

Abstract
Autophagy is an important innate safeguard mechanism for protecting an organism against invasion by pathogens. We have previously discovered that Kaposi's sarcoma-associated herpesvirus (KSHV) evades this host defense through tight suppression of autophagy by targeting multiple steps of autophagy signal transduction. Here, we report that KSHV K7 protein interacts with Rubicon autophagy protein and inhibits the autophagosome maturation step by blocking Vps34 enzymatic activity, further highlighting how KSHV deregulates autophagy-mediated host immunity for its life cycle.
TEXT
Autophagy is an important homeostatic mechanism involving the formation of double-membrane vesicles, called autophagosomes, which sequester damaged cytoplasmic organelles, protein aggregates, or invading intracellular pathogens for degradation. Conserved from Saccharomyces cerevisiae to humans, autophagy takes place through a series of steps that include vesicle initiation, nucleation, and elongation, followed by vesicle fusion with lysosomes for the cargo degradation (1). This intracellular catabolic degradation system is tightly controlled by autophagy-related genes (Atg), which can initiate or suppress steps in the autophagy pathway in order to maintain cellular homeostasis (2). The serine/threonine kinase mammalian target of rapamycin (mTOR) is an important regulator of autophagy. Under normal conditions, mTOR represses autophagy induction by phosphorylating Unc-like kinase 1 and 2 (ULK1/2) (3). Nutrient starvation conditions or treatment with the mTOR inhibitor rapamycin impedes mTOR kinase activity, leading to autophagy initiation and nucleation of a phagophore membrane. During the initiation step of autophagy, Beclin 1 forms a complex with Vps34, a class III phosphoinositide 3-kinase (C3 PI 3-kinase), which contributes to autophagosome nucleation (4). On the other hand, cellular Bcl-2 constitutively binds to Beclin 1 and blocks this autophagosome nucleation (5). During the elongation step of autophagy, light chain 3 (LC3-I) is proteolytically processed by Atg3/7 enzymes and conjugated with a lipidated phosphatidylethanolamine (PE) via a ubiquitin-like conjugation system. Lipidated LC3-II can serve as a marker for autophagosome formation since LC3-II is embedded within the lumen of the autophagosome (6). As a regulatory mechanism, cellular FLIP targets Atg3 E2 enzyme to block autophagosome elongation (7). Autophagosomes subsequently undergo a maturation step by fusion with endosomes or lysosomes. The Beclin 1/Vps34/UVRAG complex positively contributes to autophagosome maturation (8) and endocytic trafficking (9), while these processes are inhibited by Rubicon interaction (10, 11). Finally, the acidic environment in autolysosomes ultimately degrades the cargo by lysosomal hydrolysis.
Kaposi's sarcoma-associated herpesvirus (KSHV; human herpesvirus 8 [HHV-8]) belongs to the gammaherpesvirus family, which includes Epstein-Barr virus (EBV), herpesvirus saimiri (HSV), and murine gammaherpesvirus 68 (MHV-68) (12). KSHV is etiologically linked to Kaposi's sarcoma (KS) as well as two rare B-cell proliferative diseases, primary effusion lymphoma (PEL) and multicentric Castleman's disease (MCD) (13). Recent studies have broadened our understanding of the mechanisms by which herpesviruses modulate autophagy machinery and cellular innate immune responses (2, 14–19). For instance, vBcl-2 interacts with Beclin 1 complex to downregulate autophagy at the vesicle nucleation step (20), and vFLIP suppresses autophagy at the vesicle elongation step by preventing Atg3 E2 enzyme from binding and processing LC3 (7). To further explore how KSHV modulates cellular autophagy machinery at the autophagosome maturation step, we performed a yeast two-hybrid screen using the full-length Rubicon autophagy maturation inhibitor protein as bait to screen the KSHV cDNA library and found an interaction with KSHV K7 in yeast. KSHV K7 lytic protein has been shown to be able to protect cells from apoptosis by various stimuli in vitro (21). K7 also suppresses endoplasmic reticulum (ER) stress-induced apoptosis by modulating intracellular calcium efflux (22). Coimmunoprecipitation verified the efficient binding between K7 and epitope-tagged Rubicon as well as between K7 and endogenous Rubicon (Fig. 1A; see Fig. 3B). Rubicon contains an amino-terminal RUN domain (RUN), a serine-rich region (SR-N), a central coiled-coil domain (CCD), a second serine-rich region (SR-C), a helix-coiled region (HC), and a carboxy-terminal cysteine-rich region (CR) (Fig. 1B). We constructed a series of Rubicon truncation mutants fused with mammalian glutathione S-transferase (GST) to define the region responsible for the K7 interaction. GST pulldown showed that the HC region of Rubicon was sufficient for K7 association (Fig. 1C), and thus the deletion of the HC domain abolished the K7 interaction (Fig. 1D). Furthermore, confocal analysis revealed the substantial colocalization of Rubicon and K7 in HeLa cells (Fig. 1E). Altogether, these data indicated that Rubicon interacts with K7 through its HC domain.
