

Abstract
Viral vectors based on adeno-associated virus (AAV) are showing exciting promise in gene therapy trials targeting the adult liver. A major challenge in extending this promise to the pediatric liver is the loss of episomal vector genomes that accompanies hepatocellular proliferation during liver growth. Hence maintenance of sufficient transgene expression will be critical for success in infants and children. We therefore set out to explore the therapeutic efficacy and durability of liver-targeted gene transfer in the challenging context of a neonatal lethal urea cycle defect, using the argininosuccinate synthetase deficient mouse. Lethal neonatal hyperammonemia was prevented by prenatal and early postnatal vector delivery; however, hyperammonemia subsequently recurred limiting survival to no more than 33 days despite vector readministration. Antivector antibodies acquired in milk from vector-exposed dams were subsequently shown to be blocking vector readministration, and were avoided by crossfostering vector-treated pups to vector-naive dams. In the absence of passively acquired antivector antibodies, vector redelivery proved efficacious with mice surviving to adulthood without recurrence of significant hyperammonemia. These data demonstrate the potential of AAV vectors in the developing liver, showing that vector readministration can be used to counter growth-associated loss of transgene expression provided the challenge of antivector humoral immunity is addressed.
Introduction
Gene transfer vectors based on adeno-associated virus (AAV), a dependent parvovirus,1 are potent tools for liver-targeted gene delivery and show exciting promise for human therapy.2,3 To date, all clinical trials targeting the liver using AAV-mediated gene delivery have been in adult populations, yet many of the most difficult to treat disorders of liver function involve infants and children, where liver growth presents a particular challenge. In mouse and primate models, we and others have shown that hepatocellular proliferation results in loss of episomal AAV vector genomes with stable long-term transgene expression being dependent upon the subset of vector genomes that undergo genomic integration.4,5,6,7,8 Consistent with these observations, we have also shown, using the ornithine transcarbamylase-deficient spfash mouse,9 that a single dose of AAV vector delivered to young adult mice confers life-long correction of the underlying metabolic phenotype, while similar treatment in the newborn period confers only transient benefit.10 In the current study, we therefore set out to explore whether vector redelivery during liver growth might overcome this limitation.
Rather than using the spfash mouse which has a mild phenotype that allows survival to adulthood in the absence of treatment, we turned to the more challenging neonatal lethal argininosuccinate synthetase (ASS) knockout mouse model.11,12 This severe urea cycle defect, commonly known as citrullinemia type 1, results in early neonatal hyperammonemia and death within 48 hours of birth. This rapid postnatal deterioration faithfully recapitulates the clinical scenario confronting clinicians treating infants with this condition and related urea cycle defects. As an initial intervention affected pups were injected in the immediate newborn period via the intraperitoneal route with an AAV vector encoding the murine ASS cDNA under the transcriptional control of a liver-specific promoter. The development of lethal hyperammonemia, however, proved too rapid, relative to the kinetics of onset of transgene expression, necessitating in utero gene delivery in late gestation.13 Vector delivery at 16 days of gestation, 2–3 days in advance of parturition, extended survival to ~3 weeks of age with death accompanied by severe hyperammonemia. Vector readministration in the immediate newborn period and at 14 and 28 days initially conferred only modest increments in survival to a maximum of 33 days. Antivector antibodies passively acquired in milk by suckling pups from inadvertently vector-exposed dams were subsequently shown to explain the poor efficacy of postnatal vector readministration and was overcome by crossfostering in utero-treated pups to vector-naive dams. Using this strategy, recurrent postnatal vector readministration proved effective and allowed survival to adulthood without clinically significant hyperammonemia.
These data show that AAV vectors are capable of rescuing a lethal neonatal onset liver-based metabolic phenotype, and that recurrent vector readministration can be used to maintain clinically relevant phenotypic correction through to adulthood provided that antivector antibody responses are avoided. Importantly, these findings bode well for the extension of human clinical trials exploiting AAV for liver-targeted gene delivery to pediatric populations, thus greatly expanding the potential therapeutic utility of this exciting technology.
Results
Rapid onset of hyperammonemia in ASS−/− mice prevented effective gene therapy in the immediate newborn period
At birth, ASS−/− pups appeared healthy and were clinically indistinguishable from ASS+/− and ASS+/+ littermates. Within 8 hours, however, ASS−/− pups developed signs of illness, including reduced physical activity and pallor, progressing to coma and death within 40 hours of birth (Figure 1a). Plasma ammonia at 8 hours revealed the presence of marked hyperammonemia in ASS−/− pups consistent with rapid postnatal metabolic deterioration (Figure 1b). In an initial attempt to rescue this neonatal lethal phenotype, ASS−/− pups (n = 5) received 2.5 × 1011 vg/pup of rAAV2/8-LSP1mASS by intraperitoneal injection within 1–6 hours of birth in combination with sodium benzoate and L-arginine. This therapeutic regime failed to extend survival (data not shown), most likely because the kinetics of onset of transgene expression were too slow to counteract the rapid progression to lethal hyperammonemia.
Figure 1.

