

Abstract
Machado–Joseph disease (MJD) is a dominantly inherited ataxia caused by a polyglutamine-coding expansion in the ATXN3 gene. Suppressing expression of the toxic gene product represents a promising approach to therapy for MJD and other polyglutamine diseases. We performed an extended therapeutic trial of RNA interference (RNAi) targeting ATXN3 in a mouse model expressing the full human disease gene and recapitulating key disease features. Adeno-associated virus (AAV) encoding a microRNA (miRNA)-like molecule, miRATXN3, was delivered bilaterally into the cerebellum of 6- to 8-week-old MJD mice, which were then followed up to end-stage disease to assess the safety and efficacy of anti-ATXN3 RNAi. Despite effective, lifelong suppression of ATXN3 in the cerebellum and the apparent safety of miRATXN3, motor impairment was not ameliorated in treated MJD mice and survival was not prolonged. These results with an otherwise effective RNAi agent suggest that targeting a large extent of the cerebellum alone may not be sufficient for effective human therapy. Artificial miRNAs or other nucleotide-based suppression strategies targeting ATXN3 more widely in the brain should be considered in future preclinical tests.
Introduction
Even though major research advances have improved our understanding of neurodegenerative diseases, preventive therapy is currently lacking. The current study explores RNA interference (RNAi) as a potential route to preventive therapy for the polyglutamine (polyQ) neurodegenerative disorder Machado–Joseph disease (MJD), also known as spinocerebellar ataxia type 3.
The nine known polyQ disorders are caused by a CAG repeat expansion in the disease gene that encodes an abnormally long stretch of polyQ in the respective disease protein. MJD, one of the most common polyQ diseases, is a dominantly inherited form of spinocerebellar degeneration. The expanded CAG repeat in MJD resides in the ATXN3 gene, which encodes the deubiquitinating enzyme ataxin 3 (ATXN3).1,2 In MJD, the CAG repeat, normally 12–44 triplets in length, becomes expanded to between ~60 and 87 triplets.3,4 The mutant protein causes selective neuronal degeneration despite being widely expressed. The major symptoms of MJD are progressive ataxia and eye movement abnormalities which are thought primarily to reflect dysfunction and degeneration of the cerebellum and brainstem. Many additional clinical features of MJD reflect neuropathologic changes in other brain regions including the basal ganglia, thalamus, substantia nigra, and spinal cord.5,6,7
ATXN3 normally helps regulate the stability and activity of diverse proteins in various cellular pathways.1,8 Mutant (expanded) ATXN3 is prone to form insoluble aggregates and to undergo proteolysis which generates polyQ-containing fragments that further promote aggregation.9,10 The disease mechanism in MJD is thought to be a toxic gain-of-function although partial loss of ATXN3 function may also contribute.1 Mutant ATXN3 has been reported to trigger several pathogenic cascades1,11,12 but the critical molecular events driving MJD pathogenesis remain unresolved. Several pathways have been targeted in the pursuit of therapy for MJD but none has yet advanced to human clinical trials.9,13,14,15,16,17,18
In the absence of a well-defined central pathogenic pathway in MJD, silencing strategies that act far upstream in the pathogenic cascade by directly targeting mutant ATXN3 or ATXN3 transcripts hold promise as potential therapy. Supporting the feasibility of this disease gene silencing strategy is the absence of an overt phenotype in Atxn3 knockout mice,19 implying that reducing ATXN3 levels will not itself lead to deleterious effects. RNAi-mediated silencing of ATXN3 transcripts with lentivirus encoding short hairpin RNAs (shRNA) has been shown to reduce degeneration in acute rat models of MJD,20,21 and to rescue motor phenotype and neuropathology in transgenic mice expressing a carboxyl-terminal ATXN3 fragment in Purkinje cells.22,23 Other nucleotide-based suppression strategies including peptide nucleic acids and antisense oligonucleotides have also successfully decreased ATXN3 levels in MJD fibroblasts.24,25 These prior studies, however, are limited by one or more factors including the fact that the model systems used for testing RNAi did not closely mirror the actual human disease, the nucleotide-based reagent would not readily be transferrable to human trials, and/or efficacy was assessed only over a short period in an acute disease model.
Here, we test RNAi as a therapy for MJD in the first controlled, long-term clinical trial employing a mouse model that closely approximates the human disease environment. Specifically, we investigated whether chronic RNAi treatment to suppress mutant ATXN3 in the cerebellum of transgenic mice expressing the full-length human disease gene18,26 could delay or revert pathological and motor features of disease. To silence mutant ATXN3, we chose an artificial microRNA (miRNA)-like design that targets the 3′-untranslated region of ATXN3, which is fully described in an accompanying paper in this issue.27 This approach was chosen because artificial miRNAs act as efficient RNAi shuttles with improved safety compared with conventional shRNAs.28,29 Viral-mediated RNAi delivery at 6–8 weeks of age effectively reduced mutant ATXN3 levels in the cerebellum. While reducing levels of mutant ATXN3 in the cerebellum did not improve survival or motor function in treated mice, viral-delivered miRATXN3 resulted in successful, long-term ATXN3 suppression and appeared safe. The results encourage continued pursuit of this strategy as a potential route to therapy for MJD and related neurodegenerative diseases.
Results
Homozygous YACMJD84.2 mice display early-onset motor deficits, supporting their use in preclinical trials
The YACMJD transgenic mouse lines express the full-length human ATXN3 gene harboring either a normal length CAG repeat (CAG15, Q15) or an expanded repeat at the high end of the human disease range (CAG84, Q84).26 We previously reported that homozygous YACMJD84.2 transgenic mice display motor deficits as early as 6 weeks of age.18 The YACMJD mouse models are ideally suited for the RNAi trial reported here because they express the very target we would hope to suppress in a human clinical trial, the full-length human disease gene.
Before initiating the RNAi trial, we sought to better characterize the motor phenotype of hemizygous and homozygous YACMJD84.2 mice (termed wt/Q84 and Q84/Q84, respectively). Both wt/Q84 and Q84/Q84 mice show reduced weight gain compared with nontransgenic (wt/wt) littermates (Figure 1a). Hemizygous wt/Q84 mice display a mild motor phenotype first detectable at 75 weeks of age (reduced locomotor and exploratory activity; Figure 1e,f). In contrast, homozygous Q84/Q84 mice die prematurely (mean survival: 39 weeks; Figure 1b) and develop early motor impairment manifested by increased latency to traverse a balance beam, progressive limb dragging on the beam, and decreased locomotor and exploratory activity on an open-field test (Figure 1c–f).
Figure 1.
