

Abstract
Objective:
The objective of this study was to develop a novel 1 month depot paclitaxel (PTX) microspheres that give a sustained and complete drug release.
Materials and Methods:
PTX loaded microspheres were prepared by o/w emulsion solvent evaporation technique using the blends of poly(lactic-co-glycolic acid) (PLGA) 75/25, polycaprolactone 14,000 and polycaprolactone 80,000. Fourier transform infrared spectroscopy was used to investigate drug excipient compatibility. Compatible blends were used to prepare F1-F6 microspheres, the process was characterised and the optimum formulation was selected based on the release. Optimised formulation was characterised for solid state of the drug using the differential scanning calorimetry (DSC) studies, surface morphology using the scanning electron microscopy (SEM), in vivo drug release, in vitro in vivo correlation (IVIVC) and anticancer activity. Anticancer activity of release medium was determined using the cell viability assay in Michigan Cancer Foundation (MCF-7) cell line.
Results:
Blend of PLGA with polycaprolactone (Mwt 14,000) at a ratio of 1:1 (F5) resulted in complete release of the drug in a time frame of 30 days. F5 was considered as the optimised formulation. Incomplete release of the drug resulted from other formulations. The surface of the optimised formulation was smooth and the drug changed its solid state upon fabrication. The formulation also resulted in 1-month drug release in vivo. The released drug from F5 demonstrated anticancer activity for 1-month. Cell viability was reduced drastically with the release medium from F5 formulation. A 100% IVIVC was obtained with F5 formulation suggesting the authenticity of in vitro release, in vivo release and the use of the formulation in breast cancer.
Conclusions:
From our study, it was concluded that with careful selection of different polymers and their combinations, PTX 1 month depot formulation with 100% drug release and that can be used in breast cancer was developed.
Keywords: Blends, complete release, microspheres, paclitaxel, polycaprolactone, polylactide-co-glycolide
INTRODUCTION
A greater attention has been recently bestowed upon the development of sustained and controlled drug delivery systems.[1,2] These dosage forms are very attractive for a variety of reasons. By now, for a large number of diseases, a large number of drugs are available. However, the effectiveness of these drugs is often limited because of the side-effects or need of invasive administration in a clinical setting.[3] The advantages with sustained or controlled delivery system are reduction in the dosing frequency, achievement of local delivery, dose reduction, side-effect reduction or uniform drug delivery.[4,5,6] There is a rejuvenated interest to develop these systems for a variety of drugs. In this study, we aimed to develop a novel controlled release parenteral depot microsphere formulation for paclitaxel (PTX), a drug useful in the treatment of breast cancer.
PTX, a poly-oxygenated naturally occurring diterpenoid isolated from the bark of the Pacific yew tree, is a microtubule-stabilizing agent.[7] It demonstrated promise to treat breast cancer. It has a low therapeutic index. It is highly lipophilic and practically insoluble in water. As it is always associated with toxic side-effects, it is used only when the potential benefits are more than the treatment risks. The generally accepted dose is 200-250 mg/m2 as a 3- or 24-h infusion, every 3 weeks. Due to its hydrophobic nature, currently, it is administered in a clinical set up using a 1:1 (v/v) mixture of ethanol and Cremophor EL, diluted in water for injection prior to infusion.[8,9,10,11] This excipient has several drawbacks which include: Instability, filtering requirements, use of non-plasticised containers, side-effects (e.g., severe hypersensitivity reactions). Low solubility, low bioavailability, side-effects of PTX therapy, need for the use of Cremophor have resulted in the development of a delivery system, which reduces systemic toxicity and increases safety and efficacy. This has led to the development of novel polymeric drug delivery systems for both systemic and local delivery of PTX, such as injectable pastes, micelles, liposomes, nanospheres, cyclodextrins complexes, emulsions, macromolecular adducts and microsphere formulations.[7] An albumin based nanoparticular formulation for PTX demonstrated significant promise over the existing clinical formulation and has been recently approved for clinical administrations as Abraxane®.[12] Another controlled release formulation that demonstrated significant promise is a biodegradable microsphere formulation. Several groups developed PTX microspheres for a variety of clinical and therapeutical applications.[13,14,15,16,17] Compared to other delivery systems investigated for PTX, microspheres have added advantages. Microspheres can improve treatment efficacy while reducing toxicity. The microspheres continue to protect the encapsulated agent after administration, permitting delivery by routes that might otherwise not be feasible. Tailored release can be achieved with microspheres by judicious selection of the polymers. Stable formulations can be developed with the use of appropriate excipients. Local and site specific drug delivery can be achieved with microspheres. In some specific cases, the size of the microspheres can be adjusted to retard the removal from the site of injection say for example the peritoneal cavity, if tumour seeding in the peritoneal cavity is treated with PTX microspheres.[18] The microspheres release encapsulated molecules over extended time intervals, from days to several months. Compared to biodegradable implants, microspheres do not result in fibrous tissue formation at the site of injection and also the drug release has been demonstrated to be more consistent.[19] Because of its short half-life, poor water solubility and therapeutic use in chronic disease, PTX has been considered to be an ideal candidate for the design of controlled release microsphere formulations.