Fig 1.

KSHV K7 interacts with Rubicon. (A) Flag-Rubicon was transfected with vector or K7-V5 into HEK293T cells. Whole-cell lysates (WCL) were subjected to immunoprecipitation (IP) with Flag or V5 antibodies, followed by immunoblotting (IB) with antibodies as indicated. (B) Rubicon structure and its binding partners. (C) K7-V5 was transfected with vector or GST-tagged Rubicon mutants into HEK293T cells. WCL were subjected to GST pulldown (GST-PD), followed by IB with indicated antibodies. (D) K7-V5 was transfected with Flag-Rubicon or an HC deletion mutant into HEK293T cells. WCL were subjected to IP with anti-Flag antibody, followed by IB with Flag and V5 antibodies. (E) K7-V5 and Flag-Rubicon or the HC deletion mutant (ΔHC) were cotransfected into HeLa cells. Cells were fixed and stained with Flag and V5 antibodies at 36 h posttransfection, followed by confocal microscopy.
Fig 3.

K7 promotes Rubicon interactions with the Beclin 1/UVRAG/Vps34 complex. (A) HEK293T cells were cotransfected with K7-V5 with vector, Flag-Beclin 1, Flag-UVRAG, or Flag-Vps34. WCL were used for IP with Flag antibody, followed by IB with indicated antibodies. (B) WCL from HeLa-vector or HeLa-K7 stable cells were used for IP with Flag antibody, followed by IB with the indicated antibodies. (C) HEK293T cells were cotransfected with Rubicon-V5 and K7-GFP, Flag-Beclin 1, Flag-UVRAG, or Flag-Vps34 combinations as indicated. WCL were used for IP with Flag antibody, followed by IB with the indicated antibodies. (D) WCL from HeLa-vector or HeLa-K7 stable cells were used for IP with Rubicon antibody, followed by IB with the indicated antibodies.
Because Rubicon inhibits the autophagosome maturation step of autophagy, we first examined whether KSHV K7 plays a role in the autophagosome maturation step. Upon autophagy induction, soluble microtubule-associated protein light chain 3 (LC3-I) is converted to a lipidated form (LC3-II) that recruits the autophagic substrate p62/SQSTM to the autophagosome, and finally, p62/SQSTM protein undergoes degradation during autophagosome maturation (6). To monitor the effect of K7 expression on the conversion of LC3-I to LC3-II and p62 degradation, we generated HeLa cell lines containing vector or stably expressing K7 (Fig. 2A). To induce autophagy activation, HeLa cells were treated with rapamycin and probed for LC3-I/LC3-II and p62 levels (Fig. 2B). Rapamycin treatment increased LC3-II levels and decreased p62 levels in vector-containing HeLa cells, whereas it increased LC3-II levels without affecting p62 levels in K7-expressing HeLa cells (Fig. 2B), indicating that K7 expression has no effect on LC3 conversion (a marker for autophagosome formation) but blocks p62 degradation (a marker for autophagosome maturation). To further test whether K7 blocked the autophagosome maturation step, we monitored the formation of autophagosomes and autolysosomes using a fluorescent fusion protein, mRFP-GFP-LC3 (ptf-LC3) (where mRFP and GFP are monomeric red fluorescent protein and green fluorescent protein, respectively). Because GFP is more sensitive than RFP to the acidic environment of lysosomes, the tandem RFP-GFP-LC3 protein labels autophagosomes as yellow (GFP/RFP) puncta and autolysosomes as red (RFP) puncta, and thus, quantifications of yellow and red puncta represent the effect on autophagosome maturation (23). When this reporter was expressed in stable HeLa cell lines, the numbers of red puncta decreased approximately 2-fold in K7-expressing cells compared to those in vector control cells (Fig. 2C), suggesting that K7 has a role as a negative regulator of autophagosome maturation.
