

Abstract
Context:
To date, many findings reveal that most of the modern drugs have the ability to interact with herbal drugs.
Aims:
This study was conducted to determine the inhibitory effects of mitragynine on cytochrome P450 2C9, 2D6 and 3A4 activities.
Methods and Material:
The in vitro study was conducted using a high-throughput luminescence assay.
Statistical Analysis:
Statistical analysis was conducted using one-way ANOVA and Dunnett's test with P < 0.05 vs. control. The IC50 values were calculated using the GraphPad Prism® 5 (Version 5.01, GraphPad Software, Inc., USA).
Results:
Assessment using recombinant enzymes showed that mitragynine gave the strongest inhibitory effect on CYP2D6 with an IC50 value of 0.45±0.33 mM, followed by CYP2C9 and CYP3A4 with IC50 values of 9.70±4.80 and 41.32±6.74 μM respectively. Positive inhibitors appropriate for CYP2C9, CYP2D6, and CYP3A4 which are sulfaphenazole, quinidine and ketoconazole were used respectively. Vmax values of CYP2C9, CYP2D6 and CYP3A4 were 0.0005, 0.01155 and 0.0137 μM luciferin formed/pmol/min respectively. Km values of CYP2C9, CYP2D6, and CYP3A4 were 32.65, 56.01, and 103.30 μM respectively. Mitragynine noncompetitively inhibits CYP2C9 and CYP2D6 activities with the Ki values of 61.48 and 12.86 μM respectively. On the other hand, mitragynine inhibits CYP3A4 competitively with a Ki value of 379.18 μM.
Conclusions:
The findings of this study reveal that mitragynine might inhibit cytochrome P450 enzyme activities, specifically CYP2D6. Therefore, administration of mitragynine together with herbal or modern drugs which follow the same metabolic pathway may contribute to herb-drug interactions.
Keywords: Cytochrome P450, herb-drug interactions, luminescence, mitragynine
INTRODUCTION
Mitragyna speciosa Korth. can be found in the tropical and sub-tropical regions of Asia. This Rubiaceae family plant which is known as ketum or biak-biak in Malaysia and kratom in Thailand has been used as a folk medicine since early times. In general, two varieties of ketum had been identified, one with red veins in the leaf and the other one with green veins. Mitragyna speciosa with red veins exhibits stronger biological activities than the variety with green veins.[1] Ketum is currently being used by drug users in two situations, either for reducing opium addiction or as opium replacement when opium is unavailable.[2] Mitragynine [Figure 1] had been obtained as a major constituent in Mitragyna speciosa leaves with 66% of the total alkaloids in the Thai species and 12% of the total alkaloids in the Malaysian species. Other compounds which are present in this plant are paynantheine, speciogynine, 7-hydroxymitragynine and speciociliatine.[3]
Figure 1.
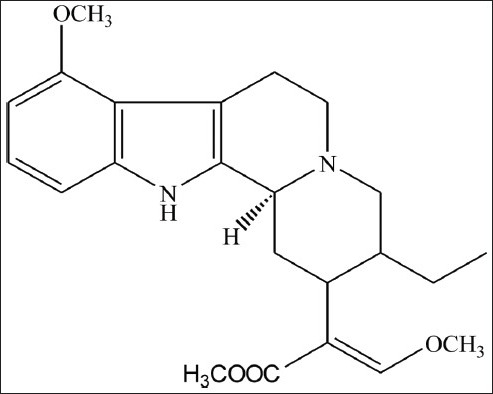
Chemical structure of mitragynine
Previous findings showed action of an antinociceptive through the supraspinal opioid receptors when mitragynine was administered. This activity was mediated by μ and δ opioid receptors.[4,5] In Thailand, the usage of this plant in drug treatment program and drug detoxification are due to these findings[6] There are proof that mitragynine is identical to codeine in term of antitussive and as analgesic drugs. In antinociceptive activity, Mitragyna speciosa and morphine were known to be more potent than mitragynine.[3,7]
Cytochrome P450 enzymes are the multi-genes family of heme-containing enzymes that are responsible for the oxidative metabolism of many xenobiotics including carcinogens, chemicals and most therapeutic drugs.[8] Any chemical compounds which do not belong to the common composition of the human body are known as xenobiotics. These compounds enter the body through diet, air and medication. Therapeutic drugs as well as xenobiotics are eliminated from the body via the process of metabolism.[9]
The basis of drug metabolism is to increase the water solubility of a lipophilic compound which makes them more water-soluble, hence readily excreted in urine or bile.[10] Most drug compounds are metabolized by the multi-gene family of heme-containing enzymes known as cytochrome P450 enzymes (CYPs). To date, more than 70% of all therapeutic drugs are metabolized mainly by CYP 3A4, 2D6, 1A2, 2C9 and 2C19.[11]
The effect of Mitragyna speciosa methanolic extract on CYP450 enzyme activities using a luminescent assay was investigated.[12] Since mitragynine is the major alkaloid in the extract, we aimed to investigate if the effects seen in Mitragyna speciosa methanolic extract were caused primarily by mitragynine. One important in vitro model for studying the drug effects on cytochrome P450 enzyme activity is recombinant enzymes. The recombinant expressed enzymes are frequently used to determine the risk of drug-drug interactions related to enzyme inhibition and for drug clearance prediction.[13] The most commonly used expression systems are the baculovirus systems. The recombinant expressed enzyme system is the simplest in vitro model. This model produced in endoplasmic reticulum of eukaryotic host cell and contains individual human cytochrome P450 enzyme and P450 reductase (and cytochrome b5 for CYP2C9, CYP2C19 and CYP3A4).[14] Therefore, the aim of the present study was to investigate the inhibitory effect of mitragynine on cytochrome P450 2C9, 2D6 and 3A4 activities using recombinant CYP450 enzymes.
