

Abstract
Background
It is unclear whether levels of high-density lipoprotein cholesterol (HDL-C) or apolipoprotein A-I (apoA-I) remain inversely associated with cardiovascular risk among patients who achieve very low levels of low-density lipoprotein cholesterol (LDL-C) on statin therapy. It is also unknown whether a rise in HDL-C or apoA-I after initiation of statin therapy is associated with a reduced cardiovascular risk.
Methods and results
We performed a meta-analysis of 8 statin trials in which lipids and apolipoproteins were determined in all study participants at baseline and at 1-year follow-up. Individual patient data were obtained for 38,153 trial participants allocated to statin therapy, of whom 5387 suffered a major cardiovascular event. HDL-C levels were associated with a reduced risk of major cardiovascular events (adjusted hazard ratio 0.83, 95%CI 0.81–0.86 per 1 standard deviation increment), as were apoA-I levels (HR 0.79, 95%CI 0.72–0.82). This association was also observed among patients achieving on-statin LDL-C levels < 50 mg/dL. An increase of HDL-C was not associated with reduced cardiovascular risk (HR 0.98, 95%CI 0.94–1.01 per 1 standard deviation increment), whereas a rise in apoA-I was (HR 0.93, 95%CI 0.90–0.97).
Conclusions
Among patients treated with statin therapy, HDL-C and apoA-I levels were strongly associated with a reduced cardiovascular risk, even among those achieving very low LDL-C. An apoA-I increase was associated with a reduced risk of major cardiovascular events, whereas for HDL-C this was not the case. These findings suggest that therapies that increase apoA-I concentration require further exploration with regard to cardiovascular risk reduction.
Keywords: high-density lipoprotein cholesterol, apolipoprotein, meta-analysis, cardiovascular outcomes
Introduction
Low levels of high-density lipoprotein cholesterol (HDL-C) are a well-established risk factor for coronary heart disease (CHD).1,2 A similar association has consistently been shown for the concentration of apolipoprotein A-I (apoA-I), the major protein constituent of these lipoproteins. HDL-C levels were also shown to be inversely associated with risk of cardiovascular events among people with low levels of low-density lipoprotein cholesterol (LDL-C).3 HDL-C levels have also been shown to be an important risk factor for cardiovascular events among patients treated with statins and even among those treated with high-dose statins.4,5 As a consequence, HDL-C and apoA-I have become important targets in the quest for novel anti-atherogenic agents.
However, the hypothesis that HDL-C and apoA-I directly confer biological protection against atherosclerosis has never been proven. The same is true for the hypothesis that raising HDL-C or apoA-I levels will result in reduced CHD risk. In fact, several recent lines of evidence have questioned HDL-C and apoA-I as relevant therapeutic targets. First, a recent study showed that some genetic variants that raise HDL-C levels are not associated with a proportionally lower risk of myocardial infarction.6 Second, a JUPITER subanalysis has shown that HDL-C and apoA-I had virtually no predictive value among patients treated with potent statin therapy who reached very low levels of LDL-C.7 Third, data from population studies and from a meta-analysis have suggested that changes in HDL-C levels after initiation of lipid modifying therapy are not independently associated with CHD risk.8,9 Fourth, several recent trials testing HDL raising therapies have shown unexpected results. The Atherothrombosis Intervention in Metabolic Syndrome with Low HDL/High Triglycerides: Impact on Global Health Outcomes (AIM-HIGH) trial, which evaluated the efficacy and safety of niacin, was stopped prematurely because of lack of efficacy.10 The trial evaluating the cholesteryl ester transfer protein (CETP) inhibitors torcetrapib was stopped prematurely due to excess mortality in the torcetrapib arm, whereas the trial evaluating the CETP inhibitor dalcetrapib was terminated prematurely on the grounds of futility.11, 12 These data combined have raised serious doubts about the efficacy of HDL targeted therapy for the prevention of cardiovascular events.
We hypothesized that among statin-treated patients, higher levels of HDL-C and apoA-I are indeed associated with lower cardiovascular risk, even among those achieving very low LDL-C levels. Second, we hypothesized that patients whose HDL-C or apoA-I levels increased after the initiation of statin therapy had a lower risk of cardiovascular events compared to those whose HDL-C or apoA-I did not change or decreased. We tested these hypotheses by performing a meta-analysis of individual patient data from 8 large statin trials.
Methods
Design and literature search
The design of this meta-analysis has been described previously.13 Briefly, the literature was searched to identify all randomized controlled trials that assigned study participants in at least 1 of the study groups to statin therapy. Trials in which total cholesterol, LDL-C, HDL-C, triglycerides, and apolipoproteins A-I and B were measured at baseline and during statin therapy in the entire study population were selected. Trials with a mean follow-up for cardiovascular events shorter than 2 years and those including fewer than 1000 participants were excluded. The literature search strategy has been described previously.13 Investigators from eligible trials were contacted and asked to provide individual patient data. The requested patient characteristics included sex, age, smoking status, body mass index, diabetes mellitus, systolic and diastolic blood pressure, fasting glucose, total cholesterol, LDL-C, HDL-C, triglycerides and apolipoprotein A-I and B at baseline and at 1-year follow-up, study medication, and history of stable coronary artery disease, myocardial infarction, percutaneous coronary intervention, or coronary artery bypass grafting. The following outcomes (and time to event) were also collected: fatal and nonfatal myocardial infarction, fatal other CHD, hospitalization for unstable angina, fatal and nonfatal stroke, peripheral artery disease, and congestive heart failure. Quality of the included trials was assessed by the Delphi score.14 This meta-analysis followed the Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA) guidelines.15 A PRISMA checklist was provided at submission.
