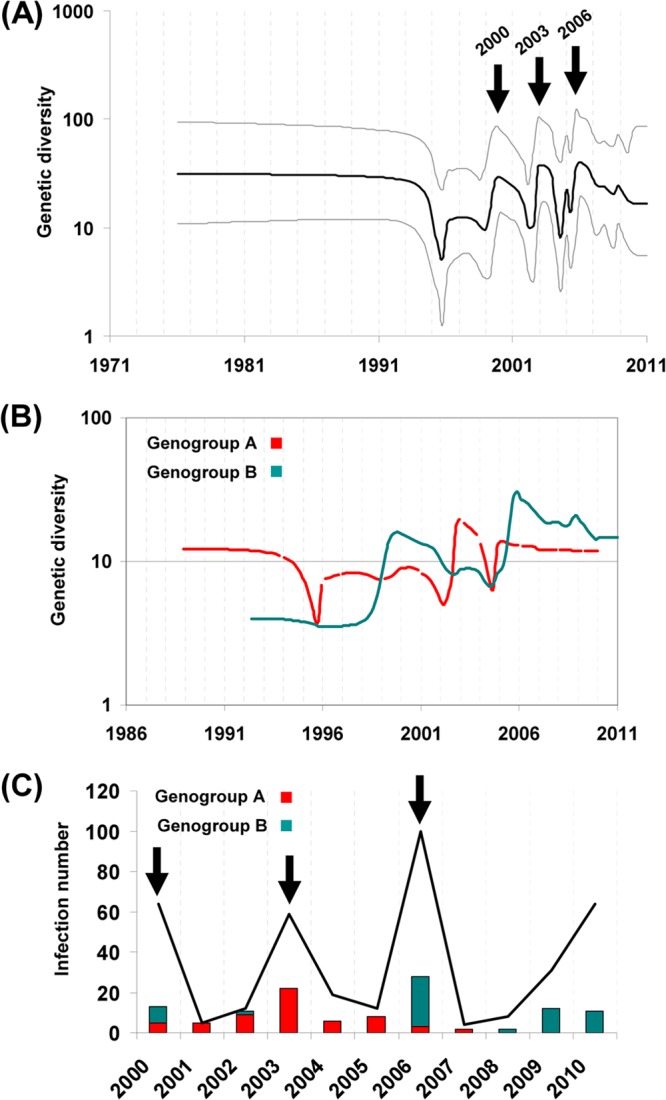
Global population dynamics reconstructed for the CVB5 genogroups. (A) Bayesian skyline plot estimated with the entire 1DVP1 gene data set of 295 sequences indicating the variation of the genetic diversity (Neτ, where Ne is the effective population size and τ is the generation time [41]), a measurement of the number of effective infections over time, as estimated with the CVB5 sequence sample. Black line, median posterior value; gray lines, 95% HPD intervals. The y axis depicts the value of Neτ on a logarithmic scale. (B) Superimposed Bayesian skyline plots estimated separately with the 1DVP1 gene sequences of the two CVB5 genogroups. The pattern shows the differences and the alternation between the two genogroups over time. (C) Surveillance data for CVB5 infections (black line) and strains (histogram bars) detected in France between 2000 and 2010 and the subgenogroups prevailing over the period.
Selection analyses with the 1DVP1 gene-encoding sequences.
The finding of a global pattern of epidemics caused alternately by viruses from two lineages lends weight to the hypothesis of immune selection between two antigenic variants. To test the hypothesis, we used independent maximum likelihood methods (42) implemented in the HyPhy program available in the Datamonkey facility (http://www.hyphy.org) to investigate the patterns of selection of individual codon sites in the two genogroups. We identified no statistical evidence of positive selection for either VP1 codon either by the consensus of the methods or by the individual tests. In fact, evidence of negative selection with all the methods was found in nearly all codon sites in the 1D gene of genogroups A and B. The results were also confirmed with the FUBAR method (43) by the detection of 280/281 sites with evidence of purifying selection at a posterior probability of ≥0.9. Earlier data for EVs (17, 22) were in line with our results, indicating unambiguously that the VP1 protein evolved under highly conservative selection pressure, a situation in which the historical occurrences of adaptive evolution are not easily detected (48).
Estimation of nonsynonymous substitutions along the CVB5 phylogenetic backbone.