MATERIALS AND METHODS
Chemicals and Reagents: This study was carried out from September 2010 to July 2011 at the Centre for Drug Research, Universiti Sains Malaysia, Malaysia. This assay was carried out using the P450-Glo™ Screening Systems from Promega, USA. It contained recombinant human cytochrome P450 2C9, 2D6 and 3A4 enzymes in the microsomes produced by baculovirus-infected insect cells, membrane fraction without any cytochrome P450 enzyme activities, the luminogenic cytochrome P450 substrates [6’deoxyluciferin (Luciferin-H), ethylene glycol ester of luciferin 6’ methyl ether (Luciferin ME-EGE) and luciferin 6’ benzyl ether (Luciferin-BE)], an nicotinamide adenine dinucleotide phosphate (NADPH) regeneration system, 1M potassium phosphate buffer, pH 7.4, luciferin detection reagent and luciferin-free water. Positive inhibitors which are sulfaphenazole, quinidine and ketoconazole were purchased from Sigma Chemicals (St. Louis, USA), ACROS Organics and U.S. Pharmacopeia respectively. Ethanol 96% was purchased from AR Grade Qrëc™. Acetonitrile was purchased from Fisher Scientific (Loughborough, UK). Mitragynine was isolated at the Centre for Drug Research, Universiti Sains Malaysia (USM), Malaysia.
Preparation of mitragynine
Mitragynine was weighed and dissolved fully in 96% ethanol. 15mM of mitragynine stock solution was prepared followed by a serial dilution for required concentration. The final percentage of ethanol was ensured to be less than 2% to avoid any inhibition caused by solvent.
Enzyme assay
The luminescence assay was carried out as described in P450-Glo™ Screening Systems by Promega, USA. The range of mitragynine and positive inhibitors chosen was 0.02-200μM. The assay was carried out in 96 white flat-bottom plates (Greiner Bio-one, USA). 6’deoxyluciferin (Luciferin H), ethylene glycol ester of luciferin 6’methyl ether (Luciferin ME-EGE) and luciferin 6’benzyl ether (Luciferin-BE) were used as substrates for CYP2C9, CYP2D6 and CYP3A4 respectively. The mixture was pre-incubated for 15 min after addition of mixture reaction (membrane with enzyme, substrate and 1M potassium phosphate buffer) into plates containing various concentrations of the mitragynine. NADPH regeneration system was added into each well to initiate the reaction, followed by 30 min incubation for CYP2C9 and CYP3A4. For CYP2D6, 45 min incubation time was needed. After that, luciferin detection reagent was added to stop the reaction and the luminescence measurement was taken. Incubation of 20 min was performed to stabilize the luminescence. Luminescence was detected using microplate reader and values were displayed as relative light unit (RLU).
Determination of enzyme kinetic parameters
Maximum velocity, Vmax, and Michaelis-Menten constant, Km, for CYP2C9, CYP2D6 and CYP3A4 were assessed from nonlinear regression plots of velocity versus substrate concentrations. Experiments were conducted with increasing concentration of substrates until there was no more increase in velocity. Km is half of the Vmax. For inhibition constant, Ki and the mode of inhibition determination, a series of experiments were conducted. Experiments were carried out with 4 to 5 concentrations of specific substrate with a range of inhibitor concentrations (0–100μM).