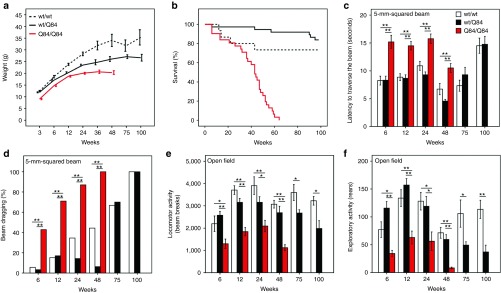
Homozygous YACMJDQ84.2 transgenic mice (Q84/Q84) display a robust, early-onset phenotype. Comparison of phenotypic features in nontransgenic (wt/wt), hemizygous (wt/Q84), and homozygous (Q84/Q84) mice. (a) Q84/Q84 mice (red) show a decrease in body weight gain compared with wt/wt (dashed black) and wt/Q84 (black) littermates (note: nonlinear X axis). (b) Kaplan–Meier survival curves show reduced survival of Q84/Q84 mice (wt/wt n = 15; wt/Q84 n = 37; Q84/Q84 n = 31). (c,d) Early motor impairment of Q84/Q84 mice on the 5-mm-squared beam walking test, manifested by (c) increased latency to traverse the beam (bars represent the mean of two consecutive trials on day 4 of testing ± SEM) and (d) progressive hind limb dragging (bars represent frequency of dragging on day 4). (e,f) Q84/Q84 mice display decreased locomotor (beam breaks, e) and exploratory activity (number of rears, f) on the open-field test during 30 minutes (bars represent mean ± SEM). Test groups used in motor tests ranged from n = 6–15 for each specific age. Statistical significance of *P < 0.05 and **P < 0.005 is indicated.
By 8 weeks of age, wt/Q84 mice and, more markedly, Q84/Q84 mice recapitulate neuropathological features of MJD: progressive intranuclear accumulation of ATXN3 in neurons and the accumulation of aggregates in various brain regions known to be affected in MJD, including the deep cerebellar nuclei (DCN) (Figure 2a). No quantifiable loss of DCN neurons or Purkinje cells, however, is observed in wt/Q84 or Q84/Q84 transgenic mice at 24 weeks (Figure 2b,c). In contrast to Q84 mice, YACMJD15.4 mice (wt/Q15) expressing nonpathogenic ATXN3 do not develop motor deficits, strong intranuclear ATXN3 accumulation, or aggregate formation even late in life (Supplementary Figure S1).
Figure 2.

Homozygous Q84/Q84 transgenic mice recapitulate Machado–Joseph disease neuropathology in the cerebellum. (a) Immunohistochemistry with anti-ATXN3 antibody (1H9) reveals neuronal nuclear accumulation of ATXN3 and aggregates in wt/Q84 and Q84/Q84 mice in the deep cerebellar nuclei (DCN) with stronger and earlier accumulation in homozygous mice. Scale bar = 25 µm. (b) NeuN-positive neurons in the DCN (lateral, intermediate, and medial nuclei) are preserved in 24-week-old wt/Q84 and Q84/Q84 mice. Bars represent the mean neuronal density ± SEM of NeuN-positive cells (five sections per animal, three animals per group). (c) Absence of Purkinje neuronal loss in wt/Q84 and Q84/Q84 transgenic mice at 24 weeks. Bars represent the mean number ± SEM of Calbindin-positive cells in the primary fissure of single plane images (three sections per animal, three animals per group).
In summary, homozygous Q84/Q84 transgenic mice expressing the full-length human ATXN3 gene recapitulate several key pathological hallmarks of MJD and display an early-onset, readily quantifiable motor phenotype. These findings support the use of Q84/Q84 mice in preclinical trials for MJD, such as the RNAi study reported here.
Human ATXN3 is selectively reduced in the cerebellum of MJD mice infused with miRATXN3
Based on the analyses above, we selected Q84/Q84 mice to perform a preclinical trial of viral-delivered RNAi to suppress expression of human mutant ATXN3. We chose to deliver an artificial miRNA mimic targeting human ATXN3 (miRATXN3)27 (Figure 3a) to the cerebellum because it is one of the most affected brain regions in MJD.6,7,30 Preventive RNAi therapy in humans likely would be administered early in the course of symptoms. Accordingly, we selected 6–8 weeks of age as the time for RNAi delivery, an age when Q84/Q84 mice already have robust intranuclear accumulation of mutant ATXN3 and show early signs of a motor phenotype without cerebellar neuronal loss.
Figure 3.

miRATXN3-mediated suppression of human ATXN3 expression in cerebellum of Q84/Q84 mice. (a) Diagram of the microRNA-like construct, miRATXN3, which targets a sequence in the 3′-untranslated region of human ATXN3 transcripts. (b) Schematic of the trial of viral-delivered miRATXN3 to the cerebellum of Q84/Q84 mice. (c) Experimental and control groups included in the trial. (d) Specific reduction of human ATXN3 mRNA in miRATXN3-injected Q84/Q84 cerebella, assessed by quantitative real-time reverse transcription-PCR (n = 3). ΔΔCT values of human and mouse ATXN3 transcripts were normalized to Gapdh mRNA levels and are shown as % fold-change relative to miRMis-treated cerebella (±SEM). *Statistical significance of P < 0.05. (e) Immunoblot for ATXN3 (1H9) shows reduced human mutant ATXN3 (ATXN3Q84) levels in cerebella of miRATXN3-treated Q84/Q84 mice compared with all control groups. Three mice (1–3) were analyzed per group, 10 weeks after viral injection. Relative viral transduction efficiency is suggested by hrGFP expression levels. (f) Quantification of immunoblots shown in e. Human ATXN3Q84 and endogenous murine Atxn3 bands were quantified by densitometry and normalized for Gapdh levels. Bars represent the average ATXN3 level (±SEM) relative to Q84/Q84 vehicle-infused mice. Statistical significance of *P < 0.05 and **P < 0.005 is indicated. (g) Immunohistochemistry with anti-ATXN3 antibody 1H9 reveals depletion of ATXN3-positive labeling in the deep cerebellar nuclei and cerebellar lobules of Q84/Q84 miRATXN3. Scale bar = 500 µm. AAV, adeno-associated virus; hrGFP, humanized Renilla green fluorescent protein.
Recombinant adeno-associated virus (AAV, 2/1 serotype) was used to deliver miRATXN3 to the DCN of Q84/Q84 mice. Two additional recombinant AAV vectors were injected as controls: U6-miRMis (a scrambled sequence) and CMV-hrGFP (lacking any miRNA). All three recombinant AAV express the reporter protein hrGFP to label transduced neurons. Virus was stereotaxically injected bilaterally into the medial and lateral cerebellar nuclei of 6- to 8-week-old Q84/Q84 transgenic mice (Q84/Q84 miRATXN3, Q84/Q84 miRMis and Q84/Q84 hrGFP, respectively) (Figure 3b,c). Two additional groups controlling for the infusion itself were injections of vehicle alone into wt/wt and Q84/Q84 mice (Figure 3c). All groups of mice were analyzed for motor function immediately before the injections, then longitudinally at 12, 24, 36, and 48 weeks of age, and allowed to survive to the end stage of disease (Figure 3b).
To assess the efficacy of viral transduction and ATXN3 suppression, a subset of animals were euthanized 10 weeks after viral delivery (Figure 3b). Though there was variable transduction coverage from animal to animal, as expected for stereotaxic injections, the cerebellum was widely transduced by the virus, both in the injected DCN and in the cerebellar lobules (Supplementary Figures S2 and S3). Some brainstem nuclei and thalamic nuclei were also transduced but to a much lower extent (Supplementary Figure S2).