Rapid onset of hyperammonemia in ASS-deficient pups. Time-mated heterozygous dams were monitored every 4 hours from 18 days of gestation (E18) to determine the approximate time of parturition. Tail tips were harvested at birth for genotyping. All pups received daily doses of sodium benzoate and L-arginine. (a) The survival of ASS−/− pups (n = 6, dotted line) was then compared with ASS+/+ controls (n = 4, unbroken line). (b) In a separate experiment ASS+/+, ASS+/−, and ASS−/− pups were culled at 8 hours of age and plasma ammonia levels determined (n = 3–4 pups per group). Error bars represent SEM, ASS+/+ versus ASS−/− P = 0.0005, ASS+/+ versus ASS+/− not significant. ASS, argininosuccinate synthetase.
Prenatal gene therapy and recurrent postnatal vector readministration extended survival of ASS−/− pups, but not to adulthood
To overcome the rapid postnatal onset of lethal hyperammonemia, we pursued a strategy involving in utero gene delivery at 16 days of gestation (E16) followed by recurrent postnatal vector readministration as necessary for survival. Each fetus received 2.5 × 1011 vg of rAAV2/8-LSP1mASS by intraperitoneal injection. In utero treatment alone extended survival of ASS−/− pups to a maximum of 22 days. The same dose of vector readministered at birth (D0), D0 and 14 days (D14) or D0, D14 and 28 days (D28) provided marginal increments in survival, but even pups receiving a total of four doses did not survive beyond 33 days (Figure 2a). All pups received daily doses of L-arginine to ameliorate extrahepatic deficiency of this amino acid, a urea cycle intermediate, and were culled when signs of illness became evident. At the time of culling, plasma ammonia levels and liver ASS enzymatic activity were measured (Figure 2b,c). In all instances, plasma ammonia levels were pathologically elevated, while liver ASS enzymatic activities were universally low, with only slight increments associated with the receipt of additional vector doses.
Figure 2.

Survival was extended in ASS−/− mice after prenatal injection with rAAV2/8-LSP1mASS. ASS−/− pups were injected prenatally at E16 with 2.5 × 1011 vg of rAAV2/8-LSP1mASS. At birth, litter sizes were reduced to four pups per dam. Vector was then readministered at D0, D14 and/or D28 (n = 3 to four pups per group). All pups, including untreated controls (n = 3), received daily doses of L-arginine. (a) The survival curve shows the days on which ASS−/− pups were culled due to the development of clinical signs of illness. (b) Plasma ammonia and (c) liver ASS activity were measured at the time of culling. Error bars represent SEM, *P < 0.05 versus ASS+/+ controls. ASS, argininosuccinate synthetase.
High anti-rAAV2/8 antibody levels were detected in pups after in utero gene delivery
We next explored the possibility that an antivector immune response might underlie the failure of vector readministration to substantially extend survival beyond that observed after in utero gene delivery alone. In a similarly configured experiment, healthy ASS+/+ fetuses (E16) received 2.5 × 1011 vg of a vector encoding enhanced green fluorescent protein (rAAV2/8-LSP1eGFP) by intraperitoneal injection followed by no further treatment or postnatal vector readministration at D0 or D0 and D14 or D14 alone. Additional control groups received a single vector dose at D0 or D14. Pups were culled at D21 for analysis of enhanced green fluorescent protein (eGFP) expression in liver lysates and antivector antibodies in serum. In pups receiving a single dose of vector at E16, D0 or D14, expression of eGFP at D21 was highest in mice treated at D14 (Figure 3a). This result is consistent with greater loss of episomal vector genomes, as a consequence of liver growth, in pups treated at earlier time points.5 Importantly, the efficacy of gene transfer in pups receiving vector at D14 was reduced by prior vector exposure at E16 and/or D0, consistent with the possibility of an antivector immune response initiated by in utero treatment (Figure 3a). In support of this possibility, the highest antivector antibody titers were seen in pups that had been exposed to vector in utero and correlated with lower levels of eGFP expression (Figure 3a,b).
Figure 3.

Neutralizing antibodies to AAV capsid blocked vector readministration in ASS+/+ pups after prenatal vector delivery. Cohorts of ASS+/+ mice were injected with 2.5 × 1011 vg of rAAV2/8-LSP1eGFP at E16 (n = 16), E16 and D0 (n = 6), E16, D0 and D14 (n = 8), E16 and D14 (n = 9), D0 (n = 11) and D14 (n = 6). The control group consisted of uninjected pups (n = 8). At D21, mice were culled. (a) Livers were collected to quantitate GFP protein. (b) Blood was collected from each mouse and antibodies to the vector were analyzed in serum by ELISA. Error bars represent SEM, *P < 0.05 versus uninjected controls. AAV, adeno-associated virus; abs, absorbance; ASS, argininosuccinate synthetase; GFP, green fluorescent protein; vg, vector genome.