We quantified the sequence variability of the VP1 protein among the CVB5 isolates (i.e., the variability among the leaves in the 1D gene phylogeny) to compare the diversity of the two genogroups. The entropy analysis (Fig. 4A) showed a greater number of variable amino acid sites in genogroup A (n = 14) than in genogroup B (n = 3). More than 50% of the variable sites in genogroup A clustered between amino acid positions 75 and 108. This portion of the VP1 protein includes two β sheets (B and C) and two amino acid loops, those connecting β sheets B and C and β sheets C and D (Fig. 4B). We found phylogenetic evidence of an evolutionary role in the 1DVP1 phylogeny for 15/17 variable sites (see below and Table 3). The evolutionary pathway of the VP1 protein was investigated by estimating the ancestral amino acid states of nodes within the 1DVP1 phylogeny using the ancestral state reconstruction method implemented in HyPhy (42). The method allowed the reconstruction of nonsynonymous substitutions across the lineages of the two CVB5 genogroups. Nineteen nonsynonymous changes were reconstructed along the backbone of the 1DVP1 phylogeny (Table 3). Within this set of 19 substitutions, 12 occurred at nodes specifying the tree topology of genogroup A, including nodes on the secondary branches specifying the individual subgenogroups (Fig. 1). The other set of seven substitutions occurred only on basal branches within the tree topology of genogroup B (Fig. 1). Excluding reversions at VP1 sites 273 and 279, amino acid changes at 10 codon sites (positions 13, 19, 75, 85, 95, 108, 248, 272, 273, and 275) were reconstructed at 27 different nodes, indicating parallel substitutions (Table 3). At seven sites, the substitutions eventually resulted in similar amino acid changes in different lineages of genogroups A and B: R13P, Y75F, D85N, S95N, M108L, G273S, and T275T. Parallel nonsynonymous substitutions resulted in different amino acid residues among lineages at codon sites G19E/S, M108L/I, E272K/D, and G273S/H/A. Parallel substitutions at codon sites 19, 95, 108, 272, and 273 occurred in different lineages of the two genogroups.
Fig 4.

Amino acid changes in the VP1 capsid protein of CVB5. (A) VP1 protein variability of genogroups A and B. Variability was calculated as the entropy for each amino acid position. The difference in entropies between the sequences of the two genogroups was calculated. Significant differences (P ≤ 0.05) are shown in red. (B) The alignment shows the VP1 amino acid sequences reconstructed at key ancestral nodes for the major CVB5 lineages and the homologous sequences of SVDV and CVB3. The CVB5 sequences were obtained with the ASR method (48). The sequence reconstructed for the root of the CVB5 tree is used as the reference, and dots indicate amino acid similarities with the reference. CVB5-B nodes 1 and 2 are indicated in Fig. 1. Helices and strands determined in the 3D structures of SVDV (52, 53) and CVB3 (54), assigned manually, are labeled and represented by different symbols: ∼ and #, respectively. Residues located on the external surface of particles of both SVDV and CVB3 are shown by asterisks. The SVDV antigenic sites are surrounded by dashed lines and include residues 83 and 84 (site 1), residue 256 (site IV), and residue 273 (site 3A) (50–52).
Table 3.
Evolutionarily important and parallel amino acid changes reconstructed for the CVB5 lineages with the 1DVP1 gene genealogya
| VP1 amino acid residueb |
Ancestral residuec | Amino acid change(s) within CVB5 lineages and subgenogroupsd |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Position | Location | A1 | A234 | A2 | A3 | A4 | B012 | B0 | B1 | B2 | |
| 7 | N terminus | V | V7I | I7V (4) | |||||||
| 13 | N terminus | R | R13P | R13P | |||||||
| 19 | N terminus | G | G19E | G19S | |||||||
| 45 | N terminus | A | A45S | ||||||||
| 75 | βB strand (+) | Y | Y75F | F75Y | |||||||
| 85 | B-C loop, Ag site | D | D85N | ||||||||
| 89 | βC strand (+) | F | F89I | ||||||||
| 90 | βC strand (+) | A | A90G | ||||||||
| 95 | C-D loop | S | S95N (2) | S95N | S95N | S95N | |||||
| 99 | C-D loop | V | V99A | ||||||||
| 108 | αA-βD junction | M | M108L | M108L, M108I | M108L | ||||||
| 156 | E-F loop (+) | V | V156I | ||||||||
| 180 | βG1 strand | M | M180I | ||||||||
| 248 | βI strand | V | V248I (2) | ||||||||
| 272 | C terminus (+) | E | E272K | E272D | |||||||
| 273 | C terminus (+), Ag site | G | G273S | G273S | G273H | G273A, S273G | |||||
| 275 | C terminus (+) | T | T275A | T275A | |||||||
| 276 | C terminus (+) | D | D276E | ||||||||
| 279 | C terminus (+) | T | T279A | A279T | A279T | ||||||
Nonsynonymous substitutions were determined at key nodes of the CVB5 phylogeny with the ancestral state reconstruction (ASR) method with the Datamonkey web facility (42).
