

Abstract
3,3′-Diindolylmethane (DIM), an indole derivative from vegetables of the Brassica genus, has antiproliferative activity in breast cancer cells. Part of this activity is thought to be due to DIM inhibition of Akt signaling, but an upstream mechanism of DIM-induced Akt inhibition has not been described. The goals of this study were to investigate the kinetics of inhibition of Akt by physiologically relevant concentrations of DIM and to identify an upstream factor that mediates this effect. Here we report that DIM (5–25 μM) inhibited Akt activation from 30 minutes to 24 hours in tumorigenic MDA-MB-231 cells but did not inhibit Akt activation in non-tumorigenic preneoplastic MCF10AT cells. DIM inhibited hepatocyte growth factor (HGF)-induced Akt activation by up to 46%, cell migration by 66% and cell proliferation by up to 54%, but did not inhibit induction of Akt by epidermal growth factor or insulin-like growth factor-1. DIM decreased phosphorylation of the HGF receptor, c-Met, at tyrosines 1234 and 1235, indicating decreased activation of the receptor. This decrease was reversed by pretreatment with inhibitors of p38 or calcineurin. Our results demonstrate the important role of HGF and c-Met in DIM’s anti-proliferative effect on breast cancer cells and suggest that DIM could have preventive or clinical value as an inhibitor of c-Met signaling.
Keywords: 3,3′-diindolylmethane; breast cancer; Akt; hepatocyte growth factor; c-Met
1. Introduction
Diet contributes to approximately 35%, with a range of 10–70%, of cancer incidence [1–2]. The reported inverse correlation of consumption of certain fruits and vegetables with cancer risk suggests that these foods and their bioactive components are a promising area of study for cancer prevention or treatment [3–4]. Of particular interest are vegetables of the Brassica genus like broccoli, cabbage, or kale, which contain numerous bioactive compounds, including glucosinolates like glucobracissin [5–6]. When Brassica vegetables are cut or chewed, glucobracissin is exposed to the enzyme myrosinase, which catalyzes the breakdown of glucobracissin to indole-3-carbinol (I3C), which itself has protective activity in a variety of cancers [7–8]. Upon ingestion, I3C encounters the acidic environment of the stomach, and is rapidly converted into condensation products; the major product is 3,3′-diindolylmethane (DIM).
Pharmacokinetic studies using rodents show that concentrations of 32–200 μM DIM can be achieved in tissues following ingestion, with the highest levels in the liver, followed by the kidney, lung, heart, and brain [9]. Based on the maximal DIM levels achievable in rodents and on maximum tolerated doses of DIM in humans, Howells and colleagues estimated that concentrations of up to 50 μM DIM can be considered physiologically relevant [10], consistent with our own estimates of physiologically relevant concentrations that can be achieved in humans through consumption of Brassica vegetables [11]. DIM is antitumorigenic against carcinogen-induced mammary cancer in Sprague-Dawley rats [12] and against human breast cancer xenografts in nude mice [13]. DIM induces a G1 cell cycle arrest [14] and increases apoptosis in MDA-MB-231 and MCF-7 breast cancer cells via decreased Bcl-2 protein, increased Bax, and decreased Bax/Bcl-2 binding [15]. Thus, DIM may be a promising alternative for breast cancer prevention or treatment because of its efficacy in vitro and in vivo and because it is widely available in common vegetables, has low toxicity, and is bioavailable.
Signaling in multiple pathways in breast cancer cells is altered by DIM. One of these pathways, Akt signaling, is frequently dysregulated in breast cancer, making Akt an attractive target of nutritional preemption or therapeutic treatments [16]. Akt is activated by diverse stimuli in cells, including the growth factors epidermal growth factor (EGF), insulin-like growth factor 1 (IGF-1), and hepatocyte growth factor (HGF) [17]. Inhibition of Akt by DIM has been reported in cells of multiple cancer types [18]. In breast cancer cells, this inhibition has many cellular consequences, including inhibition of the apoptosis inhibitor NF-κB, induction and nuclear localization of the cell cycle inhibitor p27kip, induction of apoptosis, and enhanced sensitivity to the chemotherapeutic agent taxotere [20–22]. While many outcomes of Akt inhibition by DIM in breast cancer cells have been described, mechanisms by which DIM inhibits Akt remain unresolved. Furthermore, several studies describing this inhibition use very high concentrations (50–100 μM) of DIM that are unlikely to be achieved in vivo through dietary or pharmacological means. Moreover, these studies report effects on Akt phosphorylation after 24–72 hours, despite the known rapid kinetics of Akt activation and signaling in cancer cells [23]. The present study was designed to further characterize the inhibitory effects of DIM on Akt signaling in breast cancer cells using physiologically relevant concentrations of the indole, and to determine the role of primary upstream activators in these effects.