Previous studies indicated that the preparation of microspheres is feasible for PTX using biodegradable polymers such as polylactide-co-glycolides and can be used in clinical conditions. However, studies indicated that some problems still remain.[18] Especially, PTX is very hydrophobic and thus complete release of the drug at the site of injection cannot be achieved and complete therapeutic effectiveness is not demonstrated. Further, there could be deleterious toxic effects due to the burst release of the drug that still remain in the microsphere at the site of injection. For example, Demetrick et al., (2000) developed a controlled drug delivery system for PTX using poly (L-lactic acid) to provide local delivery to the peritoneal cavity and noticed that total amount of PTX released was approximately 25% of total PTX content of microspheres.[18] Therefore, it was inferred that a substantial amount of PTX remained within the matrix after the burst and the first phase of controlled release. Several other studies demonstrated similar results. As a reason attempts were investigated to achieve complete, improved and desired release of PTX from microsphere formulations. For example, Ruan and Feng (2003) developed PTX microspheres using a new kind of copolymer poly(lactic acid)-poly(ethylene glycol)-poly(lactic acid) (PLA-PEG-PLA) and compared it's in vitro release with poly(lactic-co-glycolic acid) (PLGA) microspheres.[20] It was noticed that approximately only 20% drug was released from PLGA microspheres, whereas 49.6% sustained release of drug from PLA-PEG-PLA microspheres was achieved within 1 month. Another method that can be used to achieve complete and desired release of PTX from microspheres is with the use of blend of polymers. Blend of PLGAs and polycaprolactones in the form of PTX films resulted in a totally different kind of drug release compared to that of the films formed using pure polymers.[21] This strategy can be used to achieve time specific and complete release of the drug from the microspheres formed using the blend of these polymers. Microspheres prepared from blends of these polymers have been previously used for different purposes because of their advantage.[22] Therefore, the objective of this study was to develop a 1 month depot microsphere formulation for PTX that can lead to complete release of the drug using blends of polylactide-co-glycolide (75:25) and polycaprolactone of different molecular weights and further characterise the optimised formulation for various in vitro/in vivo properties.
MATERIALS AND METHODS
Materials
Drug and chemicals used in the study were of analytical grade and procured either as gift samples or purchased. PTX was a gift sample from Relisys Medical Devices Pvt. Ltd., Hyderabad. PLGA (75:25), polycaprolactone (M.Wts 14,000 and 80,000) were purchased from Sigma-Aldrich Ltd. Dichloromethane, polyvinylalcohol were purchased from SD Fine Chemicals Ltd. A UV-V is Spectrophotometer from Fisher scientific was used in the analysis of the samples. A Perkin-Elmer fourier transform infrared spectroscopy (FTIR) spectrophotometer was used to determine drug-polymer chemical compatibility. Differential scanning calorimetry (DSC) used to characterise the solid state of the drug in the formulation was from Shimadzu. A JSM-5200 scanning electron microscope (SEM) was used to study the surface morphology of the microspheres. All the other equipment used electronic balance, shaking water bath, high speed centrifuge were all from standard sources.
Methods
Drug-excipient compatibility studies
The drug excipient compatibility was determined with the help of FTIR. An aliquot of 5 mg of the sample was thoroughly mixed with 100 mg potassium bromide IR powder and compacted under vacuum at a pressure of about 12 Psi for 3 min. The resultant disc was mounted in a suitable holder in Perkin Elmer IR spectrophotometer and the IR spectrum was recorded from 4000 cm−1 400 cm−1 in a scan time of 12 min. This studies were performed for the physical mixtures of (a) PTX, (b) PLGA, (c) PTX with PLGA, (d) PCL-14,000, (f) PTX with PCL 14,000, (g) PTX loaded polymer blend (PLGA with PCL 14,000), (h) PTX with PCL 80,000. To prepare the sample, either 5 mg drug or 5 mg drug and 5 mg polymer combination was used.
Preparation of the PTX biodegradable polymeric microspheres
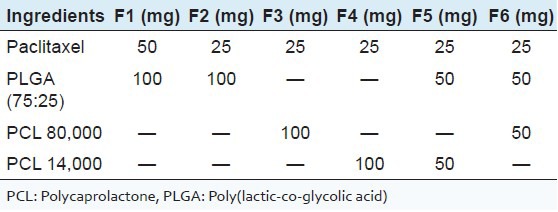
PTX polymeric microparticles were prepared using an emulsion solvent evaporation method. Briefly, to prepare the organic phase, 25 or 50 mg of drug and 100 mg polymer/polymer mixtures were dissolved in 5 ml dichloromethane [Table 1]. A 2% polyvinylalcohol (PVA) solution was used as the aqueous phase. Microspheres were prepared by slowly injecting the organic phase into the aqueous solution of PVA, which was placed on a magnetic stirrer. This results in o/w emulsion. The magnetic stirrer was maintained at a required speed for 3-4 h to evaporate the organic solvent. Finally, the particles formed were filtered and then kept in dessicator overnight for the absorption of moisture. Placebo microspheres were also prepared using the same method without incorporating the drug in the organic phase.