The artificial miRATXN3 mimic effectively suppressed ATXN3 in the cerebellum, reducing human ATXN3 transcripts and mutant ATXN3 protein (ATXN3Q84) levels to 54 and 59% of control levels, respectively (Figures 3d–g and 4a). The suppression by miRATXN3 appeared specific to human ATXN3 transcripts and protein, as endogenous mouse Atxn3 transcript and protein levels were unchanged (Figure 3d,f). Purkinje neurons and DCN neurons transduced with miRATXN3 showed a clear depletion of ATXN3 when compared with nontransduced neurons or neurons transduced with control viruses (Figure 4b,c).
Figure 4.
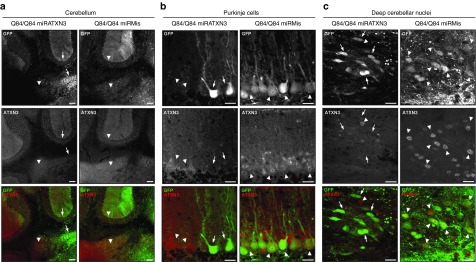
Cerebellar neurons transduced with the AAV2/1-miRATXN3-hrGFP virus show depletion of ATXN3 10 weeks after delivery. (a) Confocal z-stack images of coronal sections of cerebellum show decreased ATXN3 labeling (1H9 antibody) in miRATXN3-transduced (GFP-positive) Purkinje cell layer and deep cerebellar nuclei (DCN) (arrows) compared with nontransduced (GFP-negative) areas or neurons transduced with AAV2/1-miRMis-hrGFP (arrowheads). Scale bar = 100 µm. (b) miRATXN3-treated Purkinje cells (arrows) show depletion of ATXN3 staining, in contrast to GFP-negative and control virus-transduced cells (arrowheads). Scale bar = 20 µm. (c) Similarly, miRATXN3-positive DCN neurons (arrows) show reduction levels of ATXN3 compared with controls (arrowheads). Scale bar = 20 µm.
Assessing safety of miRATXN3 expression in the cerebellum of MJD mice
The safety of AAV2/1-miRATXN3-hrGFP delivery and the resultant human ATXN3 reduction in the cerebellum was assessed by evaluating markers of toxicity 10 weeks after injection. Cerebellar sections from all treatment groups showed a similar, low baseline level of GFAP-positive astrocytes that did not correlate with miRATXN3-mediated reduction of human ATXN3 (Figure 5a). In addition, no signs of inflammation were observed in miRATXN3-treated cerebella, as staining for the microglial marker Iba1 was similar in all mouse groups, except for occasional focal labeling along injection tracts (Figure 5b).
Figure 5.
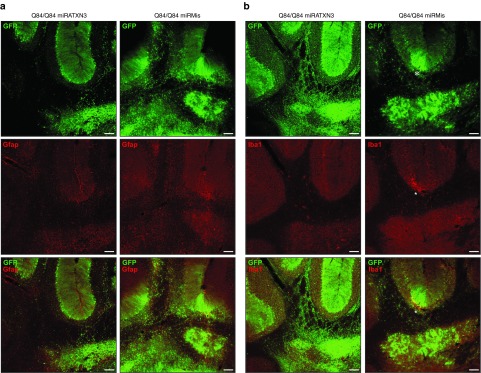
Cerebellar delivery of adeno-associated virus 2/1-miRATXN3-hrGFP virus does not lead to signs of gliosis or inflammation 10 weeks after infusion. Confocal immunofluorescence images of cerebellum show similar levels of the (a) astrocytic marker GFAP and (b) the microglial marker Iba1 in viral-transduced areas of Q84/Q84 miRATXN3 and Q84/Q84 miRMis mice. *Iba1 staining is focally increased adjacent to injection sites. Scale bar = 100 µm.
In addition, sustained expression of miRATXN3 did not result in overt neuronal toxicity since the distribution and density of NeuN-positive cells in the cerebella of miRATXN3-treated mice appeared identical to that of Q84/Q84 control groups (Figure 6a). Consistent with this observation, cerebellar NeuN levels assessed by immunoblot were similar in all infused Q84/Q84 groups (Figure 6b,c). Counting of viral-transduced Purkinje neurons (which do not stain for NeuN) also showed similar numbers from Q84/Q84 mice expressing miRATXN3 or Q84/Q84 miRMis (Figure 6d,e).
Figure 6.

Cerebellar viral delivery of miRATXN3 mimic is not neurotoxic 10 weeks postinjection. (a) Neuronal labeling (NeuN) shows similar patterns in the cerebella from Q84/Q84 miRATXN3 and Q84/Q84 miRMis mice. Scale bar = 100 µm. (b) Immunoblotting for NeuN shows similar levels in the cerebella of all infused Q84/Q84 groups. Three mice (1–3) were used per group. (c) Quantification of the immunoblots shown in b. NeuN bands were quantified by densitometry and normalized to Gapdh levels. Bars represent the average NeuN level (±SEM) relative to Q84/Q84 vehicle-infused mice. No statistical significance was found between groups. (d) Single-plane confocal micrographs of viral-transduced (GFP-positive) cerebellar lobules (primary fissure) immunostained for Calbindin show similar number of Purkinje neurons in miRATXN3-injected and miRMis-injected Q84/Q84 mice. Scale bar = 50 µm. (e) Purkinje cell count using images (exemplified in d) of three sections per mouse (three animals per group) reveals no significant differences between Q84/Q84 miRATXN3 and Q84/Q84 miRMis mice.
miRATXN3 treatment in Q84/Q84 mice does not ameliorate motor deficits
The above studies establish that miRATXN3 delivery effectively reduces levels of human mutant ATXN3 in the cerebellum without evidence of adverse effects. A critical question for potential therapy is whether this ATXN3 suppression alters motor deficits in Q84/Q84 mice. Accordingly, we assessed the effect of reducing human mutant ATXN3 levels in Q84/Q84 mice on motor performance and other phenotypic measures. All injected mouse groups were evaluated longitudinally. Cerebellar injections alone led to no adverse effects: wt/wt and Q84/Q84 mice injected with vehicle showed similar results in weight gain, survival, and motor performance compared with their corresponding noninjected groups (Figures 1 and 7). All viral-infused Q84/Q84 mouse groups showed indistinguishable weight (Figure 7a), survival curves (average age of death: 58 weeks) (Figure 7b), latency to traverse a squared 5-mm beam (Figure 7c), hind limb dragging (Figure 7d), and locomotor and exploratory activities on the open field (Figure 7e,f). Importantly, Q84/Q84 mice injected with miRATXN3 showed no improvement in the various phenotypic measures despite the robust reduction in cerebellar ATXN3 levels. The increased survival of all Q84/Q84 groups in our trial compared with the earlier phenotypic assessment (Figure 1) may reflect the repeated motor assessments of all mice in the trial, which did not take place in the earlier assessment and which could itself have life-prolonging effects.
Figure 7.