2. Materials and Methods
2.1. Cell culture
MDA-MB-231 cells were purchased from American Type Culture Collection (ATCC) and were grown in Dulbecco’s Modified Eagle Medium (DMEM, Invitrogen) supplemented to 4.0 g/L glucose, 3.7 g/L sodium bicarbonate, and 10% FBS (Omega Scientific) in a humidified incubator at 37 °C and 5% CO2. MCF10AT cells were purchased from ATCC and were grown under similar conditions except they were maintained in DMEM/F12 (Invitrogen) with 10% FBS, 20 ng/mL epidermal growth factor (Sigma), 0.5 μg/mL hydrocortisone (Sigma), 10 μg/mL insulin (Invitrogen), 100 ng/mL cholera toxin (Sigma), and 1% Pen/Strep (Invitrogen). Cells were passaged regularly before reaching 80% confluence, and cells used in experiments were at less than 25 passages. Prior to all experiments, monolayers were washed with PBS and the medium was changed to either medium with 1% FBS or serum-free medium for 24 hours. DIM (LKT Laboratories) or the vehicle control DMSO (Acros Organics) was added for the indicated times, followed by HGF (Sigma) treatment for various time points when indicated. Cyclosporin A and sodium orthovanadate were purchased from Sigma and SB202190 was purchased from Tocris. All other cell culture supplies were purchased from Fisher Scientific.
2.2. Western blot analysis
To prepare whole cell lysates, monolayers were scraped directly in protein loading buffer (10% glycerol, 5% 2 mercaptoethanol, 10% SDS, 0.125 M Tris HCl pH 6.7, 0.15% bromophenol blue), sonicated for 15 s, and heated to 99.9 °C for 5 min. Equal amounts of protein were separated by SDS-PAGE and transferred to Immobilon P membranes (Millipore). Membranes were blocked in 5% nonfat dry milk in Tris-buffered saline + Tween-20 (TBST) for 1 h and then probed with rabbit anti-Akt (sc-8312), mouse anti-GSK-3α/β (sc-7291), mouse anti-tubulin (sc-5274) from Santa Cruz Biotechnology, rabbit anti-phospho Akt Ser473 (#4058), rabbit anti-phospho GSK-3α/β (#9331), rabbit anti-phospho Met Tyr1234/1235 (#3077) from Cell Signaling Technology, or rabbit anti-Met from Abcam (ab51067) overnight at 4 °C. After washing three times in TBST, membranes were incubated in 5% nonfat dry milk in TBST containing the appropriate secondary antibody-HRP conjugates (Santa Cruz Biotechnology), washed again, and exposed to Western Lighting Plus-ECL enhanced chemiluminescence substrate (Perkin Elmer) for 1 min. Immunoreactive proteins were detected using CL-Xposure film (Thermo Scientific). Bands were quantified using ImageJ software (Bethesda, MD). Band intensities of indicated phosphorylated proteins were normalized to the intensities of their respective total proteins and were expressed as percent of control for each experiment.
2.3. Wound healing assay
Cell migration was assessed as described [13] with modifications. MDA-MB-231 cells were grown to confluence in 24-well plates. A sterile pipette tip was used to scratch the monolayer and create a gap of constant width. Monolayers were then washed three times with PBS to remove floating cells and debris. Serum-free medium with or without HGF (60 ng/mL) and with increasing concentrations of DIM was added to the cells for 16 h. Cells were fixed with 10% formalin/PBS and stained with crystal violet in 10% formalin/PBS. Wounds were photographed using a Nikon Coolpix990 digital camera connected to the Nikon Eclipse TS100 microscope at a 100X magnification. Artificial lines delineating the outline of the wound were drawn on images of the original wounds and then overlaid on final wound images. Cells that migrated across the lines in each of two images from duplicate wells of three independent experiments were counted.
2.4. Thymidine incorporation assay
Cell proliferation was determined by a thymidine incorporation assay as described [24], with modifications. At 50% confluence, MDA-MB-231 cells in 12-well plates were washed with PBS, incubated in serum-free medium overnight, and then treated with varying concentrations of DIM for 4 h and HGF (60 ng/mL) for 1 h. After the incubation, cells were washed and fresh serum-free medium containing 10 μCi of [3H]-thymidine (Perkin Elmer) was added to each well and allowed to incubate at 37 °C for 4 h. The medium was removed and cells were washed three times with ice-cold 10% trichloroacetic acid. Cells were lysed in 0.3 N NaOH, and [3H]-thymidine incorporation into DNA was determined by scintillation counting of the lysates.
2.5. Statistics
Protein expression, wound healing, and [3H]-thymidine incorporation data were analyzed using one-way ANOVA and Fisher’s least significant difference test or Tukey’s honest significance post-test for multiple comparisons using GraphPad Prism Software. Levels of significance are noted for p < 0.05 and p < 0.001.
3. Results
3.1. DIM inhibits Akt activation by fetal bovine serum in MDA-MB-231 cells
Increasing concentrations of DIM decreased Akt phosphorylation at Ser473 at 24 h without affecting total Akt levels (Fig. 1A). These results are consistent with decreased activation of Akt, since Akt must be phosphorylated at Ser473 to be fully active. DIM at 25 μM also decreased Akt phosphorylation in a time-dependent manner, with inhibition appearing at as early as 30 min and with a maximal and sustained effect reached by 4 h (Fig. 1B). For this reason, we chose 4 h as our standard time point for DIM treatment. At 4 h, DIM also inhibited Akt activation in a concentration-dependent manner in MDA-MB-231 cells (Fig. 1C) without affecting total Akt levels. Not all cells were equally sensitive to DIM, as DIM did not decrease Akt phosphorylation in non-tumorigenic, pre-neoplastic MCF10AT cells (Fig. 1D).