Table 1.
Compositions used in the preparation of different microsphere formulations
Evaluation of microspheres
Particle size, encapsulation and in vitro drug release
The mean particle size and the shape were determined by using an optical microscope. In this method, size of 25 particles was determined by using the stage micrometer and the average size was determined. Percentage practical yield was calculated. Yield was determined as the weight of the microspheres recovered with respect to sum of all the starting material. The percentage yield of prepared PTX microspheres was determined using the formulae:
Percentage yield = (practical yield/theoretical yield) × 100
Percentage encapsulation was determined using the UV-V is assay method based standard curve developed using the drug spiked placebo particles. A 10 mg of each batch of microspheres were dissolved in 5 ml of dichloromethane in a test-tube, shaken well and was left open overnight for the evaporation of dichloromethane. Then, 10 ml of methanol was added to the remnants in the test-tube, stirred well, filtered and then analysed for the drug content in UV spectroscopy at 227 nm. Efficiency of drug entrapment for each batch was calculated in terms of percentage drug entrapment as per the following formula:
PDE = (Practical drug content/theoretical drug content) × 100
To determine in vitro release, a method previously published in the literature was used.[23] Briefly, each batch of PTX microspheres containing 3.5 mg of the drug were taken in microtubes and then suspended in 1 ml distilled water. These microtubes were then placed in horizontal shaking water bath. The entire set up was maintained at 37°C in a water bath. At the end of a specific time points, these microtubes were centrifuged at a speed of 5000 rpm for 10 min. After centrifugation of samples, 1 ml of supernatant solution was withdrawn, filtered and the drug content was analysed. The amount of PTX released was analysed by UV spectrophotometer at the λmax value 227 nm using the distilled water as the blank. Meanwhile, the volume withdrawn was replenished with an equal volume of fresh distilled water in the microtubes, the particles resuspended and placed again in the water bath to continue the release study. The release study was performed for 1 month.
Surface morphology
In order to examine the particle surface morphology and shape, SEM was used. Dry PTX microspheres were spread over a slab. The sample was shadowed in a cathodic evaporator with gold layer 20 nm thick. Photographs were taken using a JSM-5200 SEM (Tokyo, Japan) operated at 20 kV. Pictures of PTX microspheres were taken by random scanning of slab.
Solid state characterization of the drug in the microspheres
Thermal properties of the powder samples were investigated using the differential scanning Calorimetry. Approximately, 10 mg of the sample was analysed in an open aluminium pan and heated at scanning rate of 10°C/min between 0°C and 300°C. Empty pan was used as the standard reference material. The thermograms of PTX, physical mixture of blend of PLGA and PCL 14,000, physical mixture of drug with the blend of polymers PLGA and PCL 14,000, PTX microspheres and Placebo microspheres were obtained.
In vivo drug release and in vitro in vivo correlation (IVIVC) studies
In vivo drug release study was conducted in rats after getting approval from an Ethical Committee constituted for this project in Vaagdevi College of Pharmacy, Warangal, Andhra Pradesh, India. The approval number is vcop/2012/6/1. After the quarantine period, rats were divided into two groups. Each groups contained six rats. One group was administered solution via i.v. route at an equivalent dose that is injected in the form of biodegradable microspheres. Other group was injected with biodegradable microspheres intraperitoneally. In vivo data were obtained for only the optimized formulation; this was considered to be the final formulation developed in this study. From the in vitro release data, F5 was considered to be the optimum formulation. A 500 μl of this suspension containing 3.5 mg of the drug were injected into each rat. In case of i.v. administration 3.5 mg of PTX was dissolved in 1:1 combination of Cremophor EL and ethanol. A volume of 1 ml of the formulation was injected. Blood samples (0.5 ml) were collected from retro orbital sinus of rat eye under anaesthesia at intervals of 1, 3, 6, 12 and 24 h in case of i.v. solution. For microparticular system along with the above time intervals samples were also collected periodically till 40 days. The blood samples so collected were added to a series of graduated micro centrifuge tubes containing 0.3 ml of sodium citrate solution (4% w/v in water). All the samples were centrifuged at 3000 rpm for 10 min and plasma was separated into other micro centrifuge tube by using the micro pipette and stored in the deep freeze. The drug was extracted from the plasma by adding 500 μl of ethyl acetate, and vortexed on cyclo mixer for 20 min. The organic phase was separated and collected into another micro centrifuge tube and allowed to air dry by keeping the lid of the tube open for 24 h. These dried tubes were stored in deep freeze until high pressure liquid chromatography (HPLC) analysis was performed. HPLC analysis samples were reconstituted with 50 μl of mobile phase (acetonitrile:water, 60:40) and analysed at 227 nm wavelength. In Vivo release profile was then constructed.