Statistical analysis
Results were represented as the mean ± SEM of the three replicates with two independent experiments (n=6). Statistical analysis was conducted using one-way ANOVA and Dunnett's test with P < 0.05 vs. control. The IC50 values were calculated using the GraphPad Prism® 5 (Version 5.01, GraphPad Software, Inc., USA) by plotting the log concentration of the mitragynine versus percentage inhibition of cytochrome P450 enzyme activities. Kinetic parameters, Ki and mode of inhibition were determined by linear regression using Microsoft Excel. Vmax and Km values were determined using nonlinear regression plots using GraphPad Prism® 5 (Version 5.01, GraphPad Software, Inc., USA). Lineweaver-Burk plots were used to determine the mode of inhibition graphically. Double-reciprocal plots of slope from Lineweaver-Burk plots versus inhibitor concentrations were used to determine the Ki values. Experiments were carried out in triplicates.
RESULTS
Mitragynine gave potent inhibitory effect on CYP2D6 with an IC50 value of 0.45 ± 0.33 μM [Table 1]. On the other hand, mitragynine showed moderate inhibitory effect on CYP2C9 and CYP3A4, with IC50 values of 9.70 ± 4.80 and 41.32 ± 6.74 μM respectively. The inhibition ranking order based on IC50 values is CYP2D6 > CYP2C9 > CYP3A4 and the inhibitory effect of mitragynine on CYP2C9, CYP2D6, and CYP3A4 is illustrated in Figure 2. Figure 2 shows that mitragynine started inhibiting CYP2D6 significantly at the concentration of 0.02 μM and gave almost 90% of inhibition at 200 μM. On the other hand, CYP2C9 and CYP3A4 showed about 60% of inhibition at 200 μM. Positive inhibitors, which are sulfaphenazole, quinidine, and ketoconazole were used to validate this assay. The IC50 values of sufaphenazole, quinidine and ketoconazole were 0.26 ± 0.15, 0.024 ± 0.004 and 0.11 ± 0.06 μM respectively [Table 1].
Table 1.
IC50 values of mitragynine
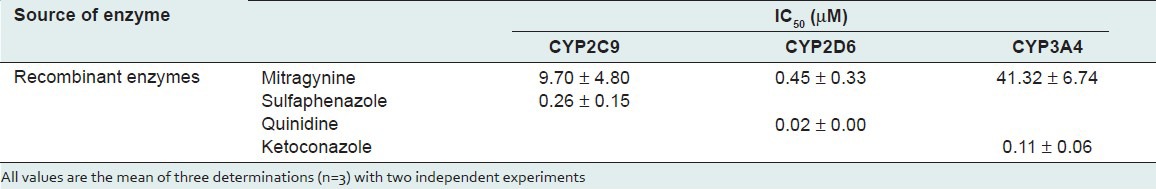
Figure 2.
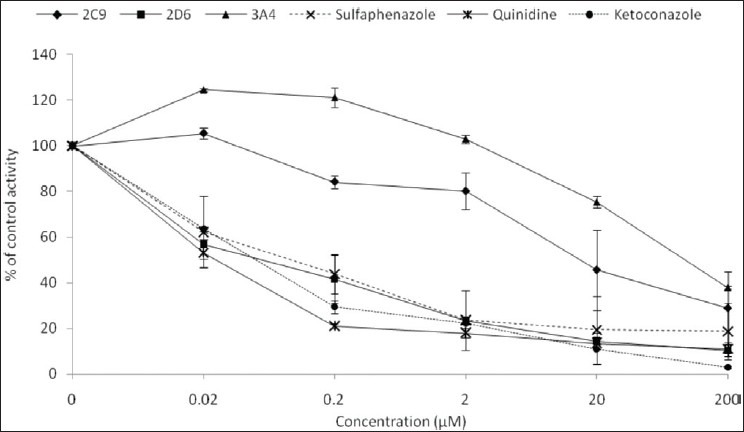
Inhibitory effect of mitragynine, sulfaphenazole, quinidine and ketoconazole on human cytochrome P450 2C9, 2D6 and 3A4 using recombinant enzymes. Each data represents the value of mean ± SEM for three determinations (n=3) of two independent experiments