Fig. 1.

DIM inhibits Akt activation in MDA-MB-231 cells but not in non-tumorigenic MCF10AT cells. (A) Western blot analysis of phosphorylated and total Akt protein from cells incubated in 1% FBS overnight and then treated with increasing concentrations of DIM for 24 h. (B) Western blot analysis of phosphorylated and total Akt protein from cells treated with 25 μM DIM for 0–8 h. (C) and (D) Same as A except MDA-MB-231 cells (C) or MCF10AT cells (D) were treated with DIM for 4 h. Tubulin was used as a loading control. Histograms depict phospho-Akt band density normalized to total Akt band density expressed as percent of control. Bars represent mean ± SEM of 3 independent experiments. *, p<0.05.
3.2. DIM inhibits Akt activation downstream of HGF, but not EGF or IGF-1, and inhibits phosphorylation of the HGF receptor
To define a mechanism for DIM’s inhibition of Akt activation in invasive breast tumor cells, we sought to determine whether DIM inhibits Akt activation by individual growth factors. EGF, IGF-1, and HGF all strongly induced Akt phosphorylation, as demonstrated by the increases in levels of phospho-Akt Ser473 by 25 ng/mL EGF and IGF-1 (Fig. 2A, bands 3 and 5) and 40–80 ng/mL HGF (Fig. 2B, bands 2, 4, and 6). DIM did not inhibit Akt activation by 25 ng/mL of either EGF or IGF-1 (Fig. 2A). However, DIM did decrease levels of HGF-induced phospho-Akt Ser473, by 24±6, 15±6, and 46±6% when HGF was added at 40, 60, or 80 ng/mL, respectively (Fig. 2B). Treatment with any of these growth factors or with DIM did not affect total Akt levels.
Fig. 2.

DIM inhibits Akt activation by HGF and decreases phosphorylation of c-Met. (A) MDA-MB-231 cells were incubated overnight in serum-free medium, treated with DIM for 4 h, and then epidermal growth factor (EGF, 25 ng/mL) or insulin-like growth factor-1 (IGF-1, 25 ng/mL) for 10 min. (B) Cells were incubated overnight in serum-free medium, treated with DIM for 4 h, and HGF (40 ng/mL) for 10 minutes. Total cell lysates were collected and analyzed by Western blotting for phospho-Akt Ser473 and total Akt. (C) Western blot analysis of phospho-c-Met Tyr1234/1235 and total c-Met from cells incubated in serum-free medium overnight and then treated with DMSO or 25 μM DIM for 4 h, followed by HGF treatment for 0–30 min. Tubulin was used as a loading control. Histograms depict phospho-Akt or phospho-c-Met band density normalized to total Akt or total c-Met band density expressed as percent of control. Bars represent mean ± SEM of 3 independent experiments. *, p<0.05; n.s., not statistically significant.
HGF activates downstream signaling by binding to c-Met and inducing homodimerization and trans-autophosphorylation of tyrosines Y1234 and Y1235 in the activation loop of c-Met’s kinase domain. These tyrosine phosphorylations are required for full activation of c-Met’s kinase activity [25]. To investigate whether DIM inhibits activation of c-Met by HGF, we determined the degree of tyrosine phosphorylation of c-Met at Tyr1234/1235 after treatment with DMSO or 25 μM DIM for 4 hours and 40 ng/mL HGF for 0–30 minutes via Western blotting (Fig. 2C). We observed phosphorylation of c-Met after one minute and increased phosphorylation levels after 5, 10, 20, and 30 minutes, with the strongest phosphorylation observed at 30 minutes. At 5, 10, 20, and 30 minutes, DIM significantly decreased phosphorylation of c-Met by 49±9, 19±9, 39±9, and 63±9%, respectively, and total c-Met levels did not change.
3.3. DIM inhibits phosphorylation of the Akt substrate GSK-3α/β downstream of serum and HGF
DIM produced similar inhibitory effects on phosphorylation of GSK-3α/β, a substrate of Akt, following treatment with 1% FBS (Fig. 3A) and 40 ng/mL HGF (Fig. 3B) in concentration-dependent manners. DIM at 25 μM significantly decreased GSK-3α/β by 35±9% when induced by FBS and by 24±7% when induced by HGF. Levels of total GSK-3α/β were not affected by DIM treatment. These results indicate that physiologically relevant concentrations of DIM inhibit short-term Akt activity, and that the HGF pathway is one pathway involved in this effect.
Fig. 3.