IVIVC was established according to Drewe and Guitard basing on degree A.[24] The parameters compared were cumulative absorption profile to that of in vitro dissolution profile, i.e., correlation of the amount of drug dissolved to that of respective fraction of dose absorbed (T50). Cumulative amount of the drug absorbed was calculated using Wagner-Nelson method approximating the kinetics of the drug to one compartment open model. According to Wagner-Nelson method, the cumulative amount of drug released from the microspheres into the systemic circulation in a rat was calculated using the equation given below:
Ab/Ab∞ = (Cp + K[AUC]0t)/K[AUC]0∞
Where Ab is the cumulative amount released at any time, Ab∞ is the dose administered, Cp is the plasma concentration at any time t, K is the elimination rate constant and area under the curve (AUC) is the area under the curve. K was also determined in this study in another set of rats where PTX was administered via i.v. route.
In vitro cytotoxicity studies
In vitro cytotoxicity studies were performed as previously described in the literature.[16] Human breast carcinoma MCF-7 (procured from National Cell Science Centre, Pune, India) cells were grown in monolayer and then used in the studies. From F5 formulation, the release media was collected sterile filtered and MTT assay was conducted with this filtrate. The prepared formulation F5 microspheres were subjected for cell lines studies. The release media was collected for over a period of 1 month. Release media collected from drug suspension and from the placebo microspheres were used as controls. Cell viability was assessed using the MTT assay.[16]
Statistical analysis
All experiments were performed six times and the data were expressed as mean ± SD and Tukey's post hoc test was performed to analyse the significance of difference between different groups using the statistical analysis software package SPSS (Version 16.0, IBM, USA).
RESULTS
FTIR results were obtained with pure drug and various blends. The spectrum of PTX drug has shown characteristic peaks at 1715 cm−1 (ketone group, C = Ostretching vibrations are seen at 1715 cm−1), 3441.12 cm−1 (OH group, broad), 1647.26 cm−1 (amide group, CONH2), 709.83 cm−1 (C-H aromatic ring), 1244.13 cm−1 (C-O group). Some bands of PTX are not prominent in the physical mixture of drug-loaded with the polymer blend since these are identical to those of polymer blend (PLGA + PCL 14,000) and appear at almost the same wave numbers. The spectra of PTX loaded polymer blend were not characteristically different from the spectra of the PTX. The peaks appearing at 3441.12, 3838.47, 1647.26, 1602.90, 1541, 491 and 472 cm−1 for PTX were also appearing in PTX–loaded polymer blend, indicating the chemical stability of the drug in the blend. Six microsphere formulations were prepared with the biodegradable polymers PLGA 75/25, PCL 14,000 PCL 80,000 and with their blends. The process development was characterised. Upon fabrication of batch F1, during filtration it was noticed that a large proportion of the drug was not incorporated into the microspheres. Thus, in the subsequent batches the batch size was changed by reducing the drug level taken to fabricate the microspheres in order to reduce the precipitation of the drug. In F2, upon reduction in the drug amounts, drug precipitation was not observed, but the particles were irregular in shape and randomly dispersed. In F3, PCL-80,000 was used as polymer. Many irregular shaped particles of smaller and bigger sizes as well as clusters were noticed. In F4, PCL-14,000 was employed. The particles were fine, but many powder like particles were dispersed in between the microspheres, suggesting incomplete drug encapsulation. In the next two batches, the blend of polymer PLGA with PCL was used. In F5, PLGA with PCL-14,000 blend was used. Particles appeared to be clear and spherical. The next batch was formulated to see whether the particles further improvement in particles can be achieved. In F6, the polymer blend PLGA with PCL-80,000 was used and it was noticed that the particle size increased when compared to F5. The particles were not clear and very few spherical particles were found. The average particle size of each batch for formulations F1, F2, F3, F4, F5, F6 are 12, 10.4, 15.6, 12.4, 7.2, 15.6, μm respectively. F5 formulation demonstrated optimum results. The results of percentage drug entrapment efficiency are shown in the Table 2. The percentage entrapment efficiency was found to be 5.9% to 28.2%. The plots of cumulative percentage drug release versus time for all the six formulations were drawn and represented graphically as shown in Figure 1. Microspheres demonstrated prolonged and sustained release of PTX. The formulations F5 showed complete release (100%) of the entrapped drug in 30 days and F3 showed a minimum of 17.12% cumulative drug release during the same time. F5 was considered to be the optimum formulation. F5 formulation was characterised for solid state analysis by DSC and for surface morphology by SEM in the DSC curves displayed in Figure 2, melting endotherm of pure PTX was found to be 224.4°C. There was no peak detected in the temperature ranges of 150-250°C for blank microspheres and the optimised formulation. Surface morphology of the optimized microsphere formulation was examined by SEM [Figure 3]. Microspheres were smooth and spherical.
Table 2.
