

Abstract
Thrombin is the central protease in the cascade of blood coagulation proteases. The structure of thrombin consists of a double β-barrel core surrounded by connecting loops and helices. Compared to chymotrypsin, thrombin has more extended loops that are thought to have arisen from insertions in the serine protease that evolved to impart greater specificity. Previous experiments showed thermodynamic coupling between ligand binding at the active site and distal exosites. We present a combined approach of molecular dynamics (MD), accelerated molecular dynamics (AMD), and analysis of the residual local frustration of apo-thrombin and active-site-bound (PPACK-thrombin). Community analysis of the MD ensembles identified changes upon active site occupation in groups of residues linked through correlated motions and physical contacts. AMD simulations, calibrated on measured residual dipolar couplings, reveal that upon active site ligation, correlated loop motions are quenched, but new ones connecting the active site with distal sites where allosteric regulators bind emerge. Residual local frustration analysis reveals a striking correlation between frustrated contacts and regions undergoing slow time scale dynamics. The results elucidate a motional network that probably evolved through retention of frustrated contacts to provide facile conversion between ensembles of states.
Introduction
Thrombin is the central protease in the cascade of blood coagulation proteases. Structurally, thrombin consists of a double β-barrel core surrounded by connecting loops and helices. Genetic analysis of the clotting factor genes demonstrates that the clotting proteases of the chymotrypsinogen superfamily have evolved as a result of several gene duplications, exon shuffling, and intron sliding events. Prothrombin has a unique exon organization and is thought to be the ancestral gene of the clotting factor family.1 The extended active site loops in thrombin are thought to have arisen from insertions in the serine protease that evolved to impart greater specificity.1,2 Thrombin is produced in a low-activity zymogen form that requires proteolytic cleavage to attain full activity. This cleavage event results in small overall changes to the molecular architecture but results in a large change in dynamics wherein one β-barrel becomes more dynamic and the other becomes less dynamic.3 The result is a more perfectly formed active site for rapid proteolytic cleavage activity. Despite the highly specific nature of thrombin activity, in association with allosteric modulators, the substrate specificity is tuned to activate either procoagulant or anticoagulant substrates.4 In addition, allostery is key to thrombin regulation,5,6 and misregulation can lead to bleeding disorders or thrombosis. Although, traditionally, allostery was defined as occurring among subunits in a multisubunit system such as hemoglobin,7 the phenomenon of altered activity resulting from binding of a regulatory molecule on the opposite side of a monomeric enzyme is now also recognized as a form of allostery.8,9
Several experimental and computational approaches have hinted that the solution structure of thrombin is a broad and malleable dynamic ensemble.10,11 H/D exchange mass spectrometry showed that d-Phe-Pro-Arg chloromethylketone (PPACK) occupation of the active site not only protected the active site loops but also propagated to decreased exchange in several regions of the protein distant from the active site.12 Thrombin has two binding sites distal to the active site; exosite 1 is where thrombomodulin binds, and exosite 2 is where heparin binds. Isothermal titration calorimetry (ITC) experiments showed alteration of thermodynamic parameters of ligands binding to thrombin exosites when the active site was occupied.13 Binding of active site ligands altered the balance of enthalpic and entropic contributions to binding of exosite 1 ligands and vice versa.14 Such thermodynamic compensation phenomena are more likely if the allosteric mechanism is entropic rather than enthalpic, suggesting that differences in the dynamic properties of the system affect the ligand binding mechanism.15,16 Indeed, X-ray crystallography shows no significant changes in the thrombin structure upon ligand binding, providing further evidence that it may exist as a malleable dynamic ensemble.
NMR studies and MD simulations remain the most direct approaches to investigating protein dynamics.17 A recent NMR/MD study on PPACK-thrombin revealed a large degree of dynamic motions, particularly in the active site loops, spanning time scales from picoseconds to milliseconds.10 A computational exploration revealed a strong dynamic component in the allosteric regulation of thrombin by TM.11 In the work presented here, we combine conventional MD, NMR-calibrated accelerated MD (AMD), and analysis of the residual local frustration to further explore the dynamics of thrombin, with particular interest in changes that occur upon active site ligation. The combined approach allows us to analyze a broad range of motional time scales.
Methods
Molecular Dynamics and Community Analysis
Atomic coordinates for thrombin were obtained from the protein data bank 1.9 Å X-ray crystal structure [PDB ID: 1PPB].18 The active site inhibitor was removed for the apo-thrombin calculations. Both systems were placed at the center of a periodically repeating box, and the simulation cell size was defined such that the distance between the edge of the simulation box and the surface of the solute was at least 12 Å. All simulations were performed in explicit solvent, and three Cl– counterions were introduced to obtain cell neutrality. Six 20 ns conventional MD simulations were performed using a different random seed generator for the Maxwellian distribution of atomic velocities following standard energy minimization and equilibration procedures. Periodic boundary conditions and a time step of 2 fs were employed. Bonds involving protons were constrained using the SHAKE algorithm. Electrostatic interactions were treated using the Particle Mesh Ewald (PME) method19 with a direct space sum limit of 10 Å. The ff99SB force field20 was used for the solute residues, and the TIP3P water force field21 was employed for the solvent molecules. In the case of PPACK-thrombin simulations, an in-house gaff force field was generated for the PPACK inhibitor. The conventional MD simulations were analyzed in the community analysis and also constituted the starting point for the AMD simulations. These simulations also provided the average (unbiased) dihedral angle energy, ⟨V0(dih)⟩ and total energy ⟨V0(tot)⟩ values used to define the acceleration parameters in the AMD simulations described below.