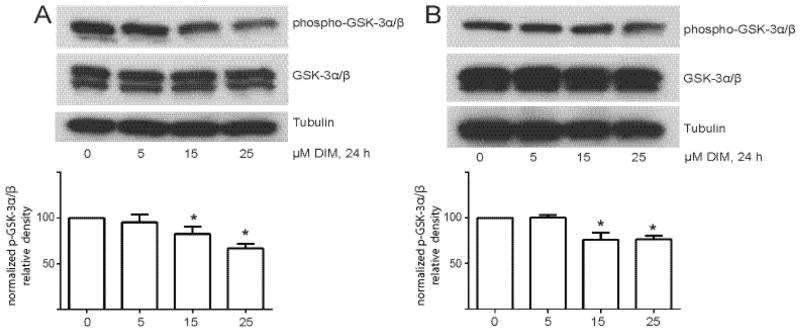
DIM decreases Akt activity in cells. (A) Cells were incubated in medium containing 1% FBS overnight and then treated with increasing concentrations of DIM for 4 h. (B) Cells were serum-starved overnight and then treated with increasing concentrations of DIM for 4 h and then 40 ng/mL HGF was added for 10 min. In both experiments, total cell lysates were collected and analyzed by Western blotting for phospho and total GSK-3α/β, and the loading control tubulin. Histograms depict phospho- GSK-3α/β band density normalized to total GSK-3α/β band density expressed as percent of control. Bars represent mean ± SEM of 3 independent experiments. *, p<0.05.
3.4. DIM inhibits HGF-induced cell motility
Figure 4A shows a representative picture of wounded MDA-MB-231 cells in serum-free medium with no HGF. On average 113±6 cells migrated to close the wound. When 60 ng/mL HGF was added, significantly more (211±8, p = 0.05) cells migrated across the wound borders, partially closing the wound. Cells treated with 5–15 μM DIM showed a 14±19 and 47±1% decrease in HGF-induced migration, respectively (Fig. 4B). DIM at 25 μM produced the most striking effect, with inhibition of migration by 66±3% below maximal levels (p<0.001).
Fig. 4.

DIM inhibits HGF-activated cell motility. (A) Confluent monolayers of MDA-MB-231 cells were scratched with a sterile pipette tip, creating gaps of fixed width. Floating cells and debris were washed away with PBS, and serum-free medium or serum-free medium containing HGF (60 ng/mL) and 0, 5, 15, or 25 μM DIM was added. After 16 h, the wounds were photographed. Photographs are representative of 2 independent fields from duplicate wells. (B) Cells that migrated across the original wound borders were counted. Results are representative of three independent experiments. *, p < 0.05; **, p< 0.001.
3.5. DIM inhibits HGF-induced cellular proliferation
Figure 5 shows that 60 ng/mL significantly (p<0.05) increased the uptake of [3H]-thymidine by cells compared to cells in serum-free medium by 47±1%, and that DIM strongly inhibited this uptake in a concentration-dependent manner. Treatment of MDA-MB-231 cells with 5 and 15 μM DIM led to a 34±15% and 47±20% inhibition of the HGF-induced response. Incubation of cells with 25 μM DIM produced an inhibition of thymidine uptake to 54±17% below control levels and 7% below levels seen with serum-free medium alone, indicating that DIM strongly inhibits HGF-induced cellular proliferation.
Fig. 5.
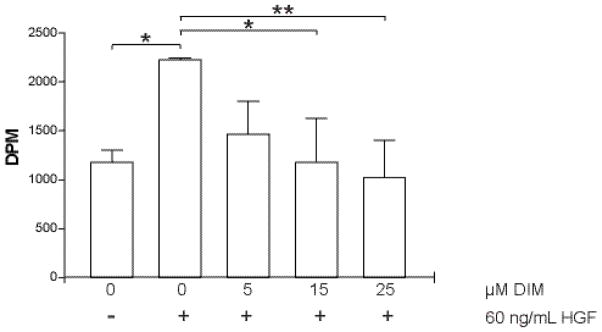
DIM inhibits HGF-stimulated cellular proliferation. Cells were serum-starved overnight and then treated with DMSO or DIM for 4 hours. HGF (60 ng/mL) was added to cells for 1 hour and the cells were then washed with PBS. Fresh serum-free medium containing [3H]-thymidine was added for an additional 4 hours. Excess [3H]-thymidine was removed and cells were lysed in 0.3 N NaOH. Incorporation of [3H]-thymidine into DNA, as a measure of proliferation, was determined by scintillation counting of the lysates. *, p < 0.05; **, p< 0.001.
3.6. DIM may inhibit c-Met phosphorylation via activation of calcineurin or p38 signaling, but not general protein tyrosine phosphatase activation
The MAP kinase p38 and calcineurin are important mediators of some of DIM’s effects in breast cancer cells [26–27]. To determine the possible involvement of these mediators, and protein tyrosine phosphatases in DIM’s effect of decreased c-Met tyrosine phosphorylation, we pretreated cells with inhibitors of each of these proteins before treatment with DIM and HGF. DIM decreased c-Met phosphorylation induced by HGF by 30±10%, and pretreatment of cells with 100 μM sodium orthovanadate (SOV), a protein tyrosine phosphatase inhibitor, did not reverse the DIM-induced decrease in c-Met phosphorylation (36±17%) (Fig. 6A).
Fig. 6.