Entrapment efficiency of different batches of microspheres
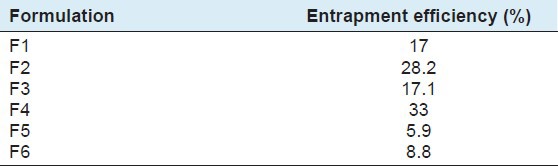
Figure 1.
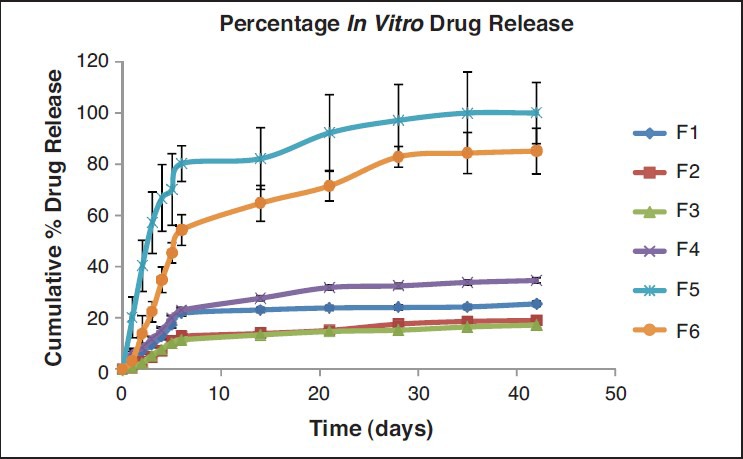
In Vitro release profile of paclitaxel from the different microsphere formulations (key: F1: ♦; F2: ▪; F3: ▴; F4: X; F5: +; F6:•)
Figure 2.
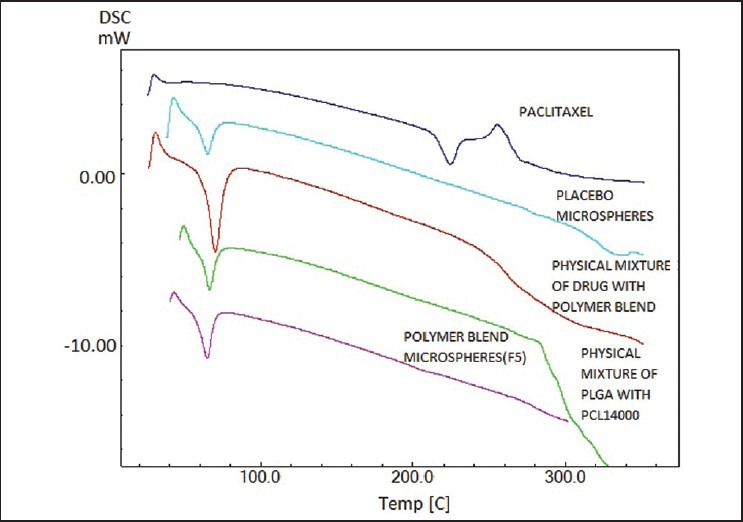
Thermograms of paclitaxel, placebo microspheres, physical mixture of drug with polymer blend, physical mixture of poly(lactic-co-glycolic acid) with PCL 14,000, polymer blend microspheres (F5)
Figure 3.

Scanning electron microscopy pictures of paclitaxel microspheres from F5 batch
HPLC method adopted in the study was successfully employed. The retention time of the drug was 8 min. Extraction efficiency of the drug was 80%. A linear standard curve was obtained between 0.015-2 mcg/ml. An elimination rate constant of 1.5/h was obtained with PTX administered via the i.v. route. The optimum formulation resulted in 1 month and complete release in vivo [Figure 4]. Further, IVIVC was also established. IVIVC was found to be 100% with the optimum formulation [Figure 5]. The release media from the optimized formulation demonstrated anticancer activity in breast cancer cells for over a period of 1 month. The data are shown in Figure 6 while that of placebo did not show any effects. The effects with suspension formulation did not last for more than 2 days.
Figure 4.
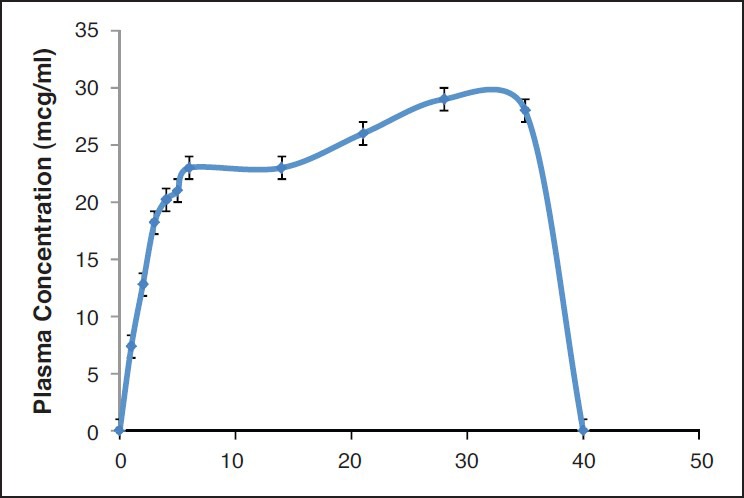
Plasma concentration time after one time administration of optimized microsphere formulation
Figure 5.