Allosteric networks were characterized using a community network analysis approach previously applied to investigate allostery in tRNA/protein complexes and other protein systems.22−24 This approach constructs a dynamic contact map consisting of a network graph in which each residue is treated as a “node”, connected by edges to other nodes when two residues are deemed to be “in contact” throughout the majority of the simulation. The dynamic contact map is subsequently decomposed into communities (ie. clusters of residues) of highly intraconnected but loosely interconnected nodes using the Girvan–Newman algorithm.25 Central to this method is the calculation of edge “betweenness”, the number of shortest paths that cross an edge. The edge betweenness is calculated for all edges, and the edge with the greatest betweenness is removed. This process is repeated, and a modularity score is tracked to identify the division that results in the optimal community structure. Network graph calculations were performed using the python module NetworkX.25
AMD Simulations and Analysis
AMD simulations were performed as described previously using an in-house modified version of the AMBER 10 code.10,11 A “dual boost” AMD approach is used,26 in which two acceleration potentials are applied simultaneously to the system; the first acceleration potential is applied to the torsional terms only, and a second, weaker acceleration is applied across the entire potential. This dual boost AMD protocol represents a unified approach facilitating the efficient sampling of both the torsional degrees of freedom and slow diffusive motions in the solute. In total, six independent dual boost AMD simulations were performed for 10 000 000 steps (the equivalent of 20 ns of MD) for each system. The physical conditions, force fields, and all other simulation parameters employed were identical to those described for the conventional MD simulations. The specific acceleration parameters used in this study were [Eb(dih) – ⟨V0(dih)⟩] = [4 kcal/mol × no. of solute residues] and α(dih) = [0.8 kcal/mol × no. of solute residues] for the torsional acceleration and [Eb(tot) – ⟨V0(tot)⟩] = α(tot) = [0.16 kcal/mol × no. of atoms in the simulation cell] for the background total acceleration. These acceleration parameters had been previously identified as the optimal acceleration parameters for the reproduction of experimental RDCs in PPACK-thrombin, accessing configurational dynamics on time scales up to 10s–100s of microseconds.10 For each AMD simulation, a corrected canonical ensemble was obtained by reweighting each point in the configuration space on the modified potential by the strength of the Boltzmann factor of the bias energy, exp[βΔV(rt(i))] at that particular point, and the bias potential block averaging method was employed to remove statistical noise errors.27
The internal dynamics present in the different AMD simulations of apo-thrombin and PPACK-thrombin were assessed by calculating order parameters, S2, from the free-energy weighted AMD ensembles. Members of each ensemble were superimposed onto the backbone atoms (N, Cα, C′) of all heavy chain residues for the appropriate average structure, and order parameters, S2 were calculated as
where μi are the Cartesian coordinates of the normalized internuclear vector of interest. Others have shown that S2 values calculated from standard MD simulations in this way were in excellent agreement with experimental S2 values calculated using the Lipari–Szabo autocorrelation function approach.28 The order parameters presented here are averaged over all six AMD trajectories for each system.
Residue-by-residue cross-correlations for the free-energy-weighted AMD ensembles were calculated using the generalized cross-correlation approach applied to all backbone Cα atomic coordinates based on the mutual information method developed by the Grubmüller group29 using the g_correlation module in GROMACS 3.3.3.30
Residual Local Frustration Analysis
An algorithm for determining residual local frustration, that is, whether a contact between amino acid residues is energetically optimized or not in the folded state, was developed by the Wolynes and Komives groups some time ago.31 This algorithm assesses residue–residue interactions by systematically perturbing the identity of individual residues and evaluating the resulting total energy change. For the work presented here, we used the “configurational frustration” index, in which the decoy set involves randomizing not just the identities but also the distance and densities of the interacting amino acids i, j. This scheme effectively evaluates the native pair with respect to a set of structural decoys that might be encountered in the folding process. After constructing a histogram of the energy of the decoys and comparing the distribution to the native energy, cutoffs are implemented to identify minimally frustrated or highly frustrated residues. Energetically favorable contacts between residues are minimally frustrated, whereas highly frustrated contacts are energetically unfavorable in the native state. Depictions of the contacts on structural models typically show minimally frustrated contacts in green and highly frustrated contacts in red. The average of the frustration scores over all of the contacts made by a particular residue are also plotted in a per-residue format. A webserver is now available for performing these computations.32 For the work presented here, the minimally and highly frustrated contacts are depicted on the lowest-energy structure from the Boltzmann-reweighted ensemble of structures from the AMD simulations. To compute the average per-residue frustration, we clustered the Boltzmann reweighted ensemble and averaged the frustration scores of all contacts made by each residue in the representative structure from the three most populated clusters. It is interesting to note that the residual local frustration varied between members of the ensemble, and the error bars on the residual local frustration plots represent one standard deviation.