Lipids, apolipoproteins, and outcome definition
Lipid and apolipoprotein levels at baseline and at 1-year follow-up were obtained from the participating trials. The 1-year time point was chosen because this was the first uniform time point when apolipoproteins were measured in all participating trials. Cholesterol levels reported in mmol/L were converted to mg/dL by multiplying by 38.7, and triglycerides levels reported in mmol/L were converted to mg/dL by multiplying by 88.5. Time to first major cardiovascular event was the outcome measure used in this analysis. Follow-up started at the 1-year timepoint so events occurring within the first year were not included in this analysis. Major cardiovascular event was a composite of fatal or nonfatal myocardial infarction, fatal other CHD, hospitalization for unstable angina, and fatal or nonfatal stroke. All included trials were approved by an institutional review committee and all study participants of the respective trials gave informed consent.
Statistical analysis
Baseline characteristics, levels of lipids and apolipoproteins at baseline and during participation in the trial, as well as absolute changes and percentage changes between on-trial and baseline levels were calculated for each trial separately. Change in HDL-C level was calculated as on-trial HDL-C level at 1 year minus baseline HDL-C level. Change in apoA-I level was calculated as on-trial apoA-I level at 1 year minus baseline apoA-I level. Study participants allocated to placebo were excluded. Cox proportional hazards models were used to quantify the association between on-statin HDL-C or apoA-I levels and time to the occurrence of first major cardiovascular event. Hazard ratios (HRs) and corresponding 95% confidence intervals (95%CI) were calculated by quintiles of on-statin HDL-C or apoA-I using those in the bottom quintile as reference category. A trend test was used to test for linear trend across quintiles. We also analyzed risk per 1 standard deviation of on-statin HDL-C or apoA-I levels. In addition, we analyzed risk per 1 standard deviation increment of on-statin HDL-C or apoA-I levels. Proportional hazards models were adjusted for trial, sex, age, smoking (current versus not), diabetes mellitus, systolic blood pressure, body mass index, glucose, and on-statin non-HDL-C. Additional analyses with alternative adjustment were performed after replacing non-HDL-C with either LDL-C or apolipoprotein B. Analyses were not additionally adjusted for prevalent cardiovascular disease since all trials enrolled either 0% or 100% patients with prevalent disease, so adjustment for trial implies adjustment for prevalent cardiovascular disease. Hazard ratios and corresponding 95%CI were also calculated by quintiles of HDL-C and apoA-I in the subgroups of people with on-statin LDL cholesterol ≥ 130 mg/dL, 100–130 mg/dL, 70–100 mg/dL, 70–50 mg/dL, and < 50 mg/dL. Forest plots were constructed and interaction terms were calculated to assess whether adjusted hazard ratios per 1 standard deviation increment of on-statin HDL-C or apoA-I differed significantly between subgroups based on baseline characteristics, lipid levels, or trial. Hazard ratios and corresponding 95% confidence intervals were also calculated by quintiles of change (between baseline and 1-year on-trial) in HDL-C or apoA-I in study participants allocated to statin therapy. For these analyses, study participants were excluded if either baseline or 1-year HDL-C or apoA-I levels were missing. Major cardiovascular events occurring between the baseline and 1-year measurements were not considered. Proportional hazards models by HDL-C quintiles were adjusted for trial, sex, age, smoking (current versus not), diabetes mellitus, systolic blood pressure, body mass index, glucose, non-HDL-C change, baseline HDL-C, and baseline non-HDL-C. The equivalent analysis by apoA-I quintiles were adjusted for the same variables except for baseline HDL-C which was replaced by baseline apoA-I, Additional analyses were performed after alternative adjustment replacing non-HDL-C with either LDL-C or apolipoprotein B.
The Cochran Q statistic and the I2 statistic were used to quantify statistical heterogeneity across studies with regard to the association between either HDL-C or apoA-I levels and risk of major cardiovascular events. The I2 statistic is derived from the Q statistic ([Q–df/Q]*100) and provides a measure of the proportion of the overall variation attributable to between-study heterogeneity.16 The potential for publication bias was addressed by drawing funnel plots and visual assessment. Proportionality of hazards over time was graphically checked by plotting the cumulative hazards over time for all quartiles against each other. All reported p-values are 2-sided. A p-value < 0.05 was considered statistically significant. Statistical analyses were performed using SPSS (version 17.0, Chicago, Illinois, USA).
Results
Literature search
The results of the literature search are shown in Supplementary Figure 1, and have been published previously.13 Briefly, 8 trials fulfilled all inclusion criteria: the Scandinavian Simvastatin Survival Study (4S),17 the Air Force/Texas Coronary Atherosclerosis Prevention Study (AFCAPS-TexCAPS),18 the Long-term Intervention with Pravastatin in Ischaemic Disease (LIPID) trial,19 the Collaborative Atorvastatin Diabetes Study (CARDS),20 the Treating to New Targets (TNT) trial,21 the Incremental Decrease in Endpoints through Aggressive Lipid Lowering (IDEAL) trial,22 the Stroke Prevention by Aggressive Reduction in Cholesterol Levels (SPARCL) trial,23 and the Justification for the Use of Statins in Prevention: an Intervention Trial Evaluating Rosuvastatin (JUPITER).24 Individual patient data were obtained from all 8 trials. Study characteristics of these 8 trials are shown in Supplementary Table 1. Trials were of high quality with a median Delphi score of 9 (range 6–9). Heterogeneity between trials with regard to the association with risk of major cardiovascular events was low for HDL-C (Q=9.93, P=0.19, I2=29%, and apoA-I (Q= 6.30, P = 0.51, I2= 0) Visual assessment of funnel plots did not suggest strong evidence for bias. The proportionality assumptions were satisfied.