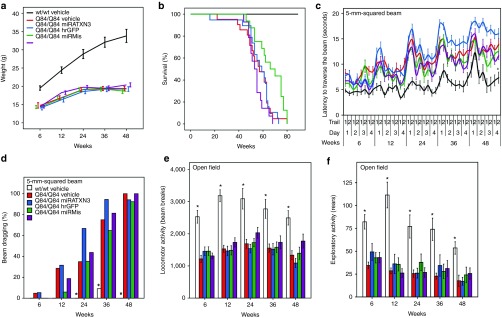
Longitudinal phenotypic evaluation shows similar results in miRATXN3-treated Q84/Q84 mice as in control mice. (a) All injected Q84/Q84 groups show decreased weight gain compared with wt/wt vehicle mice (note: nonlinear X axis). (b) Kaplan–Meier survival curves show similar survival rates in all groups of infused Q84/Q84 mice. (c) Similar early motor impairment in all Q84/Q84 mouse groups on 5-mm beam walking test, shown by increased latency traversing the beam compared with wt/wt vehicle control group (data points represent mean time to traverse the beam per trial/day/week ± SEM). (d) All Q84/Q84 groups show similar progressive hind limb dragging during the beam cross compared with wt/wt mice receiving vehicle (bars = frequency of dragging on day 4). (e) All groups of Q84/Q84 infused mice display decreased locomotor activity compared with wt/wt mice receiving vehicle (bars represent mean ± SEM). (f) Exploratory activity on open-field test is decreased in all Q84/Q84 mouse groups compared with wt/wt mice receiving vehicle (bars represent mean ± SEM). For d, *statistically significant difference between wt/wt vehicle mice and all Q84/Q84 mouse groups (P < 0.01). For c, e, and f, *statistically significant difference between wt/wt vehicle mice and all Q84/Q84 mouse groups using two-way analysis of variance repeated measures test (post hoc by Tukey test): (c) F4,64 = 11.150 for effect of injection, P < 0.001; (e) F5,70 = 11.755 for effect of injection, P < 0.001; (f) F5,70 = 8.779 for effect of injection, P < 0.001. No statistical difference was found between Q84/Q84 infused mouse groups for any motor function test. Mouse # per group: wt/wt vehicle, 11; Q84/Q84 vehicle, 21; Q84/Q84 miRATXN3, 19; Q84/Q84 miRMis, 16; and Q84/Q84 hrGFP, 17.
In summary, we noted no deleterious effect of miRATXN3 infusion in the cerebellum of Q84/Q84 mice, sustained ATXN3 suppression in the cerebellum did not increase survival or rescue aspects of motor impairment in Q84/Q84 mice.
miRATXN3 viral transduction is effective until end stage of disease
Effective RNAi therapy for chronic neurodegenerative diseases will require sustained target gene suppression. Importantly, the miRATXN3-expressing virus resulted in high transduction efficacy in the cerebellum over time. Whole cerebella of miRATXN3-infused Q84/Q84 mice analyzed at end stage of life (9–10 months after injection) continued to show a reduction of human mutant ATXN3 (ATXN3Q84) levels to 40%, as well as a smaller reduction in mouse endogenous Atxn3 to 78% of the corresponding levels in vehicle-infused mice (n = 9) (Figure 8a,b). ATXN3 immunostaining of cerebellar sections from end-stage mice confirmed persistent ATXN3 suppression in cells treated with miRATXN3 (Figure 8c).
Figure 8.
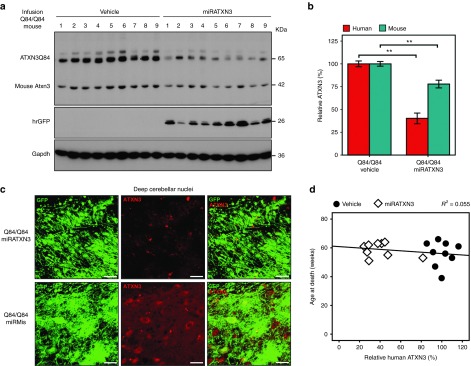
Viral-delivered miRATXN3 remains effective at reducing human ATXN3 until disease end stage. (a) Immunoblot for ATXN3 (1H9 antibody) shows ATXN3 depletion in whole cerebellum of Q84/Q84 mice receiving miRATXN3 mice at end of life compared with all control groups. Nine mice (1–9) were analyzed per group. Viral transduction efficiency per mouse is suggested by individual hrGFP expression levels. (b) Quantification of immunoblot shown in a. Human ATXN3Q84 and mouse Atxn3 were quantified by densitometry and normalized for Gapdh levels. Bars represent average ATXN3 levels (±SEM) relative to Q84/Q84 vehicle-infused mice. **Statistical significance of P < 0.005. (c) Confocal z-stack images of coronal sections of mouse cerebella at end stage showing decreased ATXN3 immunofluorescence (1H9 antibody) in miRATXN3-transduced (GFP-positive) cells in the deep cerebellar nuclei compared with cells transduced with adeno-associated virus 2/1 (AAV2/1)-miRMis-hrGFP. Scale bar = 20 µm. (d) No correlation was found between age-at-death and levels of human mutant ATXN3 in the cerebellum of Q84/Q84 mice. Black circles and white squares represent Q84/Q84 mice injected with vehicle or AAV2/1-miRATXN3-hrGFP, respectively. Pearson correlation R2 = 0.055, P = 0.359.
A beneficial effect of RNAi-mediated suppression of ATXN3 on disease phenotype might have been missed if the efficiency of viral delivery varied greatly among injected mice. Thus, at the end of the experiment, we sought to correlate the final levels of ATXN3 suppression with motor behavior in individual mice. As shown in Figure 8, there was in fact some variation among injected mice in the efficiency of viral transduction and ATXN3 suppression. But when we assessed the individual motor performances of miRATXN3-infused mice, taking into account the degree of human ATXN3 suppression in the cerebellum of each mouse, we still observed no correlation between various motor parameters or age-at-death and the measured levels of human mutant ATXN3 in the cerebellum (Figure 8d).
Discussion
Preventive therapy is an unmet need in all polyglutamine neurodegenerative diseases including MJD. Although several approaches have been explored toward MJD therapy, none has yet advanced to a clinical trial due to limitations either of the therapeutic agent tested or the disease models employed. Moreover, despite the availability of many animal models of MJD,1 the field has not settled on a model that best recapitulates molecular genetic aspects of the human disease while also displaying a robust motor phenotype, supporting its use for preclinical studies. Here, we sought both to test viral-mediated RNAi as a therapeutic strategy in MJD and to validate the YACMJDQ84.2 mouse as an appropriate model for preclinical studies.
Our analysis of YACMJD84.2 transgenic mice leads us to conclude that homozygous (Q84/Q84) mice may be a particularly suitable model to perform preclinical studies of MJD. For testing RNAi and other nucleotide-based strategies, the Q84/Q84 model possesses the attractive feature of expressing full-length human ATXN3,26 precisely the disease target in humans. Our characterization revealed that Q84/Q84 mice die prematurely and develop progressive, early-onset motor deficits and neuropathological hallmarks reminiscent of MJD. Because the Q84/Q84 mouse mirrors numerous aspects of human MJD and displays a readily measurable phenotype, it may represent the best available animal model for future preclinical studies.