Fig 2.

K7 inhibits autophagosome maturation. (A) HeLa cells were transfected with a pIRES-puro empty vector or a pIRES-K7-Flag-puro construct. Cells were selected using 1.5 μg/ml puromycin for 2 weeks and subjected to IP with Flag antibody, followed by IB with Flag and actin antibodies to confirm the stable expression of K7. (B) HeLa-vector or HeLa-K7 stable cells were treated with 2 μM rapamycin, and WCL were collected at the indicated time points and subjected to IB. (C) HeLa-vector or HeLa-K7 stable cells were transfected with a ptf-LC3 construct and treated with 2 μM rapamycin at 24 h posttransfection. Cells were fixed after 4 h of rapamycin treatment and subjected to confocal microscopy analysis. A total of 100 cells were analyzed for quantification of autophagosomes (yellow puncta) and autolysosomes (red puncta) per cell.
Rubicon is a subunit of the Beclin 1/UVRAG/Vps34 autophagy complex (10, 11, 24). To investigate whether KSHV K7 blocks autophagosome maturation through its interaction with Rubicon, we examined the K7 interaction with the Rubicon/Beclin 1/Vps34 autophagy complex. K7 effectively bound to exogenously expressed Beclin 1, UVRAG, and Vps34 (Fig. 3A) as well as endogenous Rubicon, Beclin 1, and UVRAG (Fig. 3B). Because the interaction of Rubicon with the Beclin 1/UVRAG/Vps34 autophagy complex blocks Vps34 lipid kinase activity (24), we next determined whether K7 affected Rubicon interactions with the Beclin 1/UVRAG/Vps34 complex. Indeed, K7 expression led to increased levels of Rubicon interactions with Beclin 1, UVRAG, and Vps34 either in a transient expression (Fig. 3C) or in a stable expression (Fig. 3D), indicating that KSHV K7 binds to Rubicon, promoting Rubicon's interactions with the Beclin 1/UVRAG/Vps34 autophagy complex.
As the PX domain of p40(phox) specifically binds to phosphatidylinositol 3-phosphate (PtdIns-3-P) produced by Vps34 lipid kinase, the p40(phox) PX-eGFP fusion protein (where eGFP is enhanced GFP) was used as a noninvasive probe to measure intracellular PtdIns-3-P levels and distributions (25). p40(phox) PX-eGFP formed numerous punctate structures in HeLa-vector cells that appeared to be the PtdIns-3-P-rich vesicles (Fig. 4A). The intensity and number of p40(phox) PX-eGFP-stained vesicles were considerably lower in K7-expressing HeLa cells than in vector-containing HeLa cells (Fig. 4A), suggesting that K7 inhibits PtdIns-3-P production. To further confirm the K7-mediated inhibition of Vps34 enzymatic activity, we expressed K7 alone or K7 together with Rubicon and then measured the effects on Vps34 activity using an in vitro lipid kinase and PtdIns-3-P production assay. As shown previously (24), UVRAG expression readily increased Vps34 enzymatic activity (Fig. 4B). While individual expression of either Rubicon or K7 slightly reduced Vps34 activity, coexpression of K7 and Rubicon efficiently suppressed Vps34 activity (Fig. 4B), indicating that K7 interaction with Rubicon suppresses Vps34 kinase activity.
Fig 4.