Baseline characteristics and events
Baseline characteristics of the study participants are shown in Supplementary Table 2. Levels of lipids and apolipoproteins at baseline and at 1-year on-trial, as well as the absolute and percentage changes between baseline and on-trial levels are shown in Supplementary Table 3. A total of 38,153 study participants were randomized to a statin arm and had a complete set of lipid and apolipoprotein levels during statin treatment available. During 155,573 person-years follow-up, 158 (0.4%) study participants developed a fatal myocardial infarction and 1678 (4.4%) developed a non-fatal myocardial infarction. Fatal other CHD occurred in 615 study participants (1.6%) and fatal or nonfatal stroke occurred in 1029 study participants (2.7%). A total of 2806 participants (7.4%) were hospitalized for unstable angina. A total of 6286 major cardiovascular events occurred in 5387 study participants (event rate 14.1%). Of these, 4577 experienced 1 event, 728 experienced 2 events, 75 experienced 3 events, and 7 experienced 4 events.
On-statin HDL cholesterol and apolipoprotein A-I levels and risk of events
The risk of major cardiovascular events was strongly and inversely associated with levels of on-statin HDL-C and apoA-I levels (Table 1). Patients in the top quintile of on-statin HDL-C had a multivariable adjusted hazard ratio of 0.65 (95%CI 0.59–0.71) for major cardiovascular events compared to those in the bottom quintile (p for linear trend across quintiles < 0.001). The multivariable adjusted hazard ratio per 1 standard deviation HDL-C increment was 0.83 (95%CI 0.81–0.86). For those in the top quintile of apoA-I, the multivariable adjusted hazard ratio was 0.53 (95%CI 0.48–0.59, p for linear trend < 0.001). The hazard ratio per 1 standard deviation apoA-I increment was 0.79 (95%CI 0.77–0.82). Analyses in which non-HDL-C was replaced as an adjustment variable by either LDL-C or apoB yielded similar results (data not shown). The strong and inverse association between on-statin HDL-C level and risk of major cardiovascular events was similar in the subgroups of patients achieving on-statin LDL-C > 130 mg/dL, 130-100 mg/dL, 100-70 mg/dL, 70-50 mg/dL, and < 50 mg/dL (Figure 1A). There was also a strong inverse association between apoA-I quintiles and risk of cardiovascular events in each of the subgroups of patients achieving on-statin LDL-C > 130 mg/dL, 130-100 mg/dL, 100-70 mg/dL, 70-50 mg/dL, and < 50 mg/dL (Figure 1B). Analyses where non-HDL-C was replaced as an adjustment variable by either LDL-C or apoB yielded similar results (data not shown).
Table 1.
On-statin HDL cholesterol and apolipoprotein A-I levels and risk of major cardiovascular events
| HDL cholesterol quintiles
|
p* | per 1 SD | p | |||||
|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | ||||
|
| ||||||||
| Mean, mg/dL | 32 ± 3 | 39 ± 2 | 45 ± 2 | 52 ± 2 | 68 ± 12 | |||
| Range, mg/dL | < 36 | 37 – 42 | 43 – 47 | 48 – 57 | > 57 | |||
| Events / total | 1495/7872 | 1124/7317 | 1124/7658 | 916/7783 | 728/7523 | |||
| Event rate | 19.0% | 15.4% | 14.7% | 11.8% | 9.7% | |||
| Hazard ratio | 1.00 | 0.84 | 0.80 | 0.70 | 0.65 | < 0.001 | 0.83 | < 0.001 |
| 95%CI | (0.77–0.90) | (0.74–0.87) | (0.64–0.76) | (0.59–0.71) | (0.81–0.86) | |||
|
| ||||||||
|
Apolipoprotein A-I quintiles
|
||||||||
| 1 | 2 | 3 | 4 | 5 | ||||
|
| ||||||||
| Mean, mg/dL | 112 ± 9 | 130 ± 4 | 142 ± 3 | 157 ± 6 | 190 ± 21 | |||
| Range, mg/dL | < 121 | 122 – 136 | 137 – 149 | 150 – 168 | > 169 | |||
| Events / total | 1500/7548 | 1230/7773 | 1009/7271 | 1015/7968 | 633/7593 | |||
| Event rate | 19.9% | 15.8% | 13.9% | 12.7% | 8.3% | |||
| Hazard ratio | 1.00 | 0.79 | 0.69 | 0.66 | 0.53 | < 0.001 | 0.79 | < 0.001 |
| 95%CI | (0.73–0.85) | (0.64–0.75) | (0.61–0.72) | (0.48–0.59) | (0.77–0.82) | |||
Risk of major cardiovascular events shown as number of events / total number of study participants, event rates, and hazard ratios with corresponding 95% confidence intervals. Hazard ratios were adjusted for trial, sex, age, smoking, diabetes mellitus, systolic blood pressure, body mass index, glucose, and non-HDL cholesterol. Analyses were performed by quintiles of HDL cholesterol or apolipoprotein A-I levels using those in the bottom quintile as reference, and also by 1 standard deviation increment.
p-values for trend test across quintiles. HDL = high-density lipoprotein.
Figure 1.