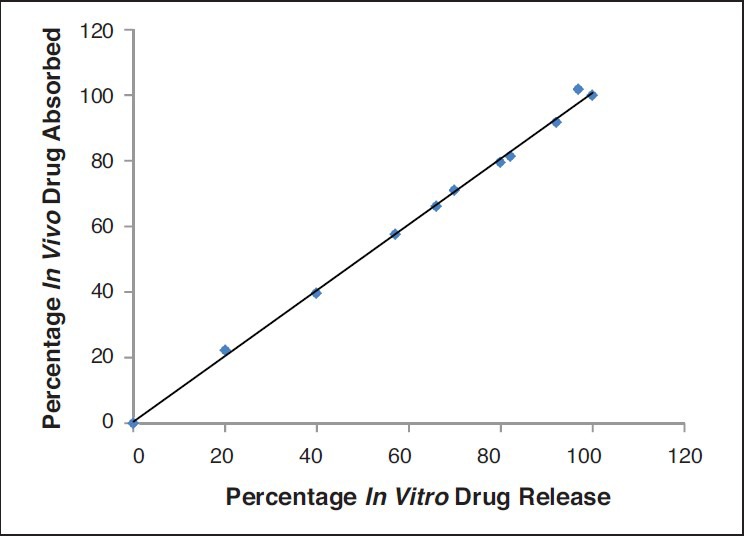
In Vitro in vivo correlation of drug release from optimized microspheres
Figure 6.
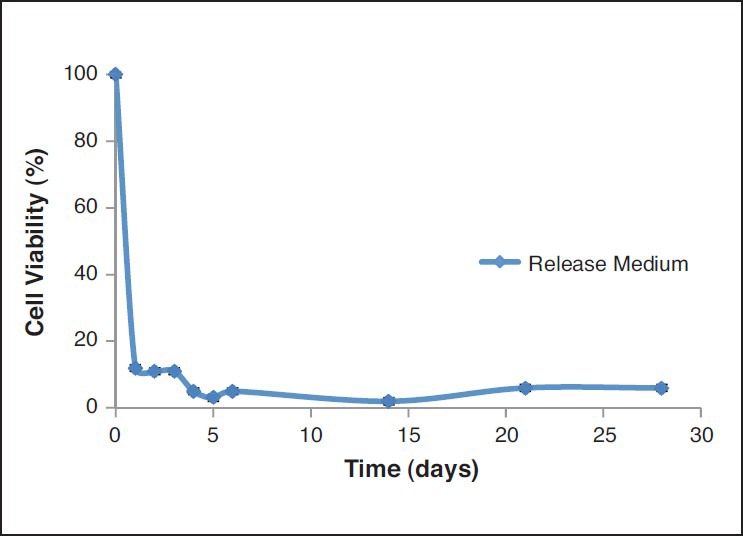
Cell viability of the release medium from F5 formulation during 30 days
DISCUSSION
FTIR has been performed to determine the drug polymer compatibility and to select the suitable blends so as to formulate the microspheres without any drug-polymer interaction. FTIR spectral data could confirm the drug: Excipient interaction/chemical stability of PTX in the drug loaded polymer blend. All the three polymers PLGA (75/25), polycaprolactone 14,000 and polycaprolactone 80,000 retained the chemical integrity of the drug. Thus, microspheres of PTX were prepared with these polymers individually or using their mixture. The blends retained the integrity of drug and as a reason these polymers were selected for further studies. Different microsphere formulations F1-F6 were prepared. The process development suggested that F5 was the best formulation for further studies. In the case of batches, which had unencapsulated drug, the particles were recovered using sieving. A maximum of 28.2% drug entrapment efficiency was obtained in the PTX microspheres of formulation F2 in which the amount of drug incorporated was 25 mg and the PLGA concentration was 100 mg. It was noticed that the formulation F5, which was the preferred formulation based on the process development has shown the lowest drug entrapment efficiency which may be because of low solubility of drug in the polymer blend. In vitro drug release from PTX microspheres was determined. It was noticed that PTX release rate decreased in the order: PLGA: PCL, PLGA, PCL. From formulation F5, drug release may be due to diffusion, but not due to polymer degradation. As the formulation (F5) prepared using a blend of PLGA with PCL 14,000 at a ratio 1:1 resulted in complete release of drug over a period of 30 days, it was considered as the optimized formulation. Although F5 had low entrapment, the aim of the study was to develop a 1 month depot microspheres of PTX and that can result in complete release and in our future studies we would improve this formulation further to improve the entrapment efficiency. However, as the objectives of the study were already met, we continued further studies with F5. DSC studies were performed to understand the nature of the encapsulated drug in the matrix. The physical state of PTX in the polymer matrix would also influence its release characteristics. The absence of drug peak may be due to conversion of PTX from crystalline state to amorphous. SEM pictures indicated that the microspheres were spherical and had a smooth surface.