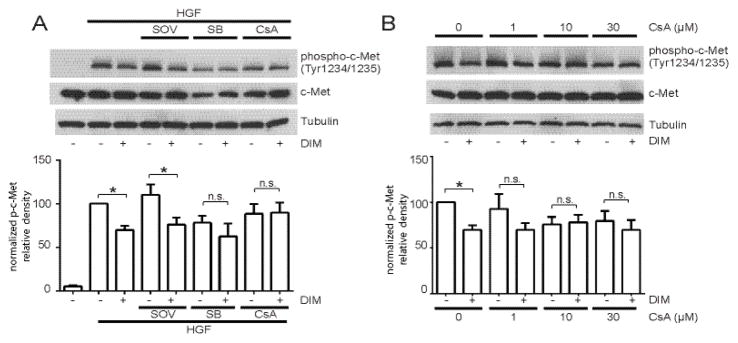
Involvement of tyrosine phosphatases, p38, and calcineurin in DIM’s effects on c-Met phosphorylation. (A) MDA-MB-231 cells were serum-starved overnight, then pretreated with 100 μM SOV, 10 μM SB or 10 μM CsA for 30 minutes, followed by DMSO or 25 μM DIM for 4 h and 40 ng/mL HGF for 10 min. Because total c-Met levels changed with some pretreatments, band intensities are normalized to tubulin and not total c-Met. (B) Same as (A) except cells were pretreated with increasing concentrations of CsA before addition of DIM and HGF. In both experiments, total cell lysates were collected and analyzed by Western blotting for phospho-c-Met Tyr1234/1235, total c-Met, and tubulin. Histograms depict phospho-c-Met band density normalized to tubulin band density expressed as percent of control. Bars represent mean ± SEM of 3 independent experiments. *, p<0.05; n.s., not statistically significant.
While DIM decreased c-Met phosphorylation in cells without pretreatment, this response was attenuated in cells pretreated with the p38 inhibitor SB202190 (SB) and the calcineurin inhibitor cyclosporin A (CsA) (Fig. 6A). While SB and CsA pretreatment non-significantly decreased c-Met phosphorylation by 21±12% and 12±11%, respectively (Fig. 6A), DIM did not further decrease c-Met phosphorylation. Further exploration of the possible involvement of calcineurin in DIM’s effect via a CSA dose response showed that DIM did not decrease c-Met phosphorylation in cells pre-treated with 1–30 μM CsA (Fig. 6B). CsA at 1 μM had no effect on total or phosphorylated c-Met while 10 and 30 μM CsA alone non-significantly decreased c-Met phosphorylation by 23±11 and 21±11%, respectively. Regardless, DIM did not further decrease phospho-c-Met levels when cells were pre-treated with 1, 10, or 30 μM CsA.
4. Discussion
We report here for the first time that DIM inhibits short- and long-term Akt activation by HGF in MDA-MB-231 breast cancer cells by inhibiting HGF/c-Met signaling. In addition, DIM strongly inhibits HGF-induced cell migration and proliferation. These effects are seen using concentrations of DIM well into the range that is considered physiologically relevant [10–11].
DIM was effective in inhibiting Akt activation in the tumorigenic MDA-MB-231 cell line, but had no effect in the preneoplastic non-tumorigenic MCF10AT cell line, demonstrating cancer cell specificity. Similar selective effects on cancer vs. non-cancer cells are observed with other bioactive food components, including allyl sulfides from garlic and curcumin from turmeric [28–29].
HGF binding to its receptor, c-Met, activates signaling through the Akt and Ras pathways. The net result of Akt activation by HGF in cancer cells is an invasive growth phenotype characterized by increased proliferation, survival, and motility of the cancer cells [30–32]. Cell lines that overexpress HGF and/or c-Met are tumorigenic in nude mice, and down regulation of these proteins decreases the tumorigenic potential of the cells [33–34]. HGF and c-Met are frequently overexpressed in breast cancer, and c-Met overexpression is a strong and independent predictor of poor prognosis [31, 35]. Patients with high HGF levels in tumor extracts have significantly shorter disease-free and overall survival times compared to patients with low HGF levels. [36]. Our studies show that DIM inhibits HGF/c-Met signaling, suggesting opportunities for nutritional preemption of various stages of tumorigenesis.
Current clinical strategies to inhibit HGF/c-Met signaling in existing tumors include development of monoclonal antibodies against c-Met or HGF, c-Met or HGF antagonists, c-Met kinase inhibitors, or compounds that inhibit downstream effectors of c-Met signaling [37]. Our results suggest that DIM is a promising treatment possibility for breast cancers that have dysregulated HGF/c-Met signaling. These strong effects were seen at physiologically relevant concentrations of DIM, suggesting that DIM could be effective with low toxicity, and it would be worthwhile to explore synergistic effects of DIM and other inhibitors of c-Met signaling to allow clinicians to use lower concentrations of other more toxic compounds.
Other bioactive food components also decrease c-Met tyrosine phosphorylation or c-Met signaling in various systems. Orally administered pomegranate fruit extract resulted in decreased phospho-c-Met levels in lungs of benzo(a)pyrene-treated A/J mice [38]. The flavonols quercetin, myricetin, and kaempferol decrease HGF-induced c-Met phosphorylation, Akt phosphorylation, cell migration, and cell morphology changes in DAOY human medulloblastoma cells [39]. In MDA-MB-231 cells, berbamine decreases FBS-induced c-Met phosphorylation [40], and green tea catechins inhibit HGF-induced c-Met activation and signaling [41]. Because diverse compounds with varying structures can all inhibit c-Met signaling by decreasing phosphorylation of the receptor, this suggests that a general cellular stress response to anticancer phytochemicals may be involved.
