

Abstract
Mutations in the FKRP gene are associated with a wide range of muscular dystrophies from mild limb-girdle muscular dystrophy (LGMD) 2I to severe Walker–Warburg syndrome and muscle-eye-brain disease. The characteristic biochemical feature of these diseases is the hypoglycosylation of α-dystroglycan (α-DG). Currently there is no effective treatment available. In this study, we examined the adeno-associated virus serotype 9 vector (AAV9)-mediated gene therapy in the FKRP mutant mouse model with a proline to leucine missense mutation (P448L). Our results showed that intraperitoneal administration of AAV9-FKRP resulted in systemic FKRP expression in all striated muscles examined with the highest levels in cardiac muscle. Consistent with our previous observations, FKRP protein is localized in the Golgi apparatus in myofibers. Expression of FKRP consequently restored functional glycosylation of α-DG in the skeletal and cardiac muscles. Significant improvement in dystrophic pathology, serum creatine kinase levels and muscle function was observed. Only limited FKRP transgene expression was detected in kidney and liver with no detectable toxicity. Our results provided evidence for the utility of AAV-mediated gene replacement therapy for FKRP-related muscular dystrophies.
Introduction
The Fukutin-related protein (FKRP) gene encodes a predicted type II membrane protein consisting of a hydrophobic N-terminal transmembrane domain, a stem region, and a putative catalytic C-terminal domain with a D×D motif, a conserved structure found in many glycotransferases.1 FKRP mutations are associated with muscular dystrophies of highly variable clinical severity, ranging from common and milder limb-girdle muscular dystrophy (LGMD2I), to severe forms of congenital muscular dystrophy (MDC1C) with or without involvement of central nervous system, Walker–Warburg syndrome and muscle-eye-brain disease.2,3,4,5 These diseases share a common biochemical defect in the glycosylation of α-dystroglycan (α-DG) in muscle tissues commonly referred to as dystroglycanopathies. Other genes involved in dystroglycanopathies include fukutin, POMGnT1, POMT1, POMT2, and LARGE.6 There is an evidence that POMT1, POMT2, and POMGnT are able to initiate the first two steps of a unique O-mannosylation pathway of glycosylation to α-DG.7,8 More recently, LARGE has been shown to act as a bifunctional glycosyltransferase, that has both xylosyltransferase and glucuronyltransferase activities, producing repeating units of [–3-xylose–α1,3-glucuronic acid-β1–].9 However, the function of both fukutin and FKRP in the glycosylation of α-DG remain elusive.
α-DG is a central component of the dystrophin-associated glycoprotein complex, linking the actin cytoskeleton of the muscle fibers with extracellular ligands, such as laminin α2, perlecan, biglycan, neurexin, and agrin. This linkage is important for the integrity of the muscle fiber structure.10,11,12 The function of α-DG and its interaction with extracellular matrix proteins largely depends on O-linked glycosylation in the mucin-like domain.10,13 Defective α-DG glycosylation as the result of FKRP mutations is thought to lead to disruption of the linkage between the subsarcolemmal cytoskeleton and the extracellular matrix, which in turn leads to muscle degeneration. In addition, functionally glycosylated α-DG plays an important role in the development of the central nervous system.14,15
Currently there is no effective treatment for FKRP-related diseases. Over the past decades, adeno-associated virus (AAV)-mediated gene therapy has been extensively investigated in different disease models and has emerged as one of the most promising approaches as a delivery vector, owing to its high tropism for skeletal and cardiac muscles and nonpathogenicity in humans.16,17,18,19,20,21,22 Several AAV serotypes have been tested in preclinical studies with long-term success in targeting muscles through a systemic approach.17,18,19 New progress in clinic trials for sarcoglycan deficient LGMD2D, LGMD2C and other diseases has offered realistic hope for AAV vector-mediated gene therapy to achieve therapeutic results in the clinics.23,24,25
Cardiomyopathy is a common feature in FKRP-related muscular dystrophies and becomes an increasingly important consideration for treatment development. Cardiac involvement includes dilated cardiomyopathy,26 focal myocardial fibrosis,27 and cardiac failure.26,28,29 The severity of cardiac disease, however, may not be related to the severity of skeletal muscle involvement.30 Exploration and optimization of AAV serotypes have also led to the identification of highly effective AAV serotypes with cardiac muscle tropism permitting effective delivery of therapeutic transgene to affected cardiac muscles.20,21,22 Among the AAV serotypes, AAV9 has higher tropism to cardiac muscle, thus could be the choice for treating FKRP-related cardiomyopathy.20,21
Recently we have created mouse models containing reported FKRP mutations in patients with LGMD2I and MDC1C. Our analyses showed that knock-out of the FKRP was embryonic lethal. Knock-in of different missense mutations including P448L resulted in muscular dystrophy with variable severity ranging from early death to a mild dystrophic phenotype with later onset of muscle degeneration.31 These observations provide direct evidence to support the important role of FKRP in glycosylation of α-DG and correlation between genotype and phenotypic variations in clinics. These mouse models are also valuable for developing and testing experimental therapies for the diseases. In this study, we examined the efficacy of AAV9-mediated FKRP gene therapy in the P448L mutant mice. While the heterozygotes of the P448L mutation are phenotypically normal, the homozygotes showed lack of functional glycosylated α-DG and early disease onset (around 3–4 weeks of age) with prominent muscle pathology and impaired skeletal muscle functions. Here we show that systemic delivery of AAV9-FKRP at the age of 3–4 weeks produced high levels of FKRP expression in all skeletal muscles examined, and most prominently, in the cardiac muscle. This was associated with the restoration of functional glycosylation of α-DG up to near normal levels in cardiac muscle, amelioration of the skeletal muscle pathology, and significantly improved function. The treatment did not cause detectable toxicity in the liver or kidney. Our study indicates that AAV9-mediated gene therapy represents an effective strategy for FKRP-related muscular dystrophy.