Although in vitro studies help in the design of suitable microsphere formulations, optimal design of controlled release systems requires a thorough understanding of pharmacokinetics and pharmacodynamics of drug with and without the delivery system. Further, IVIVC is helpful in the product development. The development of an IVIVC is a dynamic process starting from the very early stages of the development program through the final step. The comprehension of IVIVC of delivery is essential to better tailor the delivery system. In vitro release studies demonstrated that optimised formulation resulted in 1 month and complete release of the drug from the microspheres. However, simple in vitro release may not indicate the same effects in vivo. Therefore, we also conducted studies to determine in vivo release. The optimum formulation also resulted in 1 month and complete release in vivo. Further, IVIVC was also established. This correlation was 100% suggesting that results from in vitro studies are valid. Further, the pharmacodynamics were determined in a cell culture model. Inhibitory concentration (IC50) of PTX in MCF-7 cells was found to be 90 ± 3 ng/ml. The release media from the PTX microspheres (F5) resulted in greater cell growth inhibition compared to that of the release media from placebo microspheres. During the entire release period, cytotoxicity was noted. This could be attributed to the released PTX from the microspheres. The cytotoxicity against MCF-7 cell lines was affected significantly by the released amount of PTX. The results indicate that PTX release from microspheres was sustained and effective in inhibiting the cell growth. The results are shown in the Figure 6 Since 100% IVIVC was obtained and the release media demonstrated anticancer activity in breast cancer cells, this simple in vitro experiment could indicate that the formulation can effectively be used in the treatment of breast cancer. Complicated breast cancer animal models were thus avoided. Thus, all these studies indicate that a 1 month completely releasing depot formulation for PTX that can be used in the treatment of breast cancer was successfully developed. A blend of polymers can thus be used to develop a suitable formulation as per the needs and can improvise the problems existing with the use of individual polymers. Rather than synthesising a new and expensive polymer, a simple blend of polymers can thus be successfully used to obtain the desired release. This strategy was proved helpful in the development of improved and novel microsphere formulation for PTX that can be used in breast cancer.
CONCLUSIONS
PTX is used in the treatment of various cancers including breast cancer. A sustained release dosage form for PTX has added advantages. In this study, PTX microspheres that can release the drug for 1 month were successfully prepared using blends of polylactide-co-glycolide and polycaprolactone. It can be concluded that with careful selection of different polymers and their combinations, PTX 1 month depot formulation, which completely releases the drug can be developed. The formulation can be used successfully in the treatment of breast cancer.
Footnotes
Source of Support: Nil
Conflict of Interest: None declared
REFERENCES
- 1.Path S, Dara P, Yamsani SK, Thadkapally R, Aukunuru J. Development and evaluation of oral elementary osmotic pump tablets for ropinirole hydrochloride. Indian Drugs. 2012;49:23–30. [Google Scholar]
- 2.Anampally S, Aukunuru J. Development of granisetron transdermal films: In vitro and ex vivo characterization. Indian Drugs. 2010;47:21–8. [Google Scholar]
- 3.Anampally S, Mankala SK, Gogu PK, Aukunuru J. Development of ropinirole (free base) transdermal patch us ing blends of hydroxypropyl methylcellulose/eudragits and it's in vitro/in vivo characterization. A J Pharm Res Health Care. 2011;3:89–98. [Google Scholar]
- 4.Anuradha CA, Aukunuru J. Preparation, characterisation and in vivo evaluation of bis-demethoxy curcumin analogue (BDMCA) nanoparticles. Trop J Pharm Res. 2010;9:51–8. [Google Scholar]
- 5.Gaddam N, Aukunuru J. Systemic delivery of diclofenac sodium after topical application of gels incorporated with drug-loaded solid lipid nanoparticles (SLN) J Pharm Res Health Care. 2010;2:177–87. [Google Scholar]
- 6.Konatham S, Reddy B, Aukunuru J. Enhanced liver delivery and sustained release of curcumin with drug loaded nanoparticles after intravenous administration in rats. A J Pharm Res Health Care. 2011;3:99–108. [Google Scholar]