Passive antibody transfer in milk-impeded redelivery of vector to suckling pups
The observation of high antivector antibody titers in pups treated with vector in utero raised the possibility that inadvertent maternal exposure might have stimulated an immune response with subsequent passive transfer of antivector antibodies. The time between in utero vector treatment at E16 and birth 2–3 days later was too short to invoke a transplacental antibody transfer mechanism. We therefore hypothesized that antivector antibodies might be acquired by pups from the mother's milk during suckling. A crossfostering experiment was therefore designed to address this question (Figure 4a). Newborn pups from vector-naive dams were fostered to vector-injected dams and vice versa. At D14, half of the pups in each group were injected with vector, and all were culled at D21 for analysis of serum antivector antibody and liver eGFP levels. Pups from vector-injected dams fostered to vector-naive dams had no detectable antivector antibody at D21 unless exposed to vector at D14. In contrast, pups from vector-naive dams fostered to vector-injected dams had readily detectable antivector antibody levels at D21 irrespective of vector exposure at D14, a result definitively confirming passive transfer in milk, albeit indirectly (Figure 4b). Moreover, the levels of passively transferred antivector antibody in pups fostered to vector-injected dams were sufficient to block vector-mediated gene transfer at D14 as indicated by the analysis of eGFP expression levels in liver lysate (Figure 4c).
Figure 4.

Maternal antibodies from milk blocked vector transduction in suckling pups. (a) Pregnant ASS+/+ females were injected with 1012 vg of rAAV2/8-LSP1eGFP at E16. At birth, pups from uninjected dams were crossfostered to injected dams and vice versa. At D14, pups were left untreated (Group A, n = 6; Group D, n = 7) or injected with 2.5 × 1011 vg rAAV2/8-LSP1eGFP (Group B, n = 9; Group C, n = 6). (b) At D21, mice were culled and antivector antibodies analyzed in serum by ELISA. (c) Livers were also collected to quantitate GFP protein. Error bars represent SEM, *P < 0.0001 versus group A. ASS, argininosuccinate synthetase; eGFP, enhanced green fluorescent protein; rAAV, recombinant adeno-associated virus; vg, vector genome.
Successful rescue of the neonatal lethal ASS−/− phenotype
Having established that antivector antibodies passively acquired by suckling pups from vector-exposed dams are sufficient to block postnatal vector delivery, we reconfigured our earlier described ASS−/− phenotype rescue experiment. The key change was to foster newborn pups treated in utero to vector-naive dams. To further enhance the probability of success, we also changed the capsid serotype for vector readministration beyond the newborn period and weaned treated mice onto low protein chow (9% wt/wt). As previously, daily injections of L-arginine were given. Consistent with our earlier findings, ASS−/− pups treated with vector at E16 and D0 did not survive beyond D28, while pups receiving vector readministration at D14, or D14 and D28 all survived to adulthood, albeit with slower growth kinetics than wild-type littermates (Figure 5a,b). Young adult ASS−/− mice that received three doses of vector had variable and persistently elevated plasma ammonia levels, while ASS−/− mice receiving four doses had plasma ammonia and glutamine levels close to wild-type mice (Figure 5c and Table 1). The corresponding ASS enzymatic activities in liver lysates for mice receiving three and four doses of vector were 11.5 and 15.3% of wild-type levels, respectively (Figure 5d). Immunohistochemical analysis of ASS expression in liver sections from mice in the four doses cohort revealed that the proportion of hepatocytes expressing ASS averaged ~22% and were distributed across the hepatic lobule with a bias toward localization in perivenous regions (Figure 6).
Figure 5.

Survival to adulthood was achieved in ASS−/− pups after prenatal gene delivery, recurrent vector readministration using a different capsid serotype, crossfostering and dietary protein restriction. ASS−/− pups were injected at E16 and D0 with 2.5 × 1011 vg of rAAV2/rh10-LSP1mASS. All injected pups were then crossfostered to vector-naive dams and received daily doses of L-arginine. The vector was packaged using the serotype 8 capsid and injected into mice at D14 (n = 3) and D28 (n = 3). Uninjected age-matched ASS+/+ controls were also included (n = 3). Pups were weaned onto a low protein chow (9% wt/wt). (a) The survival curve shows the days on which ASS−/− pups were culled due to signs of illness. (b) The growth of vector-treated mice was monitored daily over the course of the experiment. (c) Plasma ammonia and (d) liver ASS activity were measured at the time of illness or at the experiment endpoint on D56. Error bars represent SEM, *P < 0.0001 versus ASS+/+ controls. ASS, argininosuccinate synthetase; vg, vector genome.
Table 1. Biochemical phenotype of vector-treated adult ASS−/− mice.
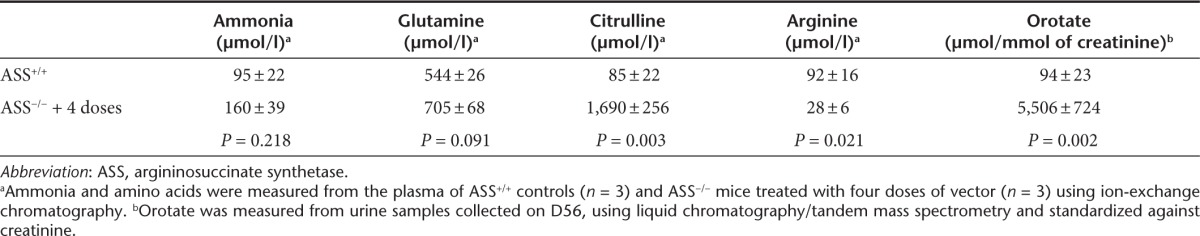
Figure 6.