Results
AAV vector construction and in vitro verification of FKRP expression
The AAV9 vector and chicken β-actin promoter coupled with the CMV enhancer (referred to as CMV-CB promoter) was used for effective transgene expression.18 The mouse Fkrp coding sequence was codon-optimized for high expression (Supplementary Figure S1, online). The AAV9-CMV-CB-FKRP vector (referred to as AAV9-FKRP) was initially examined in C2C12 myoblasts and differentiated myotubes. Strong expression of FKRP was detected by immunofluorescence staining with the antibody FKRP829 (also called FKRPSTEM). The strongest signals in the myoblasts were localized in the peri-nuclear areas although weak punctate signals were also found in other parts of the cytoplasm (Figure 1a). Consistent with our previous observation with plasmid-mediated FKRP expression and recent findings in human rectus muscle tissue,32,33 these signals colocalized with GM130, a cis-Golgi marker. In differentiating myotubes, FKRP staining appeared as punctate dots within the entire cytoplasm and surrounding the nuclei where the signals also colocalized with GM130 (Figure 1b). Colocalization of the Myc-tagged FKRP and GM130 was confirmed by double staining (Supplementary Figure S2, online).
Figure 1.

FKRP expression in C2C12 myoblasts (a) and a single myotube (b) detected by immunocytochemistry with the antibodies to FKRP (red signal) and the Golgi marker GM130 (green signal). The two signals colocalize with close proximity (Merged). Blue, DAPI nuclear staining. Scale bar, 20 µm.
FKRP expression and localization in skeletal muscle by local injection
We next examined the AAV9-FKRP expression in skeletal muscle by intramuscular injection of 5 × 1010 vector genomes (v.g.) into tibialis anterior (TA) muscle of the P448L mutant mice aged 4–6 weeks. In the untreated mice, functionally glycosylated α-DG was barely detectable in the muscles by both immunostaining and western blot. One week after vector injection, strong FKRP expression was detected with the FKRPSTEM antibody in the majority of myofibers of the injected muscle. FKRP immunoreactivity was detected in an array of puncta regularly distributed within the sarcoplasm. A high number of puncta with relatively stronger signals were also detected near the sarcolemma (Figure 2). The polyclonal antibody to Myc-tag revealed the same patterns as those detected by the FKRPSTEM antibody. Consistent with our earlier observation, the signals either overlapped or closely colocalized with the signal of GM130 in the cis-Golgi apparatus (Figure 2). A few fibers with FKRP immunoreactivity throughout the entire sarcoplasm were also detected (Supplementary Figure S3, online). Endogenous FKRP was not detected by the antibody. Expression of FKRP was confirmed by western blot (Figure 3). No signal representing endogenous FKRP was clearly detected in the muscles of untreated animals (Supplementary Figure S3, online). In contrast to the controls, the treated muscles of FKRP mutant mice showed strong signals with the IIH6 antibody that recognizes functionally glycosylated α-DG (Figure 2). These results indicate that FKRP expression by AAV-9 mediated gene delivery is able to restore functional glycosylation of α-DG in muscles.
Figure 2.

FKRP expression and functional glycosylation of α-DG in tibialis anterior (TA) muscle of the P448L mutant mice after AAV-FKRP treatment. Upper panel: FKRP was detected with the FKRPSTEM antibody (left) and the Golgi apparatus detected with antibody GM130 (middle). Merged is the overlay of FKRPSTEM with GM130 (right). Lower panel: FKRP was detected with the FKRPSTEM (left) and functionally glycosylated α-DG detected with the IIH6 (middle). Control, TA muscle from untreated control P448L mutant mouse stained with IIH6 (right). Blue, DAPI nuclear staining. Scale bar, 60 µm.
Figure 3.
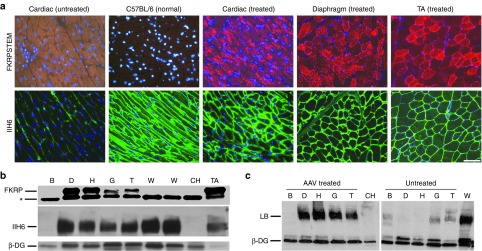
Expression of FKRP and functionally glycosylated α-DG. (a) Expression of FKRP and functionally glycosylated α-DG detected with antibody FKRPSTEM (upper panel, red) and IIH6 (lower panel, green) respectively, after AAV9-FKRP treatment. Untreated, muscles from untreated FKRP mutant mice. C57BL/6 (Normal), cardiac muscle. Treated, muscles from AAV9-FKRP treated FKRP mutant mice. Scale bar, 60 µm. (b) Detection of FKRP expression and functionally glycosylated α-DG by western blots with IIH6 and laminin binding (LB) assay in AAV9-FKRP (AAV) treated P448L mutant mice. Lanes: B, brain; D, diaphragm; H, heart; G, gastrocnemius; T, tibialis anterior; CH, heart from control P448L mutant mouse; W, cardiac muscles from wild-type control C57BL/6 mice; TA, tibialis anterior muscle after local injection of AAV9-FKRP. *Nonspecific protein within tissues. Protein loading of 40 µg was applied from each sample.
Systemic treatment with AAV9-FKRP restores functional α-dystroglycan in all skeletal muscles examined
We next examined the systemic effect of AAV9-FKRP treatment by intraperitoneal (i.p.) injection to the P448L mutant mice aged 3–4 weeks. The mice were given a single dose of 5 × 1011 v.g. and examined 4 months later. FKRP expression was first assessed by immunostaining (Figure 3a). Skeletal muscles including TA, quadriceps, gastrocnemius, and diaphragm, all showed relatively homogeneous expression of the FKRP protein in the majority of muscle fibers detected with the FKRPSTEM antibody. These results were confirmed with the rabbit antibody against Myc-tag. Similar to the patterns observed in the TA muscles after local treatment, FKRP formed small puncta that predominantly localized near to, but not in the sarcolemma. Once again, the signals of FKRP colocalized well with the signal of GM130. A variable proportion (less than 15%) of myofibers in all muscles was found without clearly detectable signal for FKRP expression (Supplementary Figure S4, online). Sporadically, myofibers (less than 1%) with FKRP overexpression covering the entire cytoplasm were also observed in all muscles. Variation of FKRP expression in different muscles was demonstrated by western blots (Figure 3b). FKRP expression was generally higher in the diaphragm than those in the other skeletal muscles, likely due to the i.p. delivery route which allows a direct contact of the diaphragm with the viral vectors.