The 15 positions indicated in bold were subject to amino acid changes. These changes occurred along the backbone or the secondary branches of the CVB5 phylogeny, and they determined the evolutionary pathway of lineages. The structural features indicated were reported for other enteroviruses, in particular, swine vesicular disease virus (SVDV) and coxsackievirus B3 (CVB3), as described earlier (52, 53, 54). +, amino acid positions exposed at the virion surface (see also figure 4B); Ag, antigenic.
The ancestral states of the amino acid residues reconstructed for the root of the CVB5 phylogeny. All the substitutions indicated are given relative to the ancestral state.
The amino acid changes were reconstructed at different nodes within CVB5 subgenogroups A1 to A4 and B0 to B2. The changes are also given at two important nodes, of which one (named A234) is common to subgenogroups A2, A3, and A4 and the other (B012) is common to all subgenogroups B. The majority of amino acid changes were estimated once in the phylogeny, with three exceptions, as indicated by the numbers in parentheses.
Relationships between evolutionary changes in the CVB5 phylogenetic backbone and antigenic features.
The antigenic sites and the virus 3D structure are unknown in CVB5, but the antigenic sites of the surface proteins of swine vesicular disease virus (SVDV), a close relative of human CVB5 (49), have been investigated in great detail by analyses of monoclonal antibody-resistant mutants (50, 51). Seven neutralization sites named 1, 2A, 2B, 3A, 3B, 3C, and IV have been located in structures exposed at the SVDV surface, of which sites 1, 3A, and IV are located in VP1. The 3D structure of SVDV and of the closely related EV type coxsackievirus B3 (CVB3) has been determined (52–54). We identified antigenically significant amino acid residues in the VP1 proteins of CVB5 lineages by sequence comparison with SVDV and CVB3 (Fig. 4). A number of nonsynonymous changes that had been selected during the transmission of CVB5 strains determined the evolutionary pathway of genogroups A and B, as they were found in or near two antigenic sites described in other picornaviruses. The evolution of subgenogroups A2, A3, and A4 was determined by selection of amino acid variations within or near SVDV antigenic sites 1 (D85N) and 3A (E272K and G273S). The evolution of genogroup B was shaped by changes selected at three residues within or near antigenic site 3A (G273S, D276E, and T279A). Antigenic site 1 is located within the B-C loop of VP1, a structure exposed near the 5-fold axis of the EV particles (6, 52, 54). Antigenic site 3A is made up of residues within the VP1 C terminus, which is located on the virus top surface. The VP1 C terminus flanks the outer surface of protein VP2 and partly supports its main projection. The amino acid change V156I in CVB5 genogroup B also occurred in an exposed residue that does not belong to a known antigenic site in SVDV. The lineages of genogroup B exhibit four major antigenic variations compared with the sequences of the A2 and A3 subgenogroups.
DISCUSSION
The data presented in this study are, to our knowledge, the first detailed description of population dynamics of two genogroups of EVs estimated in an investigation combining analyses of phylogenies and selection. The distinct phylodynamic patterns of the two CVB5 genogroups are remarkably reminiscent of the phylogenetic differences between influenza A and B viruses (55). The phylodynamic pattern of CVB5, particularly that of genogroup B, is caused by epochal evolution, as described earlier in influenza A virus (47), but the CVB5 pattern arises as a consequence of sporadic positive immune selection of circulating strains, which is different from the pattern of influenza A virus (56). Here, the 1DVP1 gene genealogy indicated that genogroup A (tMRCA = 1908) exhibited substantial conservation of ancestral states in molecular characteristics. It was also marked by an increased diversification into multiple lineages in a recent period: subgenogroups A1 (tMRCA = 1992), A3 (tMRCA = 1994), and A4 (tMRCA = 2001). The occurrence of a second genogroup B in 1941 represented a significant population split in CVB5, which suggested that the occurrence of amino acid changes in the VP1 capsid protein allowed the novel strain to evade the host immunity induced by its predecessor. Afterwards, the two genogroups coexisted through distinct evolutionary histories, as estimated by the trees inferred with the 1DVP1 gene and 3CD locus. Multiple consistent lineages were inferred from the 3CD genealogy, allowing a fine division of CVB5 into subgenogroups.
Main epidemiological factors of CVB5 transmission in Europe.
The population genetic history of CVB5 essentially depicts the spread of infections in Europe from the early 1990s to 2011 and shows three main transmission features. First, the spread of CVB5 strains resulted in a complex epidemic pattern across European countries. Taking into account the uncertainties of tMRCA estimates, the CVB5 epidemics occurred 4 to 11 years after the estimated divergence dates. As the temporal occurrence of epidemics is not directly related to the divergence date of the virus involved, this indicates that the availability of a large number of susceptible individuals is a major factor (57). The distinctive temporal increases in genetic diversity inferred for the two genogroups were consistent with the infection rates reported by public health agencies in France (this study) and the United States (13). For instance, the EV report for the United States indicated a singular CVB5 epidemic pattern with sharp increases at 3- to 6-year intervals. Our data also emphasize the ability of coalescence-based models (41) to depict the complex epidemic patterns of EV infections from genetic data, as reported earlier for other widespread RNA viruses (58).