Note: Thrombin has several numbering schemes; the chymotrypsin numbering scheme is used in the text because it is used in PDB files. It is denoted in this paper with the subscript CT. This numbering scheme creates problems for the data presentation in this paper; therefore, sequential numbering (of the light chain or A-chain followed by the heavy chain) is used in the plots, and sequential residue numbers are given in parentheses throughout the paper for reference.
Results
Community Network Analysis
We performed a set of six independent 20 ns conventional MD simulations for both apo-thrombin and PPACK-thrombin. During the equilibration procedure, a rather large conformational transition in the active site loops was observed for apo-thrombin that involved a reorientation of both the γ-loop (178–195) and the Na+ binding loop (264–271), forming a more “open” active site pocket.
A community network analysis approach22,24 was applied to identify groups of residues undergoing correlated motions in PPACK-thrombin and apo-thrombin. Representative community network analyses obtained from conventional MD simulations are shown in Figure 1. The flow of information in the physical network of the protein was measured by the edge betweenness, defined as the number of shortest paths that pass through the edge in the network, and is a direct measure of the strength of intercommunity communication within the network (black lines in Figure 1a and b). PPACK ligation causes consolidation of the community structure including the two active site communities most proximal to the PPACK binding site (green and brown, Figure 1). The community that includes part of the Na+ binding loop in apo-thrombin (Figure 1c, brown) consolidates with the active site serine195CT (241), the 70s loop (98–113), residues 191–194CT of the γ-loop (237–240), and the N-terminal residues 17–19CT (38–40) in the PPACK-liganded form (brown, Figure 1d). PPACK ligation also acts to consolidate the N-terminal β-barrel, which is formed by two separate communities in apo-thrombin and forms a large community that also contains the 30s loop (55–62) and part of the 60s loop (82–94) (orange, Figure 1d). In summary, the community analysis revealed consolidation of the Na+ binding site, the base of the γ-loop, and the N-terminus of the heavy chain into one community and most of the active site loops into a second community upon PPACK binding. These two communities, which are strongly connected in PPACK-thrombin, unite the residues required for proteolytic catalysis. The A-chain community also becomes more strongly connected to the active site and the community containing the 70s loop. On the basis of the substantial consolidation observed in the community analysis upon PPACK binding, we set out to examine whether there were concomitant changes in dynamics.
Figure 1.
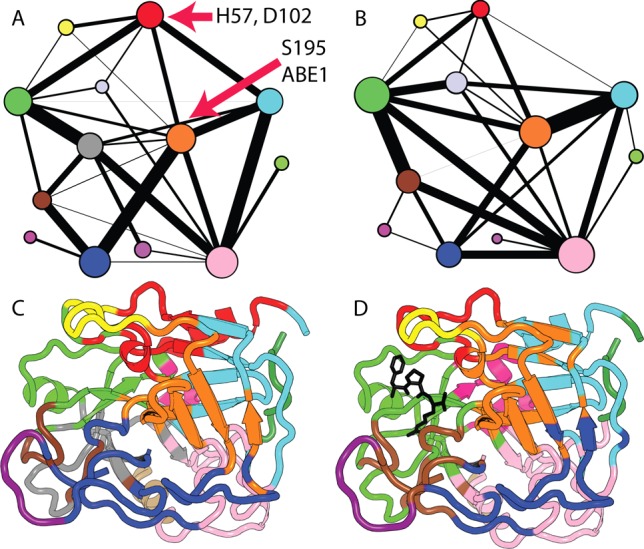
Community analysis of apo-thrombin (A,C) and PPACK-thrombin (B,D). The two-dimensional view of the communities in panels (A) and (B) depicts the relative size of the communities (based on the number of residues) as colored circles of varying sizes with the thickness of the connecting lines representing the relative interconnectivity among communities. Panels (C) and (D) are structural representations of communities superimposed on PDB 1PPB. Community definitions are provided in Table 1, Supporting Information.