In support of this hypothesis, we observed that p38 and calcineurin, both of which are stress signaling factors, may have a role in DIM’s mechanism of action on c-Met signaling. It is interesting to note that treatment with SB and CsA to inhibit p38 and calcineurin, respectively, decreased c-Met activation slightly but non-significantly, as demonstrated by decreased levels of phospho-c-Met Tyr1234/1235 (Fig. 6A). This is consistent with a report showing that increased intracellular calcium inhibits c-Met tyrosine phosphorylation independent of PKC in GTL-16 gastric carcinoma cells [42], and also suggests other potential mechanisms of regulation of c-Met activity that have not yet been reported and are worthy of further study. Regardless of the decrease in HGF-induced activation of c-Met in SB- and CsA-pretreated cells, however, treatment with DIM did not further inhibit c-Met activation. This indicates that inhibition of p38 or calcineurin may abolish DIM’s effects. Future experiments should further explore the relationship of p38, calcineurin, c-Met, and DIM, as well as study c-Met inhibition by DIM in an in vivo model.
Taken together, our results indicate that DIM strongly and selectively inhibited Akt activation, migration, and cellular proliferation in tumor-derived breast cancer cells by a mechanism that involves the blockade of c-Met activation by HGF. Given the role of the HGF/c-Met/Akt pathway in breast tumorigenesis, DIM is a promising compound for prevention or treatment of breast cancer.
Acknowledgments
Financial support: Grant CA 69056 from the National Institutes of Health, fellowship support for H.L.N from California Breast Cancer Research Program 14GB-0142
The authors thank Dale Leitman and Wally Wang for helpful discussions during the course of the work and John Milner for critical review of the manuscript.
Footnotes
Conflicts of interest: None
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Doll R, Peto R. The causes of cancer: quantitative estimates of avoidable risks of cancer in the United States today. Oxford; New York: Oxford University Press; 1981. [PubMed] [Google Scholar]
- 2.WCRF/AICR. Food, Nutrition, Physical Activity and the Prevention of Cancer: a Global Perspective. Washington, DC: 1997. [Google Scholar]
- 3.Negri E, Lavecchia C, Franceschi S, Davanzo B, Parazzini F. Vegetable and Fruit Consumption and Cancer Risk. International Journal of Cancer. 1991;48:350–4. doi: 10.1002/ijc.2910480307. [DOI] [PubMed] [Google Scholar]
- 4.Terry P, Wolk A, Persson I, Magnusson C. Brassica vegetables and breast cancer risk. JAMA. 2001;285:2975–7. doi: 10.1001/jama.285.23.2975. [DOI] [PubMed] [Google Scholar]
- 5.Kristal AR, Lampe JW. Brassica vegetables and prostate cancer risk: a review of the epidemiological evidence. Nutr Cancer. 2002;42:1–9. doi: 10.1207/S15327914NC421_1. [DOI] [PubMed] [Google Scholar]
- 6.Higdon JV, Delage B, Williams DE, Dashwood RH. Cruciferous vegetables and human cancer risk: epidemiologic evidence and mechanistic basis. Pharmacol Res. 2007;55:224–36. doi: 10.1016/j.phrs.2007.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kim YS, Milner JA. Targets for indole-3-carbinol in cancer prevention. J Nutr Biochem. 2005;16:65–73. doi: 10.1016/j.jnutbio.2004.10.007. [DOI] [PubMed] [Google Scholar]
- 8.Bradlow HL. Review. Indole-3-carbinol as a chemoprotective agent in breast and prostate cancer. In Vivo. 2008;22:441–5. [PubMed] [Google Scholar]
- 9.Anderton MJ, Manson MM, Verschoyle R, Gescher A, Steward WP, Williams ML, et al. Physiological modeling of formulated and crystalline 3,3′-diindolylmethane pharmacokinetics following oral administration in mice. Drug Metab Dispos. 2004;32:632–8. doi: 10.1124/dmd.32.6.632. [DOI] [PubMed] [Google Scholar]
- 10.Howells LM, Moiseeva EP, Neal CP, Foreman BE, Andreadi CK, Sun YY, et al. Predicting the physiological relevance of in vitro cancer preventive activities of phytochemicals. Acta Pharmacologica Sinica. 2007;28:1274–304. doi: 10.1111/j.1745-7254.2007.00690.x. [DOI] [PubMed] [Google Scholar]