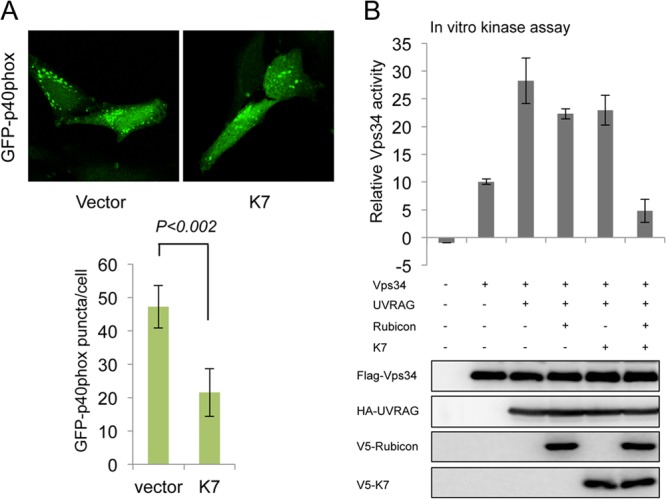
K7 inhibits Vps34 enzymatic activity. (A) HeLa-vector or HeLa-K7 stable cells were transfected with the p40(phox) PX-eGFP construct (GFP-p40phox). At 24 h posttransfection, cells were fixed, followed by confocal microscopy analysis. A total of 100 cells were analyzed for quantification of the p40(phox) PX puncta per cell. (B) Flag-Vps34 was coexpressed in HEK293T cells with hemagglutinin (HA)-UVRAG, V5-Rubicon, and/or V5-K7. WCL were subjected to IP with anti-Flag antibody and assayed for PI3K kinase activity by in vitro kinase assay (Echelon Biosciences Inc., Salt Lake City, UT). Vps34, UVRAG, Rubicon or K7 expressions were examined by IB with the indicated antibodies.
Having demonstrated a negative effect of K7 on autophagosome maturation, we next examined the role of K7 in KSHV lytic replication. Because we failed to generate antibody to detect K7 expression in the context of the KSHV genome, we introduced the 3×Flag epitope tag into the K7 C terminus using the new KSHV bacterial artificial chromosome BAC16 and were able to detect expression of K7 during lytic replication and K7 interaction with endogenous Rubicon (Fig. 5A and B). Next, we also introduced specific point mutations into the K7 start codon to generate the K7 knockout (KO) BAC16-GFP virus and then examined the effect of K7 on viral lytic replication (Fig. 5A). Wild-type (WT), K7-Flag, and KO BAC16-GFP viruses were reconstituted in iSLK cells, which contain a doxycycline-inducible RTA expression system stably integrated in the cellular chromosome (26). Upon doxycycline treatment, RTA expression is sufficient to initiate the lytic replication phase and culminates in the release of infectious virion particles from the cells. Following 4 days of doxycycline treatment, cell-free supernatants of iSLK cells were transferred to 293A cells and infectious virus was quantified by fluorescence-activated cell sorter (FACS) analysis of GFP expression at 24 h postinfection (Fig. 5C). The levels of infectious viruses for the WT and K7-Flag derivative viruses were comparable, but the K7 KO virus showed a slight reduction of GFP-positive numbers (Fig. 5C). When viral protein expression levels were analyzed in iSLK cells by immunoblotting, the K7 KO virus-infected cells showed small reductions of the K3 and K8 protein levels compared to the WT BAC16 virus-infected cells, whereas these K3 and K8 protein levels were recovered by the complementation of K7 in iSLK-BAC16-K7 KO cells (Fig. 5D and E). However, as shown in Fig. 5D, the K7 KO virus-infected cells showed a lower level of p62 during lytic replication than the WT virus-infected cells. In summary, our results indicate that the KSHV K7 interacts with Rubicon and this interaction facilitates Rubicon function to block the autophagosome maturation. However, since vBcl-2 and vFLIP effectively mitigate autophagy-mediated innate immunity during the KSHV life cycle, the absence of the K7 gene might lead to only a minor effect on viral lytic replication and viral production in culture. Nevertheless, this indicates that KSHV has uniquely evolved to carry vBcl-2, vFLIP, and K7 genes that inhibit various steps of the autophagy pathway, ultimately contributing to the virus life cycle (Fig. 5F).
Fig 5.