Residual levels of ASS protein in the livers of vector-treated ASS−/− mice. Livers from (a) control ASS+/+ and (b) vector-treated ASS−/− (E16, D0, D14 and D28) mice were examined using immunohistochemistry for ASS expression (dark brown) and colocalized with glutamine synthetise (purple) at D56. Sections were counterstained with hematoxylin. Bars indicate 100 μm. (c) ASS-positive cells were counted from 10 randomly selected fields of view at ×200 original magnification in liver sections of three mice that had received four vector doses and expressed as a percentage of the total number of hepatocytes in the fields examined. ASS, argininosuccinate synthetase.
Analysis of plasma amino acids and urinary orotate levels in mice surviving to adulthood after receiving four doses of vector showed persistence of a mild metabolic phenotype, with significantly elevated plasma citrulline, decreased plasma arginine and increased urinary orotate (Table 1). Interestingly, a sparse fur, abnormal skin and hair (ash) phenotype was also evident (Figure 7), reminiscent of that observed in ornithine transcarbamylase-deficient spfash mice.9
Figure 7.

A sparse fur phenotype persisted in vector-treated ASS−/− mice. ASS−/− mice treated with three or four doses of vector exhibited a sparse fur phenotype. (a) Serial photographs of a representative mouse show transient fur growth from D14 to D42, although this disappeared by D56. Photomicrographs of fur samples from (b) a treated ASS−/− and (c) a normal ASS+/+ mouse are shown. Bars indicate 20 μm. ASS, argininosuccinate synthetase.
Discussion
Gene transfer vectors based on AAV are proving to be exceptionally powerful gene transfer tools for both basic science and therapeutic applications in man and animals. Underpinned by ongoing technological advances in AAV vectorology,14,15,16 it is now possible to produce recombinant viral particles in clinically relevant quantities, and to tailor vector tropism to defined target tissues and cell types by encapsidating the prototypic AAV type 2 recombinant genome in selected capsids from other AAV serotypes. A major challenge now is to better understand the complex host-vector interactions that will be encountered in human therapeutic use and to explore these in the context of disease and organ-specific applications.
A potentially significant limitation in the therapeutic use of AAV vectors is the loss of episomal vector genomes from actively dividing target cell populations.4,5,6 In the juvenile mouse liver, for example, one to two doublings of liver mass over 14 days is associated with an ~40-fold reduction in vector copy number consistent with a mechanism that involves both dilution and active degradation.5 Longer-term persistence of expression is dependent upon the smaller subset of vector genomes that undergo genomic integration. The same phenomenon has been reported in infant Rhesus monkeys,17 and can be confidently anticipated in the human pediatric context where the first doubling of liver mass occurs within 6 months of birth and the second within 15 months.18 The associated decline in AAV-mediated transgene expression in the context of gene therapy to the infant human liver is therefore likely to be substantial and may result in loss of therapeutic efficacy if the underlying number of cells with stably integrated vector genomes is below the therapeutic threshold for the particular disease being treated.
In this study, we report successful rescue of the neonatal lethal phenotype of the ASS knockout mouse and survival to adulthood using a strategy involving serial vector readministration. Key challenges included the rapidity of onset of profound hyperammonemia, the maintenance of protective levels of transgene expression during liver growth, and the risk of antivector immune responses limiting the efficacy of vector readministration. To overcome the first of these challenges, it proved necessary to treat pups with vector in utero during late gestation, an intervention that alone extended survival to ~3 weeks. In human infants with urea cycle defects presenting in the newborn period, such an intervention would not be necessary as hyperammonemia can be brought under initial control using strategies such as veno-venous hemofiltration. Vector readministration to pups in the newborn period alone or in combination with further vector dosing at 14 and 28 days initially proved relatively ineffective with only a modest extension of survival to a maximum of 33 days. Further experimentation demonstrated that pups were passively acquiring antivector antibodies in milk from dams inadvertently exposed to vector during in utero treatment of their pups in late gestation. Crossfostering of in utero-treated pups to vector-naive dams avoided this source of neutralizing antivector humoral immunity, and extended survival of pups receiving vector readministration at 14 days or 14 and 28 days to adulthood. Only pups receiving vector readministration at both 14 and 28 days showed reliable control of hyperammonemia.