The preclinical trial reported here is the first to evaluate the ability of long-term RNAi suppression of mutant ATXN3 to rescue disease features in a genetically accurate animal model. In earlier studies, RNAi suppression of ATXN3-encoding transcripts with lentiviral-delivered shRNAs mitigated striatal degeneration in an acute rat model of MJD,20,21 and cerebellar injections of shRNA-encoding lentivirus that selectively targets mutant ATXN3 transcripts20 rescued neuropathology and motor deficits in transgenic mice.22 The rodent models used in these earlier studies, however, do not recapitulate important elements of human MJD: they are either acute expression models20,21 or express an engineered, toxic fragment of mutant ATXN3 only in Purkinje cells.22,23 In contrast, the current trial tests long-term treatment in Q84/Q84 mice with the advantages noted above.
One advantage of the current study is the use of a miRNA-like molecule to target human ATXN3, the development of which is fully described in an accompanying article in this same issue.27 Although shRNAs have been tested against many disease targets, shRNA molecules can be neurotoxic, possibly reflecting inefficient processing into mature small interfering RNA molecules.28,29 Artificial miRNAs are processed more efficiently, leading to both effective target gene silencing and improved cellular safety.28,29 Our miRNA mimic effectively targets a sequence in the 3′-untranslated region of ATXN3 (miRATXN3) as described in the accompanying paper.27
In the current RNAi trial, we investigated whether long-term treatment with this miRNA-like reagent to suppress expression of expanded ATXN3 could ameliorate pathological and motor phenotypes. The tested molecule, miRATXN3, effectively sustained suppression of mutant human ATXN3 in the cerebellum to 59 and 40% of control levels at 18 weeks of age and end-stage disease, respectively. In cerebellar regions expressing miRATXN3, we observed a corresponding decrease in ATXN3 intranuclear accumulation, which may contribute to MJD pathogenesis.1 miRATXN3-mediated suppression appeared selective for human ATXN3 over endogenous murine Atxn3 at 18 weeks, but over time, there was partial loss of selectivity as murine Atxn3 was also reduced by ~20% at the end of life. This age-dependent loss of selectivity could reflect a cumulative, small off-target effect on mouse Atxn3 transcripts or the consequence of protein interactions between human ATXN3 and murine Atxn3. Although the 3′-untranslated region sequence targeted by miRATXN3 may not exist in all ATXN3 transcripts,31,32 the major human ATXN3 transcripts expressed in cerebellum are clearly silenced by miRATXN3 because ATXN3 immunostaining with the 1H9 antibody, which cross-reacts with murine Atxn3, shows markedly reduced signal in cerebellar neurons expressing miRATXN3.
Cerebellar viral delivery of miRATXN3 proved to be safe, as has been observed with artificial miRNAs for other polyQ diseases.28,29,33,34 Long-term treatment of Q84/Q84 with viral-delivered miRATXN3 did not alter life expectancy as treated mice showed similar survival rates as control mice. In addition, no signs of increased astrogliosis, microglial activation, or decrease in NeuN levels and Purkinje neuron number were found in RNAi-treated mice.
Despite the apparent safety and effective long-term silencing of human mutant ATXN3, longitudinal assessment of motor function failed to show improvement in motor deficits of treated Q84/Q84 mice. Given the roughly 50% reduction in mutant ATXN3 in the cerebellum of miRATXN3-treated mice, we anticipated that the motor phenotype in homozygous Q84/Q84 mice would be lessened, approaching the much milder, delayed motor impairment observed in hemizygous wt/Q84 mice which express ATXN3 at ~50% the level of Q84/Q84 mice. The failure of miRATXN3 treatment to improve motor deficits in Q84/Q84 mice despite the apparent widespread delivery of miRATXN3 virus to the cerebellum (Supplementary Figure S3) suggests that the motor impairment of Q84/Q84 mice, and very likely persons with MJD, is not solely due to cerebellar dysfunction. Other regions of the posterior fossa including the brainstem, as well as deeper regions of the basal ganglia, may contribute to motor dysfunction.5,6,7 In this trial, we did observe modest miRATXN3 delivery to pontine and thalamic nuclei, but to a much lower extent than in the cerebellum. Thus, 50% reduction of mutant ATXN3 confined to the cerebellum is not sufficient to improve motor impairment in Q84/Q84 mice. An alternative explanation for the lack of motor improvement is that intervention at 6–8 weeks might be later than ideal; by that time Q84/Q84 mice show motor and pathological signs accompanied by neuronal dysfunction18 that might not easily be reverted. Future silencing strategies for MJD should consider earlier and/or broader delivery approaches.
In conclusion, this preclinical study of RNAi as a potential therapeutic approach for MJD confirms the long-term efficacy of a miRNA-like RNAi agent in suppressing human mutant ATXN3. The particular tested RNAi molecule, miRATXN3, was well tolerated when delivered broadly to the cerebellum of Q84/Q84 transgenic mice. The results support further preclinical testing of miRATXN3 toward potential human application. Although RNAi-mediated suppression of ATXN3 in a large extent of the cerebellum did not improve or delay the motor deficits of MJD transgenic mice, artificial miRNAs or other nucleotide-based gene silencing reagents delivered at early stages of disease and more broadly to the brain continue to hold promise as potential routes to therapy for MJD and other dominantly inherited neurodegenerative diseases.
Materials and Methods
Animals. All animal procedures were approved by the University of Michigan Committee on the Use and Care of Animals. YACMJD transgenic mice were generated in a C57BL/6 background strain and have been maintained on the same background for 6 years. Mice were housed in cages with a maximum number of five animals and maintained in a standard 12-hour light/dark cycle with food and water ad libitum. Genotyping was performed using DNA isolated from tail biopsy at the time of weaning, as previously described.26 YACMJD84 transgenic mice hemizygosity/homozygosity was determined by quantitative real-time PCR using iQ SYBR Green Supermix (Bio-Rad, Hercules, CA) by amplification of a fragment of the ATXN3 transgene with primers 5′GAATATTTTAGCCCTGTGGAATT and 5′AGTTTAAAATCAGTACCTGTAAAAACG, and normalizing for a genomic fragment of the mouse Actb gene amplified with primers 5′GAACGGACACCATGGGCGGG and 5′GTGTGTCCCCAAGCCCCACG. The reactions were performed in an iCycler iQ real-time PCR detection system (Bio-RAD). Genotypes of all studied mice were confirmed using DNA extracted from tails collected postmortem. Animals were euthanized 10 weeks after viral infusion or at end stage of life, anesthetized with ketamine/xylazine, and perfused transcardially with either phosphate-buffered saline (for RNA and protein studies) or 4% paraformaldehyde (for immunohistochemistry). Brains perfused with phosphate-buffered saline were immediately placed on dry ice and stored at −80 °C.