AMD Simulations
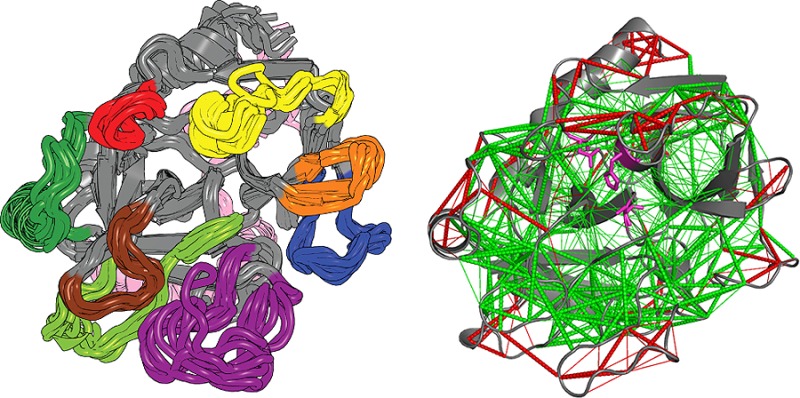
Residual dipolar couplings (RDCs), which report on an ensemble average over all orientations of the magnetic dipole interaction vector up to the chemical shift coalescence limit, provide useful experimental data for determining the ensemble of structures that best represents the dynamic properties of a protein.33,34 When the experimentally derived RDCs are compared to RDCs that are back-calculated from ensembles of structures generated from AMD simulations, the acceleration level that provides the most realistic representative structural ensemble can be identified. We previously demonstrated that the RDCs measured on PPACK-thrombin did not agree well with the available crystal structures (R2 = 0.72). Agreement was only marginally improved (R2 = 0. 80) when the RDCs were back-calculated from an ensemble of structures obtained by conventional MD.10 However, remarkable agreement was obtained between the experimental RDCs and the RDCs back-calculated from the ensemble of structures obtained from an AMD simulation at the optimal acceleration level (R2 = 0.92).10 The ensembles obtained from such AMD simulations of both PPACK-thrombin and apo-thrombin are shown in Figure 2, with the loops colored according to the scheme shown below the structure.
Figure 2.
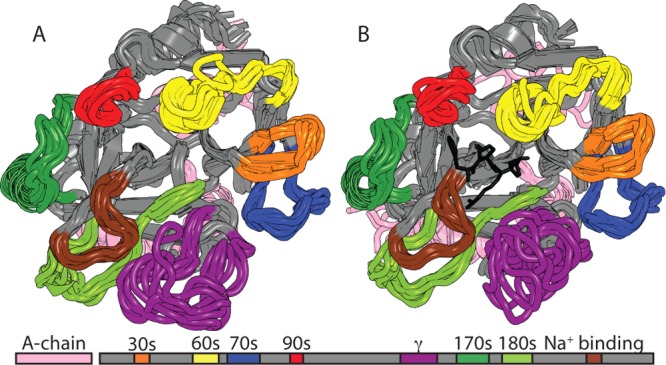
Ensemble of the 10 representative structures of the RMSD clusters from the Boltzmann reweighted structures from the RDC-calibrated AMD for apo-thrombin (left) and PPACK-thrombin (right). The loops are colored according to the schematic under the structures.
For PPACK-thrombin, order parameters (S2) from conventional MD simulations agreed extremely well with those measured by NMR relaxation experiments that are limited to the picosecond–nanosecond time regime by the molecular tumbling time (∼17 ns).10 However, the fact that ensembles obtained from AMD were required for good agreement with the RDC measurements suggested that motions on longer time scales are contributing to the solution structure. Therefore, order parameters for the N–H bond vectors were calculated from the AMD ensembles (SAMD2). A comparison of SAMD for apo-thrombin11 to those obtained previously for PPACK-thrombin10 is shown in Figure 3a. Most of the active site loops in both forms are highly flexible, yet a marked decrease in flexibility is observed upon active site ligation with PPACK (Figure 3b). As expected, PPACK ligation caused significant ordering (ΔS2 > 0.1) of residues that directly contact the PPACK Arg side chain. The loops that surround the active site also experience significant ordering, including the 60s loop (82–94), the γ-loop, and the 180s loop (225–239). Some regions distal to the PPACK also showed significant ordering upon PPACK ligation, including the A-chain, the 30s loop, residue 221CT (269) of the sodium binding loop, and residues in the C-terminal helix (Figure 3C).
Figure 3.
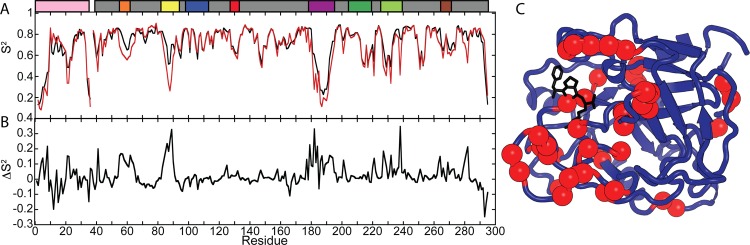
(A) Order parameters calculated from AMD simulations of apo-thrombin (red) and PPACK-thrombin (black). (B) Differences in order parameters between the forms: ΔS2 = SPPACK2 – Sapo from AMD order parameters. (C) Those residues with a ΔS2 > +0.1 are marked with red spheres on the structure of thrombin (PDB code 1PPB), indicating stabilization upon active site occupation by PPACK. The schematic of important surface loops is provided above the graph.