- 11.Chang X, Firestone GL, Bjeldanes LF. Inhibition of growth factor-induced Ras signaling in vascular endothelial cells and angiogenesis by 3,3′-diindolylmethane. Carcinogenesis. 2006;27:541–50. doi: 10.1093/carcin/bgi230. [DOI] [PubMed] [Google Scholar]
- 12.Chen I, McDougal A, Wang F, Safe S. Aryl hydrocarbon receptor-mediated antiestrogenic and antitumorigenic activity of diindolylmethane. Carcinogenesis. 1998;19:1631–9. doi: 10.1093/carcin/19.9.1631. [DOI] [PubMed] [Google Scholar]
- 13.Chang X, Tou JC, Hong C, Kim HA, Riby JE, Firestone GL, et al. 3,3′-Diindolylmethane inhibits angiogenesis and the growth of transplantable human breast carcinoma in athymic mice. Carcinogenesis. 2005;26:771–8. doi: 10.1093/carcin/bgi018. [DOI] [PubMed] [Google Scholar]
- 14.Hong C, Kim HA, Firestone GL, Bjeldanes LF. 3,3′-Diindolylmethane (DIM) induces a G(1) cell cycle arrest in human breast cancer cells that is accompanied by Sp1-mediated activation of p21(WAF1/CIP1) expression. Carcinogenesis. 2002;23:1297–305. doi: 10.1093/carcin/23.8.1297. [DOI] [PubMed] [Google Scholar]
- 15.Hong C, Firestone GL, Bjeldanes LF. Bcl-2 family-mediated apoptotic effects of 3,3′-diindolylmethane (DIM) in human breast cancer cells. Biochem Pharmacol. 2002;63:1085–97. doi: 10.1016/s0006-2952(02)00856-0. [DOI] [PubMed] [Google Scholar]
- 16.Morgensztern D, McLeod HL. PI3K/Akt/mTOR pathway as a target for cancer therapy. Anticancer Drugs. 2005;16:797–803. doi: 10.1097/01.cad.0000173476.67239.3b. [DOI] [PubMed] [Google Scholar]
- 17.Fan S, Ma YX, Wang JA, Yuan RQ, Meng Q, Cao Y, et al. The cytokine hepatocyte growth factor/scatter factor inhibits apoptosis and enhances DNA repair by a common mechanism involving signaling through phosphatidyl inositol 3′ kinase. Oncogene. 2000;19:2212–23. doi: 10.1038/sj.onc.1203566. [DOI] [PubMed] [Google Scholar]
- 18.Banerjee S, Kong D, Wang Z, Bao B, Hillman GG, Sarkar FH. Attenuation of multi-targeted proliferation-linked signaling by 3,3′-diindolylmethane (DIM): from bench to clinic. Mutat Res. 2011;728:47–66. doi: 10.1016/j.mrrev.2011.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kunimasa K, Kobayashi T, Kaji K, Ohta T. Antiangiogenic effects of indole-3-carbinol and 3,3′-diindolylmethane are associated with their differential regulation of ERK1/2 and Akt in tube-forming HUVEC. J Nutr. 2010;140:1–6. doi: 10.3945/jn.109.112359. [DOI] [PubMed] [Google Scholar]
- 20.Rahman KW, Sarkar FH. Inhibition of nuclear translocation of nuclear factor-{kappa}B contributes to 3,3′-diindolylmethane-induced apoptosis in breast cancer cells. Cancer Res. 2005;65:364–71. [PubMed] [Google Scholar]
- 21.Wang Z, Yu BW, Rahman KM, Ahmad F, Sarkar FH. Induction of growth arrest and apoptosis in human breast cancer cells by 3,3-diindolylmethane is associated with induction and nuclear localization of p27kip. Mol Cancer Ther. 2008;7:341–9. doi: 10.1158/1535-7163.MCT-07-0476. [DOI] [PubMed] [Google Scholar]
- 22.Rahman KM, Ali S, Aboukameel A, Sarkar SH, Wang Z, Philip PA, et al. Inactivation of NF-kappaB by 3,3′-diindolylmethane contributes to increased apoptosis induced by chemotherapeutic agent in breast cancer cells. Mol Cancer Ther. 2007;6:2757–65. doi: 10.1158/1535-7163.MCT-07-0336. [DOI] [PubMed] [Google Scholar]
- 23.Zhang L, Lee KC, Bhojani MS, Khan AP, Shilman A, Holland EC, et al. Molecular imaging of Akt kinase activity. Nat Med. 2007;13:1114–9. doi: 10.1038/nm1608. [DOI] [PubMed] [Google Scholar]
- 24.Vivar OI, Lin CL, Firestone GL, Bjeldanes LF. 3,3′-Diindolylmethane induces a G(1) arrest in human prostate cancer cells irrespective of androgen receptor and p53 status. Biochem Pharmacol. 2009;78:469–76. doi: 10.1016/j.bcp.2009.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Longati P, Bardelli A, Ponzetto C, Naldini L, Comoglio PM. Tyrosines1234-1235 are critical for activation of the tyrosine kinase encoded by the MET proto-oncogene (HGF receptor) Oncogene. 1994;9:49–57. [PubMed] [Google Scholar]
- 26.Gong Y, Sohn H, Xue L, Firestone GL, Bjeldanes LF. 3,3′-Diindolylmethane is a novel mitochondrial H(+)-ATP synthase inhibitor that can induce p21(Cip1/Waf1) expression by induction of oxidative stress in human breast cancer cells. Cancer research. 2006;66:4880–7. doi: 10.1158/0008-5472.CAN-05-4162. [DOI] [PubMed] [Google Scholar]