The levels of gene transfer, in terms of both ASS enzymatic activity in liver lysates and the proportion of transduced hepatocytes, were also evaluated in order to better predict the likely threshold for therapeutic benefit in human infants. In the two treatment cohorts of ASS knockout mice surviving to adulthood, the mean ASS activity in liver lysates were 11.5 and 15.3% of wild-type values after three and four doses of vector, respectively. The incremental difference between these values was associated with improved control of plasma ammonia levels, yet even with 15% wild-type ASS activity, treated mice retained a recognizable phenotype. This included slow growth, sparse fur, akin to that seen in ornithine transcarbamylase-deficient spfash mice, and residual biochemical features of ASS deficiency, including orotic aciduria, elevated plasma glutamine and citrulline and reduced arginine levels. In the same four-dose cohort, the mean proportion of hepatocytes stably expressing ASS activity was ~22%. The phenomenon of metabolic zonation,19,20 whereby urea cycle enzymes are more highly expressed in the periportal zones of the hepatic lobule is important to the further interpretation of these results. While the AAV serotype 8 capsid facilitates efficient liver-targeted gene delivery in mice, higher levels of transgene expression are seen in the perivenous relative to the periportal zones,21,22,23 a less than optimal pattern for urea cycle enzymes. With a more physiological distribution of ASS enzymatic activity across the hepatic lobule better phenotype correction is therefore likely to be achieved with lower mean ASS activities from fewer hepatocytes. Encouraging in this regard is recent evidence that the type 8 capsid confers a predominantly periportal pattern of transgene expression in non-human primates,22 suggesting similar favorable performance properties in the human liver for the treatment of urea cycle defects.
The capacity of antivector antibodies, passively acquired in milk, to markedly limit the efficacy of vector readministration to suckling pups, to our knowledge has not been previously reported in the gene transfer context. This phenomenon is not without precedent, however, as mice vaccinated with Moloney murine leukemia virus have been reported to pass humoral immunity to pups through milk with resultant protection from infection.24 Similarly, maternally derived antibodies passively acquired in milk have been shown to reduce chimerism after in utero hematopoietic stem cell transplantation.25 Collectively, these observations highlight the importance of avoiding the development of antivector humoral immune responses in gene therapy applications where vector redelivery is required to maintain therapeutic benefit. The fact that vector redelivery to pups was effective once maternally derived antivector humoral immunity was excluded is consistent with our previously published data5 showing that juvenile mice of the ages used do not mount sufficiently strong humoral responses against AAV capsid proteins to completely block vector redelivery, although efficiency is reduced. With this in mind in configuring the current study, we employed a strategy that involved the initial use of the rh10 capsid for treatment in utero and birth and the serotype 8 capsid for redelivery at 14 and 28 days. Whether changing the capsid serotype for vector redelivery beyond the newborn period contributed to the success of the current study remains to be established.
Successful rescue of neonatal and juvenile lethal phenotypes in mice, using AAV-mediated gene transfer, has been reported for several diseases including methylmalonic acidemia,26,27 proprionic acidemia,28 and Crigler Najjar type I syndrome.29 In each of these examples, the liver was a target for gene delivery and a single dose of AAV given in the immediate newborn period was sufficient for survival into adulthood. This requirement for only a single dose of vector contrasts markedly with our finding that rescue of neonatal lethal ASS deficiency and survival to adulthood necessitates vector readministration. There are two possible explanations for these differing findings. First, the proportion of gene-modified hepatocytes required to achieve phenotype correction in ASS deficiency may be higher, with the number of hepatocytes carrying integrated vector genomes following neonatal treatment being insufficient to control disease as the contribution made to ASS expression by episomal genomes is lost during liver growth. As discussed earlier, metabolic zonation of urea cycle enzymes, is almost certainly a contributing factor. The second possibility, at least for the reports involving proprionic acidemia28 and Crigler Najjar type I,29 is an extrahepatic contribution to phenotype correction. In each of these studies, ubiquitously active promoters were used to express proprionyl coenzyme A carboxylase and uridine diphosphoglucuronosyltransferase 1A1, respectively, raising the possibility that transgene expression outside the liver may have exerted an effect. This is made plausible by the physiological expression of these enzymes in multiple tissues.30,31
Irrespective of the exact explanation for these interdisease differences in the need for vector readministration after early neonatal treatment, it is clear that loss of episomal AAV vector genomes from replicating hepatocytes during liver growth presents a formidable challenge to the long-term curative treatment of genetic/metabolic liver disease in infants and children using AAV-mediated gene therapy. Data presented in this report demonstrate that vector readministration is effective in the mouse, although human application is likely to require additional strategies to evade or suppress unwanted antivector immune responses. Other possibilities include the use of strategies to increase the proportion of AAV vector genomes undergoing genomic integration, such as the inclusion of rDNA elements in the vector genome.32,33
Materials and Methods
Animal experiments. All experimental procedures were evaluated and approved by the institutional Animal Care and Ethics Committee. The ASS mice (B6;129S7-Ass1tm1Bay/J) were purchased from the Jackson Laboratory (Bar Harbor, ME). Animals were housed in a temperature controlled environment with 12-hour light/dark cycles. Age-matched mice were given free access to water and maintained on standard rodent chow (12117; Specialty Feeds, Australia) or, where stated, weaned to low protein chow (9% wt/wt, SF04-059; Specialty Feeds, Glen Forrest, Australia). ASS−/− mice were given daily injections of sodium benzoate (0.1 g/kg) (Special Products, Weybridge, UK) and/or L-arginine (1 g/kg) (Phebra, Lane Cove, Australia). All injections were performed intraperitoneally unless otherwise stated. To determine the genotype of mice, tail DNA was extracted using the Extract-N-Amp Tissue PCR Kit (XNAT2R; Sigma-Aldrich, St Louis, MO). Mice were genotyped by PCR as described.34 For in utero injection of vector at E16, pregnant females were anesthetised using isoflurane inhalation anesthesia. Buprenorphine (0.01 mg/kg) (Reckitt Benckiser, West Ryde, Australia) was given by subcutaneous injection as an analgaesic. A laparotomy was performed to expose the uterus and each pup was injected with vector in a volume of 5 μl. The abdominal incision was then closed with sutures. A subcutaneous injection of ampicillin was given and mice were maintained on carprofen (0.14 ml/250 ml) given orally in drinking water for 7 days. Pups were generally born on E19, which was designated as D0.