Correlated Motion Analysis
To identify residues undergoing correlated motions on longer time scales, AMD simulations were performed that were optimized based on previous work comparing experimental RDCs to those back calculated from AMD simulations carried out at different acceleration levels.10 The analysis of apo-thrombin revealed correlated motions between the active site loops, exosite 1, and other distal sites. In particular, the entire γ-loop appears to undergo strongly correlated motions with the A-chain residues 1H–1DCT and 12–14CCT (1–5 and 20–25), the catalytic triad, H57CT, D102CT, S195CT (79, 135, 241), the 60s loop, the 70s loop, the 90s loop (127–133), the surface strand under the 70s loop (145–151), the 170s loop (204–219), and the 180s loop (Figure 4, lower triangle). Whereas these correlated motions appeared to involve the entire γ-loop in apo-thrombin, only the tip of the γ-loop (residues 146–149ECT (182–190)) appears to be undergoing the same set of motions in PPACK-thrombin (Figure 4, upper triangle). In apo-thrombin, the 170s and, to a lesser extent, the 180s loop, which are strongly correlated to the γ-loop, are also correlated with the A-chain and catalytic residues. Upon PPACK ligation, most of these correlated motions are lost. In apo-thrombin, the 30s loop and the 60s loop are weakly correlated, but the 30s loop is not correlated to the 70s and 90s loops. Correlated motions between the 30s and 60s loops are stronger in PPACK-thrombin, and these extend to the 70s and 90s loops (Figure 4).
Figure 4.
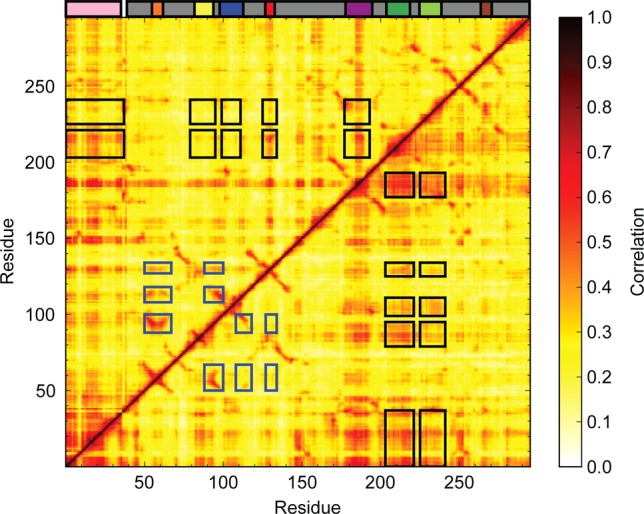
Analysis of correlated motions performed on the AMD trajectories of PPACK-thrombin (top triangle) and apo-thrombin (bottom triangle). The motions range from 0.0 (no correlation, white) to 1.0 (completely correlated, black). The schematic diagram indicating the location of surface loops is inserted above the correlated motions plot. The black boxes indicate correlations of the 170s and 180s loops with the A-chain and surface loops that are stronger in apo-thrombin than in PPACK-thrombin. The blue boxes indicate correlations of the 30s loop with the 60s, 70s, and 90s loops that are stronger in PPACK thrombin than in apo-thrombin.
Residual Frustration Correlating with Longer Time Scale Dynamics
We applied a previously derived algorithm to identify the residual local frustration in representative structures from RMSD clusters of the Boltzmann reweighted AMD simulation results.31 According to the principle of minimal frustration,35 contacts made in the folded native state should be minimally frustrated, meaning that they are energetically favorable. We previously showed that while most contacts made in the native state are, indeed, minimally frustrated, some 10–15% of contacts are energetically unfavorable (i.e., highly frustrated) in the native state. These highly frustrated contacts map to functional sites and are thought to have been preserved in evolution. Both apo- and PPACK-thrombin show regions of high frustration in many of the surface loops (Figure 5). To discover whether regions of high frustration also map to dynamic regions, we compared the average residual frustration across representative structures from the three most populated RMSD clusters derived from the AMD simulations to the order parameters. The order parameters derived from conventional MD simulations (Sns2) agree very well with order parameters derived from NMR relaxation experiments on thrombin10 and reveal the disorder resulting from motions in the nanosecond time regime. The order parameters derived from the RDC-calibrated AMD simulations (SAMD) reveal the disorder resulting from motions on longer time scales. The Sns2 did not correspond well to the regions of high residual frustration; however, the correspondence with the SAMD is remarkable (Figure 6). These results highlight that regions of high residual frustration may have evolved to allow slow time scale motions to occur with relative energetic ease, as previously suggested by Wolynes and colleagues.36
Figure 5.
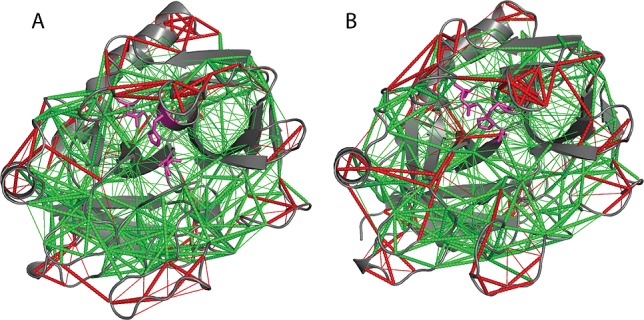
Analysis of residual local frustration31 in the lowest-energy structure from the RDC-calibrated AMD ensemble of apo-thrombin (left) and PPACK-thrombin (right). The contacts that are minimally frustrated are shown in green, and the contacts that are highly frustrated are shown in red. Thin lines represent water-mediated contacts. The active site catalytic residues are shown in magenta.
Figure 6.