FKRP expression correlated well with the levels of functionally glycosylated α-DG in the AAV9-FKRP treated mice. To compare the levels of FKRP expression to those of functionally glycosylated α-DG within the same myofibers, serial sections were stained with both IIH6 and FKRPSTEM antibodies. Nearly all fibers that showed detectable levels of FKRP expression were stained clearly with IIH6 antibody as well (Figure 3, Supplementary Figure S4, online). By contrast, most myofibers without detectable FKRP displayed little to no signals with the IIH6. A few IIH6-positive fibers without detectable FKRP expression were also observed. However, such fibers were also detected in untreated FKRP mutant mice (Supplementary Figure S4, online). Signal intensity for functionally glycosylated α-DG was not higher in the fibers with strong FKRP signal covering the entire cytoplasm than that in the majority of myofibers containing only punctate signals for FKRP. This suggests that highly overexpressed FKRP could not further enhance the functional glycosylation of α-DG. The overall levels of functionally glycosylated α-DG detected by western blots ranged from 30 to 70% of normal levels with the higher levels detected in the diaphragm (Figure 3b). The functionality of the α-DG was supported by a laminin binding assay with intensity of immunoreactivity similar to that in normal muscle (Figure 3c).
FKRP expression significantly improves skeletal muscle pathology and functions
Restoration of high levels of functional α-DG 4 months after AAV9-FKRP treatment unambiguously improved pathology in all skeletal muscles examined. Histological evaluation by hematoxylin and eosin (H&E) staining showed that muscle fibers (>95%) were highly uniform in size. Small regenerating myofibers with diameters less than 30 µm became rare (less than 3%) in the treated muscles whereas up to 25% was detected in the controls (Figure 4). At the same time, hypertrophic fibers (with sizes larger than normal fibers of the equivalent muscle) decreased in AAV-FKRP treated samples when compared with the untreated FKRP mutant mice (Supplementary Figure S5, online). Myofibers with central nucleation (a marker of muscle degeneration and regeneration) were significantly reduced (less than 15%) in all muscles of the treated mice compared with the control muscles (greater than 60%). Focal muscle degeneration and infiltrations were frequently observed in the untreated control muscles, but absent in the treated muscles (Figure 4, Supplementary Figure S6, online). Improvement in muscle pathology was most conspicuous in the diaphragm. The untreated P448L mutant mice showed large areas of muscle degeneration and regeneration with groups of small-caliber fibers and mononucleate infiltrates in the extracellular matrix (Figure 4a). By contrast, the diaphragms of AAV9-FKRP treated mice were highly homogeneous in fiber size with little infiltrate as shown in Figure 4a. The percentage of central-nucleated fibers was also reduced significantly from greater than 50% in the control samples to less than 5% in the treated samples (Figure 4b).
Figure 4.

Histological analysis of muscle tissues. (a) Histological analysis of muscle tissues from control P448L mutant (upper panel) and AAV9-FKRP treated P448L mutant mice (lower panel). Hematoxylin and Eosin (H&E) staining. (b) Percentage of centrally nucleated fibers (CNF) (left panel) and percentage of regenerating fibers (RF) with small calibers (≤30 µm in diameter; right panel). n = 5, *Significant, two-tailed t-test, *P ≤ 0.05. Scale bar, 60 µm. Dia, diaphram; Gas, gastrocnemicus; Qua, qudriceps; TA, tibialis anterior.
We also evaluated skeletal muscle function with grip force tests. Both the forelimb and hindlimb maximum grip force strength showed significant improvement in the AAV9-FKRP treated P448L mutant mice, reaching 80 and 90% normal levels (C57 mice as 100%) whereas the force was within 70 and 75% normal levels in the untreated mutant mice, respectively (Figure 5a, b). Improved muscle functions was strongly supported by a significant reduction in serum levels of creatine kinase, with average of 700 U/l compared with above 4,000 U/l in the untreated group (Figure 5c).
Figure 5.

Grip force measurement and serum tests. (a) Hindlimb and (b) forelimb grip force measurement. Grip force strength is normalized for body weight. P448L, P448L mutant mice without AAV treatment. AAV-treated, P448L mutant mice treated with AAV9-FKRP. WT, C57BL/6 normal control. (c) Serum tests. *Significant difference was observed between the AAV-treated FKRP mutant mice when compared with untreated FKRP mutant mice (n = 5, two-tailed t-test, *P ≤ 0.05). CK (creatine kinase, kU/l), creatinine (mg/l), urea nitrogen (mg/ml), total bilirubin (mg/l), alanine transaminase (ALT) (U/dl), alkaline phosphatase (ALP) (U/dl) and γ-glutamyltransferase (GGT) (U/l).
High levels of FKRP expression and functional glycosylation of α-DG in cardiac muscle
P448L mutant mice also lack functional glycosylated α-DG in cardiac muscle. As expected, AAV9-CMV-CB vector achieved a higher FKRP expression in the cardiac muscle when compared with that of the skeletal muscles. More than 95% of cardiac muscle fibers showed strong punctate signals for FKRP detected by antibodies to both FKRP and the Myc-tag (Supplementary Figure S4, online), which colocalized with the signal for the Golgi marker GM130 (Supplementary Figure S7, online). The signals were clearly higher in the cardiac muscle than that in the skeletal muscles (except diaphragm) (Figure 3b). Consistently, functionally glycosylated α-DG became evident in nearly all cardiac muscle fibers as detected by IIH6 antibody (Figure 3a). The intensity of IIH6 signal reached near normal levels by western blots and the same size band of α-DG bound laminin as well, indicating its functionality.
Real-time PCR analysis of FKRP transgene and endogenous FKRP expression
To determine the amount of FKRP required for the restoration of functional glycosylation of α-DG, we measured the levels of FKRP transcript by real-time quantitative polymerase chain reaction (qPCR). Primers were designed for transcripts specific to endogenous FKRP and the FKRP transgene (Life Technologies, Applied Biosystems, Foster City, CA). The results showed that levels of wild-type FKRP mRNA were higher in cardiac muscle and diaphragm, almost twofold greater than those in TA muscles. The mRNA levels of mutant endogenous FKRP in the muscles were however considerably lower than the levels of the wild-type diaphragm, heart and TA muscle (about 40, 60, and 60%, respectively) (Figure 6a). Interestingly, the levels of mRNA of the FKRP transgene were higher than that of the mutant endogenous FKRP in the diaphragm, but were lower in the heart and especially in the TA muscle (Figure 6a). The mRNA levels from the transgene were consistent with the levels of the vector-expressed FKRP protein in the three muscles detected by both immunofluorescence staining and western blots.
Figure 6.

Detection of FKRP transcript in muscles and protein in liver and kidney. (a) Quantitative PCR for the detection of endogenous and transgene FKRP expression. P448L FKRP, endogenous mutant FKRP; WTFKRP, normal FKRP expressed in C57BL/6 mice; transgene, AAV9-FKRP vector-expressed FKRP. The Copies (normalized) indicates the relative copy number to the control GAPDH. n = 3. (b) Detection of FKRP in liver and kidney from AAV9-FKRP-treated mouse. (c) α-DG and β-DG detection in liver and kidney by western blots. K, kidney; L, liver; P448L Cont, untreated P448L mouse; P448L-AAV9, AAV9-FKRP-treated mouse.