A second feature of our investigation is the interepidemic circulation of CVB5 in Europe, as reflected by the maintenance of low levels of genetic diversity between the estimated expansion stages and monophyletic clusters including European viruses sampled over long periods. Multiple factors are associated with the long-term circulation of virus strains (59). In CVB5, the maintenance of sustained interindividual transmission chains over countries, shown by the extensive geographical mixing in phylogenetic trees, is a likely consequence of the social and economic exchanges between European countries (60). We earlier reported the circulation of EV strains in the winter season (61), a factor which can also contribute to maintenance of virus diffusion. Despite substantial epidemic shifts and troughs, the overall genetic diversity of genogroup B increased from 1991 to 2011. For the earlier time period, we cannot exclude the possibility that the dynamics of genogroup B was affected by sampling bias. The phylodynamic pattern may also indicate a low transmission rate of genogroup B virus strains across European countries until the late 1990s as a consequence of herd immunity against genogroup A viruses over decades of earlier circulation. Of note, the two genogroups exhibited similar interepidemic genetic diversity by the end of the study period.
A third important feature of CVB5 transmission is the cocirculation in Europe of virus strains of multiple subpopulations, which resulted in an alternation of the prevailing genogroup involved in the epidemic waves. The temporal occurrence and the alternation of genogroups suggest the involvement of a nonrandom determinant. The phylodynamic patterns of other RNA viruses characterized by strong temporal structure and rapid strain turnover (59) indicate partial cross immunity between strains. This hypothesis is also suggested by multistrain epidemiological models for echoviruses (62).
Molecular bases of the CVB5 evolutionary pathways in the VP1 protein.
We found contrasting patterns of nonsynonymous substitutions in the evolutionary dynamics of the two CVB5 genogroups. First, these substitutions operated on different codon sets, with the consequence that amino acid changes were distributed in distinct parts of the VP1 protein between the two genogroups. The only common evolutionary event was the amino acid change G273S (at the VP1 C terminus). This pattern suggested that amino acid toggling at specific positions played a limited role in CVB5 evolution and transmission over time, in contrast to earlier data reported for EV71 subgenogroup C1 (21). Second, the nonsynonymous substitutions were not distributed uniformly across the tree topologies of genogroups A and B, and these variations are unlikely to result from substantial differences in their nucleotide substitution rates. In population genetics, the nonsynonymous substitutions that usually shape the main backbone lineages are particularly important because they represent mutations fixed during the evolutionary pathway (60). Accordingly, the strains which have the genetic features of the backbone lineage compose the successful virus population effectively propagating over time, in contrast to populations at the terminal lineages of a phylogenetic tree (see below), and positive selection is more likely to be detected among these strains. The surviving lineage of genogroup A was shaped by amino acid changes at nine codon sites, which concurred to produce changes in the VP1 capsid protein (V7I, A45S, Y75F, N85D, F89I, A90G, V99A, E272K, and G273S). Each of the individual subgenogroups A was characterized by at least one of these changes. The backbone lineage of genogroup B is determined by nonsynonymous substitutions at seven sites; five are located in the second half of the protein (V156I, M180I, G273S, D276E, and T279A), and three of these cluster in the C terminus and are exposed on the virus surface. Notably, all the corresponding nonsynonymous substitutions were reconstructed in the oldest part of the backbone in this genogroup, i.e., 50 to 70 years ago, which indicates that the most recent part of the lineage has evolved only through the accumulation of synonymous substitutions. However, nonsynonymous substitutions become relatively more common among the terminal lineages—short-lived virus populations—of the CVB5 phylogenetic tree. Our analysis of the amino acid variability in the CVB5 VP1 protein is in agreement with this known feature of RNA virus evolution, suggesting that most nucleotide substitutions are deleterious in terminal lineages (63). In addition, we provide consistent evidence that genogroup A displays a higher diversity than genogroup B because of a greater number of variable amino acid sites. Overall, our data indicate that genogroup A is still evolving at neutralizing antigenic epitopes, while genogroup B has been experiencing a long antigenic stasis over the last 50 years.