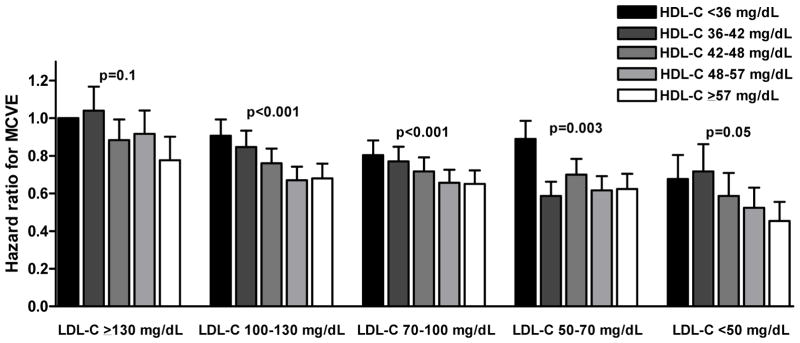
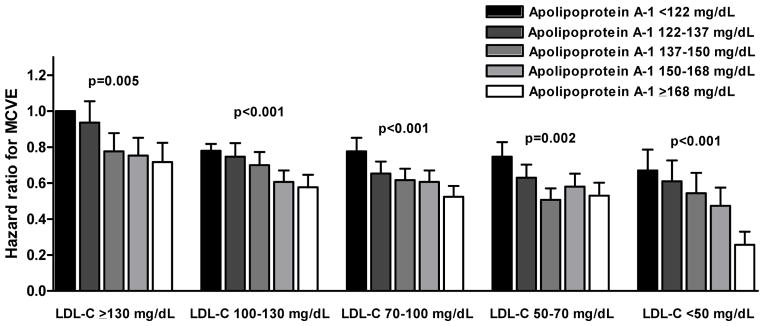
Figure 1A. Cardiovascular risk by HDL cholesterol quintiles and LDL cholesterol categories
Figure 1B. Cardiovascular risk by apolipoprotein A-I quintiles and LDL cholesterol categories
Bars indicate multivariate adjusted hazard ratios for risk of major cardiovascular events by HDL cholesterol or apolipoprotein A-I quintiles and LDL cholesterol categories. Hazard ratios were adjusted for trial, sex, age, smoking status, diabetes mellitus, systolic blood pressure, body mass index, and glucose. Patients with on-statin LDL-C > 130 mg/dL and in the lowest HDL-C or apolipoprotein A-I quintile were used as reference category. P-values for trend test across HDL-C or apolipoprotein A-I quintiles. MCVE = major cardiovascular events; HDL-C = high-density lipoprotein cholesterol; LDL-C = low-density lipoprotein cholesterol.
Figure 2A shows the multivariable adjusted hazard ratios and 95%CI for major cardiovascular events per 1 SD increment of HDL-C and apoA-I level in various subgroups based on the presence or absence of patient or treatment characteristics. There were no statistically significant interaction terms by any of the analyzed subgroups for either HDL-C or apoA-I. There was, however, statistical evidence that the association between HDL-C and risk of major cardiovascular events differed between the various trials (p for interaction = 0.044; Figure 2B). For apoA-I, this interaction term was non-significant.
Figure 2.

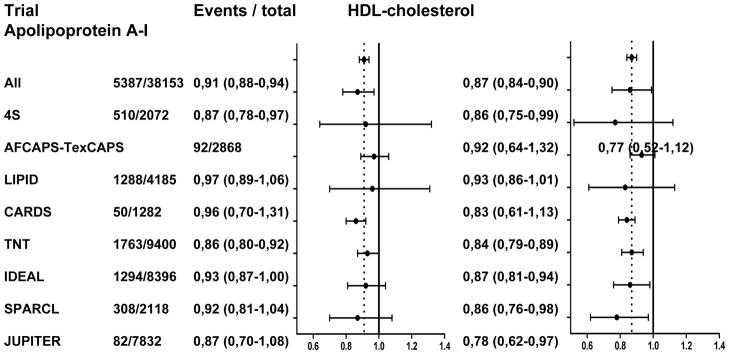
Figure 2A. Association between on-statin HDL cholesterol or apolipoprotein A-I and risk of major cardiovascular events by baseline characteristics
Figure 2B. Association between on-statin HDL cholesterol or apolipoprotein A-I and risk of major cardiovascular events stratified by trial
Data presented for each subgroup or trial are the number of major cardiovascular events / total number of study participants in that subgroup or trial, hazard ratio and corresponding 95% confidence interval for risk of major cardiovascular events per 1 standard deviation increase of HDL cholesterol, and equivalent hazard ratio for apolipoprotein A-I. Graphics represent hazard ratios (dots) and corresponding 95% confidence intervals (horizontal lines). Vertical dotted line represents point estimate for all participants combined. Hazard ratios were adjusted for trial, sex, age, smoking status, diabetes mellitus, systolic blood pressure, body mass index, glucose, and non-HDL cholesterol. HDL = high-density lipoprotein, BMI = body mass index, LDL-C = low-density lipoprotein cholesterol, CHD = coronary heart disease, TG = triglycerides
Change in HDL cholesterol and apolipoprotein A-I and risk of major cardiovascular events
Both baseline and on-trial HDL-C and apoA-I levels (and thus the possibility to calculate change in HDL-C and apoA-I) were available for 37,747 study participants enrolled in a statin arm, of whom 3902 developed a major cardiovascular event after the 1-year HDL-C and apoA-I measurements. The changes in HDL-C and apoA-I levels between baseline and the 1-year on-trial time point are shown in Supplementary Table 3. The correlation-coefficient between baseline HDL-C and change in HDL-C was −0.198 (p<0.001). The correlation coefficient between baseline apoA-I and change in apoA-I was −0.312 (p<0.001).
Patients in the top quintile of HDL-C change had a multivariable adjusted hazard ratio of 0.98 (95%CI 0.87–1.09) for risk of major cardiovascular events compared to those in the bottom quintile (p for linear trend across quintiles = 0.7, Table 2). The multivariable adjusted hazard ratio for cardiovascular events per 1 standard deviation HDL-C change was 0.98 (95%CI 0.84–1.01). Patients in the top quintile of apoA-I change had a hazard ratio of 0.83 (95%CI 0.74–0.93, p for linear trend = 0.001). The multivariable adjusted hazard ratio for cardiovascular events per 1 standard deviation apoA-I change was 0.93 (95%CI 0.90–0.97). Analyses in which non-HDL-C was replaced as an adjustment variable by either LDL-C or apoB yielded similar results (data not shown). Analyses that additionally included patients enrolled in placebo arms of the included trials showed similar results (data not shown).
Table 2.