Stereotaxic mouse cerebellar viral delivery. Stereotaxic administration of AAV2/1 virus or vehicle was performed on 6- to 8-week-old wild-type or Q84/Q84 transgenic mice placed under anesthesia using a mixture of O2 and Isoflurane (dosage 4% for induction, 1.5% maintenance). Mice received bilateral intracerebellar injections (two sites/hemisphere) of AAV2/1 virus diluted in Lactated Ringer's solution (Hospira, Lake Forest, IL; total of four injections per cerebellum). For each injection, ~2.0 × 1013 vg/ml of virus (0.5–2 µl) was delivered to the medial or lateral cerebellar nucleus at an infusion rate of 0.5 µl/minute using a 10-µl Hamilton syringe (BD, Franklin Lakes, NJ) retrofitted with a glass micropipette. One minute after the infusion was completed, the micropipette was retracted 0.3 mm and allowed to remain in place for 4 minutes prior to complete removal from the mouse brain. Anterior–posterior and medial–lateral coordinates were calculated from bregma and the dorsal–ventral coordinates were calculated from the dural surface. These measurements were made on an experimentally determined flat skull.
Motor evaluation. YACMJD84.2 and YACMJD15.4 mice assessed in the initial motor function characterization were evaluated at 6, 12, 24, 48, 75, and 100 weeks (±1 week). All groups were generated using littermate male and female mice. Q84/Q84 mice and wt/wt littermate control mice used in the miRATXN3 trial were tested at 6 (preinfusion), 12, 24, 36, and 48 weeks (±1 week). Weight was determined for each evaluation time point.
Motor coordination and balance was tested by assessing the ability of mice to traverse a 5-mm-wide, 44-cm-long Plexiglas square beam, from a clear platform 20 × 20 cm to an enclosed black box 20 × 20 × 20 cm, at a 53 cm height. Mice crossed the beam for two trials on each of 4 days of testing. Time to traverse the beam was recorded for each trial with a maximum cutoff of 20 seconds with falls scored as 20 seconds. Hind limb dragging during the beam traversal was also recorded.
Locomotor and exploratory activities were assessed by placing the mice in a photobeam activity system open-field apparatus (San Diego Instruments, San Diego, CA) during 30 minutes. Locomotor activity corresponds to the total distance traversed by the mice, which is calculated from the total number of beam brakes. Exploratory activity was measured by the total number of rears.
Quantitative real-time reverse transcription-PCR. Total RNA from mouse cerebellum was obtained by an initial extraction using Trizol Reagent (Invitrogen, Eugene, OR) followed by a purification using the RNeasy mini kit (Qiagen, Gaithersburg, MD) following the manufacturer's instructions. Reverse transcription of 1.5 µg of total RNA per sample was performed using the iScript cDNA synthesis kit (Bio-RAD). Human ATXN3, mouse Atxn3, and mouse Gapdh (housekeeping) transcript levels were accessed by quantitative real-time PCR as mentioned above using 1 µl of total cDNA per sample and the pair of primers (5′CGATCCTATAAATGAAAGATCA/5′CTGTTGTAATTGAGCCAAGAAA), (5′TGATCCTATAAACGAAAGATCC/5′TTGCTGTAATTGAGCCAAGAAT), and (5′ TCTGACGTGCCGCCTGGAGA/5′GAGAGCAATGCCAGCCCCGG), respectively. Relative gene expression was determined using the ΔΔ CT method, normalizing for Gapdh mRNA levels.
Western blotting. Protein lysates from cerebellum were obtained by lysis in radioimmunoprecipitation assay buffer containing protease inhibitors (Complete; Roche Diagnostics, Indianapolis, IN), followed by sonication and centrifugation. The supernatants were collected, total protein concentration was determined using the BCA method (Pierce, Rockford, IL) and then stored at −80 °C. Fifty microgram of total proteins per sample were resolved in 10% sodium dodecyl sulfate–polyacrylamide gel electrophoresis gels, and corresponding polyvinylidene difluoride membranes were incubated overnight at 4 °C with monoclonal primary antibodies: mouse anti-ATXN3 (1H9) (1:2,000, MAB5360; Millipore, Billerica, MA), mouse anti-GAPDH (1:3,000, MAB374; Millipore), or rabbit anti-NeuN (1:2,000, ABN78; Millipore). Bound primary antibodies were visualized by incubation with a peroxidase-conjugated anti-mouse or anti-rabbit secondary antibody (1:10,000; Jackson Immuno Research Laboratories, West Grove, PA) followed by treatment with the ECL-plus reagent (Western Lighting; PerkinElmer, Waltham, MA) and exposure to autoradiography films. Band intensities were quantified using the Quantity One Software analysis (Bio-Rad).
Immunohistochemistry and neuronal count. Brains from mice perfused with 4% paraformaldehyde were postfixed overnight at 4 °C in the same fixative, immersed in 30% sucrose/phosphate-buffered saline, and sectioned on a sledge microtome (SM200R; Leica Biosystems, Buffalo Grove, IL). Free-floating 30-µm coronal sections were collected and stored in cryoprotectant solution at −20 °C. Brain sections processed for immunohistochemistry were initially subjected to antigen retrieval and incubated using the Vector MOM immunodetection kit (Vector Laboratories, Burlingame, CA) whenever a mouse primary antibody was used. For bright-field microscopy, sections were incubated with mouse anti-ATXN3 (1H9) (1:1,000, MAB5360; Millipore) or mouse anti-NeuN (1:1,000, MAB377; Millipore) that were detected by incubation with a biotinylated anti-mouse antibody followed by avidin–biotin complex coupled to horseradish peroxidase (Vector Laboratories) and by DAB substrate (Vector Laboratories). Bright-field micrographs were obtained using an Olympus BX51 microscope (Olympus, Center Valley, PA). For double or single immunofluorescence, sections were incubated with mouse anti-ATXN3 (1H9) (1:1,000, MAB5360; Millipore), rabbit anti-NeuN (1:1,000, ABN78; Millipore), rabbit anti-GFAP (1:500, Z0334; Dako, Carpinteria, CA), rabbit anti-Iba1 (1:250; Wako, Richmond, VA), or mouse anti-Calbindin (1:1,000, C9848; Sigma, St Louis, MO), and then incubated with the corresponding secondary Alexa Fluor 568 and/or 647 antibodies (1:1,000; Invitrogen). All sections for immunofluorescence were then stained with 4,6-diamidino-2-phenylindole dihydrochloride (Sigma), mounted with Prolong Gold Antifade Reagent (Invitrogen), and imaged using a FV500 Olympus confocal microscope.
Sections stained for Calbindin were imaged in the confocal microscope and single-plane images were obtained from GFP-positive (viral transduced) neurons in midline cerebellar sections. Purkinje cell counts were obtained by determining the total number of neurons in the region of the folium within 100 µm of the primary fissure in three sections from each mouse (n = 3).18
Neuronal density of DCN (comprising lateral, intermediate, and medial nuclei) was estimated by unbiased stereology using the newCast Visiopharm software (Visiopharm, Hoersholm, Denmark) and a DP71 Olympus camera (Olympus) attached to an Olympus BX51 microscope. The DCN region was delineated in five coronal sections from one hemisphere per mouse (n = 3 per group) and stained for NeuN, with an interdistance of 120 µm. The optical fractionator method was used to estimate the number of NeuN-positive cells counted in these sections at a final magnification of 600×.