FKRP expression in kidney, liver, and toxicity assessment
We also examined FKRP transgene expression and levels of functionally glycosylated α-DG in the liver and kidney not known to be implicated in the pathophysiology of the dystroglycanopathies. Previous studies have reported high efficiency of AAV9 gene transfer to the hepatocytes. However, we detected FKRP expressed only in a small proportion (less than 5%) of the hepatocytes by immunohistochemistry (Figure 6b). No FKRP was clearly detected by immunohistochemistry in the kidney. Western blot analysis revealed that FKRP expression was nearly undetectable in both of these organs. Functionally glycosylated α-DG was not detected in the livers of all normal control mice, untreated and AAV9-FKRP treated FKRP mutant mice by western blot with IIH6 antibody (Figure 6c). In contrast, glycosylated α-DG was detected in the kidney of normal mice, but clearly reduced in the untreated FKRP mutant mice, suggesting that defects in FKRP affect the functional glycosylation of α-DG. The levels of expression increased considerably in the AAV9-FKRP treated mutant mice although this was still lower than that in normal mice (Figure 6c). These results suggest that a small amount expression of vector-derived FKRP, although not clearly detectable, might have functional significance. Interestingly, β-DG was clearly detected in the liver tissues, but barely detectable in the kidneys of the mice from all groups (normal mice, AAV-treated, and untreated FKRP mutant mice) despite the same quantity loading (40 µg) of tissue sample (Figure 6c). One possible explanation is that the kidney expresses very low levels of DG protein, but a large proportion of α-DG was functionally glycosylated. In contrast, there are high levels of DG expression with little functional glycosylation in the liver. However, differential glycosylation of α-DG between the two organs could not be excluded. Serum tests showed that levels of alkaline phosphatase, creatinine, BUN, and ALT, indicators for liver and kidney functions, were all comparable with those seen in normal C57BL/6 mice. No significant difference in body weight was observed between the AAV9-FKRP treated and untreated groups during the period of treatment after the i.p. delivery of the AAV9-FKRP (Supplementary Figure S8, online).
Discussion
AAV-mediated gene therapy has been shown to be highly effective in correcting disease phenotypes of several animal models of muscular dystrophies with deficiencies in dystrophin and the sarcoglycans.17,19,23 In general AAV is amongst one of the safest and least immunogenic gene vectors, although cellular immunity to the vector34 or novel transgene product35 has been reported in a few cases from numerous human gene therapy clinical studies. In this study, we chose the AAV9 serotype as we recently reported that AAV9 produced high levels of transgene expression in skeletal muscles with a higher tropism in cardiac muscle.36 The chicken β-actin promoter coupled with the CMV enhancer was used for effective transgene expression which might be further enhanced by codon-optimization of the cDNA (Supplementary Figure S1, online). We now show for the first time that this vector is able to effectively and persistently render the expression of FKRP in the mouse model of FKRP P448L mutation found in patients. This leads to the restoration of functionally glycosylated α-DG in all skeletal and cardiac muscles with significant improvement in muscle pathology and function, providing direct evidence for the therapeutic potential of AAV9-mediated gene therapy for FKRP-related muscular dystrophies. These results also further confirm that FKRP is essential for functional glycosylation of α-DG and FKRP mutations are the causal factors for the LGMD2I.
Cardiomyopathy is a common feature in FKRP-related muscular dystrophies and becomes an increasingly important consideration for treatment development. AAV9 with higher tropism to cardiac muscle over several other AAV serotypes could be the choice for treating FKRP-related cardiomyopathy.20,21 This is now further confirmed in the current study. FKRP expression both at the mRNA and protein levels is higher in the cardiac muscle than in all skeletal muscles examined in our study. Consequently, this resulted in the highest expression of functionally glycosylated α-DG, reaching the levels similar to that in normal cardiac muscle. While we could not verify the therapeutic effect on cardiac muscle functions due to mild pathological changes in the cardiac functions within the age group of the model mouse, one would expect a significant functional rescue if similar efficiency in FKRP expression and functional glycosylation of α-DG is achieved in hearts with cardiomyopathy due to FKRP mutations. A long-term therapeutic effect of AAV9-mediated FKRP delivery to the cardiac muscle is being currently conducted.
There are potential disadvantages of using AAV9 for systemic gene therapy for muscular dystrophy. One is the reported high levels of transduction of transgene in liver. Since FKRP mutations do not apparently affect liver function and FKRP function in organs other than muscles remains poorly understood, unwanted overexpression of FKRP in the liver might pose risk of side effects. However, this concern could be overcome by the use of recombinant AAV9 vector coupled with tissue specific promoter and microRNA-targeted transgene mRNA degradation.36 In the current study, although we did not use muscle- and heart-specific promoter in AAV9 for FKRP expression, we detected no broad expression of FKRP in the liver, but only in a small proportion (<5%) of the hepatocytes with levels no higher than that in muscles. The limited expression of FKRP protein to a small proportion of hepatocytes is apparently not consistent with some early reports that AAV9 displayed homogenous transgene expression under the control of CMV-CB promoter in liver. One possible explanation is the nature of the transprotein expressed by the AAV vector. In our previous mini-dystrophin gene therapy study, the AAV9 vector with CMV promoter produced high levels of dystrophin expression in muscle, but very low levels in the liver, despite the presence of high copy number of viral DNA. Several other possible explanations have been proposed to explain the variation in expression of transgenes in liver, including route-dependent and gender-dependent variation in levels of transgene expression.37
Importantly, the expression of FKRP did not induce the expression of functional glycosylation of α-DG in the liver. Furthermore, no clear liver toxicity was observed from serum enzyme tests and histology. Although FKRP expression was minimal in the kidney which expresses functionally glycosylated α-DG,38 this amount could still be beneficial as the treated kidney improved the lower levels of glycosylation of α-DG in mutant mice towards normal levels. These results suggest that AAV9 together with muscle specific promoter could constitute an effective, largely muscle specific vector system with preference for cardiac muscle to treat FKRP-related dystroglycanopathies with cardiomyopathy.