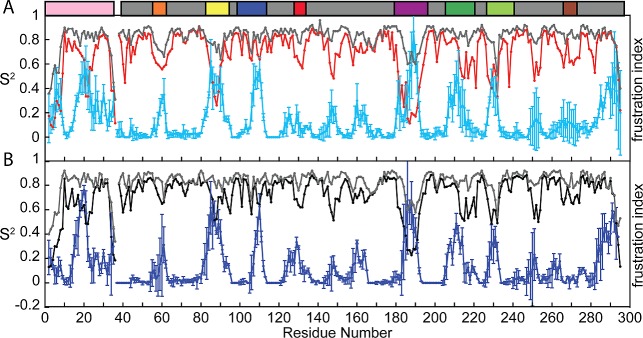
Comparison of the order parameters reflecting nanosecond time scale motions versus longer time scale motions (SAMD2), with the average per residue fraction of highly frustrated contacts for the three lowest-energy structures from the AMD simulation. (A) The S2ns (gray) and S2AMD (red) for apo-thrombin are compared to the average fraction of highly frustrated contacts (cyan). (B) The Sns (gray) and SAMD2 (black) for PPACK-thrombin are compared to the average fraction of highly frustrated contacts (blue). The schematic of important surface loops is provided above the graph.
Discussion
We used a combination of community network analysis, RDC-calibrated AMD simulations, and analysis of residual frustration to explore the dynamic ensemble of thrombin in solution from the nanosecond to the microsecond time regime. Comparative analysis of apo-thrombin and the active-site-ligated thrombin systems identified differences in the dynamic fluctuations and changes in correlated motions upon active site ligation.
Analysis of thrombin complexed with the relatively small substrate analogue PPACK showed a substantial rearrangement of the community structure. PPACK binding consolidated the two active site communities most proximal to the PPACK binding site as well as the Na+ binding loop with the active site serine, the 70s loop, part of the γ-loop, and part of the A-chain. Thus, substrate binding is predicted to consolidate the catalytic residues. Interestingly, this consolidation resulted in more residual local frustration near the active site (Figure 5).
The generalized cross-correlation analysis predicted that many of the thrombin surface loops are undergoing correlated motions (Figure 4). The analysis of residual local frustration revealed that all of these loops are highly frustrated in the native structure of thrombin (Figure 5). The striking correlation between the long timescale dynamics and the residual local frustration suggests that evolution has selected for energetically unfavorable contacts within the surface loops in order to facilitate larger amplitude slower motions. Upon PPACK binding, many of the loops retain dynamics and also remain highly frustrated.
The surface loops exhibiting correlated motions are not necessarily structurally proximal, hinting at how allosteric regulation by binding of ligands at sites distal to the active site might occur. Upon PPACK binding, the active site community consolidates with the 70s loop where fibrinogen binds to stimulate its own cleavage and where thrombomodulin binds to alter substrate specificity. Indeed, previous experiments have demonstrated a thermodynamic coupling between the 70s loop and the active site.14 Another loop that is distal to the active site but that differs between apo- and PPACK-thrombin is the γ-loop. Residues leading up to the γ-loop are highly frustrated and highly dynamic in apo-thrombin but become much less dynamic in the PPACK-bound form, with only the tip of the γ-loop remaining dynamic. Although the strongly correlated motions in apo-thrombin between the γ-loop and the 170s loop almost completely disappear upon PPACK ligation, correlations are strengthened between the tip of the loop and nearly every other loop in thrombin, including the 30s loop, the 60s loop, the 70s loop, the β-strand connecting the 60s and 70s loops, and the 90s loop. Thus, we can speculate that the reduced dynamics result from the binding of PPACK, causing these regions to sample a more restricted conformational space, and that this reduced sampling leads to stronger correlated motions extending between the active site and the surface loops involved in allosteric regulation. The order parameter analysis revealed, as expected, that active site occupation decreases the microsecond motions in the surface loops (Figure 3), consistent with amide H/D exchange experiments that showed that binding of PPACK dampened exchange in all of the surface loops including those distal to the active site.12 The shuffling of motions in the microsecond–millisecond time scale could explain how active site occupation lowers the entropic penalty for binding an exosite 1 allosteric modulator.14 Thermodynamic measurements of binding show that the allosteric pathway between these two sites operates in both directions14 and changes in order parameters in the thrombin surface loops upon PPACK binding to the active site are similar to those seen upon TM56 binding to exosite 1.11
The serine protease architecture is that of a double β-barrel fold in which the β-strands are connected by surface loops. The active site is between the two β-barrels, each of which has several β-strands connected by the surface loops. Such a β-strand architecture provides a direct connection between distal surface loops, allowing transmission of allosteric information by subtle redistribution of the dynamic ensemble.37 Such local unfolding or cracking was predicted some time ago to be the fundamental mechanism of allostery, and residual local frustration is indeed found at such sites.38 It will be interesting to see if dynamic allostery is present in other proteins with mostly β-strand structures similar to thrombin.