Cell culture. Human embryonic kidney 293 cells35 were maintained in Dulbecco's modified Eagle medium (Gibco, Invitrogen, Grand Island, NY) supplemented with 10% (vol/vol) fetal bovine serum (JRH Biosciences, Lenexa, KS) and 1% (wt/vol) L-glutamine (Gibco, Invitrogen), and maintained at 37 °C in a humidified 5% CO2-air atmosphere.
Virus production. Recombinant AAV vectors were produced in human embryonic kidney 293 cells packaging either pLSP1mASS or pLSP1eGFP, and pseudo-serotyped with the AAV8 or AAVrh10 capsid (courtesy of James M. Wilson, University of Pennsylvania, Philadelphia, PA) as previously described.5 Vector genomes were titered by real-time quantitative PCR as previously described.36
Blood and urine biochemistry. For amino acids, quantitation plasma was deproteinized by ultrafiltration before addition of an internal standard, then analyzed by ion-exchange chromatography with postcolumn ninhydrin detection on a Biochrom 30 amino acid analyzer (Biochrom, Cambridge, UK). Plasma ammonia and urinary orotic acid were analyzed as previously described.36
Measurement of antibodies to the vector by ELISA. For extraction of serum, blood was collected using either the tail vein or cardiac puncture method into uncoated collection tubes. Samples were allowed to clot at room temperature for 1 hour before being centrifuged at 300g at 4 °C for 20 minutes. The serum was removed and frozen at –80 °C. Antibodies to the rAAV vectors were measured by ELISA as previously described.5
Immunohistochemistry. Formalin-fixed liver sections were dewaxed and rehydrated in xylene and an ethanol gradient. Antigen retrieval was performed using 10 mmol/l sodium citrate buffer pH 7.4, followed by blocking with 0.3% hydrogen peroxidize for 30 minutes. Endogenous avidin and biotin were blocked using the avidin-biotin kit (Vector Laboratories, Burlingame, CA), following the manufacturer's instructions. Sections were then blocked with 10% donkey serum and stained with an antibody against ASS (2.5 µg/ml, ab77590; Abcam, Cambridge, UK). After washing in phosphate-buffered saline with 0.05% Tween-20, samples were incubated with a biotinylated donkey anti-goat secondary antibody (1.2 µg/ml, 705-065-147; Jackson ImmunoResearch, West Grove, PA) and detected with horseradish peroxidase, using the Vectastain Elite ABC (PK-7100, Vector Laboratories) as per manufacturer's instructions. For detection of glutamine synthetase, a rabbit polyclonal anti-glutamine synthetase primary antibody (1/150 dilution, ab16802; Abcam) was used. Bound primary antibody was detected with a biotinylated donkey anti-rabbit secondary antibody. Following completion of ASS and glutamine synthetase detection, sections were counterstained with hematoxylin. Images were captured using an Olympus BX50 fluorescent microscope (Olympus, Center Valley, PA) and the ProgResCapturePro 2.6 software.
14C-radiochemical assay. Liver ASS activity was assayed as described previously with minor modifications. After the reaction was terminated, the mixture was immediately applied to 0.8 × 4 cm plastic columns (731-1550; BioRad, Gladesville, Australia) of AG 1-X8 acetate resin (140-1453; BioRad) equilibrated with 10 mmol/l Tris-acetate, pH 7.4, then rinsed with two 1 ml volumes of Tris-acetate buffer. The argininosuccinate fraction was then eluted using 6 ml of 0.1 mol/l acetic acid, with 1 ml removed for scintillation counting.
Fluorometric analysis of eGFP expression. Quantitation of eGFP was performed by fluorometric analysis as previously described.21
Statistics. Results were analyzed by one-way analysis of variance with Dunnett's post-test or two-tailed Student's unpaired t-test using GraphPad Prism (GraphPad Software, La Jolla, CA). P values ≤ 0.05 were considered statistically significant.
Acknowledgments
This work was supported by the Australian National Health and Medical Research Council (NHMRC), project grant number APP1008021. C.Y.K. was a recipient of a Children's Medical Research Institute postgraduate scholarship. We thank Margot Latham (The Children's Hospital at Westmead) for assistance with manuscript preparation. The authors declared no conflict of interest.