Kinetic parameters of mitragynine on cytochrome P450 2C9, 2D6 and 3A4 were obtained using recombinant enzymes. Four parameters have been determined—maximum velocity (Vmax), Michaelis-Menten constant (Km) and inhibitor constant (Ki) as well as the mode of inhibition. Our findings show that the corresponding Vmax values of CYP2C9, CYP2D6, and CYP3A4 were 0.00050, 0.0116, and 0.0137 μM luciferin formed/min/pmol CYP450 respectively [Table 2]. Km values of CYP2C9, CYP2D6, and CYP3A4 were 32.65, 56.01, and 103.3 μM correspondingly [Table 2]. According to the Lineweaver-Burk plots, mitragynine is a noncompetitive inhibitor of CYP2C9 [Figure 3] with the Ki value of 155.80 μM [Figure 4; Table 3]. Figure 5 shows that mitragynine is a noncompetitive inhibitor towards CYP2D6 with the Ki value of 12.86 μM [Figure 6; Table 3]. On the other hand, mitragynine is a competitive inhibitor towards CYP3A4 [Figure 7] with the Ki value of 379.18 μM [Figure 8; Table 3]. The mode of inhibition can be determined graphically using the Lineweaver-Burk plot as shown in Figures 3, 5 and 7.[15] This plot is the linearization of the Michaelis-Menten plot and is known to be very useful in analyzing the pattern of enzyme inhibition. The pattern of straight lines formed was used to differentiate the mode of inhibition. Competitive inhibition shows the series of lines converging above the x-axis whereas the noncompetitive inhibition shows the series of lines converging on the x-axis. The double reciprocal plot of Lineweaver-Burk plot versus mitragynine concentrations was plotted as shown in Figures 4, 6 and 8 to determine the Ki values. The Ki value is the value where the line intercepts at the x-axis (-Ki = - x-axis interception).
Table 2.
Vmax and Km values of mitragynine

Figure 3.
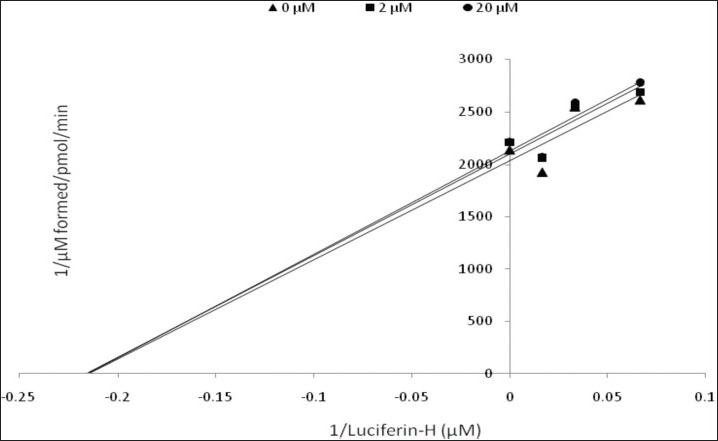
Lineweaver-Burk plot of the inhibitory effect of mitragynine on CYP2C9 activity. Each data represents the value of three determinations (n=3)
Figure 4.
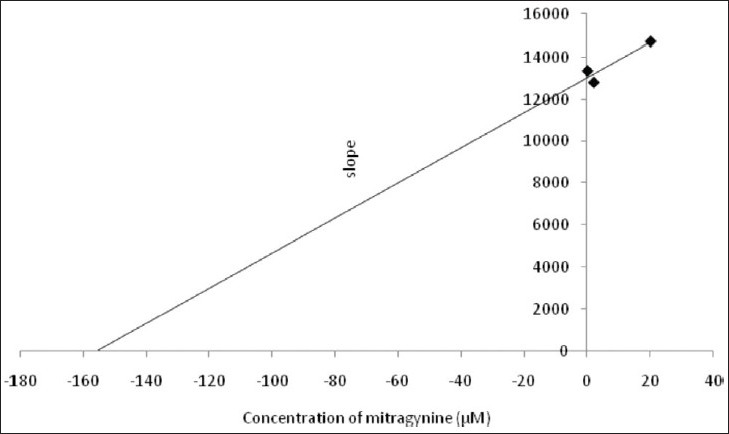
Double reciprocal plot of Lineweaver-Burk plot versus mitragynine concentration for CYP2C9
Table 3.
Ki values and mode of inhibition of mitragynine
Figure 5.