Statistical analysis. Student's t test (comparison of two groups) or one-way analysis of variance test with post hoc multiple comparisons (more than two groups) by the Tukey test were used to compare deep cerebellar and Purkinje neuron counts, gene expression levels, and protein levels. Pearson correlation was used to analyze correlations of human ATXN3 levels with mouse behavior parameters and age-at-death. Mouse motor performance revealed by latency to traverse a beam, total beam breaks, and number of rears on the open-field was analyzed using one-way analysis of variance (mice used on the initial characterization of the YACMJD transgenic mice) or two-way analysis of variance repeated measures with post hoc comparisons by the Tukey test (mice used on the RNAi trial). Beam walking dragging was analyzed using Pearson χ2 test. A P < 0.05 was considered statistically significant in all analyses. Data were analyzed using IBM SPSS Statistics 20 software (SPSS, Chicago, IL).
SUPPLEMENTARY MATERIAL Figure S1. Hemizygous YACMJD15.4 (wt/Q15) transgenic mice do not show motor deficits or MJD-typical pathological hallmarks. Figure S2. Coronal brain sections illustrating transduced regions in Q84/Q84 mice, 10 weeks after cerebellar infusion with AAV2/1-miRATXN3-hrGFP. Figure S3. Composite of coronal sections of cerebellum illustrating transduced anterior cerebellum lobules (6Cb–9Cb) in Q84/Q84 mice, 10 weeks after cerebellar infusion with AAV2/1-miRATXN3-hrGFP.
Acknowledgments
We thank the members of Paulson Lab for fruitful discussions. M. do C.C. was a recipient of fellowships from Fundação para a Ciência e a Tecnologia (FCT), Portugal (SFRH/BPD/28560/2006), and National Ataxia Foundation (NAF Research Fellowship Award 2011). V.G.S. was funded by NIH NS072158 and the Mateus Ataxia Research Fund. B.L.D. was funded by NIH P01 NS50210. E.R.-L. and H.L.P. were funded by NHI NS067111. H.L.P. was also funded for this work by NIH N038712 and the Mateus Ataxia Research Fund. The authors declared no conflict of interest.
Supplementary Material
Hemizygous YACMJD15.4 (wt/Q15) transgenic mice do not show motor deficits or MJD-typical pathological hallmarks.
Coronal brain sections illustrating transduced regions in Q84/Q84 mice, 10 weeks after cerebellar infusion with AAV2/1-miRATXN3-hrGFP.
Composite of coronal sections of cerebellum illustrating transduced anterior cerebellum lobules (6Cb–9Cb) in Q84/Q84 mice, 10 weeks after cerebellar infusion with AAV2/1-miRATXN3-hrGFP.
References
- Costa Mdo C, Paulson HL. Toward understanding Machado-Joseph disease. Prog Neurobiol. 2012;97:239–257. doi: 10.1016/j.pneurobio.2011.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams AJ, Paulson HL. Polyglutamine neurodegeneration: protein misfolding revisited. Trends Neurosci. 2008;31:521–528. doi: 10.1016/j.tins.2008.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lima M, Costa MC, Montiel R, Ferro A, Santos C, Silva C, et al. Population genetics of wild-type CAG repeats in the Machado-Joseph disease gene in Portugal. Hum Hered. 2005;60:156–163. doi: 10.1159/000090035. [DOI] [PubMed] [Google Scholar]
- Maciel P, Costa MC, Ferro A, Rousseau M, Santos CS, Gaspar C, et al. Improvement in the molecular diagnosis of Machado-Joseph disease. Arch Neurol. 2001;58:1821–1827. doi: 10.1001/archneur.58.11.1821. [DOI] [PubMed] [Google Scholar]
- Coutinho P, Andrade C. Autosomal dominant system degeneration in Portuguese families of the Azores Islands. A new genetic disorder involving cerebellar, pyramidal, extrapyramidal and spinal cord motor functions. Neurology. 1978;28:703–709. doi: 10.1212/wnl.28.7.703. [DOI] [PubMed] [Google Scholar]
- Rüb U, Brunt ER, Deller T. New insights into the pathoanatomy of spinocerebellar ataxia type 3 (Machado-Joseph disease). Curr Opin Neurol. 2008;21:111–116. doi: 10.1097/WCO.0b013e3282f7673d. [DOI] [PubMed] [Google Scholar]
- Schöls L, Bauer P, Schmidt T, Schulte T, Riess O. Autosomal dominant cerebellar ataxias: clinical features, genetics, and pathogenesis. Lancet Neurol. 2004;3:291–304. doi: 10.1016/S1474-4422(04)00737-9. [DOI] [PubMed] [Google Scholar]
- Todi SV, Paulson HL. Balancing act: deubiquitinating enzymes in the nervous system. Trends Neurosci. 2011;34:370–382. doi: 10.1016/j.tins.2011.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch P, Breuer P, Peitz M, Jungverdorben J, Kesavan J, Poppe D, et al. Excitation-induced ataxin-3 aggregation in neurons from patients with Machado-Joseph disease. Nature. 2011;480:543–546. doi: 10.1038/nature10671. [DOI] [PubMed] [Google Scholar]
- Paulson HL, Perez MK, Trottier Y, Trojanowski JQ, Subramony SH, Das SS, et al. Intranuclear inclusions of expanded polyglutamine protein in spinocerebellar ataxia type 3. Neuron. 1997;19:333–344. doi: 10.1016/s0896-6273(00)80943-5. [DOI] [PubMed] [Google Scholar]
- Matos CA, de Macedo-Ribeiro S, Carvalho AL. Polyglutamine diseases: the special case of ataxin-3 and Machado-Joseph disease. Prog Neurobiol. 2011;95:26–48. doi: 10.1016/j.pneurobio.2011.06.007. [DOI] [PubMed] [Google Scholar]
- Paulson HL. Dominantly inherited ataxias: lessons learned from Machado-Joseph disease/spinocerebellar ataxia type 3. Semin Neurol. 2007;27:133–142. doi: 10.1055/s-2007-971172. [DOI] [PubMed] [Google Scholar]
- Fujikake N, Nagai Y, Popiel HA, Okamoto Y, Yamaguchi M, Toda T. Heat shock transcription factor 1-activating compounds suppress polyglutamine-induced neurodegeneration through induction of multiple molecular chaperones. J Biol Chem. 2008;283:26188–26197. doi: 10.1074/jbc.M710521200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teixeira-Castro A, Ailion M, Jalles A, Brignull HR, Vilaça JL, Dias N, et al. Neuron-specific proteotoxicity of mutant ataxin-3 in C. elegans: rescue by the DAF-16 and HSF-1 pathways. Hum Mol Genet. 2011;20:2996–3009. doi: 10.1093/hmg/ddr203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hübener J, Weber JJ, Richter C, Honold L, Weiss A, Murad F, et al. Calpain-mediated ataxin-3 cleavage in the molecular pathogenesis of spinocerebellar ataxia type 3 (SCA3). Hum Mol Genet. 2013;22:508–518. doi: 10.1093/hmg/dds449. [DOI] [PubMed] [Google Scholar]