The outstanding effect of CVB5 in causing recurrent annual and seasonal epidemics of aseptic meningitis in Europe is due to the changing prevalence between sequential lineages of two genogroups. Immune selection on distinct neutralizing antigenic sites may play an important role in the temporal variations in virus spread. This hypothesis is lent support by the occurrence of genogroup B as a significant epidemic EV (while genogroup A was already established as a widespread pathogen) and the alternate epidemic pattern observed in Europe over the last 20 years. Both features suggest that genogroup B virus strains acquired increased properties for epidemic spread against genogroup A through partial cross immunity. The resulting antigenic differences generated partial cross immunity against the genogroups, with the evolutionary consequence that a nonoverlapping virus strain structure arose over time.
Supplementary Material
ACKNOWLEDGMENTS
We are grateful to Hartwig P. Huemer (Vienna, Austria), Agnes Farkas (Budapest, Hungary), Clara Larcher (Bolzano, Italy), Patrice Morand and Christine Morel-Baccard (Grenoble, France), Jacques Izopet and Jean-Michel Mansuy (Toulouse, France), and Stéphanie Marque-Juillet (Versailles, France) for providing us with a number of the virus samples used in this study. We are indebted to Gisela Enders, director of the Laboratory Enders & Partners, Stuttgart, Germany, for help and involvement in our European molecular epidemiology studies. We thank Denise Antona (Institut National de Veille Sanitaire, France) for providing us with data compiled from the French Network for Enterovirus Surveillance between 2000 and 2010. We acknowledge the support of Antoine Mahul (Centre Interuniversitaire de Ressources Informatiques, Aubière, France) for assistance with the most computer-intensive analyses of large sequence data sets and the technical contribution of Gwendoline Jugie, Nathalie Rodde, and Isabelle Simon for helpful assistance with virus culture and sequencing. We thank Jeffrey Watts for help with preparing the English manuscript.
Footnotes
Published ahead of print 4 September 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JVI.02075-13.
REFERENCES
- 1.Michos AG, Syriopoulou VP, Hadjichristodoulou C, Daikos GL, Lagona E, Douridas P, Mostrou G, Theodoridou M. 2007. Aseptic meningitis in children: analysis of 506 cases. PLoS One 2:e674. 10.1371/journal.pone.0000674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ho M, Chen ER, Hsu KH, Twu SJ, Chen KT, Tsai SF, Wang JR, Shi SR. 1999. An epidemic of enterovirus 71 infection in Taiwan. N. Engl. J. Med. 341:929–935 [DOI] [PubMed] [Google Scholar]
- 3.Louie JK, Hacker JK, Gonzales R, Mark J, Maselli JH, Yagi S, Drew WL. 2005. Characterization of viral agents causing acute respiratory infection in a San Francisco University Medical Center clinic during the influenza season. Clin. Infect. Dis. 41:822–828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nathanson N. 2008. The pathogenesis of poliomyelitis: what we don't know. Adv. Virus Res. 71:1–50 [DOI] [PubMed] [Google Scholar]
- 5.Zhang Y, Tan XJ, Wang HY, Yan DM, Zhu SL, Wang DY, Ji F, Wang XJ, Gao YJ, Chen L, An HQ, Li DX, Wang SW, Xu AQ, Wang ZJ, Xu WB. 2009. An outbreak of hand, foot, and mouth disease associated with subgenotype C4 of human enterovirus 71 in Shandong, China. J. Clin. Virol. 44:262–267 [DOI] [PubMed] [Google Scholar]
- 6.Hogle JM, Chow M, Filman DJ. 1985. Three-dimensional structure of poliovirus at 2.9 Å resolution. Science 229:1358–1365 [DOI] [PubMed] [Google Scholar]
- 7.Mateu MG. 1995. Antibody recognition of picornaviruses and escape from neutralization: a structural view. Virus Res. 38:1–24 [DOI] [PubMed] [Google Scholar]
- 8.Duintjer Tebbens RJ, Pallansch MA, Chumakov KM, Halsey NA, Hovi T, Minor PD, Modlin JF, Patriarca PA, Sutter RW, Wright PF, Wassilak SG, Cochi SL, Kim JH, Thompson KM. 2013. Expert review on poliovirus immunity and transmission. Risk Anal. 33:544–605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stanway G, Brown P, Christian P, Hovi T, Hyypiä T, King NJ, Knowles NJ, Lemon SM, Minor PD, Pallansch MA, Palmemberg AC, Skern T. 2005. Family Picornaviridae, p 757–778 In Fauquet CM, Mayo MA, Maniloff J, Desselberger U, Ball LA. (ed), Virus taxonomy. Eighth report of the International Committee on Taxonomy of Viruses Elsevier/Academic Press, London, UK [Google Scholar]