O-Manno-glycosylation of α-DG is essential for its interaction with extracellular ligands and mutations in nine genes have been known to disrupt glycosylation pathway for α-DG.6 The involvement of FKRP in the functional glycosylation of α-DG is evidenced by the hypoglycosylation of α-DG in LGMD2I, Walker–Warburg syndrome and MDC1C with FKRP mutations and in our newly developed FKRP mutant mice.31 However, the roles of FKRP on the process of glycosylation of α-DG have yet to be defined. It is also possible that FKRP might be involved in glycosylation of other glycoproteins. Therefore, it is important to determine whether overexpression of FKRP via AAV-mediated gene therapy might cause unforeseeable consequences. This would be especially relevant as most mutated FKRP genes still express at least partially functional FKRP proteins judged by the presence of variable amount of glycosylated α-DG in muscles. Our results from the current study showed that expression of the FKRP transgene restored the functional glycosylated α-DG with significant improvement in muscle pathology and function. Individual muscle fibers expressing extremely high levels of FKRP, with signals covering entire cytoplasmic region, were histologically normal in size and shape without any sign of degeneration. The presence of such fibers therefore suggests that overexpression of FKRP within certain levels is unlikely to produce significant cellular toxicity. These results also support the view that FKRP might have very limited function outside the glycosylation of α-DG after birth, and would be consistent with our initial results from a glyco-proteomic study. This study used LTQ-XL Orbitrap mass spectrometer to analyze the expression and glycosylation patterns of nearly 900 glycoproteins in skeletal muscles of FKRP mutant mouse models and showed that no glycoproteins from the muscle tissues, except α-DG, were apparently altered in size by the lack of expression of functional FKRP (manuscript in preparation). These data together suggest that α-DG is the principle target of sugar modifications by FKRP, which may explain the lack of side effects found after overexpression of FKRP in the P448L mice.
One potential difficulty in assessing the function of FKRP is the inability to detect endogenous FKRP protein despite the fact that exogenous FKRP proteins can be reliably detected by immunohistochemistry and western blots in both cell culture and in vivo in animal models after gene transfer with either naked plasmid32 or AAV viral vectors. The reason(s) behind this apparent discrepancy in detecting endogenous and exogenous FKRPs is not understood. One possibility is that endogenous FKRP is expressed at very low levels, beyond sensitivity of the detection systems. To assess the levels of AAV-mediated FKRP expression relative to the endogenous FKRP expression, we therefore measured mRNA levels of both FKRPs in the treated mice in comparison with normal mice by qPCR. We would expect high levels of exogenous FKRP transcript judging from the abundant exogenous FKRP protein. However, it was rather surprising to see that the amount of endogenous mutant FKRP transcripts is roughly the same as that of vector-derived transcripts in the diaphragm, and is actually higher in heart and TA muscle (approximately twofold) than that of vector-derived transcripts in the same muscles. This result might be partly due to the use of codon-optimized cDNA with higher efficiency in translation. One other possibility is that maintaining normal levels of functional glycosylated α-DG requires only very limited amount of FKRP protein. This however would be inconsistent with the requirement of considerably high levels of vector-derived FKRP protein for the restoration of functional glycosylation observed in muscles in this study. It is clear that novel approaches are required to answer this apparent conundrum.
Materials and Methods
Antibodies. The following primary antibodies were used: FKRP-829 (FKRPSTEM) rabbit polyclonal antibody raised against the stem region (amino acid residues 29–130) of the mouse FKRP;1 anti-GM130-FITC monoclonal antibody (BD Biosciences, San Jose, CA); rabbit anti-GM130 polyclonal antibody (EMD Chemicals, Darmstadt, Germany); rabbit anti-c-Myc polyclonal antibody (Sigma, Saint Louis, MO); 9E10 monoclonal anti-Myc antibody produced from hybridoma cells (NIH DSHB, Iowa City, IA); IIH6C4 (IIH6) monoclonal antibody against functionally glycosylated α-DG (Millipore, Temecula, CA); mouse anti-β-DG monoclonal antibody (DSHB, Iowa City, IA); Monoclonal anti-β-actin (Sigma, St. Louis, MO).
AAV9-CMV-CB-FKRP vector construction and rAAV production. The full length of 1482 bp mouse FKRP was codon optimized for high expression (GeneArt, Life Technologies). The sequence coding for the ten amino acid Myc-tag was linked to the C-terminal of the FKRP coding region and Kozak sequence was placed in front of the N-terminus of the FKRP coding sequence (Supplementary Figure S1, online). The synthetic fragment was cloned into pMK-RQ (KanR) using Sfil and Sfil sites. The resulting vector was named FKRP-PMK-RQ (GeneArt, Life Technologies). The NotI-SalI fragment of codon-optimized human-FKRP-Myc was cohesively ligated to the NotI and SalI restriction enzyme sites on AAV plasmid containing CMV-CB promoter36 and the final construct was named pXX-CB-FKRP-Myc. The recombinant AAV vectors were produced by following the three-plasmid cotransfection method.39 The titers of viral vector genome numbers were determined by the viral DNA dot blot method. The concentration of viral vectors was kept in the range of 2 × 1012 to 5 × 1012 vector genomes/ml and stored at −80 °C for future use. The final viral vector was named AAV9-CMV-CB-FKRP-myc (AAV9-FKRP in short).
C2C12 cell culture and AAV delivery. C2C12 myoblasts were cultured in DMEM supplemented with 10% fetal bovine serum and 4 mmol/l l-glutamine. Cells were grown to 70–80% confluence and then infected with AAV9-FKRP 5 × 1010 viral particles per ml of culture media. Cells were cultured either for 48 hours or for further 72 hours in differentiation media (4% serum only) and then prepared for examination. Cells grown on eight-well culture slides were prepared for immunocytochemistry and in six-well plates for western blot analysis. All culture media and reagents were purchased from Invitrogen/Gibco Life Sciences.