Risk of major cardiovascular events by change in HDL cholesterol and apolipoprotein A-I
| HDL cholesterol change quintiles
|
p* | per 1 SD | p | |||||
|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | ||||
|
| ||||||||
| Mean, mg/dL | −8.5 | −2.3 | +0.8 | +4.1 | +11.6 | |||
| Range, mg/dL | < −3.9 | −3.9 to −0.4 | −0.4 to +1.9 | +1.9 to 6.2 | > 6.2 | |||
| Events / total | 811/7894 | 782/7194 | 890/7784 | 793/7543 | 626/7332 | |||
| Event rate | 10.3% | 10.9% | 11.4% | 10.5% | 8.5% | |||
| Hazard ratio | 1.00 | 0.92 | 0.97 | 0.94 | 0.98 | 0.7 | 0.98 | 0.2 |
| 95%CI | (0.84–1.02) | (0.88–1.07) | (0.85–1.04) | (0.87–1.09) | (0.94–1.01) | |||
|
| ||||||||
|
Apolipoprotein A-I change quintiles
|
||||||||
| 1 | 2 | 3 | 4 | 5 | ||||
|
| ||||||||
| Mean, mg/dL | −26 | −8 | +1 | +10 | +27 | |||
| Range, mg/dL | < −12 | −12 to −2 | −2 to +6 | +6 to +16 | >16 | |||
| Events / total | 729/7478 | 854/7751 | 818/7193 | 796/7623 | 705/7702 | |||
| Event rate | 9.7% | 11.0% | 11.4% | 10.4% | 9.2% | |||
| Hazard ratio | 1.00 | 0.96 | 0.94 | 0.89 | 0.83 | 0.001 | 0.93 | 0.001 |
| 95%CI | (0.87–1.06) | (0.85–1.05) | (0.79–0.99) | (0.74–0.93) | (0.90–0.97) | |||
Data are based on statin arms only. Data presented are number of major cardiovascular events / total number of individuals, and hazard ratios and corresponding 95% confidence intervals. Hazard ratios were adjusted for trial, sex, age, smoking, diabetes mellitus, systolic blood pressure, body mass index, glucose, non-HDL-C change, baseline HDL-C, and baseline non-HDL-C.
p-values for trend test across quintiles. HDL indicates high-density lipoprotein.
Discussion
We performed a meta-analysis of randomized controlled trials to evaluate the association between on-statin HDL-C and apoA-I levels and risk of major cardiovascular events. We observed that among statin-treated patients, on-trial HDL-C and apoA-I levels were each strongly and inversely associated with the risk of cardiovascular events. These associations were also observed among patients achieving very low LDL-C levels. In addition, we observed that study participants whose apoA-I increased after initiation of statin therapy had a lower risk of cardiovascular events compared to those whose apoA-I levels decreased. This was not the case for HDL-C.
Our observation that HDL-C and apoA-I levels are strongly and inversely associated with risk of cardiovascular events is consistent with results from the Pravastatin Pooling Project which showed that among people taking usual-dose pravastatin, HDL-C levels were inversely associated with risk of coronary events.4 Consistently, a TNT subanalysis has shown that the risk of cardiovascular events was lower among patients in the top versus bottom HDL-C quintile, at least among those assigned to atorvastatin 10 mg.5 Among those assigned to aggressive lipid-lowering with high-dose atorvastatin 80 mg (mean on-treatment LDL-C level 77 mg/dL; 2.0 mmol/L), this association did not reach statistical significance. Consistently, a JUPITER subanalysis suggested that HDL-C levels were associated with reduced cardiovascular risk among patients in the placebo arm, but that this association was lost among people on rosuvastatin 20 mg achieving very low LDL-C (median on-treatment level 1.42 mmol/L).7 It is unclear whether the loss of statistical significance among those randomized to rosuvastatin was related to the low number of cardiovascular events or represented a true lack of association. The current meta-analysis compiled over 12,500 study participants reaching LDL-C < 70 mg/dL, of whom over 1100 developed a major cardiovascular event during follow-up. In fact, 4375 participants even reached LDL-C < 50 mg/dL, and the number of events in this subgroup was 194. Both HDL-C and apoA-I levels were strongly and inversely associated with the risk of major cardiovascular events in the subgroups with very low LDL-C levels. The inconsistency between the TNT and JUPITER findings and the current meta-analysis is most likely explained by the difference in statistical power.
A second topic of debate, highly relevant for the potential of HDL-C and apoA-I as therapeutic targets, is the question whether increases in these parameters are associated with a reduced risk of cardiovascular events. We show that a rise in HDL-C level was not associated with a lower risk of major cardiovascular events independent of established risk factors. Interestingly however, we did observe an association between a rise in apoA-I and a lower risk of major cardiovascular events. Our HDL-C results are not consistent with a report from the Framingham Offspring Study, which showed that changes in HDL-C after initiation of lipid modification therapy were significantly and inversely associated with cardiovascular risk.25 Our results are consistent with recent data from the EPIC-Norfolk and Rotterdam population studies which suggested that the association between changes in HDL-C levels after initiation of lipid modifying therapy and risk of CHD was mostly explained by established risk factors.8 The most important difference between these population studies and the current meta-analysis is the number of cardiovascular events; 60 in the Framingham Offspring Study and 79 in the EPIC-Norfolk and Rotterdam studies versus 5291 in the current meta-analysis. Our results are also consistent with a previous meta-analysis of trials testing lipid-modifying interventions which showed that a change in HDL-C levels explained almost no variability in cardiovascular outcomes.9 It should be noted however that our meta-analysis was based on individual patient data, which overcomes the inherent problems of a study level meta-analysis such as the one by Briel et al. The finding that an increase in apoA-I is associated with a reduced cardiovascular risk independent of established risk factors reinforces the rationale for apoA-I as a target for anti-atherogenic therapy. However, HDL metabolism is complex and a large number of potential targets have been identified.26 Our current analysis does not provide clues as to which of these players in HDL metabolism should be targeted in order to increase apoA-I and reduce cardiovascular risk. Nevertheless, we might suggest that increasing apoA-I production at the mRNA level such as achieved with RVX208, infusion of apoA-I containing HDL particles such as CER-001, CSL-112, or MDCO-216, or reinfusion of delipidated prebeta HDL might enjoy better prospects than other strategies that only increase the cholesterol content of HDL particles.27–30