References
- Muzyczka N. Use of adeno-associated virus as a general transduction vector for mammalian cells. Curr Top Microbiol Immunol. 1992;158:97–129. doi: 10.1007/978-3-642-75608-5_5. [DOI] [PubMed] [Google Scholar]
- Manno CS, Pierce GF, Arruda VR, Glader B, Ragni M, Rasko JJ, et al. Successful transduction of liver in hemophilia by AAV-Factor IX and limitations imposed by the host immune response. Nat Med. 2006;12:342–347. doi: 10.1038/nm1358. [DOI] [PubMed] [Google Scholar]
- Nathwani AC, Tuddenham EG, Rangarajan S, Rosales C, McIntosh J, Linch DC, et al. Adenovirus-associated virus vector-mediated gene transfer in hemophilia B. N Engl J Med. 2011;365:2357–2365. doi: 10.1056/NEJMoa1108046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Zhu T, Qiao C, Zhou L, Wang B, Zhang J, et al. Adeno-associated virus serotype 8 efficiently delivers genes to muscle and heart. Nat Biotechnol. 2005;23:321–328. doi: 10.1038/nbt1073. [DOI] [PubMed] [Google Scholar]
- Cunningham SC, Dane AP, Spinoulas A, Logan GJ, Alexander IE. Gene delivery to the juvenile mouse liver using AAV2/8 vectors. Mol Ther. 2008;16:1081–1088. doi: 10.1038/mt.2008.72. [DOI] [PubMed] [Google Scholar]
- Wang L, Wang H, Bell P, McMenamin D, Wilson JM. Hepatic gene transfer in neonatal mice by adeno-associated virus serotype 8 vector. Hum Gene Ther. 2012;23:533–539. doi: 10.1089/hum.2011.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKay TR, Rahim AA, Buckley SM, Ward NJ, Chan JK, Howe SJ, et al. Perinatal gene transfer to the liver. Curr Pharm Des. 2011;17:2528–2541. doi: 10.2174/138161211797247541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattar CN, Nathwani AC, Waddington SN, Dighe N, Kaeppel C, Nowrouzi A, et al. Stable human FIX expression after 0.9G intrauterine gene transfer of self-complementary adeno-associated viral vector 5 and 8 in macaques. Mol Ther. 2011;19:1950–1960. doi: 10.1038/mt.2011.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doolittle DP, Hulbert LL, Cordy C. A new allele of the sparse fur gene in the mouse. J Hered. 1974;65:194–195. doi: 10.1093/oxfordjournals.jhered.a108500. [DOI] [PubMed] [Google Scholar]
- Cunningham SC, Spinoulas A, Carpenter KH, Wilcken B, Kuchel PW, Alexander IE. AAV2/8-mediated correction of OTC deficiency is robust in adult but not neonatal Spf(ash) mice. Mol Ther. 2009;17:1340–1346. doi: 10.1038/mt.2009.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patejunas G, Bradley A, Beaudet AL, O'Brien WE. Generation of a mouse model for citrullinemia by targeted disruption of the argininosuccinate synthetase gene. Somat Cell Mol Genet. 1994;20:55–60. doi: 10.1007/BF02257486. [DOI] [PubMed] [Google Scholar]
- Deignan JL, Cederbaum SD, Grody WW. Contrasting features of urea cycle disorders in human patients and knockout mouse models. Mol Genet Metab. 2008;93:7–14. doi: 10.1016/j.ymgme.2007.08.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipshutz GS, Gruber CA, Cao Y, Hardy J, Contag CH, Gaensler KM. In utero delivery of adeno-associated viral vectors: intraperitoneal gene transfer produces long-term expression. Mol Ther. 2001;3:284–292. doi: 10.1006/mthe.2001.0267. [DOI] [PubMed] [Google Scholar]
- Xiao X, Li J, Samulski RJ. Production of high-titer recombinant adeno-associated virus vectors in the absence of helper adenovirus. J Virol. 1998;72:2224–2232. doi: 10.1128/jvi.72.3.2224-2232.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell AM, Nicolson SC, Warischalk JK, Samulski RJ. AAV's anatomy: roadmap for optimizing vectors for translational success. Curr Gene Ther. 2010;10:319–340. doi: 10.2174/156652310793180706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asokan A, Schaffer DV, Samulski RJ. The AAV vector toolkit: poised at the clinical crossroads. Mol Ther. 2012;20:699–708. doi: 10.1038/mt.2011.287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Bell P, Lin J, Calcedo R, Tarantal AF, Wilson JM. AAV8-mediated hepatic gene transfer in infant rhesus monkeys (Macaca mulatta). Mol Ther. 2011;19:2012–2020. doi: 10.1038/mt.2011.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saphir O. Hoeber-Harper: New York; 1958. Autopsy diagnosis and technique. [Google Scholar]
- Jungermann K, Sasse D. Heterogeneity of liver parenchymal cells. Trends in Biochemical Sciences. 1978;3:198–202. [Google Scholar]
- Jungermann K. Zonation of metabolism and gene expression in liver. Histochem Cell Biol. 1995;103:81–91. doi: 10.1007/BF01454004. [DOI] [PubMed] [Google Scholar]