- Simões AT, Gonçalves N, Koeppen A, Déglon N, Kügler S, Duarte CB, et al. Calpastatin-mediated inhibition of calpains in the mouse brain prevents mutant ataxin 3 proteolysis, nuclear localization and aggregation, relieving Machado-Joseph disease. Brain. 2012;135 Pt 8:2428–2439. doi: 10.1093/brain/aws177. [DOI] [PubMed] [Google Scholar]
- Chou AH, Chen SY, Yeh TH, Weng YH, Wang HL. HDAC inhibitor sodium butyrate reverses transcriptional downregulation and ameliorates ataxic symptoms in a transgenic mouse model of SCA3. Neurobiol Dis. 2011;41:481–488. doi: 10.1016/j.nbd.2010.10.019. [DOI] [PubMed] [Google Scholar]
- Shakkottai VG, do Carmo Costa M, Dell'Orco JM, Sankaranarayanan A, Wulff H, Paulson HL. Early changes in cerebellar physiology accompany motor dysfunction in the polyglutamine disease spinocerebellar ataxia type 3. J Neurosci. 2011;31:13002–13014. doi: 10.1523/JNEUROSCI.2789-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitt I, Linden M, Khazneh H, Evert BO, Breuer P, Klockgether T, et al. Inactivation of the mouse Atxn3 (ataxin-3) gene increases protein ubiquitination. Biochem Biophys Res Commun. 2007;362:734–739. doi: 10.1016/j.bbrc.2007.08.062. [DOI] [PubMed] [Google Scholar]
- Alves S, Nascimento-Ferreira I, Auregan G, Hassig R, Dufour N, Brouillet E, et al. Allele-specific RNA silencing of mutant ataxin-3 mediates neuroprotection in a rat model of Machado-Joseph disease. PLoS ONE. 2008;3:e3341. doi: 10.1371/journal.pone.0003341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alves S, Nascimento-Ferreira I, Dufour N, Hassig R, Auregan G, Nóbrega C, et al. Silencing ataxin-3 mitigates degeneration in a rat model of Machado-Joseph disease: no role for wild-type ataxin-3. Hum Mol Genet. 2010;19:2380–2394. doi: 10.1093/hmg/ddq111. [DOI] [PubMed] [Google Scholar]
- Nóbrega C, Nascimento-Ferreira I, Onofre I, Albuquerque D, Hirai H, Déglon N, et al. Silencing mutant ataxin-3 rescues motor deficits and neuropathology in Machado-Joseph disease transgenic mice. PLoS ONE. 2013;8:e52396. doi: 10.1371/journal.pone.0052396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torashima T, Koyama C, Iizuka A, Mitsumura K, Takayama K, Yanagi S, et al. Lentivector-mediated rescue from cerebellar ataxia in a mouse model of spinocerebellar ataxia. EMBO Rep. 2008;9:393–399. doi: 10.1038/embor.2008.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu J, Gagnon KT, Liu J, Watts JK, Syeda-Nawaz J, Bennett CF, et al. Allele-selective inhibition of ataxin-3 (ATX3) expression by antisense oligomers and duplex RNAs. Biol Chem. 2011;392:315–325. doi: 10.1515/BC.2011.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu J, Matsui M, Gagnon KT, Schwartz JC, Gabillet S, Arar K, et al. Allele-specific silencing of mutant huntingtin and ataxin-3 genes by targeting expanded CAG repeats in mRNAs. Nat Biotechnol. 2009;27:478–484. doi: 10.1038/nbt.1539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cemal CK, Carroll CJ, Lawrence L, Lowrie MB, Ruddle P, Al-Mahdawi S, et al. YAC transgenic mice carrying pathological alleles of the MJD1 locus exhibit a mild and slowly progressive cerebellar deficit. Hum Mol Genet. 2002;11:1075–1094. doi: 10.1093/hmg/11.9.1075. [DOI] [PubMed] [Google Scholar]
- Rodríguez-Lebrón E, do Carmo Costa M, Molina-Luna K, Peron TM, Fischer S., and, Boudreau RL, et al. Silencing mutant ATXN3 expression resolves molecular phenotypes in SCA3 transgenic mice. Mol Therapy (this issue) 2013. [DOI] [PMC free article] [PubMed]
- Boudreau RL, Martins I, Davidson BL. Artificial microRNAs as siRNA shuttles: improved safety as compared to shRNAs in vitro and in vivo. Mol Ther. 2009;17:169–175. doi: 10.1038/mt.2008.231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McBride JL, Boudreau RL, Harper SQ, Staber PD, Monteys AM, Martins I, et al. Artificial miRNAs mitigate shRNA-mediated toxicity in the brain: implications for the therapeutic development of RNAi. Proc Natl Acad Sci USA. 2008;105:5868–5873. doi: 10.1073/pnas.0801775105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coutinho P, Sequeiros J. Clinical, genetic and pathological aspects of Machado-Joseph disease. J Genet Hum. 1981;29:203–209. [PubMed] [Google Scholar]
- Goto J, Watanabe M, Ichikawa Y, Yee SB, Ihara N, Endo K, et al. Machado-Joseph disease gene products carrying different carboxyl termini. Neurosci Res. 1997;28:373–377. doi: 10.1016/s0168-0102(97)00056-4. [DOI] [PubMed] [Google Scholar]
- Ichikawa Y, Goto J, Hattori M, Toyoda A, Ishii K, Jeong SY, et al. The genomic structure and expression of MJD, the Machado-Joseph disease gene. J Hum Genet. 2001;46:413–422. doi: 10.1007/s100380170060. [DOI] [PubMed] [Google Scholar]
- McBride JL, Pitzer MR, Boudreau RL, Dufour B, Hobbs T, Ojeda SR, et al. Preclinical safety of RNAi-mediated HTT suppression in the rhesus macaque as a potential therapy for Huntington's disease. Mol Ther. 2011;19:2152–2162. doi: 10.1038/mt.2011.219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boudreau RL, McBride JL, Martins I, Shen S, Xing Y, Carter BJ, et al. Nonallele-specific silencing of mutant and wild-type huntingtin demonstrates therapeutic efficacy in Huntington's disease mice. Mol Ther. 2009;17:1053–1063. doi: 10.1038/mt.2009.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Hemizygous YACMJD15.4 (wt/Q15) transgenic mice do not show motor deficits or MJD-typical pathological hallmarks.
Coronal brain sections illustrating transduced regions in Q84/Q84 mice, 10 weeks after cerebellar infusion with AAV2/1-miRATXN3-hrGFP.
Composite of coronal sections of cerebellum illustrating transduced anterior cerebellum lobules (6Cb–9Cb) in Q84/Q84 mice, 10 weeks after cerebellar infusion with AAV2/1-miRATXN3-hrGFP.