Animals and AAV vector delivery methods. FKRP P448L mutation has been associated with MDC1C.2,30 Mutant mice were generated by inGenious Targeting Laboratory (Stony Brook, NY). The targeting vector was engineered with a 1343C>T point mutation, resulting in an amino acid change from proline to leucine at position 448. The homozygote mutant mice containing a Neo cassette showed severe muscular dystrophy with brain and eye involvement and were unable to breed.31 The Neo cassette was therefore removed via crossbreeding with C57BL/6 FLP mice (inGenious Targeting Laboratory, Inc.) to create homozygote Fkrpp448L Neo- mice. This strain of homozygote mutant mice (referred as P448L mutant) has moderate skeletal muscle dystrophy essentially the same as the FKRP P448L Neo+ homozygotes reported previously,31 but lacks a clear defect in the central nervous system and shows normal breeding activity with near normal life span. There was no obvious pathology in cardiac muscle in the young adult mice. The homozygote FKRPP448L Neo-mice were therefore used in this study. C57BL/6 wild-type mice aged 4 weeks were used as controls. Experiments were approved by the Institutional Animal Care and Use Committee, Carolinas Medical Center. P448L mutants at the age of 3–4 weeks were administrated with AAV9-FKRP 5 × 1011 viral particles via single i.p. injection. Treated and control mice were euthanized 4 months after the AAV treatment. Blood and muscle samples were collected and stored for further analysis.
Histological analysis and fiber size determination. Skeletal and cardiac muscles were snapped frozen in liquid nitrogen chilled isopentane. Sections of 5 µm thickness were cut from the frozen tissues and stained with hematoxylin and eosin (H&E). Sections were then examined using an Olympus BX51 microscope.
Fiber size of TA and quadriceps was determined using the MetaMorph Basic Offline software. H&E images with original 20× magnification were subjected to Arithmetic processing to remove background noise and clearly identify muscle fibers. Processed images were then used to outline individual fibers as well as remove all other color from the image. This results in each fiber being outlined in black with a white interior and ensures that every fiber was accounted for in the final analysis. The outlined image was then calibrated appropriately using the software's calibrate distances tool. An image that was 1360 × 1040 pixels with a 20× magnification is ~0.4988 µm/pixel. Once calibrated the fibers were then threshold by excluding dark objects. Using the integrated morphometry analysis tool the equivalent radius of each fiber was determined and the data exported into excel for analysis. Fibers contacting the edge of the image were excluded from data analysis. The diameter of each fiber was calculated from the equivalent radius. During image processing the size of the image, pixel height, and width, were kept the same. Fibers with diameter smaller than 30 µm and larger than 80 µm were selected as subgroups as they were rarely detected in normal muscles.
Immunohistochemical analysis. For the detection of FKRP, Myc-tag, and Golgi marker GM130, cells and frozen sections were first fixed with 4% PFA for 15 minutes followed by incubation with 0.5% Triton X-100 for 5 minutes. The sections were then blocked with 8% bovine serum albumin and 10% fetal bovine serum diluted in phosphate-buffered saline for 30 minutes followed by incubation with primary antibody at room temperature. FKRP was detected with the FKRPSTEM antibody at 1:2,500 dilutions for 2 hours. The cis-Golgi marker GM130 was detected with fluorescence-labeled GM130 at 1:50 dilution for 6 hours. Myc-tag was detected with monoclonal antibody 9E10 at 1:15 dilution for 2 hours and polyclonal rabbit anti-Myc at 1:250 dilutions for 2 hours. Functionally glycosylated α-DG was detected with IIH6 as described previously.31 The primary antibodies were visualized with AlexaFluor488 or 594-conjugated goat anti-mouse or rabbit IgGs at 1:500 dilutions in phosphate-buffered saline for 1 hour. All antibodies were diluted in phosphate-buffered saline containing 10% fetal bovine serum and all steps were performed at room temperature unless specified. Immunofluorescence results were visualized using an Olympus BX51/BX52 fluorescence microscope and images were captured using the Olympus DP70 Digital Camera System (OPELCO, Dulles, VA).
Protein extraction, western blot analysis, and laminin overlay assay. Protein extraction and Western blot analysis were performed as reported previously.31 A laminin overlay assay was performed as reported previously.31,40 Laminin overlay assay demonstrates the ability of functionally glycosylated α-DG to bind the laminin, thus the functionality of α-DG.
Quantitative PCR analysis. RNA was isolated using TRIzol reagent (Invitrogen, Carlsbad, CA) and reverse transcribed with iScript cDNA synthesis kit (Bio-Rad, Hercules, CA). Quantitative PCR was performed on an ABI Prism 7500 Fast Real-Time PCR System using TaqMan Probe-Based Detection (Applied Biosystems, Foster City, CA). RNA samples were treated with RNase-free DNase before addition of Taqman Gene Expression Assays and Taqman Gene Expression Master Mix. All probes were custom designed from Applied Biosystems (the FKRP endogenous mutant assay ID is AIHR130; the FKRP transgene/optimized assay ID is AJ20SP0). GAPDH (assay ID is Mm03302249-g1) was used as an internal control. All reactions were performed in triplicates. Data were analyzed using RealTime StatMiner, version 4.0 (Integromics, Philadelphia, PA).
Grip force test. Grip strength was assessed with a grip strength meter consisting of horizontal forelimb mesh and an angled hindlimb mesh (Columbus Instruments, Columbus, OH). Five successful forelimb and hindlimb strength measurements were recorded in 2 minutes. The maximum values of each day over a 5-day period were used for subsequent data analysis. Grip strength measurements were normalized to body weight and expressed as kilogram force.41,42
Statistical analysis. All the results were expressed as mean ± SEM and the data were analyzed using two-tailed “t” test. A P value of <0.05 was considered as the level of significance.
SUPPLEMENTARY MATERIAL Figure S1. Codon-optimized FKRP sequences for mammalian expression. Figure S2. Co-localization of Myc-tag signal with GM130 in AAV9-FKRP treated C2C12 myoblasts. Figure S3. Detection of AAV9-mediated FKRP expression in TA muscles. Figure S4. Co-localization of FKRP expression (a, b and d detected with FKRPSTEM antibody) and functionally glycosylated α-DG (c, e and f detected with IIH6 antibody). Figure S5. Fiber size measurement. Figure S6. Histological analysis of muscle tissues from control P448L mutant mice (left column) and AAV-FKRP treated P448L mutant mice (Right column). Figure S7. Co-localization of GM130 and FKRP in cardiac muscle. Figure S8. Body weight measurement.
Acknowledgments
This work was supported by the Carolinas Muscular Dystrophy Research Endowment at the Carolinas HealthCare Foundation and Carolinas Medical Center, Charlotte, NC. The work was supported in part by the National Institute of Neurological Disorders and Stroke (1R01NS082536-01). We thank Dr Yiumo Michael Chan, Dr Xiaohua Wu, Guqi Wang, Caryn Cloer and Jianbin Li for helpful discussion of the experiment and Dr Anthony Blaeser for the morphometry analysis. We thank Dr Nury Steuerwald, Molecular Biology and Microarray Core Facilities of Cannon Research Center, Carolinas Medical Center for conducting qPCR experiments. We also thank Dr Charles Vannoy for careful editing of the manuscript. The authors declare no conflict of interest.