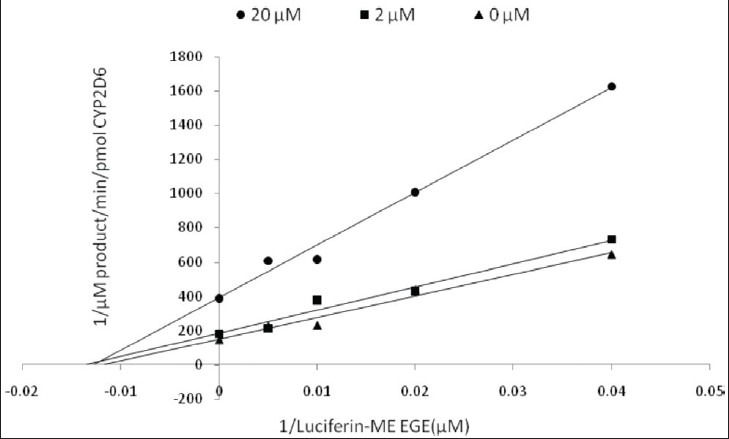
Lineweaver-Burk plot of inhibitory effect of mitragynine on CYP2D6 activity. Each data represents the value of three determinations (n=3)
Figure 6.
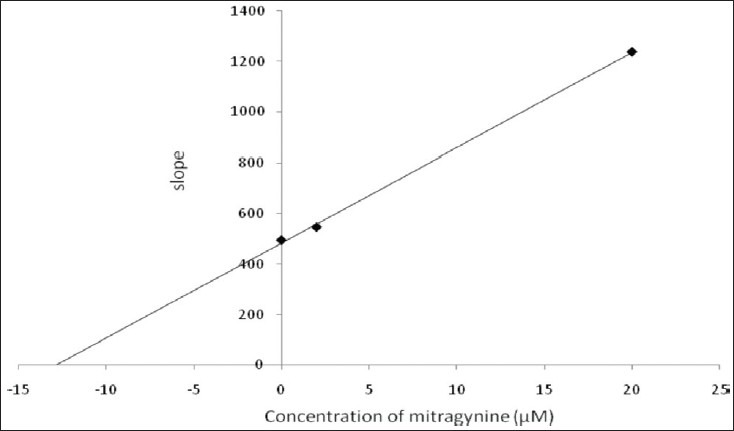
Double reciprocal plot of Lineweaver-Burk plot versus mitragynine concentration for CYP2D6
Figure 7.
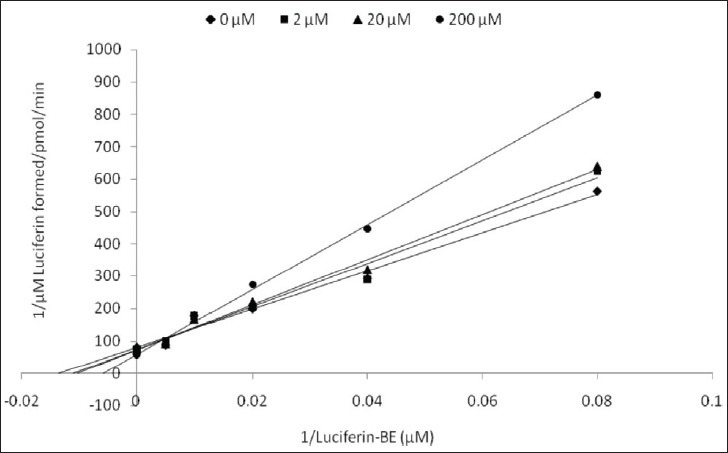
Lineweaver-Burk plot of inhibitory effect of mitragynine on CYP3A4 activity
Figure 8.
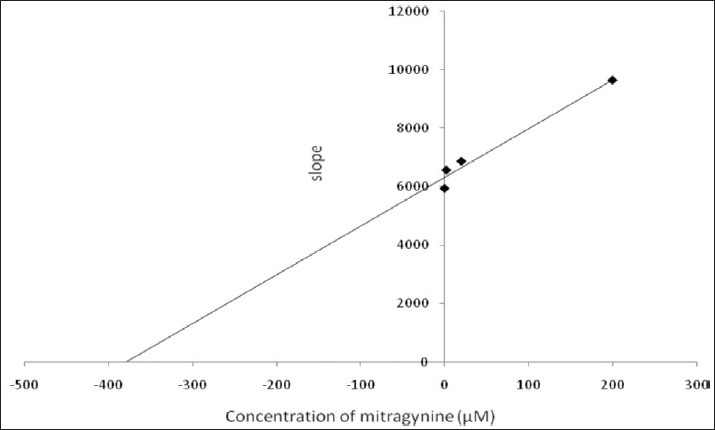
Double reciprocal plot of Lineweaver-Burk plot versus mitragynine concentration for CYP3A4
DISCUSSION
This study investigated the inhibitory effect of mitragynine on the main cytochrome P450 enzymes, which are CYP2C9, CYP2D6, and CYP3A4 using a high-throughput luminescence assay. As of date, almost one-third of adults and more than 80% of the population in developed nations and developing countries respectively depend on herbal medicines. Their reasons for use of herbal medicines are to treat common illnesses including colds, diabetes, heart disease, inflammation and central nervous system disease as well as to maintain their health.[16] Compared to prescription drugs, the safety data of herbal medicines is inadequate due to lack of reports concerning their safety and efficacy.[17] Currently, many studies on various pharmacological aspects of Mitragyna speciosa are being conducted. One essential study is to clarify the potential of inhibition and to illustrate the interactions between conventional drugs and herbal drugs consistent with the increased usage of herbal medicine presently. Thus, this study was carried out to investigate the inhibitory activity of mitragynie, which is one of the Mitragyna speciosa constituent's.