- 10.Oberste MS, Maher K, Kennett ML, Campbell JJ, Carpenter MS, Schnurr D, Pallansch MA. 1999. Molecular epidemiology and genetic diversity of echovirus type 30 (E30): genotypes correlate with temporal dynamics of E30 isolation. J. Clin. Microbiol. 37:3928–3933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Santti J, Harvala H, Kinnunnen L, Hyypia T. 2000. Molecular epidemiology and evolution of coxsackievirus A9. J. Gen. Virol. 81:1361–1372 [DOI] [PubMed] [Google Scholar]
- 12.Podin Y, Gias EL, Ong F, Leong YW, Yee SF, Yusof MA, Perera D, Teo B, Wee TY, Yao SC, Yao SK, Kiyu A, Arif MT, Cardosa MJ. 2006. Sentinel surveillance for human enterovirus 71 in Sarawak, Malaysia: lessons from the first 7 years. BMC Public Health 6:180. 10.1186/1471-2458-6-180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Khetsuriani N, Lamonte-Fowlkes A, Oberste S, Pallansch MA, Centers for Disease Control and Prevention 2006. Enterovirus surveillance—United States, 1970-2005. MMWR Surveill. Summ. 55():1–20 [PubMed] [Google Scholar]
- 14.Antona D, Lévêque N, Chomel JJ, Dubrou S, Lévy-Bruhl D, Lina B. 2007. Surveillance of enteroviruses in France, 2000-2004. Eur. J. Clin. Microbiol. Infect. Dis. 26:403–412 [DOI] [PubMed] [Google Scholar]
- 15.Tully DC, Fares MA. 2008. The tale of a modern animal plague: tracing the evolutionary history and determining the time-scale for foot and mouth disease virus. Virology 382:250–256 [DOI] [PubMed] [Google Scholar]
- 16.Guan D, van der Sanden S, Zeng H, Li W, Zheng H, Ma C, Su J, Liu Z, Guo X, Zhang X, Liu L, Koopmans M, Ke C. 2012. Population dynamics and genetic diversity of C4 strains of human enterovirus 71 in Mainland China, 1998-2010. PLoS One 7:e44386. 10.1371/journal.pone.0044386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bailly JL, Mirand A, Henquell C, Archimbaud C, Chambon M, Charbonné F, Traoré O, Peigue-Lafeuille H. 2009. Phylogeography of circulating populations of human echovirus 30 over 50 years: nucleotide polymorphism and signature of purifying selection in the VP1 capsid protein gene. Infect. Genet. Evol. 9:699–708 [DOI] [PubMed] [Google Scholar]
- 18.van der Sanden S, van der Avoort H, Lemey P, Uslu G, Koopmans M. 2010. Evolutionary trajectory of the VP1 gene of human enterovirus 71 genogroup B and C viruses. J. Gen. Virol. 91:1949–1958 [DOI] [PubMed] [Google Scholar]
- 19.Bedford T, Cobey S, Beerli P, Pascual M. 2010. Global migration dynamics underlie evolution and persistence of human influenza A (H3N2). PLoS Pathog. 6:e1000918. 10.1371/journal.ppat.1000918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Haydon DT, Bastos AD, Knowles NJ, Samuel AR. 2001. Evidence for positive selection in foot-and-mouth disease virus capsid genes from field isolates. Genetics 157:7–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tee KK, Lam TTY, Chan YF, Bible JM, Kamarulzaman A, Tong CYW, Takebe Y, Pybus OG. 2010. Evolutionary genetics of human enterovirus 71: origin, population dynamics, natural selection and seasonal periodicity of the VP1 gene. J. Virol. 84:3339–3350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kistler AL, Webster DR, Rouskin S, Magrini V, Credle JJ, Schnurr DP, Boushey HA, Mardis ER, Li H, DeRisi JL. 2007. Genome-wide diversity and selective pressure in the human rhinovirus. Virol. J. 4:40. 10.1186/1743-422X-4-40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lewis-Rogers N, Bendall ML, Crandall KA. 2009. Phylogenetic relationships and molecular adaptation dynamics of human rhinoviruses. Mol. Biol. Evol. 26:969–981 [DOI] [PubMed] [Google Scholar]
- 24.Tracy S, Gauntt C. 2008. Group B coxsackievirus virulence. Curr. Top. Microbiol. Immunol. 323:49–63 [DOI] [PubMed] [Google Scholar]
- 25.Wikswo ME, Khetsuriani N, Fowlkes AL, Zheng X, Peñaranda S, Verma N, Shulman ST, Sircar K, Robinson CC, Schmidt T, Schnurr D, Oberste MS. 2009. Increased activity of coxsackievirus B1 strains associated with severe disease among young infants in the United States, 2007-2008. Clin. Infect. Dis. 49:e44–e51 [DOI] [PubMed] [Google Scholar]