- 7.Marupudi NI, Han JE, Li KW, Renard VM, Tyler BM, Brem H. Paclitaxel: A review of adverse toxicities and novel delivery strategies. Expert Opin Drug Saf. 2007;6:609–21. doi: 10.1517/14740338.6.5.609. [DOI] [PubMed] [Google Scholar]
- 8.Gelderblom H, Mross K, ten Tije AJ, Behringer D, Mielke S, van Zomeren DM, et al. Comparative pharmacokinetics of unbound paclitaxel during 1-and 3-hour infusions. J Clin Oncol. 2002;20:574–81. doi: 10.1200/JCO.2002.20.2.574. [DOI] [PubMed] [Google Scholar]
- 9.Gelderblom H, Verweij J, Nooter K, Sparreboom A. Cremophor EL: The drawbacks and advantages of vehicle selection for drug formulation. Eur J Cancer. 2001;37:1590–8. doi: 10.1016/s0959-8049(01)00171-x. [DOI] [PubMed] [Google Scholar]
- 10.Gelderblom H, Verweij J, van Zomeren DM, Buijs D, Ouwens L, Nooter K, et al. Influence of Cremophor El on the bioavailability of intraperitoneal paclitaxel. Clin Cancer Res. 2002;8:1237–41. [PubMed] [Google Scholar]
- 11.Sparreboom A, van Zuylen L, Brouwer E, Loos WJ, de Bruijn P, Gelderblom H, et al. Cremophor EL-mediated alteration of paclitaxel distribution in human blood: Clinical pharmacokinetic implications. Cancer Res. 1999;59:1454–7. [PubMed] [Google Scholar]
- 12.Luo C, Wang Y, Chen Q, Han X, Liu X, Sun J, et al. Advances of paclitaxel formulations based on nanosystem delivery technology. Mini Rev Med Chem. 2012;12:434–44. doi: 10.2174/138955712800493924. [DOI] [PubMed] [Google Scholar]
- 13.Achim M, Tomuta I, Vlase L, Iuga C, Moldovan M, Leucuta SE. Paclitaxel-loaded poly(lactic-co-glycolic acid) microspheres: Preparation and in vitro evaluation. J Drug Deliv Sci Technol. 2008;18:410–6. [Google Scholar]
- 14.Guilei M, Song C. PCL/poloxamer 188 blend microsphere for paclitaxel delivery: Influence of poloxamer 188 on morphology and drug release. J Appl Polym Sci. 2007;104:1895–9. [Google Scholar]
- 15.Guo K, Chu CC. Biodegradable and injectable paclitaxel-loaded poly(ester amide)s microspheres: Fabrication and characterization. J Biomed Mater Res B Appl Biomater. 2009;89:491–500. doi: 10.1002/jbm.b.31239. [DOI] [PubMed] [Google Scholar]
- 16.Hiremath J, Kusum Devi V. Preparation and in vitro characterization of paclitaxel-loaded injectable microspheres. A J Pharm. 2010;4:205–11. [Google Scholar]
- 17.Song T, Yuan X, Sun A, Wang H, Kang C, Ren Y, et al. Preparation of injectable paclitaxel sustained release microspheres by spray drying for inhibition of glioma in vitro. J Appl Polym Sci. 2010;115:1534–9. [Google Scholar]
- 18.Liggins RT, D’Amours S, Demetrick JS, Machan LS, Burt HM. Paclitaxel loaded poly(L-lactic acid) microspheres for the prevention of intraperitoneal carcinomatosis after a surgical repair and tumor cell spill. Biomaterials. 2000;21:1959–69. doi: 10.1016/s0142-9612(00)00080-6. [DOI] [PubMed] [Google Scholar]
- 19.Murthy RS. Implantable therapeutic systems. In: Jain NK, editor. Advances in Controlled and Novel Drug Delivery Systems. 1st ed. Delhi: CBS publishers and Distributors; 2001. [Google Scholar]
- 20.Ruan G, Feng SS. Preparation and characterization of poly(lactic acid)-poly(ethylene glycol)-poly(lactic acid) (PLA-PEG-PLA) microspheres for controlled release of paclitaxel. Biomaterials. 2003;24:5037–44. doi: 10.1016/s0142-9612(03)00419-8. [DOI] [PubMed] [Google Scholar]
- 21.Lao LL, Venkatraman SS, Peppas NA. Modeling of drug release from biodegradable polymer blends. Eur J Pharm Biopharm. 2008;70:796–803. doi: 10.1016/j.ejpb.2008.05.024. [DOI] [PubMed] [Google Scholar]
- 22.Mundargi RC, Srirangarajan S, Agnihotri SA, Patil SA, Ravindra S, Setty SB, et al. Development and evaluation of novel biodegradable microspheres based on poly(d,l-lactide-co-glycolide) and poly(epsilon-caprolactone) for controlled delivery of doxycycline in the treatment of human periodontal pocket: In vitro and in vivo studies. J Control Release. 2007;119:59–68. doi: 10.1016/j.jconrel.2007.01.008. [DOI] [PubMed] [Google Scholar]
- 23.D’Souza SS, DeLuca PP. Methods to assess in vitro drug release from injectable polymeric particulate systems. Pharm Res. 2006;23:460–74. doi: 10.1007/s11095-005-9397-8. [DOI] [PubMed] [Google Scholar]
- 24.Drewe J, Guitard P. In vitro-in vivo correlation for modified-release formulations. J Pharm Sci. 1993;82:132–7. doi: 10.1002/jps.2600820204. [DOI] [PubMed] [Google Scholar]