Mitragynine gave the lowest IC50 value for CYP2D6 which was 0.45 ± 0.33 μM (0.18 ± 0.13 μg/ml) followed by CYP2C9 and CYP3A4 in this study. These findings are parallel to our previous study in which Mitragyna speciosa methanolic extract showed the lowest IC50 value towards CYP2D6 (3.6 ± 0.1 μg/mL).[12] Therefore, mitragynine might be responsible for the inhibition shown in the Mitragyna speciosa methanolic extract. Mitragyna speciosa had been used traditionally since early years in Thailand and some states in Malaysia as a folk medicine to treat some diseases such as pain and fever and to boost physical stamina. Several herbal medicines were reported to possess harmful herb-drug interactions.[18,19,20] Gingko biloba extracts have been reported to exhibit interactions with modern drugs such as anti-depressant drugs (e.g., trazodone), anti-platelet drugs (e.g., warfarin), nonsteroidogenic anti-inflammatory drugs and diuretic drugs.[19,20,21]
Low IC50 values of mitragynine on CYP450 enzyme activities led us to study the effect of mitragynine on the kinetics of human cytochrome P450 2C9, 2D6, and 3A4. Four kinetic parameters were studied in order to understand as well as categorize the level of mitragynine in herb-drug interactions. Vmax which is defined as maximum velocity of the enzyme activity or rate at which the enzyme catalyzes a reaction, and Km which indicates the strength of binding between enzyme and substrate were also determined. Ki values were determined graphically using Lineweaver-Burk plots. Ki can be defined as the concentration of inhibitor required to produce half maximum inhibition. The smaller the Ki, the more potent the inhibitor is and the higher is the possibility of interactions. According to the Ki value, mitragynine shows high possibility to interact with drugs metabolized by CYP2D6. (Ki = 12.86). CYP2D6 is one of the major cytochrome P450 enzymes which is responsible for drug and xenobiotic metabolism.[22] Sertraline (Zoloft), which is an anti-depressant drug, is a potent inhibitor of CYP2D6 at a dose of 200 mg and a mild inhibitor at a dose of 50 mg.[23] As an anti-depressant drug, sertraline might be taken by Mitragyna speciosa users to release their depression. Therefore, sertraline and mitragynine might cause interactions by inhibition of CYP2D6 activity if they were to be administered concomitantly.
Commonly, inhibition can be divided into three main groups which are competitive, noncompetitive and uncompetitive. Competitive inhibition occurs when the inhibitor competes with the substrate for the enzyme active site. Competitive inhibition depends on a few factors including the substrate's concentration essential for inhibition, the substrate's affinity and the inhibitor's half-life. Macrolides, ketoconazole and cimetidine are competitive inhibitors that compete for cytochrome P450 enzymes. The metabolism of substrates cannot occur on condition that as the active site on cytochrome P450 is occupied by the inhibitor.[24] On the other hand, noncompetitive inhibition happens in situations when the substrate cannot be metabolized further by the enzyme due to damage, change and inactivation of the enzyme caused by the inhibitor.[25] Uncompetitive inhibition involves the binding of inhibitor at enzyme-substrate complex which impedes the formation of product. This inhibitor binds at other than the active site known as allosteric site. Our findings suggest that mitragynine acts as a noncompetitive inhibitor towards CYP2C9 and CYP2D6 and is a competitive inhibitor towards CYP3A4 using Lineweaver-Burk plots.