Several aspects of the design of this meta-analysis warrant comment. An important strength is the availability of individual patient data. This enabled us to perform analyses at individual patient level, which allows better statistical adjustment than a study level meta-analysis. In fact, a study level meta-analysis would have pooled patients whose HDL-C and apoA-I changes went in opposite directions, thus resulting in the cancelation of true associations. Another strength is the large number of events in this meta-analysis compared to previous studies. An important limitation is the fact that the participating trials had different sets of inclusion criteria. Differences in the distribution of baseline characteristics between trials may have affected the pooled results. It is however comforting that we did not observe any significant interaction by clinically relevant subgroups. Second, we used on-statin lipid and apolipoprotein levels measured at 1-year follow-up. This time point was chosen because it was the first uniform time point when both lipids and apolipoproteins were measured in all participating trials. As a consequence, cardiovascular events occurring in the first year of therapy and therefore the highest risk patients, are not accounted for in this analysis. Another limitation is the fact that the external validity of HDL-C measurements may be suboptimal. All trials included in the meta-analysis measured HDL-C according to the standard protocol of the Centers of Disease Control and prevention.31 However, many clinical laboratories nowadays measure HDL-C using homogeneous methods which are less well standardized. Therefore, HDL-C as routinely measured in many clinical labs may not provide an index of cardiovascular risk as accurate as that in clinical trials. Finally and most importantly, limited information was available about on-trial covariates. We should emphasize that our analysis does not provide information about the cause of the changes in HDL-C and apoA-I levels. These changes may have been partially related to statin therapy, but lifestyle changes such as weight loss, physical activity and smoking cessation could have had similar effects on HDL-C and apoA-I levels and are also associated with a reduced risk of cardiovascular events. The fact that similar associations between changes of HDL-C or apoA-I and risk of cardiovascular events were seen in patients assigned statin therapy compared to those assigned to placebo, suggests that these changes are to a large extent not statin-related but most likely caused by lifestyle modifications. Therefore, the current analysis does not allow us to draw any conclusion about the cause of changes in HDL-C or apoA-I, nor about the potential causality of the observed associations between apoA-I changes and risk of cardiovascular events.
In conclusion, this meta-analysis shows that among statin-treated patients HDL-C and apoA-I levels are strongly and inversely associated with the risk of major cardiovascular events. These associations were also observed among patients achieving very low LDL-C levels. In addition, a rise in apoA-I level was associated with cardiovascular risk such that those with the largest increases had the lowest cardiovascular risk. Such an association was not observed for HDL-C. Although these results do not provide evidence about the potential causality of the association between apoA-I increases and reduced risk of cardiovascular events, they do support further exploration of apoA-I modifying therapies for the prevention of cardiovascular events.
Supplementary Material
Acknowledgments
Funding
This study was not supported by any funding. The contributing trials were funded by their respective sponsors. Dr Hovingh is funded by a Veni grant (project number 91612122) from the Netherlands Organisation for Scientific Research (NWO). Dr Mora is funded by a NHLBI grant (number R01HL117861).
Role of the sponsors
The sponsors of the contributing trials provided the requested data. They did not play any role in the statistical analysis, interpretation of the data, or the decision to submit the manuscript.
Footnotes
Disclaimer
The views expressed in this article are those of the authors and do not necessarily reflect the position or policy of the Department of Veterans Affairs or the United States government.
Author contributions
Author Contributions: Drs Boekholdt and Kastelein had full access to all the data in the study and take responsibility for the integrity of the data and the accuracy of the data analysis.
Study concept and design: Boekholdt, Kastelein.
Acquisition of data: Pedersen, LaRosa, Welch, Amarenco, Tonkin, Sullivan, Kirby, Colhoun, Hitman, Betteridge, Durrington, Clearfield, Downs, Gotto, Ridker, Kastelein.
Analysis and interpretation of data: Boekholdt, Arsenault, Hovingh, Mora, Pedersen, LaRosa, DeMicco, Tonkin, Sullivan, Kirby, Colhoun, Hitman, Betteridge, Durrington, Clearfield, Downs, Gotto, Ridker, Kastelein
Drafting of the manuscript: Boekholdt, Arsenault, Hovingh, Mora, Kastelein
Critical revision of the manuscript for important intellectual content: Pedersen, LaRosa, DeMicco, Welch, Tonkin, Sullivan, Kirby, Colhoun, Hitman, Betteridge, Durrington, Clearfield, Downs, Gotto, Ridker.
Statistical analysis: Boekholdt, Arsenault.
Administrative, technical, or material support: DeMicco, Kastelein.
Study supervision: Boekholdt, Kastelein.