Supplementary Material
References
- Esapa CT, Benson MA, Schröder JE, Martin-Rendon E, Brockington M, Brown SC, et al. Functional requirements for fukutin-related protein in the Golgi apparatus. Hum Mol Genet. 2002;11:3319–3331. doi: 10.1093/hmg/11.26.3319. [DOI] [PubMed] [Google Scholar]
- Brockington M, Yuva Y, Prandini P, Brown SC, Torelli S, Benson MA, et al. Mutations in the fukutin-related protein gene (FKRP) identify limb girdle muscular dystrophy 2I as a milder allelic variant of congenital muscular dystrophy MDC1C. Hum Mol Genet. 2001;10:2851–2859. doi: 10.1093/hmg/10.25.2851. [DOI] [PubMed] [Google Scholar]
- Beltran-Valero de Bernabe D, Voit T, Longman C, Steinbrecher A, Straub V, Yuva Y, et al. Mutations in the FKRP gene can cause muscle-eye-brain disease and Walker-Warburg syndrome. J Med Genet. 2004;41:e61. doi: 10.1136/jmg.2003.013870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Topaloglu H, Brockington M, Yuva Y, Talim B, Haliloglu G, Blake D, et al. FKRP gene mutations cause congenital muscular dystrophy, mental retardation, and cerebellar cysts. Neurology. 2003;60:988–992. doi: 10.1212/01.wnl.0000052996.14099.dc. [DOI] [PubMed] [Google Scholar]
- Emery AE. The muscular dystrophies. Lancet. 2002;359:687–695. doi: 10.1016/S0140-6736(02)07815-7. [DOI] [PubMed] [Google Scholar]
- Martin PT. Congenital muscular dystrophies involving the O-mannose pathway. Curr Mol Med. 2007;7:417–425. doi: 10.2174/156652407780831601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manya H, Chiba A, Yoshida A, Wang X, Chiba Y, Jigami Y, et al. Demonstration of mammalian protein O-mannosyltransferase activity: coexpression of POMT1 and POMT2 required for enzymatic activity. Proc Natl Acad Sci USA. 2004;101:500–505. doi: 10.1073/pnas.0307228101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beltrán-Valero de Bernabé D, Currier S, Steinbrecher A, Celli J, van Beusekom E, van der Zwaag B, et al. Mutations in the O-Mannosyltransferase Gene POMT1 give rise to the severe neuronal migration disorder Walker-Warburg Syndrome. Am J Hum Genet. 2002;71:1033–1043. doi: 10.1086/342975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inamori K, Yoshida-Moriguchi T, Hara Y, Anderson ME, Yu L, Campbell KP. Dystroglycan function requires xylosyl- and glucuronyltransferase activities of LARGE. Science. 2012;335:93–96. doi: 10.1126/science.1214115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ervasti JM, Campbell KP. A role for the dystrophin-glycoprotein complex as a transmembrane linker between laminin and actin. J Cell Biol. 1993;122:809–823. doi: 10.1083/jcb.122.4.809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gee SH, Montanaro F, Lindenbaum MH, Carbonetto S. Dystroglycan-alpha, a dystrophin-associated glycoprotein, is a functional agrin receptor. Cell. 1994;77:675–686. doi: 10.1016/0092-8674(94)90052-3. [DOI] [PubMed] [Google Scholar]
- Sugita S, Saito F, Tang J, Satz J, Campbell K, Sudhof TC. A stoichiometric complex of neurexins and dystroglycan in brain. J Cell Biol. 2001;54:435–445. doi: 10.1083/jcb.200105003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brancaccio A, Schulthess T, Gesemann M, Engel J. Electron microscopic evidence for a mucin-like region in chick muscle alpha-dystroglycan. FEBS Lett. 1995;368:139–142. doi: 10.1016/0014-5793(95)00628-m. [DOI] [PubMed] [Google Scholar]
- Michele DE, Barresi R, Kanagawa M, Saito F, Cohn RD, Satz JS, et al. Post-translational disruption of dystroglycan-ligand interactions in congenital muscular dystrophies. Nature. 2002;418:417–422. doi: 10.1038/nature00837. [DOI] [PubMed] [Google Scholar]
- Shcherbata HR, Yatsenko AS, Patterson L, Sood VD, Nudel U, Yaffe D, et al. Dissecting muscle and neuronal disorders in a Drosophila model of muscular dystrophy. EMBO J. 2007;26:481–493. doi: 10.1038/sj.emboj.7601503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haidet AM, Mendell JR, Kaspar BK. Could gene therapy be the future for muscular dystrophy. Therapy. 2010;7:287–290. doi: 10.2217/thy.10.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowles DE, McPhee SW, Li C, Gray SJ, Samulski JJ, Camp AS, et al. Phase 1 gene therapy for Duchenne muscular dystrophy using a translational optimized AAV vector. Mol Ther. 2012;20:443–455. doi: 10.1038/mt.2011.237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Zhu T, Qiao C, Zhou L, Wang B, Zhang J, et al. Adeno-associated virus serotype 8 efficiently delivers genes to muscle and heart. Nat Biotechnol. 2005;23:321–328. doi: 10.1038/nbt1073. [DOI] [PubMed] [Google Scholar]
- Odom GL, Gregorevic P, Allen JM, Chamberlain JS. Gene therapy of mdx mice with large truncated dystrophins generated by recombination using rAAV6. Mol Ther. 2011;19:36–45. doi: 10.1038/mt.2010.205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inagaki K, Fuess S, Storm TA, Gibson GA, Mctiernan CF, Kay MA, et al. Robust systemic transduction with AAV9 vectors in mice: efficient global cardiac gene transfer superior to that of AAV8. Mol Ther. 2006;14:45–53. doi: 10.1016/j.ymthe.2006.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pacak CA, Mah CS, Thattaliyath BD, Conlon TJ, Lewis MA, Cloutier DE, et al. Recombinant adeno-associated virus serotype 9 leads to preferential cardiac transduction in vivo. Circ Res. 2006;99:e3–e9. doi: 10.1161/01.RES.0000237661.18885.f6. [DOI] [PubMed] [Google Scholar]