A daily consumption of Mitragyna speciosa solution by Mitragyna speciosa users is 3 × 250 mL to relieve opiate withdrawal symptoms. This amount of Mitragyna speciosa solution contains about 75 mg of mitragynine. Based on this amount, the potential of Mitragyna speciosa to be involved in herb-drug interaction is high.[26]
According to Krippendorff et al., 2007, drug compounds can be divided into three groups based on their probability to involve in drug-drug interactions. This possibility depends on their IC50 values. Compounds have a high probability of interacting with other drugs if their IC50 values are less than 1 μM. If their IC50 values are between 1 and 10 μM, they have moderate possibility in interaction. On the other hand, they are assumed to be safe or possess low drug-drug interactions if their IC50 values are greater than 10 μM.[27] Thus, mitragynine shows high, moderate and low potential to interact with the drugs mainly metabolized by CYP2D6, CYP2C9 and CYP3A4 respectively.
CONCLUSIONS
In conclusion, the present study demonstrates that mitragynine may inhibit cytochrome P450 2C9, 2D6 and 3A4 enzyme activities under the circumstances described above. However, further investigations are necessary to ascertain if the effects seen in vitro would be reflected in the in vivo situation. The expected interactions might occur when mitragynine and other drugs that are metabolized by the same enzyme, especially CYP2D6 are administered concomitantly. Therefore, the administration of mitragynine and other drugs which are metabolized by the same metabolic pathway might contribute to the herb-drug interactions.
Footnotes
Source of Support: Nil
Conflict of Interest: No.
REFERENCES
- 1.Chittrakarn S, Sawangjaroen K, Prasettho S, Janchawee B. Keawpradub N. Inhibitory effects of kratom leaf extract (Mitragyna speciosa Korth.) on the rat gastrointestinal tract. Ethno. 2008;116:173–8. doi: 10.1016/j.jep.2007.11.032. [DOI] [PubMed] [Google Scholar]
- 2.Suwanlert S. A study of kratom eaters in Thailand. Bull. Narcotics. 1975;27:21–7. [PubMed] [Google Scholar]
- 3.Takayama H. Chemistry and pharmacology of analgesic indole alkaloids from the rubiaceous plant, Mitragyna speciosa. Chem Pharma Bul. 2004;52:916–28. doi: 10.1248/cpb.52.916. [DOI] [PubMed] [Google Scholar]
- 4.Matsumoto K, Mizowaki M, Suchitra T, Murakami Y, Takayama H, et al. Central antinociceptive effects of mitragynine in mice: Contribution of descending noradrenergic and serotonergic systems. Eur J Pharm. 1996;317:75–81. doi: 10.1016/s0014-2999(96)00714-5. [DOI] [PubMed] [Google Scholar]
- 5.Matsumoto K, Mizowaki M, Suchitra T, Takayama H, Sakai S, Aimi N. Watanabe H. Antinociceptive action of mitragynine in mice: Evidence for the involvement of supraspinal opioid receptors. Life Sci. 1996;59:1149–55. doi: 10.1016/0024-3205(96)00432-8. [DOI] [PubMed] [Google Scholar]
- 6.Jansen KL, Prast CJ. 1988. Psychoactive properties of mitragynine (kratom) J Psych Drugs. 1988;20:455–7. doi: 10.1080/02791072.1988.10472519. [DOI] [PubMed] [Google Scholar]
- 7.Watanabe K, Yano S, Horie S, Yamamoto LT, Takayama H, et al. Pharmacological Properties of Some Structurally Related Indole Alkaloids Contained in the Asian Herbal Medicines, Hirsutine and Mitragynine, with Special Reference to their Ca2Þ Antagonistic and Opioid-Like Effects. In: Pharmacological Research on Traditional Herbal Medicines. In: Watanabe H, Shibuya NR, Farnsworth NR, editors. Amsterdam The Netherlands ISBN. Harwood Academic Publishers/Overseas Publishers Association; 1999. pp. 163–77. [Google Scholar]
- 8.Guengerich FP. Uncommon P450-catalyzed reactions. Curr Drug Metab. 2001;2:93–115. doi: 10.2174/1389200013338694. [DOI] [PubMed] [Google Scholar]
- 9.Taavitsainen P. Department of Pharmacology and Toxicology. Finland: University of Oulu, Oulu; 2001. Cytochrome P450 Isoform-Specific In vitro Methods to Predict Drug Metabolism and Interactions. [Google Scholar]