Acknowledgments
B.F. and E.A.K. gratefully acknowledge support from NIH-HL070999. B.F. was supported by the Molecular Biological Physics training grant (T32 GM08326) and a predoctoral fellowship from the American Heart Association, Western States Affiliate. P.G. acknowledges a CMG training grant [NIH- T32 GM007240], and this work was co-supported by the Center for Theoretical Biological Physics [NSF PHY-0822283]. Additional support was provided by the NIH, NSF, CTBP, HHMI, NBCR, and the San Diego Supercomputer Center.
Supporting Information Available
Table S1, community organization of thrombin residues in apo-thrombin and PPACK-thrombin. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
# B.F. and P.M.G. contributed equally to this work.
The authors declare no competing financial interest.
This paper was published ASAP on May 28, 2013. The paper was reposted on June 3, 2013 with minor revisions to the main text.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Banfield D. K.; Irwin D. M.; Walz D. A.; MacGillivray R. T. A. Evolution of Prothrombin: Isolation and Characterization of the cDNAs Encoding Chicken and Hagfish Prothrombin. J. Mol. Evol. 1994, 32, 177–187. [DOI] [PubMed] [Google Scholar]
- Suel G. M.; Lockless S. W.; Wall M. A.; Ranganathan R. Evolutionarily Conserved Networks of Residues Mediate Allosteric Communication in Proteins. Nat. Struct. Mol. Biol. 2003, 10, 59–69. [DOI] [PubMed] [Google Scholar]
- Koeppe J. R.; Komives E. A. Amide H/2H Exchange Reveals a Mechanism of Thrombin Activation. Biochemistry 2006, 45, 7724–7732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esmon C. T. Regulation of Blood Coagulation. Biochim. Biophys. Acta 2000, 1477, 349–360. [DOI] [PubMed] [Google Scholar]
- Di Cera E. Thrombin. Mol. Aspects Med. 2008, 29, 203–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lechtenberg B. C.; Johnson D. J.; Freund S. M.; Huntington J. A. NMR Resonance Assignments of Thrombin Reveal the Conformational and Dynamic Effects of Ligation. Proc. Natl. Acad. Sci. U.S.A. 2010, 107, 14087–14092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monod J.; Wyman J.; Changeux J.-P. On the Nature of Allosteric Transitions: A Plausible Model. J. Mol. Biol. 1965, 12, 88–118. [DOI] [PubMed] [Google Scholar]
- Tsai C.; del Sol A.; Nussinov R. Allostery: Absence of a Change in Shape Does Not Imply that Allostery Is Not at Play. J. Mol. Biol. 2008, 378, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kern D.; Zuiderweg E. R. P. The Role of Dynamics in Allosteric Regulation. Curr. Opin. Struct. Biol. 2003, 13, 748–757. [DOI] [PubMed] [Google Scholar]
- Fuglestad B.; Gasper P. M.; Tonelli M.; McCammon J. A.; Markwick P. R. L.; Komives E. A. The Dynamic Structure of Thrombin in Solution. Biophys. J. 2012, 103, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasper P. M.; Fuglestad B.; Komives E. A.; Markwick P. R.; McCammon J. A. Allosteric Networks in Thrombin Distinguish Procoagulant vs. Anticoagulant Activities. Proc. Natl. Acad. Sci. U.S.A. 2012, 109, 21216–21222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Croy C. H.; Koeppe J. R.; Bergqvist S.; Komives E. A. Allosteric Changes in Solvent Accessibility Observed in Thrombin upon Active Site Occupation. Biochemistry 2004, 43, 5246–5255. [DOI] [PubMed] [Google Scholar]
- Kamath P.; Huntington J. A.; Krishnaswamy S. Ligand Binding Shuttles Thrombin along a Continuum of Zymogen- and Proteinase-like States. J. Biol. Chem. 2010, 285, 28651–28658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Treuheit N. A.; Beach M. A.; Komives E. A. Thermodynamic Compensation upon Binding to Exosite 1 and the Active Site of Thrombin. Biochemistry 2011, 50, 4590–4596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper A.; Dryden D. T. F. Allostery without Conformational Change. Eur. Biophys. J. 1984, 11, 103–109. [DOI] [PubMed] [Google Scholar]
- Tzeng S.-R.; Kalodimos C. G. Protein Dynamics and Allostery: An NMR View. Curr. Opin. Struct. Biol. 2011, 21, 62–67. [DOI] [PubMed] [Google Scholar]
- Markwick P. R. L.; Nilges M. Computational Approaches to the Interpretation of NMR Data for Studying Protein Dynamics. Chem. Phys. 2011, 396, 124–134. [Google Scholar]
- Bode W.; Mayr I.; Baumann U.; Huber R.; Stone S. R.; Hofsteenge J. The Refined 1.9 A Crystal Structure of Human α-Thrombin: Interaction with d-Phe-Pro-Arg Chloromethylketone and Aignificance of the Tyr-Pro-Pro-Trp Insertion Segment. EMBO J. 1989, 8, 3467–3475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheatham T. E. III; Miller J. L.; Fox T.; Darden T. A.; Kollman P. A. Molecular Dynamics Simulations on Solvated Biomolecular Systems: The Particle Mesh Ewald Method Leads to Stable Trajectories of DNA, RNA, and Proteins. J. Am. Chem. Soc. 1995, 117, 4193–4194. [Google Scholar]