Conflict of interest disclosures
Drs Boekholdt, Arsenault and Hovingh report receipt of consultancy fees from Pfizer. Dr Mora reports receipt of research grant support through her institution from Merck Sharpe & Dohme and AstraZeneca; consultancy fees from Pfizer and Quest Diagnostics; lecture honoraria from AstraZeneca and Abbott; and travel accommodations/meeting expenses from Pfizer. Dr LaRosa reports receipt of consultancy fees from Pfizer and Amgen, and travel expenses from Pfizer. Dr Pedersen reports receipt of research grant support and lecture fees from Pfizer and Merck Sharp & Dohme and lecture fees from AstraZeneca and Roche. Dr DeMicco reports being a full-time employee of Pfizer; and having stock/stock options with Pfizer. Dr Welch reports receipt of a grant, consulting fees, travel support, payment for writing or manuscript review, and provision of writing assistance, medicines, equipment, or administrative support from Pfizer, and provision of consultancy services to Edwards, MAP, and NuPathe. Dr Amarenco reports receipt of research grant support and lecture fees from Pfizer, Sanofi, Merck, AstraZeneca, Boehringer-Ingelheim, and consultancy fees from Pfizer, BMS, Merck, Boehringer-Ingelheim, AstraZeneca, Bayer, Daiichi-Sankyo, Lundbeck, Edwards, Boston Scientific, Kowa and research grants from the French government. Dr Tonkin reports receipt of consultancy fees from Amgen, AstraZeneca, Boehringer-Ingelheim, Merck Sharpe & Dohme, Pfizer and Regeneron; and lecture honoraria from AstraZeneca, Merck Sharpe & Dohme, and Roche. Dr Colhoun reports receipt of research grant support through the EU Innovative Medicines Initiative from Roche, Pfizer, Eli Lilly, Boehringer-Ingelheim, and AstraZeneca; consultancy fees from Pfizer, sanofi-aventis, Novartis, and Eli Lilly; and lecture honoraria and travel expenses from Pfizer. Dr Betteridge reports receipt of honoraria for lectures and advisory boards for Aegerion, Amgen, AstraZeneca, Kowa, Merck Sharpe & Dohme, Roche, and Takeda. Dr Hitman has received consultancy fees and lecture honoraria from GlaxoSmithKline, Eli Lily, Pfizer, NovoNordisk, Astra Zeneca, Merck Sharp & Dohme, Takeda and OSI Pharmaceuticals and grant income from Park Davies and Eli Lily. Dr Gotto is a consultant for AstraZeneca, Janssen, KOWA, Merck, and Roche; a member of the Board of Directors for Aegerion Pharmaceuticals and Arisaph Pharmaceuticals; and a member of advisory boards for DuPont, Haptocure, VascuVis, and VateraCapital. Dr Clearfield reports provision of consulting services on advisory committees to Merck Sharp & Dohme and AstraZeneca. Dr Ridker reports receipt of research grant funding from Novartis and AstraZeneca; serving as a consultant to ISIS, Vascular Biogenics, Merck Sharpe & Dohme, Abbott, and Boehringer-Ingelheim; board membership with Merck Sharp & Dohme; receipt of a grant or pending grant to his institution from Amgen; and being listed as a coinventor on patents held by the Brigham and Women’s Hospital that relate to the use of inflammatory biomarkers in cardiovascular disease and diabetes that have been licensed to AstraZeneca and Siemens. Dr Kastelein reports receipt of lecture honoraria from Merck Sharpe & Dohme, Roche, Novartis, ISIS, Genzyme, Pfizer, Kowa, and AstraZeneca. Drs Downs, Durrington, Kirby and Sullivan reported no conflict of interest.
References
- 1.Wilson PW, Garrison RJ, Castelli WP, et al. Prevalence of coronary heart disease in the Framingham Offspring Study: role of lipoprotein cholesterols. Am J Cardiol. 1980;46:649–654. doi: 10.1016/0002-9149(80)90516-0. [DOI] [PubMed] [Google Scholar]
- 2.Emerging Risk Factors Collaboration. Major lipids, apolipoproteins, and risk of vascular disease. JAMA. 2009;302:1993–2000. doi: 10.1001/jama.2009.1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.deGoma EM, Leeper NJ, Heidenreich PA. Clinical significance of high-density lipoprotein cholesterol in patients with low low-density lipoprotein cholesterol. J Am Coll Cardiol. 2008;51:49–55. doi: 10.1016/j.jacc.2007.07.086. [DOI] [PubMed] [Google Scholar]
- 4.Sacks FM, Tonkin AM, Shepherd J, et al. Effect of pravastatin on coronary disease events in subgroups defined by coronary risk factors: the Prospective Pravastatin Pooling Project. Circulation. 2000;102:1893–1900. doi: 10.1161/01.cir.102.16.1893. [DOI] [PubMed] [Google Scholar]
- 5.Barter P, Gotto AM, LaRosa JC, et al. HDL cholesterol, very low levels of LDL cholesterol, and cardiovascular events. N Engl J Med. 2007;357:1301–1310. doi: 10.1056/NEJMoa064278. [DOI] [PubMed] [Google Scholar]
- 6.Voight BF, Peloso GM, Orho-Melander M, et al. Plasma HDL cholesterol and risk of myocardial infarction: a mendelian randomisation study. Lancet. 2012;380:572–580. doi: 10.1016/S0140-6736(12)60312-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ridker PM, Genest J, Boekholdt SM, et al. HDL cholesterol and residual risk of first cardiovascular events after treatment with potent statin therapy: an analysis from the JUPITER trial. Lancet. 2010;376:333–339. doi: 10.1016/S0140-6736(10)60713-1. [DOI] [PubMed] [Google Scholar]
- 8.Ray K, Wainwright NW, Visser L, et al. Changes in HDL cholesterol and cardiovascular outcomes after lipid modification therapy. Heart. 2012;98:780–785. doi: 10.1136/heartjnl-2011-301405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Briel M, Ferreira-Gonzalez I, You JJ, et al. Association between change in high density lipoprotein cholesterol and cardiovascular disease morbidity and mortality: systematic review and meta-regression analysis. BMJ. 2009;338:b92. doi: 10.1136/bmj.b92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.AIM-HIGH Investigators. Niacin in patients with low HDL cholesterol levels receiving intensive statin therapy. N Engl J Med. 2011;365:2255–2267. doi: 10.1056/NEJMoa1107579. [DOI] [PubMed] [Google Scholar]