Recombinant K7 KSHV construction and K7 role in viral replication and autophagy. (A) BAC16-K7-Flag and BAC16-K7-KO were generated as previous described (27). DNAs isolated from overnight culture were digested by NheI and subjected to gel electrophoresis (left). The mutations of BAC16 clones were confirmed by DNA sequencing (right). (B) HEK293T-BAC16 or HEK293T-BAC16-K7-Flag cells were induced by 12-O-tetradecanoylphorbol-13-acetate (TPA; 20 ng/ml) and sodium butyrate (0.3 mM) for 72 h, WCL were collected and used for IP and IB with the indicated antibodies. (C) iSLK-BAC16 (BAC16-WT), iSLK-BAC16-K7-Flag, or iSLK-BAC16-K7-KO cells were induced by doxycycline (1 μg/ml) for 4 days, and the supernatants were harvested and used for infection in HEK293A cells. At 24 h postinfection, cells were analyzed by fluorescence microscopy and the infectious units were quantified by FACS analysis. (D) iSLK-BAC16 or iSLK-BAC16-K7 KO cells were induced by doxycycline and WCL were collected at 0, 12, 24, and 48 h postinduction and subjected to IB with the indicated antibodies. (E) iSLK-BAC16 (WT) or iSLC-BAC16-K7 KO cells were complemented with empty vector or V5-tagged K7. At 36 h postcomplementation, these cells were induced by doxycycline and WCL were collected at 48 h postinduction and subjected to IB with the indicated antibodies. (F) Model of KSHV-mediated modulation of autophagy pathway.
ACKNOWLEDGMENTS
This work was partly supported by grant 1F32AI096698 (M.A.R.), by grants CA180779, CA082057, CA31363, CA115284, AI073099, AI083025, and HL110609, the Hastings Foundation, and the Fletcher Jones Foundation (J.U.J.), and by the GRL Program (K20815000001) of the National Research Foundation of Korea (J.U.J. and B.-H.O.).
Footnotes
Published ahead of print 11 September 2013
REFERENCES
- 1.Klionsky DJ. 2005. The molecular machinery of autophagy: unanswered questions. J. Cell Sci. 118:7–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Silva LM, Jung JU. 29 May 2013. Modulation of the autophagy pathway by human tumor viruses. Semin. Cancer Biol. 10.1016/j.semcancer.2013.05.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kim J, Kundu M, Viollet B, Guan KL. 2011. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 13:132–141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Simonsen A, Tooze SA. 2009. Coordination of membrane events during autophagy by multiple class III PI3-kinase complexes. J. Cell Biol. 186:773–782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pattingre S, Tassa A, Qu X, Garuti R, Liang XH, Mizushima N, Packer M, Schneider MD, Levine B. 2005. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell 122:927–939 [DOI] [PubMed] [Google Scholar]
- 6.Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T, Kominami E, Ohsumi Y, Yoshimori T. 2000. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 19:5720–5728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lee JS, Li Q, Lee JY, Lee SH, Jeong JH, Lee HR, Chang H, Zhou FC, Gao SJ, Liang C, Jung JU. 2009. FLIP-mediated autophagy regulation in cell death control. Nat. Cell Biol. 11:1355–1362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Liang C, Feng P, Ku B, Dotan I, Canaani D, Oh BH, Jung JU. 2006. Autophagic and tumour suppressor activity of a novel Beclin1-binding protein UVRAG. Nat. Cell Biol. 8:688–699 [DOI] [PubMed] [Google Scholar]
- 9.Liang C, Lee JS, Inn KS, Gack MU, Li Q, Roberts EA, Vergne I, Deretic V, Feng P, Akazawa C, Jung JU. 2008. Beclin1-binding UVRAG targets the class C Vps complex to coordinate autophagosome maturation and endocytic trafficking. Nat. Cell Biol. 10:776–787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Matsunaga K, Saitoh T, Tabata K, Omori H, Satoh T, Kurotori N, Maejima I, Shirahama-Noda K, Ichimura T, Isobe T, Akira S, Noda T, Yoshimori T. 2009. Two Beclin 1-binding proteins, Atg14L and Rubicon, reciprocally regulate autophagy at different stages. Nat. Cell Biol. 11:385–396 [DOI] [PubMed] [Google Scholar]