- Yang L, Jiang J, Drouin LM, Agbandje-McKenna M, Chen C, Qiao C, et al. A myocardium tropic adeno-associated virus (AAV) evolved by DNA shuffling and in vivo selection. Proc Natl Acad Sci USA. 2009;106:3946–3951. doi: 10.1073/pnas.0813207106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herson S, Hentati F, Rigolet A, Behin A, Romero NB, Leturcq F, et al. A phase I trial of adeno-associated virus serotype 1-?-sarcoglycan gene therapy for limb girdle muscular dystrophy type 2C. Brain. 2012;135 Pt 2:483–492. doi: 10.1093/brain/awr342. [DOI] [PubMed] [Google Scholar]
- Mendell JR, Rodino-Klapac LR, Rosales XQ, Coley BD, Galloway G, Lewis S, et al. Sustained alpha-sarcoglycan gene expression after gene transfer in limb-girdle muscular dystrophy, type 2D. Ann Neurol. 2010;68:629–638. doi: 10.1002/ana.22251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He B, Tang RH, Weisleder N, Xiao B, Yuan Z, Cai C, et al. Enhancing muscle membrane repair by gene delivery of MG53 ameliorates muscular dystrophy and heart failure in d-Sarcoglycan-deficient hamsters. Mol Ther. 2012;20:727–735. doi: 10.1038/mt.2012.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller T, Krasnianski M, Witthaut R, Deschauer M, Zierz S. Dilated cardiomyopathy may be an early sign of the C826A Fukutin-related protein mutation. Neuromuscul Disord. 2005;15:372–376. doi: 10.1016/j.nmd.2005.02.004. [DOI] [PubMed] [Google Scholar]
- Rosales XQ, Moser SJ, Tran T, McCarthy B, Dunn N, Habib P, et al. Cardiovascular magnetic resonance of cardiomyopathy in limb girdle muscular dystrophy 2B and 2I. J Cardiovasc Magn Reson. 2011;13:39. doi: 10.1186/1532-429X-13-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Amico A, Petrini S, Parisi F, Tessa A, Francalanci P, Grutter G, et al. Heart transplantation in a child with LGMD2I presenting as isolated dilated cardiomyopathy. Neuromuscul Disord. 2008;18:153–155. doi: 10.1016/j.nmd.2007.09.013. [DOI] [PubMed] [Google Scholar]
- Margeta M, Connolly AM, Winder TL, Pestronk A, Moore SA. Cardiac pathology exceeds skeletal muscle pathology in two cases of limb-girdle muscular dystrophy type 2I. Muscle Nerve. 2009;40:883–889. doi: 10.1002/mus.21432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercuri E, Brockington M, Straub V, Quijano-Roy S, Yuva Y, Herrmann R, et al. Phenotypic spectrum associated with mutations in the fukutin-related protein gene. Ann Neurol. 2003;53:537–542. doi: 10.1002/ana.10559. [DOI] [PubMed] [Google Scholar]
- Chan YM, Keramaris-Vrantsis E, Lidov HG, Norton JH, Zinchenko N, Gruber HE, et al. Fukutin-related protein is essential for mouse muscle, brain and eye development and mutation recapitulates the wide clinical spectrums of dystroglycanopathies. Hum Mol Genet. 2010;19:3995–4006. doi: 10.1093/hmg/ddq314. [DOI] [PubMed] [Google Scholar]
- Keramaris-Vrantsis E, Lu PJ, Doran T, Zillmer A, Ashar J, Esapa CT, et al. Fukutin-related protein localizes to the Golgi apparatus and mutations lead to mislocalization in muscle in vivo. Muscle Nerve. 2007;36:455–465. doi: 10.1002/mus.20833. [DOI] [PubMed] [Google Scholar]
- Alhamidi M, Kjeldsen Buvang E, Fagerheim T, Brox V, Lindal S, Van Ghelue M, et al. Fukutin-related protein resides in the Golgi cisternae of skeletal muscle fibres and forms disulfide-linked homodimers via an N-terminal interaction. PLoS ONE. 2011;6:e22968. doi: 10.1371/journal.pone.0022968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mingozzi F, High KA. Immune responses to AAV in clinical trials. Curr Gene Ther. 2007;7:316–324. doi: 10.2174/156652307782151425. [DOI] [PubMed] [Google Scholar]
- Mendell JR, Campbell K, Rodino-Klapac L, Sahenk Z, Shilling C, Lewis S, et al. Dystrophin immunity in Duchenne's muscular dystrophy. N Engl J Med. 2010;363:1429–1437. doi: 10.1056/NEJMoa1000228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiao C, Yuan Z, Li J, He B, Zheng H, Mayer C, et al. Liver-specific microRNA-122 target sequences incorporated in AAV vectors efficiently inhibits transgene expression in the liver. Gene Ther. 2011;18:403–410. doi: 10.1038/gt.2010.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dane AP, Wowro SJ, Cunningham SC, Alexander IE. Comparison of gene transfer to the murine liver following intraperitoneal and intraportal delivery of hepatotropic AAV pseudo-serotypes. Gene Ther. 2013;20:460–464. doi: 10.1038/gt.2012.67. [DOI] [PubMed] [Google Scholar]
- Durbeej M, Larsson E, Ibraghimov-Beskrovnaya O, Roberds SL, Campbell KP, Ekblom P. Non-muscle alpha-dystroglycan is involved in epithelial development. J Cell Biol. 1995;130:79–91. doi: 10.1083/jcb.130.1.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao X, Li J, Samulski RJ. Production of high-titer recombinant adeno-associated virus vectors in the absence of helper adenovirus. J Virol. 1998;72:2224–2232. doi: 10.1128/jvi.72.3.2224-2232.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patnaik SK, Stanley P. Mouse large can modify complex N- and mucin O-glycans on alpha-dystroglycan to induce laminin binding. J Biol Chem. 2005;280:20851–20859. doi: 10.1074/jbc.M500069200. [DOI] [PubMed] [Google Scholar]
- Spurney CF, Gordish-Dressman H, Guerron AD, Sali A, Pandey GS, Rawat R, et al. Preclinical drug trials in the mdx mouse: assessment of reliable and sensitive outcome measures. Muscle Nerve. 2009;39:591–602. doi: 10.1002/mus.21211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu B, Moulton HM, Iversen PL, Jiang J, Li J, Li J, et al. Effective rescue of dystrophin improves cardiac function in dystrophin-deficient mice by a modified morpholino oligomer. Proc Natl Acad Sci USA. 2008;105:14814–14819. doi: 10.1073/pnas.0805676105. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.