- 26.Roth B, Enders M, Arents A, Pfitzner A, Terletskaia-Ladwig E. 2007. Epidemiologic aspects and laboratory features of enterovirus infections in Western Germany, 2000-2005. J. Med. Virol. 79:956–962 [DOI] [PubMed] [Google Scholar]
- 27.Trallero G, Avellon A, Otero A, De Miguel T, Pérez C, Rabella N, Rubio G, Echevarria JE, Cabrerizo M. 2010. Enteroviruses in Spain over the decade 1998-2007: virological and epidemiological studies. J. Clin. Virol. 47:170–176 [DOI] [PubMed] [Google Scholar]
- 28.Han JF, Jiang T, Fan XL, Yang LM, Yu M, Cao RY, Wang JZ, Qin ED, Qin CF. 2012. Recombination of human coxsackievirus B5 in hand, foot, and mouth disease patients, China. Emerg. Infect. Dis. 18:351–353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rezig D, Fares W, Seghier M, Yahia AB, Touzi H, Triki H. 2011. Update on molecular characterization of coxsackievirus B5 strains. J. Med. Virol. 83:1247–1254 [DOI] [PubMed] [Google Scholar]
- 30.Baek K, Yeo S, Lee B, Park K, Song J, Yu J, Rheem I, Kim J, Hwang S, Choi Y, Cheon D, Park J. 2011. Epidemics of enterovirus infection in Chungnam Korea, 2008 and 2009. Virol. J. 8:297. 10.1186/1743-422X-8-297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gullberg M, Tolf C, Jonsson N, Mulders MN, Savolainen-Kopra C, Hovi T, Van Ranst M, Lemey P, Hafenstein S, Lindberg AM. 2010. Characterization of a putative ancestor of coxsackievirus B5. J. Virol. 84:9695–9708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bailly JL, Mirand A, Henquell C, Archimbaud C, Chambon M, Regagnon C, Charbonné F, Peigue-Lafeuille H. 2011. Repeated genomic transfers from echovirus 30 to echovirus 6 lineages indicate co-divergence between co-circulating populations of the two human enterovirus serotypes. Infect. Genet. Evol. 11:276–289 [DOI] [PubMed] [Google Scholar]
- 33.Drummond AJ, Ho SY, Phillips MJ, Rambaut A. 2006. Relaxed phylogenetics and dating with confidence. PLoS Biol. 4:e88. 10.1371/journal.pbio.0040088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Drummond AJ, Rambaut A. 2007. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol. Biol. 7:214. 10.1186/1471-2148-7-214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Suchard MA, Weiss RE, Sinsheimer JS. 2001. Bayesian selection of continuous-time Markov chain evolutionary models. Mol. Biol. Evol. 18:1001–1013 [DOI] [PubMed] [Google Scholar]
- 36.Shapiro B, Rambaut A, Drummond AJ. 2006. Choosing appropriate substitution models for the phylogenetic analysis of protein-coding sequences. Mol. Biol. Evol. 23:7–9 [DOI] [PubMed] [Google Scholar]
- 37.Kass RE, Raftery AE. 1995. Bayes factors and model uncertainty. J. Am. Stat. Assoc. 90:773–795 [Google Scholar]
- 38.Minin VN, Bloomquist EW, Suchard MA. 2008. Smooth skyride through a rough skyline: Bayesian coalescent-based inference of population dynamics. Mol. Biol. Evol. 25:1459–1471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Stadler T, Kouyos R, von Wyl V, Yerly S, Böni J, Bürgisser P, Klimkait T, Joos B, Rieder P, Xie D, Günthard HF, Drummond AJ, Bonhoeffer S, Swiss HIV Cohort Study 2012. Estimating the basic reproductive number from viral sequence data. Mol. Biol. Evol. 29:347–357 [DOI] [PubMed] [Google Scholar]
- 40.Baele G, Lemey P, Bedford T, Rambaut A, Suchard MA, Alekseyenko AV. 2012. Improving the accuracy of demographic and molecular clock model comparison while accommodating phylogenetic uncertainty. Mol. Biol. Evol. 29:2157–2167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Drummond AJ, Rambaut A, Shapiro B, Pybus OG. 2005. Bayesian coalescent inference of past population dynamics from molecular sequences. Mol. Biol. Evol. 22:1185–1192 [DOI] [PubMed] [Google Scholar]
- 42.Kosakovsky Pond SL, Frost SD. 2005. Not so different after all: a comparison of methods for detecting amino acid sites under selection. Mol. Biol. Evol. 22:1208–1222 [DOI] [PubMed] [Google Scholar]
- 43.Murrell B, Moola S, Mabona A, Weighill T, Sheward D, Kosakovsky Pond SL, Scheffler K. 2013. FUBAR: A Fast, Unconstrained Bayesian AppRoximation for inferring selection. Mol. Biol. Evol. 30:1196–1202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pupko T, Pe'er I, Shamir R, Graur D. 2000. A fast algorithm for joint reconstruction of ancestral amino acid sequences. Mol. Biol. Evol. 17:890–896 [DOI] [PubMed] [Google Scholar]