- 11.Zhong Y, Wang QJ, Li X, Yan Y, Backer JM, Chait BT, Heintz N, Yue Z. 2009. Distinct regulation of autophagic activity by Atg14L and Rubicon associated with Beclin 1-phosphatidylinositol-3-kinase complex. Nat. Cell Biol. 11:468–476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mesri EA, Cesarman E, Boshoff C. 2010. Kaposi's sarcoma and its associated herpesvirus. Nat. Rev. Cancer 10:707–719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Du MQ, Bacon CM, Isaacson PG. 2007. Kaposi sarcoma-associated herpesvirus/human herpesvirus 8 and lymphoproliferative disorders. J. Clin. Pathol. 60:1350–1357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kuang E, Fu B, Liang Q, Myoung J, Zhu F. 2011. Phosphorylation of eukaryotic translation initiation factor 4B (EIF4B) by open reading frame 45/p90 ribosomal S6 kinase (ORF45/RSK) signaling axis facilitates protein translation during Kaposi sarcoma-associated herpesvirus (KSHV) lytic replication. J. Biol. Chem. 286:41171–41182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liang Q, Deng H, Li X, Wu X, Tang Q, Chang TH, Peng H, Rauscher FJ, III, Ozato K, Zhu F. 2011. Tripartite motif-containing protein 28 is a small ubiquitin-related modifier E3 ligase and negative regulator of IFN regulatory factor 7. J. Immunol. 187:4754–4763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liang Q, Deng H, Sun CW, Townes TM, Zhu F. 2011. Negative regulation of IRF7 activation by activating transcription factor 4 suggests a cross-regulation between the IFN responses and the cellular integrated stress responses. J. Immunol. 186:1001–1010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liang Q, Fu B, Wu F, Li X, Yuan Y, Zhu F. 2012. ORF45 of Kaposi's sarcoma-associated herpesvirus inhibits phosphorylation of interferon regulatory factor 7 by IKKepsilon and TBK1 as an alternative substrate. J. Virol. 86:10162–10172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rodgers MA, Bowman JW, Liang Q, Jung JU. 31 July 2013. Regulation where autophagy intersects the inflammasome. Antioxid. Redox Signal. [Epub ahead of print.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sathish N, Zhu FX, Golub EE, Liang Q, Yuan Y. 2011. Mechanisms of autoinhibition of IRF-7 and a probable model for inactivation of IRF-7 by Kaposi's sarcoma-associated herpesvirus protein ORF45. J. Biol. Chem. 286:746–756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liang C, E X, Jung JU. 2008. Downregulation of autophagy by herpesvirus Bcl-2 homologs. Autophagy 4:268–272 [DOI] [PubMed] [Google Scholar]
- 21.Wang HW, Sharp TV, Koumi A, Koentges G, Boshoff C. 2002. Characterization of an anti-apoptotic glycoprotein encoded by Kaposi's sarcoma-associated herpesvirus which resembles a spliced variant of human survivin. EMBO J. 21:2602–2615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Feng P, Park J, Lee BS, Lee SH, Bram RJ, Jung JU. 2002. Kaposi's sarcoma-associated herpesvirus mitochondrial K7 protein targets a cellular calcium-modulating cyclophilin ligand to modulate intracellular calcium concentration and inhibit apoptosis. J. Virol. 76:11491–11504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kimura S, Noda T, Yoshimori T. 2007. Dissection of the autophagosome maturation process by a novel reporter protein, tandem fluorescent-tagged LC3. Autophagy 3:452–460 [DOI] [PubMed] [Google Scholar]
- 24.Sun Q, Zhang J, Fan W, Wong KN, Ding X, Chen S, Zhong Q. 2011. The RUN domain of rubicon is important for hVps34 binding, lipid kinase inhibition, and autophagy suppression. J. Biol. Chem. 286:185–191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vieira OV, Botelho RJ, Rameh L, Brachmann SM, Matsuo T, Davidson HW, Schreiber A, Backer JM, Cantley LC, Grinstein S. 2001. Distinct roles of class I and class III phosphatidylinositol 3-kinases in phagosome formation and maturation. J. Cell Biol. 155:19–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Myoung J, Ganem D. 2011. Generation of a doxycycline-inducible KSHV producer cell line of endothelial origin: maintenance of tight latency with efficient reactivation upon induction. J. Virol. Methods 174:12–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Brulois KF, Chang H, Lee AS, Ensser A, Wong LY, Toth Z, Lee SH, Lee HR, Myoung J, Ganem D, Oh TK, Kim JF, Gao SJ, Jung JU. 2012. Construction and manipulation of a new Kaposi's sarcoma-associated herpesvirus bacterial artificial chromosome clone. J. Virol. 86:9708–9720 [DOI] [PMC free article] [PubMed] [Google Scholar]