- Hornak V.; Abel R.; Okur A.; Strockbine B.; Roitberg A.; Simmerling C. Comparison of Multiple Amber Force Fields and Development of Improved Protein Backbone Parameters. Proteins: Struct., Funct., Bioinf. 2006, 65, 712–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorgensen W. L.; Chandrasekhar J.; Madura J. D.; Impey R. W.; Klein M. L. Comparison of Simple Potential Functions for Simulating Liquid Water. J. Chem. Phys. 1983, 79, 926–936. [Google Scholar]
- Sethi A.; Eargle J.; Black A. A.; Luthey-Schulten Z. Dynamical Networks in tRNA:Protein Complexes. Proc. Natl. Acad. Sci. U.S.A. 2009, 106, 6620–6625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivalta I.; Sultan M. M.; Lee N. S.; Manley G. A.; Loria J. P.; Batista V. S. Allosteric Pathways in Imidazole Glycerol Phosphate Synthase. Proc. Natl. Acad. Sci. U.S.A. 2012, 109, E1428–1436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VanWart A. T.; Eargle J.; Luthey-Schulten Z.; Amaro R. E. Exploring Residue Component Contributions to Dynamical Network Models of Allostery. J. Chem. Theory Comput. 2012, 8, 2949–2961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagberg A. A.; Schult D. A.; Swart P. J.. Exploring Network Structure, Dynamics, and Function Using Networkx. Proceedings of the 7th Python in Science Conference (SciPy2008), Pasadena, CA, Aug 19–24, 2008; pp 11–15.
- Hamelberg D.; de Oliveira C. s. A. F.; McCammon J. A. Sampling of Slow Diffusive Conformational Transitions with Accelerated Molecular Dynamics. J. Chem. Phys. 2007, 127, 155102–155109. [DOI] [PubMed] [Google Scholar]
- Markwick P. R. L.; McCammon J. A. Studying Functional Dynamics in Bio-Molecules Using Accelerated Molecular Dynamics. Phys. Chem. Chem. Phys. 2011, 13, 20053–20065. [DOI] [PubMed] [Google Scholar]
- Genheden S.; Diehl C.; Akke M.; Ryde U. Starting-Condition Dependence of Order Parameters Derived from Molecular Dynamics Simulations. J. Chem. Theory Comput. 2010, 6, 2176–2190. [DOI] [PubMed] [Google Scholar]
- Lange O. F.; Grubmüller H. Generalized Correlation for Biomolecular Dynamics. Proteins: Struct., Funct., Bioinf. 2006, 62, 1053–1061. [DOI] [PubMed] [Google Scholar]
- Lindahl E.; Hess B.; van der Spoel D. GROMACS 3.0: A Package for Molecular Simulation and Trajectory Analysis. J. Mol. Model. 2001, 7, 306–317. [Google Scholar]
- Ferreiro D. U.; Hegler J. A.; Komives E. A.; Wolynes P. G. Localizing Frustration in Native Proteins and Protein Assemblies. Proc. Natl. Acad. Sci. U.S.A. 2007, 104, 19819–19824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenik M.; Parra R. G.; Radusky L. G.; Turjanski A.; Wolynes P. G.; Ferreiro D. U. Protein Frustratometer: A Tool to Localize Energetic Frustration in Protein Molecules. Nucleic Acids Res. 2012, Jul 40, W348–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salmon L.; Bouvignies G.; Markwick P.; Lakomek N.; Showalter S.; Li D. W.; Walter K.; Griesinger C.; Brüschweiler R.; Blackledge M. Protein Conformational Flexibility from Structure-Free Analysis of NMR Dipolar Couplings: Quantitative and Absolute Determination of Backbone Motion in Ubiquitin. Angew. Chem., Int. Ed. 2009, 48, 4154–4157. [DOI] [PubMed] [Google Scholar]
- Cervantes C. F.; Markwick P. R.; Sue S. C.; McCammon J. A.; Dyson H. J.; Komives E. A. Functional Dynamics of the Folded Ankyrin Repeats of IκBα Revealed by Nuclear Magnetic Resonance. Biochemistry 2009, 48, 8023–8031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onuchic J. N.; Luthey-Schulten Z.; Wolynes P. G. Theory of Protein Folding: The Energy Landscape Perspective. Annu. Rev. Phys. Chem. 1997, 48, 545–600. [DOI] [PubMed] [Google Scholar]
- Hegler J. A.; Weinkam P.; Wolynes P. G. The Spectrum of Biomolecular States and Motions. HFSP J. 2008, 2, 307–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyashita O.; Onuchic J. N.; Wolynes P. G. Nonlinear Elasticity, Proteinquakes, and the Energy Landscapes of Functional Transitions in Proteins. Proc. Natl. Acad. Sci. U.S.A. 2003, 100, 12570–12575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreiro D. U.; Hegler J. A.; Komives E. A.; Wolynes P. G. On the Role of Frustration in the Energy Landscapes of Allosteric Proteins. Proc. Natl. Acad. Sci. U.S.A. 2011, 108, 3499–3503. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.