- 27.Xue L, Firestone GL, Bjeldanes LF. DIM stimulates IFNgamma gene expression in human breast cancer cells via the specific activation of JNK and p38 pathways. Oncogene. 2005;24:2343–53. doi: 10.1038/sj.onc.1208434. [DOI] [PubMed] [Google Scholar]
- 28.Ghazanfari T, Yaraee R, Rahmati B, Hakimzadeh H, Shams J, Jalali-Nadoushan MR. In vitro cytotoxic effect of garlic extract on malignant and nonmalignant cell lines. Immunopharmacol Immunotoxicol. 2011;33:603–8. doi: 10.3109/08923973.2011.551832. [DOI] [PubMed] [Google Scholar]
- 29.Calaf GM, Echiburu-Chau C, Wen G, Balajee AS, Roy D. Effect of curcumin on irradiated and estrogen-transformed human breast cell lines. Int J Oncol. 2012;40:436–42. doi: 10.3892/ijo.2011.1228. [DOI] [PubMed] [Google Scholar]
- 30.Gallego MI, Bierie B, Hennighausen L. Targeted expression of HGF/SF in mouse mammary epithelium leads to metastatic adenosquamous carcinomas through the activation of multiple signal transduction pathways. Oncogene. 2003;22:8498–508. doi: 10.1038/sj.onc.1207063. [DOI] [PubMed] [Google Scholar]
- 31.Birchmeier C, Birchmeier W, Gherardi E, Vande Woude GF. Met, metastasis, motility and more. Nat Rev Mol Cell Biol. 2003;4:915–25. doi: 10.1038/nrm1261. [DOI] [PubMed] [Google Scholar]
- 32.Comoglio PM, Boccaccio C. Scatter factors and invasive growth. Semin Cancer Biol. 2001;11:153–65. doi: 10.1006/scbi.2000.0366. [DOI] [PubMed] [Google Scholar]
- 33.Rong S, Segal S, Anver M, Resau JH, Vande Woude GF. Invasiveness and metastasis of NIH 3T3 cells induced by Met-hepatocyte growth factor/scatter factor autocrine stimulation. Proc Natl Acad Sci U S A. 1994;91:4731–5. doi: 10.1073/pnas.91.11.4731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Abounader R, Lal B, Luddy C, Koe G, Davidson B, Rosen EM, et al. In vivo targeting of SF/HGF and c-met expression via U1snRNA/ribozymes inhibits glioma growth and angiogenesis and promotes apoptosis. FASEB J. 2002;16:108–10. doi: 10.1096/fj.01-0421fje. [DOI] [PubMed] [Google Scholar]
- 35.Ghoussoub RA, Dillon DA, D’Aquila T, Rimm EB, Fearon ER, Rimm DL. Expression of c-met is a strong independent prognostic factor in breast carcinoma. Cancer. 1998;82:1513–20. doi: 10.1002/(sici)1097-0142(19980415)82:8<1513::aid-cncr13>3.0.co;2-7. [DOI] [PubMed] [Google Scholar]
- 36.Yamashita J, Ogawa M, Yamashita S, Nomura K, Kuramoto M, Saishoji T, et al. Immunoreactive hepatocyte growth factor is a strong and independent predictor of recurrence and survival in human breast cancer. Cancer Res. 1994;54:1630–3. [PubMed] [Google Scholar]
- 37.Stellrecht CM, Gandhi V. MET receptor tyrosine kinase as a therapeutic anticancer target. Cancer Lett. 2009;280:1–14. doi: 10.1016/j.canlet.2008.10.045. [DOI] [PubMed] [Google Scholar]
- 38.Khan N, Afaq F, Kweon MH, Kim K, Mukhtar H. Oral consumption of pomegranate fruit extract inhibits growth and progression of primary lung tumors in mice. Cancer Res. 2007;67:3475–82. doi: 10.1158/0008-5472.CAN-06-3941. [DOI] [PubMed] [Google Scholar]
- 39.Labbe D, Provencal M, Lamy S, Boivin D, Gingras D, Beliveau R. The flavonols quercetin, kaempferol, and myricetin inhibit hepatocyte growth factor-induced medulloblastoma cell migration. J Nutr. 2009;139:646–52. doi: 10.3945/jn.108.102616. [DOI] [PubMed] [Google Scholar]
- 40.Wang S, Liu Q, Zhang Y, Liu K, Yu P, Luan J, et al. Suppression of growth, migration and invasion of highly-metastatic human breast cancer cells by berbamine and its molecular mechanisms of action. Mol Cancer. 2009;8:81. doi: 10.1186/1476-4598-8-81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bigelow RL, Cardelli JA. The green tea catechins, (−)-Epigallocatechin-3-gallate (EGCG) and (−)-Epicatechin-3-gallate (ECG), inhibit HGF/Met signaling in immortalized and tumorigenic breast epithelial cells. Oncogene. 2006;25:1922–30. doi: 10.1038/sj.onc.1209227. [DOI] [PubMed] [Google Scholar]
- 42.Gandino L, Munaron L, Naldini L, Ferracini R, Magni M, Comoglio PM. Intracellular calcium regulates the tyrosine kinase receptor encoded by the MET oncogene. J Biol Chem. 1991;266:16098–104. [PubMed] [Google Scholar]