- 45.Yang Z, Kumar S, Nei M. 1995. A new method of inference of ancestral nucleotide and amino acid sequences. Genetics 141:1641–1650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nielsen R. 2002. Mapping mutations on phylogenies. Syst. Biol. 51:729–739 [DOI] [PubMed] [Google Scholar]
- 47.Koelle K, Cobey S, Grenfell B, Pascual M. 2006. Epochal evolution shapes the phylodynamics of interpandemic influenza A (H3N2) in humans. Science 314:1898–1903 [DOI] [PubMed] [Google Scholar]
- 48.Kosakovsky Pond SL, Murrell B, Fourment M, Frost SD, Delport W, Scheffler K. 2011. A random effects branch-site model for detecting episodic diversifying selection. Mol. Biol. Evol. 28:3033–3043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Brown F, Talbot P, Burrows R. 1973. Antigenic differences between isolates of swine vesicular disease virus and their relationships to coxsackie B5 virus. Nature 245:315–316 [DOI] [PubMed] [Google Scholar]
- 50.Borrego B, Carra E, García-Ranea JA, Brocchi E. 2002. Characterization of neutralization sites on the circulating variant of swine vesicular disease virus (SVDV): a new site is shared by SVDV and the related coxsackie B5 virus. J. Gen. Virol. 83:35–44 [DOI] [PubMed] [Google Scholar]
- 51.Borrego B, García-Ranea JA, Douglas A, Brocchi E. 2002. Mapping of linear epitopes on the capsid proteins of swine vesicular disease virus using monoclonal antibodies. J. Gen. Virol. 83:1387–1395 [DOI] [PubMed] [Google Scholar]
- 52.Verdaguer N, Jimenez-Clavero MA, Fita I, Ley V. 2003. Structure of swine vesicular disease virus: mapping of changes occurring during adaptation of human coxsackie B5 virus to infect swine. J. Virol. 77:9780–9789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Fry EE, Knowles NJ, Newman JW, Wilsden G, Rao Z, King AM, Stuart DI. 2003. Crystal structure of swine vesicular disease virus and implications for host adaptation. J. Virol. 77:5475–5486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Muckelbauer JK, Kremer M, Minor I, Diana G, Dutko FJ, Groarke J, Pevear DC, Rossmann MG. 1995. The structure of coxsackievirus B3 at 3.5 Å resolution. Structure 3:653–667 [DOI] [PubMed] [Google Scholar]
- 55.Ferguson NM, Galvani AP, Bush RM. 2003. Ecological and immunological determinants of influenza evolution. Nature 422:428–433 [DOI] [PubMed] [Google Scholar]
- 56.Smith DJ, Lapedes AS, de Jong JC, Bestebroer TM, Rimmelzwaan GF, Osterhaus AD, Fouchier RA. 2004. Mapping the antigenic and genetic evolution of influenza virus. Science 305:371–376 [DOI] [PubMed] [Google Scholar]
- 57.Woolhouse ME. 2002. Population biology of emerging and re-emerging pathogens. Trends Microbiol. 10:S3–S7 [DOI] [PubMed] [Google Scholar]
- 58.Allicock OM, Lemey P, Tatem AJ, Pybus OG, Bennett SN, Mueller BA, Suchard MA, Foster JE, Rambaut A, Carrington CV. 2012. Phylogeography and population dynamics of dengue viruses in the Americas. Mol. Biol. Evol. 29:1533–1543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Grenfell BT, Pybus OG, Gog JR, Wood JL, Daly JM, Mumford JA, Holmes EC. 2004. Unifying the epidemiological and evolutionary dynamics of pathogens. Science 303:327–332 [DOI] [PubMed] [Google Scholar]
- 60.Pybus OG, Rambaut A. 2009. Evolutionary analysis of the dynamics of viral infectious disease. Nat. Rev. Genet. 10:540–550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chambon M, Archimbaud C, Bailly JL, Henquell C, Regagnon C, Charbonné F, Peigue-Lafeuille H. 2001. Circulation of enteroviruses and persistence of meningitis cases in the winter of 1999-2000. J. Med. Virol. 65:340–347 [DOI] [PubMed] [Google Scholar]
- 62.Kamo M, Sasaki A. 2002. The effect of cross-immunity and seasonal forcing in a multi-strain epidemic model. Physica D 165:228–241 [Google Scholar]
- 63.Pybus OG, Rambaut A, Belshaw R, Freckleton RP, Drummond AJ, Holmes EC. 2007. Phylogenetic evidence for deleterious mutation load in RNA viruses and its contribution to viral evolution. Mol. Biol. Evol. 24:845–852 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.