- 10.Parkinson A. Biotransformation of Xenobiotics. Casarett and Doull's Toxicology: The Basic Science of Poisons. In: Klaassen CD, editor. New York, USA: McGraw-Hill Medicinal Publishing Division; 2001. pp. 133–224. [Google Scholar]
- 11.Arimoto R. Computational models for predicting interactions with cytochrome p450 enzyme. Curr Top Med Chem. 2006;6:1909–18. doi: 10.2174/156802606778108951. [DOI] [PubMed] [Google Scholar]
- 12.Hanapi NA, Azizi J, Ismail S, Mansor SM. Evaluation of Selected Malaysian Medicinal Plants on Phase I Drug Metabolizing Enzymes, CYP2C9, CYP2D6 and CYP3A4 Activities In Vitro. Inter J Pharm. 2010;6:494–9. [Google Scholar]
- 13.Bjornsson TD, Callaghan JT, Einolf HJ, Fischer V. Gan L. The conduct of in vitro and in vivo drug-drug interaction studies: A pharmaceutical research and manufacturers of America (PhRMA) perspective. Drug Metab. 2003;31:815–32. doi: 10.1124/dmd.31.7.815. [DOI] [PubMed] [Google Scholar]
- 14.Asseffa A, Smith SJ, Nagata K, Gillette J, Gelboin HV, Gonzalez FJ. Novel exogenous heme-dependent expression of mammalian cytochrome P450 using baculovirus. Arch Biochem Biophys. 1989;274:481–90. doi: 10.1016/0003-9861(89)90461-x. [DOI] [PubMed] [Google Scholar]
- 15.Lineweaver H, Burk D. The determination of Enzyme Dissociation Constant. J Am Chem Soc. 1934;56:658–66. [Google Scholar]
- 16.Zou L, Harkey MR, Henderson GL. Effects of herbal components on cDNA-expressed cytochrome p450 enzyme catalytic activity. Life scoences. 2002;71:1579–89. doi: 10.1016/s0024-3205(02)01913-6. [DOI] [PubMed] [Google Scholar]
- 17.Shapiro LE, Shear NH. Drug Interactions/P450. Curr Prob Dermatol. 2001;13:141–52. [Google Scholar]
- 18.Ioannides C. Pharmacokinetic interactions between herbal remedies and medicinal drugs. Xenobiotica. 2002;32:451–78. doi: 10.1080/00498250210124147. [DOI] [PubMed] [Google Scholar]
- 19.Sorensen JM. Herb-drug, food-drug, nutrient-drug, and drug-drug interactions: Mechanisms involved and their medical implications. J Altern Complementary Med. 2002;8:293–308. doi: 10.1089/10755530260127989. [DOI] [PubMed] [Google Scholar]
- 20.Brazier NC, Levine MA. Drug-herb interaction among commonly used conventional medicines: A compendium for health care professionals. Am J Ther. 2003;10:163–9. doi: 10.1097/00045391-200305000-00003. [DOI] [PubMed] [Google Scholar]
- 21.Valli G, Giardina EG. Benefits, adverse effects and drug interactions of herbal therapies with cardiovascular effects. J Am Col Cardio. 2002;39:1083–95. doi: 10.1016/s0735-1097(02)01749-7. [DOI] [PubMed] [Google Scholar]
- 22.Coleman MD. Chichester, England: John Wiley and Sons Ltd; 2005. Human Drug Metabolism: An Introduction; pp. 75–95. [Google Scholar]
- 23.Sproule BA, Otton SV, Cheung SW, Zhong XH, Romach MK, Sellers EM. CYP2D6 inhibition in patients treated with sertraline. J Clin Psychopharm. 1997;17:102–26. doi: 10.1097/00004714-199704000-00007. [DOI] [PubMed] [Google Scholar]
- 24.Virani A, Mailis A, Shapiro LE, Shear NH. Drug interactions in human neuropathic pain pharmacotherapy. Pain. 1997;73:3–13. doi: 10.1016/s0304-3959(97)00041-9. [DOI] [PubMed] [Google Scholar]
- 25.Jang GR, Benet LZ. Antiprogestin-mediated inactivation of cytochrome P450 3A4. Pharmacology. 1998;57:150–7. doi: 10.1159/000028193. [DOI] [PubMed] [Google Scholar]
- 26.Vicknasingam B, Narayanan S, Beng GT, Mansor SM. The informal use of ketum (Mitragyna speciosa) for opioid withdrawal in the northern states of peninsular Malaysia and implications for drug substitution therapy. Int J Drug Policy. 2010:283–8. doi: 10.1016/j.drugpo.2009.12.003. [DOI] [PubMed] [Google Scholar]
- 27.Krippendorff BF, Lienau P, Reichel A, Huisinga W. Optimizing Classification of Drug-Drug Interaction Potential for CYP450 Isoenzyme Inhibition Assays in Early Drug Discovery. J Biom Scr. 2007;12:92–9. doi: 10.1177/1087057106295897. [DOI] [PubMed] [Google Scholar]