- Dane AP, Cunningham SC, Graf NS, Alexander IE. Sexually dimorphic patterns of episomal rAAV genome persistence in the adult mouse liver and correlation with hepatocellular proliferation. Mol Ther. 2009;17:1548–1554. doi: 10.1038/mt.2009.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell P, Wang L, Gao G, Haskins ME, Tarantal AF, McCarter RJ, et al. Inverse zonation of hepatocyte transduction with AAV vectors between mice and non-human primates. Mol Genet Metab. 2011;104:395–403. doi: 10.1016/j.ymgme.2011.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dane AP, Wowro SJ, Cunningham SC, Alexander IE. Comparison of gene transfer to the murine liver following intraperitoneal and intraportal delivery of hepatotropic AAV pseudo-serotypes. Gene Ther. 2013;20:460–464. doi: 10.1038/gt.2012.67. [DOI] [PubMed] [Google Scholar]
- Saha K, Hollowell D, Wong PK. Mother-to-baby transfer of humoral immunity against retrovirus-induced neurologic disorders and immunodeficiency. Virology. 1994;198:129–137. doi: 10.1006/viro.1994.1015. [DOI] [PubMed] [Google Scholar]
- Merianos DJ, Tiblad E, Santore MT, Todorow CA, Laje P, Endo M, et al. Maternal alloantibodies induce a postnatal immune response that limits engraftment following in utero hematopoietic cell transplantation in mice. J Clin Invest. 2009;119:2590–2600. doi: 10.1172/JCI38979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandler RJ, Venditti CP. Long-term rescue of a lethal murine model of methylmalonic acidemia using adeno-associated viral gene therapy. Mol Ther. 2010;18:11–16. doi: 10.1038/mt.2009.247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrillo-Carrasco N, Chandler RJ, Chandrasekaran S, Venditti CP. Liver-directed recombinant adeno-associated viral gene delivery rescues a lethal mouse model of methylmalonic acidemia and provides long-term phenotypic correction. Hum Gene Ther. 2010;21:1147–1154. doi: 10.1089/hum.2010.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandler RJ, Chandrasekaran S, Carrillo-Carrasco N, Senac JS, Hofherr SE, Barry MA, et al. Adeno-associated virus serotype 8 gene transfer rescues a neonatal lethal murine model of propionic acidemia. Hum Gene Ther. 2011;22:477–481. doi: 10.1089/hum.2010.164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bortolussi G, Zentilin L, Baj G, Giraudi P, Bellarosa C, Giacca M, et al. Rescue of bilirubin-induced neonatal lethality in a mouse model of Crigler-Najjar syndrome type I by AAV9-mediated gene transfer. FASEB J. 2012;26:1052–1063. doi: 10.1096/fj.11-195461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrick JJ, Lingrel JB. cDNA cloning, mapping and expression of the mouse propionyl CoA carboxylase beta (pccb), the gene for human type II propionic acidaemia. Gene. 2001;264:147–152. doi: 10.1016/s0378-1119(00)00586-2. [DOI] [PubMed] [Google Scholar]
- Diez-Roux G, Banfi S, Sultan M, Geffers L, Anand S, Rozado D, et al. A high-resolution anatomical atlas of the transcriptome in the mouse embryo. PLoS Biol. 2011;9:e1000582. doi: 10.1371/journal.pbio.1000582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Lisowski L, Finegold MJ, Nakai H, Kay MA, Grompe M. AAV vectors containing rDNA homology display increased chromosomal integration and transgene persistence. Mol Ther. 2012;20:1902–1911. doi: 10.1038/mt.2012.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisowski L, Lau A, Wang Z, Zhang Y, Zhang F, Grompe M, et al. Ribosomal DNA integrating rAAV-rDNA vectors allow for stable transgene expression. Mol Ther. 2012;20:1912–1923. doi: 10.1038/mt.2012.164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye X, Whiteman B, Jerebtsova M, Batshaw ML. Correction of argininosuccinate synthetase (AS) deficiency in a murine model of citrullinemia with recombinant adenovirus carrying human AS cDNA. Gene Ther. 2000;7:1777–1782. doi: 10.1038/sj.gt.3301303. [DOI] [PubMed] [Google Scholar]
- Graham FL, Smiley J, Russell WC, Nairn R. Characteristics of a human cell line transformed by DNA from human adenovirus type 5. J Gen Virol. 1977;36:59–74. doi: 10.1099/0022-1317-36-1-59. [DOI] [PubMed] [Google Scholar]
- Cunningham SC, Kok CY, Dane AP, Carpenter K, Kizana E, Kuchel PW, et al. Induction and prevention of severe hyperammonemia in the spfash mouse model of ornithine transcarbamylase deficiency using shRNA and rAAV-mediated gene delivery. Mol Ther. 2011;19:854–859. doi: 10.1038/mt.2011.32. [DOI] [PMC free article] [PubMed] [Google Scholar]