- 11.Barter PJ, Caulfield M, Eriksson M, et al. Effects of torcetrapib in patients at high risk for coronary events. N Engl J Med. 2007;357:2109–2122. doi: 10.1056/NEJMoa0706628. [DOI] [PubMed] [Google Scholar]
- 12.Schwartz GG, Olsson AG, Abt M, et al. Effects of dalcetrapib in patients with a recent acute coronary syndrome. N Engl J Med. 2012;367:2089–2099. doi: 10.1056/NEJMoa1206797. [DOI] [PubMed] [Google Scholar]
- 13.Boekholdt SM, Arsenault BJ, Mora S, et al. Association of LDL cholesterol, non-HDL cholesterol, and apolipoprotein B levels with risk of cardiovascular events among patients treated with statins: a meta-analysis. JAMA. 2012;307:1302–1309. doi: 10.1001/jama.2012.366. [DOI] [PubMed] [Google Scholar]
- 14.Verhagen AP, de Vet HC, de Bie RA, et al. The Delphi list: a criteria list for quality assessment of randomized clinical trials for conducting systematic reviews developed by Delphi consensus. J Clin Epidemiol. 1998;51:1235–1241. doi: 10.1016/s0895-4356(98)00131-0. [DOI] [PubMed] [Google Scholar]
- 15.Moher D, Liberati A, Tetzlaff J, et al. Preferred reporting items for systematic reviews and meta-analyses: the PRISMA statement. PLoS Med. 2009;6:e1000097. doi: 10.1371/journal.pmed.1000097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Higgins JP, Thompson SG, Deeks JJ, et al. Measuring inconsistency in meta-analyses. BMJ. 2003;327:557–560. doi: 10.1136/bmj.327.7414.557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Scandinavian Simvastatin Survival Study Group. Randomised trial of cholesterol lowering in 4444 patients with coronary heart disease: the Scandinavian Simvastatin Survival Study (4S) Lancet. 1994;344:1383–1389. [PubMed] [Google Scholar]
- 18.Downs JR, Clearfield M, Weis S, et al. Primary prevention of acute coronary events with lovastatin in men and women with average cholesterol levels: results of AFCAPS/TexCAPS. Air Force/Texas Coronary Atherosclerosis Prevention Study. JAMA. 1998;279:1615–1622. doi: 10.1001/jama.279.20.1615. [DOI] [PubMed] [Google Scholar]
- 19.The Long-Term Intervention with Pravastatin in Ischaemic Disease (LIPID) Study Group. Prevention of cardiovascular events and death with pravastatin in patients with coronary heart disease and a broad range of initial cholesterol levels. N Engl J Med. 1998;339:1349–1357. doi: 10.1056/NEJM199811053391902. [DOI] [PubMed] [Google Scholar]
- 20.Colhoun HM, Betteridge DJ, Durrington PN, et al. Primary prevention of cardiovascular disease with atorvastatin in type 2 diabetes in the Collaborative Atorvastatin Diabetes Study (CARDS): multicentre randomised placebo-controlled trial. Lancet. 2004;364:685–696. doi: 10.1016/S0140-6736(04)16895-5. [DOI] [PubMed] [Google Scholar]
- 21.LaRosa JC, Grundy SM, Waters DD, et al. Intensive lipid lowering with atorvastatin in patients with stable coronary disease. N Engl J Med. 2005;352:1425–1435. doi: 10.1056/NEJMoa050461. [DOI] [PubMed] [Google Scholar]
- 22.Pedersen TR, Faergeman O, Kastelein JJ, et al. High-dose atorvastatin vs usual-dose simvastatin for secondary prevention after myocardial infarction: the IDEAL study: a randomized controlled trial. JAMA. 2005;294:2437–2445. doi: 10.1001/jama.294.19.2437. [DOI] [PubMed] [Google Scholar]
- 23.Amarenco P, Bogousslavsky J, Callahan A, 3rd, et al. High-dose atorvastatin after stroke or transient ischemic attack. N Engl J Med. 2006;355:549–559. doi: 10.1056/NEJMoa061894. [DOI] [PubMed] [Google Scholar]
- 24.Ridker PM, Danielson E, Fonseca FA, et al. Rosuvastatin to prevent vascular events in men and women with elevated C-reactive protein. N Engl J Med. 2008;359:2195–2207. doi: 10.1056/NEJMoa0807646. [DOI] [PubMed] [Google Scholar]
- 25.Grover SA, Kaouache M, Joseph L, et al. Evaluating the incremental benefits of raising high-density lipoprotein cholesterol levels during lipid therapy after adjustment for the reductions in other blood lipid levels. Arch Intern Med. 2009;169:1775–1780. doi: 10.1001/archinternmed.2009.328. [DOI] [PubMed] [Google Scholar]
- 26.Hovingh GK, de Groot E, van der Steeg W, et al. Inherited disorders of HDL metabolism and atherosclerosis. Curr Opin Lipidol. 2005;16:139–145. doi: 10.1097/01.mol.0000162318.47172.ef. [DOI] [PubMed] [Google Scholar]
- 27.Nicholls SJ, Gordon A, Johansson J, et al. Efficacy and safety of a novel oral inducer of apolipoprotein a-I synthesis in statin-treated patients with stable coronary artery disease a randomized controlled trial. J Am Coll Cardiol. 2011;57:1111–1119. doi: 10.1016/j.jacc.2010.11.015. [DOI] [PubMed] [Google Scholar]
- 28.Chen Z, O’Neill EA, Meurer RD, et al. Reconstituted HDL elicits marked changes in plasma lipids following single-dose injection in C57Bl/6 mice. J Cardiovasc Pharmacol Ther. 2012;17:315–323. doi: 10.1177/1074248411426144. [DOI] [PubMed] [Google Scholar]
- 29.Nissen SE, Tsunoda T, Tuzcu EM, et al. Effect of recombinant ApoA-I Milano on coronary atherosclerosis in patients with acute coronary syndromes: a randomized controlled trial. JAMA. 2003;290:2292–2300. doi: 10.1001/jama.290.17.2292. [DOI] [PubMed] [Google Scholar]
- 30.Waksman R, Torguson R, Kent KM, et al. A first-in-man, randomized, placebo-controlled study to evaluate the safety and feasibility of autologous delipidated high-density lipoprotein plasma infusions in patients with acute coronary syndrome. J Am Coll Cardiol. 2010;55:2727–2735. doi: 10.1016/j.jacc.2009.12.067. [DOI] [PubMed] [Google Scholar]
- 31.Myers GL, Cooper GR, Winn CL, et al. The Centers for Disease Control-National Heart, Lung, and Blood Institute Lipid Standardization Program: an approach to accurate and precise lipid measurements. Clin Lab Med. 1989;9:105–135. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.